

Abstract
A progressive decline in maximum heart rate (mHR) is a fundamental aspect of aging in humans and other mammals. This decrease in mHR is independent of gender, fitness, and lifestyle, affecting in equal measure women and men, athletes and couch potatoes, spinach eaters and fast food enthusiasts. Importantly, the decline in mHR is the major determinant of the age-dependent decline in aerobic capacity that ultimately limits functional independence for many older individuals. The gradual reduction in mHR with age reflects a slowing of the intrinsic pacemaker activity of the sinoatrial node of the heart, which results from electrical remodeling of individual pacemaker cells along with structural remodeling and a blunted β-adrenergic response. In this review, we summarize current evidence about the tissue, cellular, and molecular mechanisms that underlie the reduction in pacemaker activity with age and highlight key areas for future work.
Keywords: sinoatrial node, maximum heart rate, intrinsic heart rate, aging, cardiac pacemaking
INTRODUCTION
Aging has long been associated with declines in physical fitness and aerobic capacity. In Homer’s Iliad, Nestor laments that his “limbs are gnarled now, the old power’s gone” (1, p. 318). This proclamation from an old warrior highlights that while some of the effects of aging can be counteracted by exercise training, they cannot be fully mitigated.
One of the unalterable consequences of aging is a progressive decline in maximum heart rate (mHR) (Figure 1). The age-dependent decline in mHR was first published in a cross-sectional study by Sid Robinson in 1938 (2), who followed up with a longitudinal study in 1975 (3). Importantly, the decline in mHR is an inherent property of cardiac aging. Unlike other measures of cardiac fitness, such as left ventricular mass, that can be increased by exercise training (4), mHR declines with age at approximately the same rate for all individuals without regard for gender, diet, or physical fitness (5, 6). The rate of decline of mHR is commonly approximated by the formula mHR ≈ 220 – age (7), or about 1 beat per minute (bpm)/year, although the original study by Robinson and more recent studies suggest that the actual rate is 0.6–0.8 bpm/year (2, 5, 8). A similar age-dependent decline in mHR has been observed in many other species, including mice, rats, rabbits, dogs, and cats (9–14).
Figure 1.
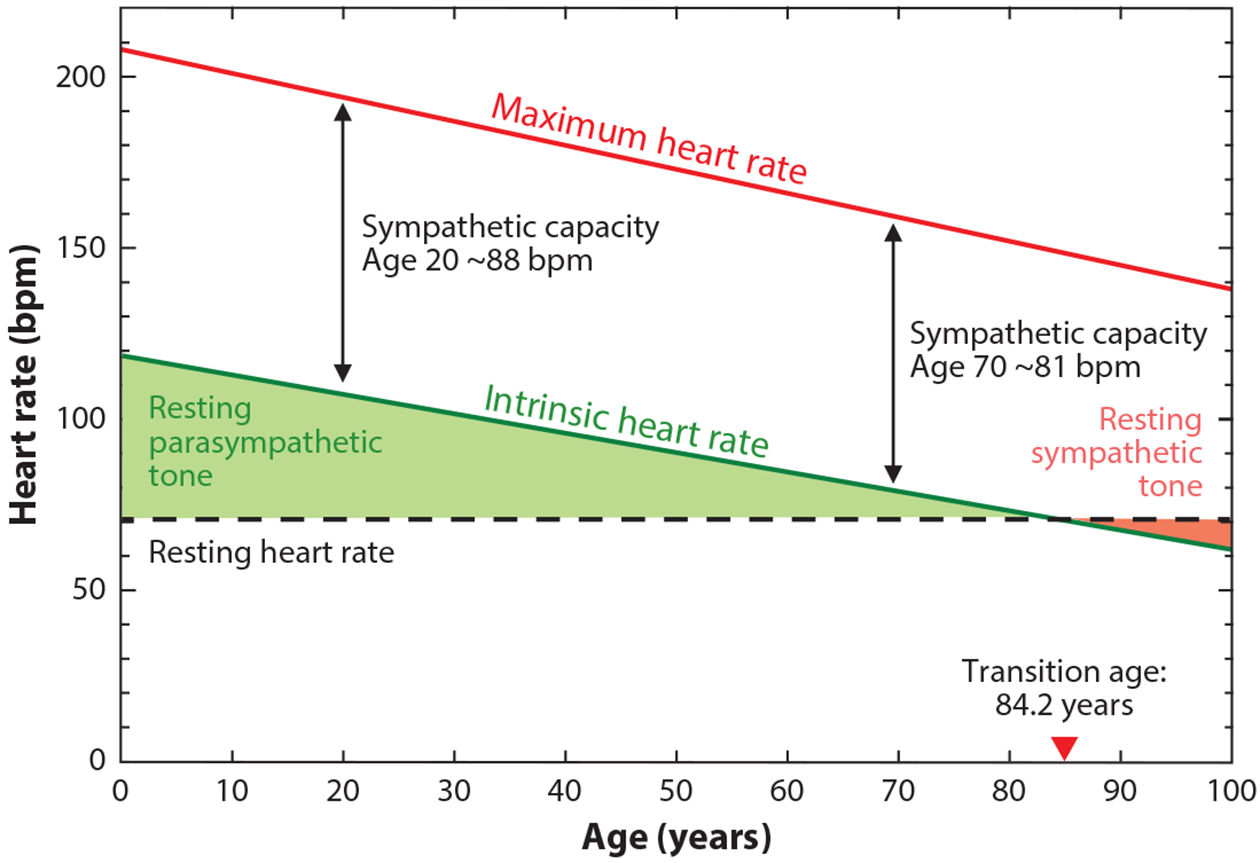
Similar declines in maximum and intrinsic heart rates with no change in resting heart rate necessitate an increase in sympathetic tone with age. Maximum heart rate (mHR; red line) is plotted as 208 – (0.7 × age); data from Reference 5. Intrinsic heart rate (green line) is plotted as 118 – (0.57 × age); data from Reference 20. Resting heart rate (dashed black line) is plotted as a constant 70 beats per minute (bpm) throughout life. The red arrowhead indicates an average transition age (84.2 years) beyond which sympathetic tone at rest (pink shading) predominates over parasympathetic tone at rest (green shading) in order to maintain resting heart rate. Sympathetic capacity (black double arrows), the difference between intrinsic and maximum heart rate, decreases slightly with age; however, the majority of the age-dependent decline in mHR is caused by the decline in intrinsic heart rate.
The decline in mHR begets an inevitable decline in the maximum rate of oxygen consumption (VO2max) and, consequently, physical performance, with age. Indeed, decreased mHR is the primary determinant of the age-dependent decline in VO2max and the main factor that limits exercise performance with age (15, 16). Moreover, of the factors that determine VO2max, mHR alone cannot be improved by exercise training. While mHR and VO2max are not important considerations for the majority of young and middle-aged individuals who do not require or utilize their full aerobic capacity, low VO2max is a leading factor that limits functional independence for many otherwise-healthy older individuals (17–19). The age-dependent decline in mHR is thus of paramount concern as the population ages because it diminishes quality of life for individuals and increases health care costs for society.
It has long been appreciated that the age-dependent decline in mHR is caused by a nearly parallel age-dependent decline in intrinsic heart rate (iHR) (20–22). iHR is the heart rate measured under autonomic blockade. It thus serves as a systemic readout of the function of the primary cardiac pacemaker, the sinoatrial node (SAN). The age-dependent decline in iHR indicates, by definition, that pacemaker activity of the SAN is compromised with age. Despite the decline in iHR, resting heart rate (rHR) is fairly constant with age because it is maintained by autonomic nervous system activity. An important corollary is that sympathetic tone must increase with age in order to maintain rHR (Figure 1). In this review, we focus on the mechanisms that cause the age-dependent loss of cardiac pacemaker function.
SINOATRIAL NODE OVERVIEW
The SAN is the pacemaker of the heart. Its eventual discovery in 1907 built on observations of numerous early scientists over the course of several centuries (for review see 23). In the eighteenth century Albrecht von Haller provided a convincing argument that the heartbeat originated within the heart itself, without requiring input from the nervous system that others had proposed (24). In the late nineteenth century, Walter Gaskell noted that the electrical signal began in the sinus auricle and was then conducted through the atrium and on to the ventricles (24). Subsequently, in 1907 Keith & Flack (25) published an extensive study detailing a special tissue within the sinus auricle—the SAN—as the site from which the heart rhythm arises.
In denervated human hearts, the SAN generates a spontaneous rhythmic electrical depolarization at a rate of approximately 100 bpm (26, 27). This depolarization propagates through the cardiac conduction system and throughout the heart to initiate each heartbeat (Figure 2a). The automaticity of the SAN is the basis of the iHR that can be measured in vivo in humans and animals under autonomic blockade (20).
Figure 2.

The cardiac conduction system and sinoatrial node myocytes. (a) The cardiac conduction system visualized in a heart from a contactin2-eGFP reporter mouse. The pacemaker depolarization initiated by the sinus node propagates through the atria to the atrioventricular (AV) node and then on to the ventricles via the His-Purkinje network. Adapted from Reference 152 under the terms of the Creative Commons Attribution (CC BY) License, http://creativecommons.org/licenses/by/4.0. (b) Sinoatrial node myocyte (SAM) morphology. SAMs are long, thin cells with few striations. Shown is a mouse SAM in the recording chamber of an electrophysiology rig. Note the patch pipette entering from the right that contacts the cell near its center. Adapted from Reference 133 under the terms of the Creative Commons Attribution-ShareAlike (CC BY-SA) License, https://creativecommons.org/licenses/by-sa/4.0. (c) Rich sympathetic innervation of SAMs. Co-immunolabeling of SAMs and sympathetic nerve fibers with antibodies against HCN4 (red) and tyrosine hydroxylase (green), respectively. Image on right shows enlargement of boxed area from image on left. E. Larson and C. Proenza, unpublished data.
The SAN is highly innervated by the autonomic nervous system, which profoundly regulates heart rate on a beat-to-beat basis to closely match the cardiac output to ever-changing metabolic demand. Efferents from the vagus nerve provide parasympathetic innervation, first synapsing in fat pad ganglia on the heart, and then projecting to the SAN where they decrease heart rate by slowing the intrinsic firing rate of the SAN. Conversely, thoracic sympathetic nerves from T1–T4 first synapse in sympathetic chain ganglia adjacent to the spinal cord and then project to the SAN where they elicit the fight-or-flight increase in heart rate. At rest, humans below the age of about 85 have a predominant parasympathetic tone that inhibits iHR to produce an average rHR of ~60–100 bpm (Figure 1) (28). During exercise or stress, sympathetic nervous system activity increases SAN firing, which in turn increases heart rate.
Within the SAN, sinoatrial node myocytes (SAMs) serve as the pacemaker cells. As noted by Keith & Flack (25), SAMs are surrounded by fibrous tissue and are in close proximity to processes from the vagus and sympathetic nerves (Figure 2c) (25). SAMs are highly specialized cells that differ considerably from the working myocytes of the atrial and ventricular myocardium in terms of function, morphology and gene expression. Compared to atrial and ventricular myocytes, SAMs are smaller in size and appear unstriated (Figure 2b), owing to fewer and poorly organized sarcomeres (29–31). SAMs are also characterized by numerous caveolae that increase the membrane surface area and likely serve to organize signaling microdomains (31). Isolated SAMs are also quite heterogeneous, which is thought to reflect a functional gradient of cellular subtypes from the center to periphery of the SAN, at least in some species (32, 33).
In the intact SAN, SAMs are electrically coupled by low-conductance (<40 pS) gap junctions formed by connexins Cx45, Cx40, and Cx30.2 (34–36). In the rest of the heart, high-conductance (>70 pS) gap junctions are formed primarily by Cx43 or Cx40 (37). The presence of lower-conductance connexins in the SAN suggests greater electrical resistance between cells (38). There is also evidence to suggest that gap junctions in the SAN are particularly sparse, which further increases electrical resistance (29, 39). This electrical isolation is thought to be necessary to protect the SAN from the hyperpolarizing influence of the much larger atrial myocardium. Computational models predict that higher electrical resistance in the central SAN coupled with a gradual decrease in resistance toward the atrial myocardium allows for the relatively small SAN to pace the atria (40). Indeed, both low-conduction velocity in the SAN and an increase in velocity at the boundary with the atria have been measured experimentally (41). A zone of transition in connexin expression between the SAN and atria has also been observed (33, 35). It is thought that these transitional areas form complex block zones and exit pathways that optimize excitation of the atria while minimizing reentry (33, 42, 43). Additionally, expression of low-conductance connexins allows for the possible formation of asymmetric gap junctions with highly rectified conductances that would allow for nearly unidirectional flow between cells (44, 45). SAMs are further isolated from the surrounding atrial myocardium by the nonconducting fibrous tissue that ensheathes them (25, 43).
Age-dependent slowing of SAN conduction is well characterized, but as is the case for many aspects of age-dependent changes in the SAN, data on changes to connexin protein expression with aging are limited (43, 46, 47). One study in guinea pigs shows a progressive loss in Cx43 protein with no change in Cx40 or Cx45 (46). The loss of Cx43 was accompanied by slowed conduction velocity through the SAN, although the underlying mechanism is not clear, given the low expression of Cx43 in SAN. In rats, the abundance of Cx30.2 transcripts in the SAN decreases with age, but whether this effect is translated to the protein level is unknown (48).
Sinoatrial Action Potentials and Aging
SAMs function as cardiac pacemaker cells by firing spontaneous action potentials (APs) that underlie the autorhythmicity of the heart. Spontaneous APs in SAMs have a characteristic waveform (Figure 3a). Compared to APs in ventricular myocytes, the upstroke of sinoatrial APs is considerably slower, and the amplitude is smaller. Sinoatrial APs also lack the transient repolarization phase, and the plateau phase is considerably less pronounced. Most importantly, sinoatrial APs do not have a stable resting membrane potential. Instead, a spontaneous depolarization during diastole brings the membrane potential to threshold to trigger the subsequent AP.
Figure 3.
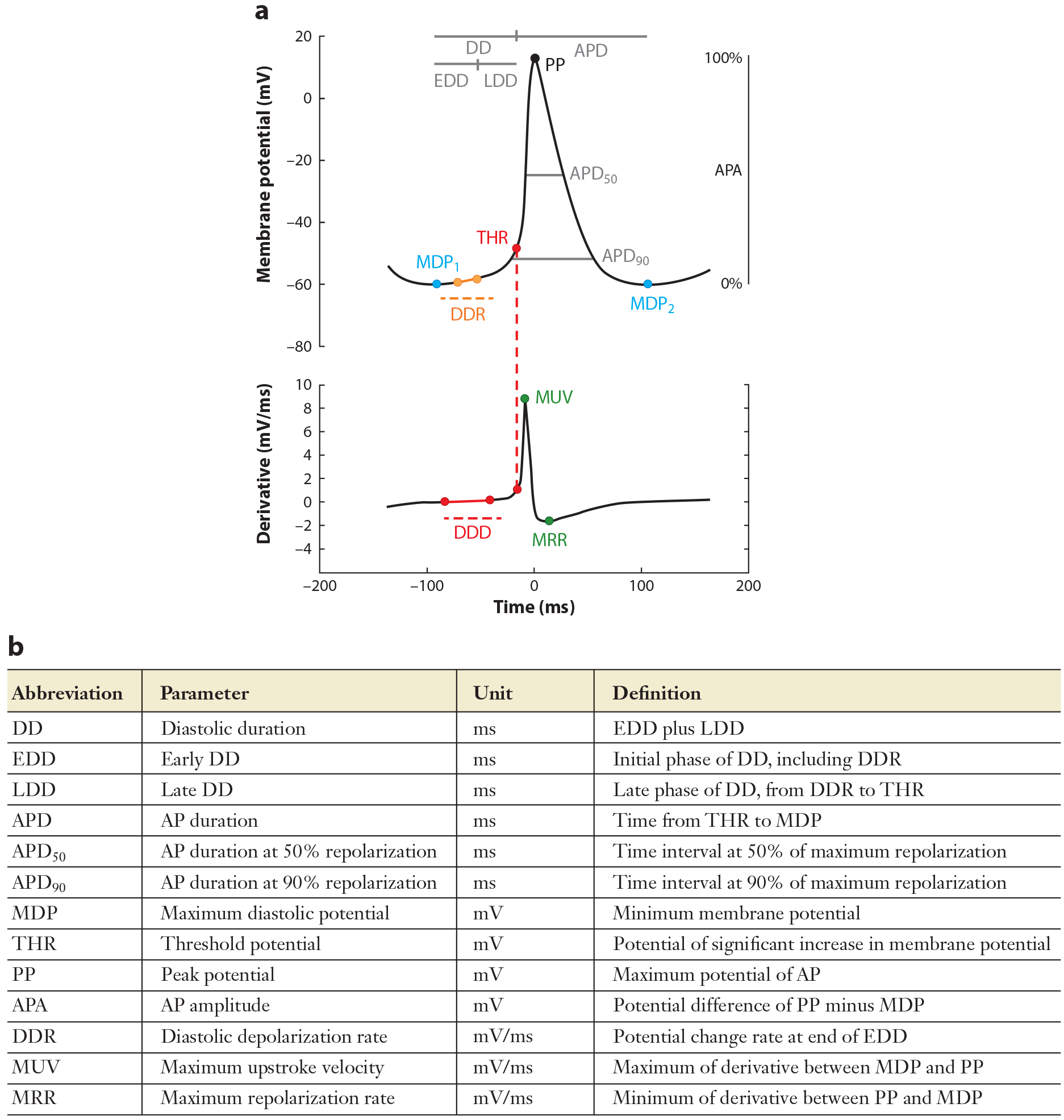
Characteristic waveform of sinoatrial node action potentials. (a) Schematic illustration of a sinoatrial action potential (top) and its first derivative (bottom) indicating key features of the action potential. (b) Summary of action potential waveform parameter descriptions. Adapted from Reference 50. Copyright 2017, Elsevier.
Age-dependent slowing of mHR is caused, at least in part, by slower firing of individual SAMs. Using transmembrane recordings of aged rabbit and cat pacemaker tissue, Alings & Bouman (10) showed that spontaneous AP firing rate decreases with age due to both increased AP duration and slowing of the diastolic depolarization rate (DDR). This was consistent with earlier work showing an age-dependent slowing of the firing rate and prolongation of AP duration in rat atrial cells within tissue preparations (9, 49). Our group and others have observed a decrease in the basal and maximal AP firing rates of isolated SAMs and SAN tissue from aged mice that mirrors the slowing of iHR and mHR (11, 13) (Figure 4a). In isolated SAMs, we found that the slower intrinsic firing rate was caused by a more negative maximum diastolic potential (MDP) and a slower DDR, while the AP duration trended toward prolongation with age (13), consistent with the previous studies (10, 11) (Figure 4b). Indeed, reanalysis of our data set from aged versus young SAMs (13) using refined parameter definitions and automated AP waveform analysis (50) revealed a significant AP prolongation in the aged cells. An exception to this trend is a study in mice that showed a slower DDR in aging, but in contrast to other studies, a shortened AP duration (47).
Figure 4.
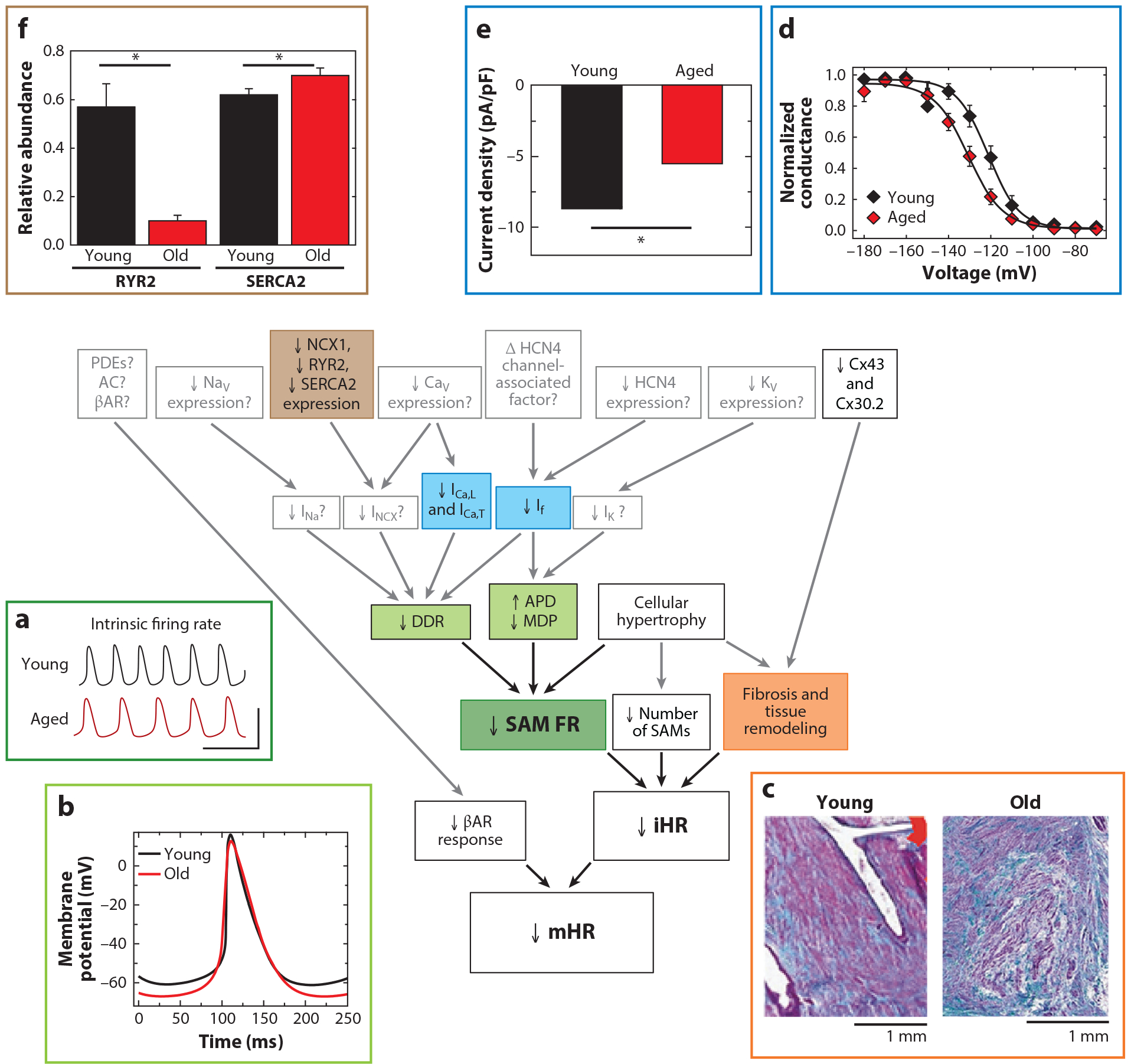
Mechanisms of the age-dependent decline in maximum heart rate (mHR). Shown are known (black boxes and arrows, color-coded to corresponding subpanels) and hypothesized (gray boxes and arrows) mechanisms associated with reduced pacemaker function in aging as described in this review. mHR is decreased primarily due to a reduction in intrinsic heart rate (iHR), as well as a reduction in the response to β-adrenergic receptor (βAR) stimulation. (a) iHR is decreased in part as a result of reduced action potential (AP) firing rate in sinoatrial myocytes (SAMs) from aged mice (red) compared to young mice (black). (b) The decreased SAM AP firing rate is caused in turn by a reduced diastolic depolarization rate (DDR), a hyperpolarized maximum diastolic potential (MDP), and an increased AP duration. The decrease in iHR is also accompanied by a reduction in the number of SAMs and fibrosis and structural remodeling of the sinoatrial node (SAN). Panels a and b adapted from Reference 13. (c) Increased diffuse fibrosis in a SAN section from an aged human heart (right), as indicated by increased blue-green staining compared to a section from a young heart (left). Adapted from Reference 153 under the terms of the Creative Commons Attribution (CC BY) License, http://creativecommons.org/licenses/by/4.0. ICa,L, ICa,T, and If currents have been demonstrated to be reduced in aged mouse SAMs. (d) Hyperpolarized voltage dependence of activation of If in excised inside-out membrane patches from SAMs from young (black) or old (red) mice. Adapted from Reference 66 under the terms of the Creative Commons Attribution-ShareAlike (CC BY-SA) License, https://creativecommons.org/licenses/by-sa/4.0. (e) Reduced ICa,L current density in SAMs from aged mice. Adapted from Reference 13. We speculate that INa, INCX, and IK may also be decreased with age (gray boxes). Reduced expression of NCX1, RYR2, SERCA2, and Cx43 transcripts and protein has been observed in a variety of species. (f) Reduced relative mRNA abundance of RYR2 and SERCA2 in SAN of old (red) compared to young (black) rats, normalized to expression in atrial muscle. Adapted with permission from Reference 48. Copyright 2011, John Wiley & Sons. We believe that aging may also reduce expression of many other channels and signaling molecules (gray boxes). Asterisks in panels e and f indicate significant age-dependent differences (p < 0.05).
IONIC REMODELING OF THE SINOATRIAL NODE WITH AGE
It is beyond the scope of this review to exhaustively describe the long and ever-growing list of ionic currents that are important for pacemaking in SAMs. For more details, the reader is referred to many excellent review articles (51–53). Here, we provide a brief overview of the major ionic currents in SAMs, followed by a more-detailed discussion of the currents that have so far been experimentally assayed in young versus aged SAMs. The sinoatrial AP waveform parameters that change with age (DDR, MDP, and AP duration) (10, 11, 13) constrain the possible underlying ionic mechanisms, indicating that the balance of currents active during these phases is altered by the natural aging process. It should be noted that the focus on ionic currents as the end effectors of changes in membrane potential does not impugn the clear roles of intracellular Ca2+ dynamics and second messengers in regulating these transmembrane conductances.
Overview of Age-Dependent Changes in Ionic Currents in Sinoatrial Node Myocytes
A unique complement of ionic currents is critical for the production of spontaneous APs in SAMs. Studies through many decades have focused primarily on the identity of the currents that cause the diastolic depolarization; comparatively less is known about currents active during other phases of the AP. The precise identities and relative amplitudes of currents active in any given cell depend on the species, the location of the cell within the SAN, and the physiological context (including short-term status such as sympathetic stimulation and longer-term processes such as aging or disease). Many inward currents contribute to the diastolic depolarization, including but not limited to the funny current (If), the Na+-Ca2+ exchange current (INCX), L- and T-type Ca2+ currents (ICa,L and ICa,T), and possibly voltage-gated Na+ currents (INa). Following the diastolic depolarization, ICa,L and ICa,T are thought to be primarily responsible for the upstroke of the AP. Major outward currents in SAMs include voltage-gated K+ currents (IKr, IKs, and Ito), Ca2+-activated K+ currents (IKCa), inward rectifiers (IKACh, IKATP, and varying amounts of IK1), and possibly If (see below).
Funny current.
The If was first discovered 40 years ago as an adrenaline-sensitive current activated by hyperpolarization in rabbit SAN tissue (54). If is produced by hyperpolarization-activated, cyclic-nucleotide sensitive (HCN) channels. There are four HCN channel isoforms (HCN1–4), of which HCN4 is the predominant isoform in the SAN of all mammals (55, 56). HCN1 and HCN2 are expressed at lower levels in the SAN in a species-dependent manner (57–61).
As the name implies, HCN channels are activated by membrane hyperpolarization. Consistent with the adrenaline sensitivity of If, the open probability of HCN4 channels is modulated by binding of cyclic nucleotides to a conserved C-terminal cyclic nucleotide–binding domain (62). Sympathetic stimulation increases cAMP in SAMs, and binding of cAMP to HCN4 channels shifts pore opening to more depolarized membrane potentials and slows channel closing. The resulting increase in inward current contributes to the sympathetic nervous system-induced increase in AP firing rate of SAMs and, consequently, heart rate (63).
Although If is best known for conducting inward current during diastole, less appreciated is the potential role of If during repolarization. HCN channels are permeable to both Na+ and K+, with a net reversal potential of approximately −30 mV. Computational models and our preliminary data suggest that the channels also pass an outward, repolarizing current during systole that may shape the AP waveform and modulate firing rate (64, 65).
Strong evidence supports a role for If in age-dependent declines in SAM firing rate. In isolated SAMs from aged mice, the voltage dependence of If is shifted to more hyperpolarized potentials, thereby reducing current available during the AP (13, 66) (Figure 4d). This hyperpolarized voltage dependence persists in excised inside-out membrane patches, thereby precluding a simple decrease in intracellular cAMP as the causative mechanism (66). Rather, the data suggest that the HCN4 channel itself or an unknown, channel-associated modulator must be altered in aging (66, 67). In addition to the shifted voltage dependence, If density is decreased in aged mouse SAMs, although this decrease is fully explained by an increase in membrane capacitance (13, 68). While a decrease in If is consistent with experiments showing reduced effects of HCN blockers in aged SAN (58, 69), there is no consensus about whether aging changes HCN channel expression: In rodents, HCN2 and HCN4 transcripts have been reported to either decrease (20, 21) or not change with age (47, 48), whereas HCN2 and HCN4 transcripts and protein were found to be upregulated in right atrial appendage from human patients (70). Regardless of mechanism, the functional reduction in If is predicted to slow pacemaking, in a manner similar to that of If blockers and HCN4 mutations (71–73).
Intracellular Ca2+ release and INCX.
A role for sarcoplasmic reticulum (SR) Ca2+ release in heart rate regulation dates back to the early 1900s. In 1912, Pilcher (74) found that applying small amounts of caffeine—unknown to him as an activator of ryanodine receptors—to dog hearts increased the heart rate. In the 1970s, oscillations in Ca2+, cAMP, and conductance were proposed to contribute to spontaneous activity in neurons and cardiac pacemaker cells (75, 76). Nearly 80 years after Pilcher (74), Rubenstein & Lipsius (77) showed in feline secondary pacemaker tissue the presence of a ryanodine-sensitive current during the late diastolic depolarization. A large body of work from many groups has established that this current is mediated by the plasma membrane Na+-Ca2+ exchanger (NCX). INCX in SAMs reflects the concerted activity of Ca2+ channels and transporters on the plasma and SR membranes—the same molecular players that mediate excitation-contraction coupling in other cardiac myocytes.
During the latter part of the diastolic depolarization, type 2 ryanodine receptors (RYR2) on the SR membrane release Ca2+ into the cytosol (78). This release is augmented by Ca2+ entry through voltage-gated Ca2+ channels on the plasma membrane that open in response to the diastolic depolarization (79). The ryanodine-sensitive current arises due to the subsequent extrusion of Ca2+ from the cell by NCX1, which exchanges three Na+ ions for each Ca2+ ion and thus generates a net inward current that contributes to the diastolic depolarization (80).
Block of RYR2 decreases the AP firing rate in SAMs, presumably through a decrease in INCX (78, 80, 81). Firing rate is also reduced by block of SERCA2A, the SR Ca2+-ATPase responsible for pumping cytosolic Ca2+ back into the SR (82, 83). Block of SERCA2A prevents the replenishment of SR Ca2+ stores, which in turn decreases the Ca2+ release through RYR2 and presumably limits INCX (83).
Age-dependent changes have been observed in a number of these Ca2+-handling proteins. At both transcript and protein levels, RYR2 is decreased in rat SAN with aging, whereas SERCA2A, RYR2, and NCX1 protein are all decreased in aged mouse SAN (11, 48) (Figure 4f). In aged mouse SAMs, Liu et al. (11) suggested that SR Ca2+ load is reduced; the amplitude and rate of rise of the Ca2+ transient were decreased and the duration of the Ca2+ transient was increased. Although the direct impact of aging on INCX in SAMs has yet to be measured experimentally, decreased SR Ca2+ release would be expected to contribute to slower AP firing rate by reducing INCX.
Voltage-gated Ca2+ currents.
In SAMs, ICa,L and ICa,T contribute to the diastolic depolarization and are largely responsible for the AP upstroke. Larson et al. (13) found that the ICa,L and ICa,T densities are lower in aged SAMs (13) (Figure 4e). As is the case for If, this decrease was fully accounted for by cellular hypertrophy without a concurrent upregulation of channel expression. In accordance, age-dependent decreases in ICa,L and Ca2+ transients have been observed in isolated human atrial tissue, canine atrial tissue, and rat ventricular tissue (11, 84–86). However, there is some variability in reported effects of aging on voltage-gated Ca2+ channel expression. While Jones et al. (86) demonstrated an age-dependent decrease in CaV1.2 protein expression in guinea pig SAN, experiments in rats showed an increase in CaV1.2 mRNA expression (48, 87). Despite the differences in expression results, the functional decreases observed in ICa,L and ICa,T densities would be predicted to contribute to slower AP firing rates in aged SAMs, both directly by reducing inward Ca2+ current and indirectly by reducing Ca2+-induced Ca2+ release and INCX, as discussed above.
Na+ currents.
Unfortunately, few data are available on age-dependent changes to Na+ channels in the SAN and their functional consequences. In fact, the role of voltage-gated Na+ channels is often overlooked in the generation of spontaneous APs in pacemaker cells: The Na+ channel blocker tetrodotoxin (TTX) slows the AP upstroke of subsidiary pacemaker cells from guinea pig (88) and cultured SAMs from rabbit (89) and slows the spontaneous beating rate of isolated mouse hearts (90). The cardiac NaV1.5 channel isoform is the site of multiple mutants that have been linked to inherited sinoatrial node dysfunction (SND) (91). And in mouse models, mutant NaV1.5 channels decrease iHR and cause SND (91).
Experiments in rats suggest an increase in NaV1.5 transcript expression with age (48), whereas in canine SAMs, INa density decreases between newborns and young dogs and again between young and adult dogs (92). However, as with ICa,L, ICa,T, and If, the decrease in INa density between young and adult canine SAMs can be accounted for by the increase in membrane capacitance (92). Yanni et al. (68) showed that in aged mice there is an increase in the area of the SAN that lacks the relatively TTX-resistant NaV1.5 sodium channel isoform and yet an increased effect of 2 μM TTX on firing rate, possibly reflecting an upregulation of TTX-sensitive Na+ channel isoforms.
K+ currents.
There are clear roles for voltage-gated, Ca2+-activated, and inward rectifier K+ channels in the repolarization and diastolic depolarization phases of the SAN AP (93–98). The delayed rectifier K+ channels KV7.1 (KCNQ1), which underlies IKs, and KV11.1 (ERG), which produces IKr, are thought to be primarily responsible for repolarization of the sinoatrial AP. While mutations in these channels are most commonly associated with long QT syndrome, the same mutations are also associated with episodic bradycardia (98–100). Furthermore, pharmacological blockade of ERG causes bradycardia in both dogs and mice (101, 102).
Recently, a mutant in the G protein–gated inwardly rectifying K+ (GIRK) channel Kir3.4, which along with Kir3.1 passes the IKACh current, was found to cause SND (103). Activation of IKACh contributes to slowed sinoatrial pacemaking in response to parasympathetic stimulation by slowing the DDR. Even in mice, which typically have predominant sympathetic drive at rest, knockout of Kir3.1 causes resting tachycardia (93), while knockout of either Kir3.1 or Kir3.4 attenuates the decreases in heart rate seen with application of parasympathetic agonists (93, 104). Similarly, Ca2+-activated K+ channels also play a role in determining the DDR and firing rate of SAMs. Both big conductance and intermediate conductance Ca2+-activated K+ channels (BK and SK4, respectively) have been found to regulate pacemaking (94–96).
Despite their importance, there is a distinct lack of functional data on K+ channels in aging SAMs. This lack of data is particularly significant given that the increase in AP duration and slowed DDR with age predict that there are likely to be changes in outward currents (10, 11). At the transcript level in aged rat SAN, some changes in delayed rectifier K+ channels have been reported: KV7.1 is increased and KV1.5, the channel that underlies IKur, is decreased (48). There is also an age-dependent decrease in Kir3.4 transcript and an increase in transcripts encoding KV1.2 and KV4.2, which produce Ito, (48). Given that Ito is increased in right atrial tissue in aged dogs, this may translate to increased sinoatrial Ito with age; however, this has yet to be confirmed experimentally (83). Unfortunately, there is no protein or current expression data yet available from aged SAMs to elucidate the functional significance of any of these transcriptional changes.
Conclusions About Electrical Remodeling of Sinoatrial Node Myocytes in Aging
Although it is clear that aging changes the expression and function of several ion channels in SAMs, a comprehensive understanding of how aging changes the ionic basis of pacemaking is not possible due to a paucity of functional data. In many cases the only existing studies are those of mRNA expression, which is often poorly correlated with protein expression (105). Many additional functional studies of age-dependent changes in ionic currents, including K+ currents, are necessary to fully understand electrical remodeling in aged SAMs.
STRUCTURAL REMODELING
As early as 1907, the SAN was noted to have a relatively high amount of fibrotic and connective tissue relative to the rest of the heart, and a further increase in fibrosis was observed in the SAN from diseased hearts (25). In an extensive review, Csepe et al. (43) note that the fibrotic tissue in the SAN may serve as an electrical insulator that, in addition to the SAN-specific gap junctions mentioned above, may protect the SAN from the larger and more hyperpolarized atrial myocardium.
A natural age-dependent increase in fibrotic tissue in the SAN has been consistently noted in humans and animal models and is strongly associated with reduced pacemaker function (106–108) (Figure 4c). Increased interstitial fibrosis and altered expression of matrix metalloproteinases correlate with SAN dysfunction and frailty in aged mice (47). In human patients, age-dependent atrial remodeling and slowed conduction velocity are associated with impaired SAN function (109) and are exacerbated in individuals with clinical SND (110), a disorder that is most prevalent in older individuals (111). Hao et al. (112) found that increased collagen, fibrosis, and TGFβ1 in the SAN were particularly pronounced in aged heterozygous SCN5A knockout mice, in association with decreased SAN function. And calsequestrin 2-null mice have increased SAN fibrosis and sinus bradycardia (113). However, in these latter cases, the genetic knockouts might also decrease SAN function by changing ionic currents; blockade of sodium channels can decrease SAN function (87–89) and calsequestrin 2 knockout also alters SR Ca2+ transients, diastolic Ca2+ levels, and spontaneous Ca2+ release patterns (113).
Despite the strong correlations, increased fibrosis in the aged SAN is not always associated with decreased pacemaker activity; severe fibrosis can occur in aged hearts with normal sinus rhythm (107). Thus, while fibrosis is associated with pacemaker anomalies, the extent to which it slows iHR in aging is unknown, in part due to the difficulty of experimentally manipulating fibrosis to reveal causal relationships.
Histological studies also show that the volume of the SAN and the number of SAMs decrease with age (106–108). Multiple labs have noted cellular hypertrophy and increased membrane capacitance of SAMs with aging (13, 68, 114). Along with the reduced volume of the SAN, this is consistent with a decrease in the number of cells. The relatively small number of SAMs makes the SAN vulnerable to loss of cells; a critical mass of SAMs is required to excite the atria. Indeed, animal models with apoptosis of SAMs have slower iHRs (115), and two-dimensional models of the SAN suggest that slower atrial rates are a direct consequence of a reduced number of SAMs (115, 116). However, it is not clear whether cellular hypertrophy of SAMs may compensate to some degree for the loss of cells. Moreover, normal sinus rhythm is maintained in some patients with only 10% of their SAMs remaining (107). Thus, as for the increase in fibrosis, a decrease in number of SAMs can be correlated with a decrease in heart rate, but the extent to which it contributes to the reduced SAN function in aging has not yet been resolved.
β-ADRENERGIC RESPONSIVITY
In addition to electrical and structural remodeling, aging also decreases mHR in part by blunting the heart rate response to sympathetic stimulation. The observation of a decreased inotropic response to catecholamines in heart muscle from aged rats (117) led to the hypothesis that aging reduces mHR by similarly decreasing the response of the SAN to β-adrenergic receptor (βAR) stimulation (14). Indeed, isoproterenol injection or maximal exercise produces smaller increases in heart rate above rHR in older human subjects (4, 118), and the βAR blocker, propranolol, causes a correspondingly smaller decrease in mHR in older individuals (119). However, as discussed above, the constant rHR with age in the context of the reduction in iHR necessitates an increase in sympathetic tone at rest that naturally decreases the capacity of the heart rate to increase above resting levels (Figure 1) (20, 120–122).
In spite of the confounds of the sympathetic response from a base of iHR versus rHR, a reduction in the absolute response to βAR stimulation with age is revealed by other measurements. For example, the dose of isoproterenol needed to increase heart rate by 25 bpm increases with age (123). Decreased βAR sensitivity is also suggested by differences in the rates of decline in iHR and mHR with age (Figure 1); while early work, citing parallel declines in iHR and mHR, concluded that there was no evidence for an age-related decrease in the mechanisms which increased heart rate above its intrinsic level (20, 21), more recent studies have revealed slight differences in the rates of decline in iHR and mHR. In humans, iHR decreases by ~0.5 bpm/year (20, 27, 124), whereas mHR decreases by ~0.7 bpm/year (2, 5). Similarly, in mice the declines in iHR and mHR differ between 3 and 32 months (~120 vs 170 bpm, respectively) (13). Overall, these data suggest that while decreases in iHR remain the primary driver of age-dependent declines in mHR, a modest role for declines in sympathetic nervous function also exists. Accordingly, Christou & Seals (22) found that the reduction in the heart rate response to isoproterenol accounts for ~17% of the decrease in mHR with age.
Molecular Basis of the Reduced β-Adrenergic Receptor Response with Age
Stimulation of βARs on SAMs activates adenylyl cyclases to increase intracellular cAMP, which activates the cAMP-dependent protein kinase (PKA). PKA phosphorylates a host of molecules in SAMs that contribute to the fight-or-flight increase in AP firing rate. As discussed above, βAR-stimulated cAMP also speeds SAM firing by binding directly to HCN channels to increase If (125).
Molecules involved in intracellular Ca2+ cycling are among the best-known targets of PKA. PKA-mediated phosphorylation of L-type Ca2+ channels increases ICa,L (126), which in turn increases Ca2+ release from the SR through RYR2 (127, 128). SR release is further augmented by PKA phosphorylation of RYR2 itself, which increases its open probability (129, 130). The resulting increase in cytoplasmic Ca2+ in turn increases extrusion by NCX (131, 132). The increase in both ICa,L and INCX is thought to contribute to the fight-or-flight increase in AP firing rate. HCN4 channels are also a site for PKA phosphorylation, which may contribute to the shift in voltage dependence of If upon βAR stimulation and thereby to the increase in SAM firing rate (133). Finally, PKA phosphorylation of the SERCA inhibitory protein, phospholamban, relieves its inhibition of SERCA to increase the rate of Ca2+ reuptake into the SR (82, 134). Continued beating of the heart requires that the increased Ca2+ influx through L-type channels must be balanced by increased extrusion by NCX, while increased SR Ca2+ release through RYR2 must in turn be balanced by increased Ca2+ reuptake through SERCA.
As mentioned above, aging changes a number of these PKA-sensitive molecules: RYR2 transcript and protein levels are decreased in aged rat SAN, whereas SERCA2A, RYR2, and NCX1 proteins are all decreased in aged mouse SAN (11, 48). Although aging decreases adenylyl cyclase activity in the ventricular myocardium (135), its activity has not been measured in aged SAN. Several groups have reported a lack of change of βAR transcripts with age in rodents (48, 136). To our knowledge, no studies have examined age-dependent protein expression of either βARs or adenylyl cyclases in isolated SAN tissue.
Studies from our lab have characterized cellular responses to stimulation of components of the βAR signaling cascade. In SAMs, stimulation of βARs with isoproterenol, stimulation of adenylyl cyclase with forskolin, and/or inhibition of phosphodiesterases with IBMX are all insufficient to restore firing rate in aged cells; firing rates increase by similar amounts in young and old SAMs in response to these manipulations but fail to overcome the slower intrinsic firing rate of aged SAMs (13, 66). At face value, these data could indicate a fundamental limit to the firing rate of aged SAMs. However, application of a high concentration of cAMP to the cytosol of SAMs restores AP firing rate in aged SAMs to youthful levels (66). These data establish that aged SAMs are capable of firing at the same rates as younger SAMs and suggest that the endogenous βAR machinery is insufficient to surmount the reduced intrinsic firing rate to maximally stimulate the cells.
Interestingly, a high concentration of IBMX was observed to restore maximum beating rate to young levels in SAN tissue strips from aged mice (11). This difference from the response of single cells could reflect an emergent tissue-level property, perhaps akin to the mechanism responsible for higher beating rates of SAN tissue compared to isolated cells from rabbits and mice (13, 137). Alternatively, it could result from IBMX activation of other cell types in the tissue or off-target effects of IBMX. Indeed, when data from aged SAMs were incorporated into an existing SAM AP model, the simulated APs reproduced the inability of βAR stimulation to rescue firing rate in aged cells; further tailoring of parameters was required to replicate the tissue-level effects (138). Ultimately, a great deal of additional experimental data will be required to formulate strong mathematical models of the effects of aging on sinoatrial cells and tissue.
CELLULAR-LEVEL CHANGES ARE THE PRIMARY DETERMINANT OF AGE-DEPENDENT DECLINES IN MAXIMUM HEART RATE
Surprisingly, age-dependent effects at the tissue and cellular levels are not additive. In both rabbits and mice, the firing rate of intact SAN tissue is higher than the firing rate of individual SAMs, indicating that there is an emergent, tissue-level property that effectively increases heart rate, with the simplest mechanism being that the fastest cell paces all others (13, 137). Age-dependent deficits in this tissue-level compensation, due to fibrosis and/or decreases in cell number, might be expected to compound with those of individual SAMs to yield a slower beating rate than that caused by either factor alone. However, this has not been observed. In fact, the changes in firing rate of isolated SAMs prior to and after isoproterenol perfusion directly mirror the change in iHR and mHR in these animals. In mice, the intrinsic AP firing rate slows by ~100 AP/min between 2 and 24 months, while iHR and the beating rate of whole nodes slow by ~60–90 bpm (11, 13, 66). The same pattern is present for the maximum AP firing rate and mHR; aging slows maximum firing rate by ~150 AP/min and mHR by ~120 bpm (13). If anything, for both intrinsic and maximum rates, there is less slowing at the tissue level than the cell level, not more as would be predicted for additive effects.
Taken together, studies of human and animal heart rates, isolated SAN tissue, and isolated SAMs suggest that age-dependent declines in mHR are driven primarily by electrical remodeling at the cellular level with smaller declines attributable to structural remodeling and reduced βAR responsivity.
CONSEQUENCES FOR HUMAN HEALTH
The age-dependent decline in cardiac pacemaker function and ensuing declines in mHR and VO2max have profound negative consequences for health span, quality of life, athletic performance, and societal health care costs. Physical exercise mitigates some effects of aging and increases health span by slowing the decline in VO2max (4, 15); however, an inevitable decline in VO2max is imposed by the age-dependent decline in mHR (2–4).
VO2max is a key determinant of functional independence and quality of life. Loss of functional independence in old age is correlated with VO2max below a threshold of 15–18 mL/(kg·min) (18). As the percentage of the population over the age of 65 continues to increase—20% of the US population will be over the age of 65 by 2035 (139)—the costs associated with this loss of independence will also increase. Age-dependent declines in mHR and VO2max also limit peak physical performance for elite athletes and weekend warriors alike. For the ever-growing and increasingly older population of recreational athletes, the age-dependent decline in VO2max limits not only performance but also the ability to participate at all.
While the age-dependent decline in pacemaker function reduces physical capacity for all people, some individuals experience more severe age-related pacemaker deficits. Clinically symptomatic SND—marked by bradyarrhythmias, alternating bradycardia and tachycardia, sinoatrial block, and dementia due to cerebral hypofusion—affects 1 in 600 people over the age of 65 (140). SND accounts for the majority of the 225,000 electronic pacemakers that are implanted each year in the United States at an annual cost of $8.3 billion (141). In addition to the increased health care costs of treating SND as the population ages, surgically implanted pacemakers also carry risks for individuals, including infection and cardiac perforation (141).
The human and societal costs of age-dependent pacemaker deficits are exacerbated by a limited number of drugs to control heart rate. One FDA-approved drug, ivabradine, is available to slow heart rate by blocking HCN channels; however, it is not specific for the sinoatrial node HCN4 channel isoform and has some off-target effects (142–144). Similarly, there are few drugs to safely increase heart rate for treatment of bradycardia. Atropine is sometimes effective at increasing heart rate, but some elderly patients with bradyarrhythmia are resistant to atropine (145, 146), consistent with the loss of parasympathetic tone at rest in the elderly (120–122).
The ideal pharmaceutical to treat SND and enhance VO2max in elderly individuals would be one that acts specifically on the SAN to increase iHR, and therefore mHR, without affecting sympathetic tone or cardiac contractility. Of the factors that contribute to the age-related decrease in mHR, cellular mechanisms that drive the loss of pacemaker function with age appear to be the first-line drug targets. Structural remodeling would be challenging to target and hard to dose in response to continued changes as the individual ages. Meanwhile, targeting the decrease in βAR responsivity poses obvious challenges for sinoatrial specificity, as most targets would also impact other cardiovascular and systemic processes. Among cellular targets, HCN4 channels are particularly attractive owing to their relatively specific expression in the SAN and the proven effectiveness of If as a target for control of heart rate (71). While ion channel blockers often act on the structurally conserved pore domain and so can be prone to off-target effects, agonists can be targeted to less-conserved allosteric domains, making them alluring targets for high-specificity drugs (147).
LIMITATIONS, GAPS IN KNOWLEDGE, AND FUTURE DIRECTIONS
The primary limitation to understanding the age-dependent slowing of mHR is a simple lack of data. Although the effects of aging on human heart rate, and even the firing rate of the SAN, are well characterized, few mechanistic data exist. Moreover, the conclusions that can be drawn from existing data sets are limited by difficulties in integrating information from different species and across different levels (molecular, cellular, tissue).
Gaps in knowledge about pacemaking exist across all levels of SAN and SAM function. At the cellular level, one of the key limitations to progress is the field’s disproportionate focus on identifying a single most important mechanism for the diastolic depolarization in SAMs (52), which has left large gaps in knowledge about currents active during other phases of the AP. The relative lack of information about K+ currents in SAMs is particularly important for aging because the AP prolongation along with reduced Ca2+ current density predicts a decrease in K+ currents in aged SAMs (10, 11, 13). Although, as discussed above, K+ currents play an important role in determining cycle length through modulation of multiple AP waveform parameters and are implicated in SND (95, 97–100, 103), there is no information to our knowledge about age-dependent changes in K+ currents in SAMs.
There is also a lack of functional data corresponding to the more numerous, and sometimes conflicting, transcript studies. For example, although expression is reduced in aged SAN for a number of Ca2+-handling proteins (11, 48, 86), there has not yet been a comparison of INCX in young versus old SAMs (in part due to difficulties inherent to experimental isolation of INCX). Even in the case of currents for which age-dependent changes have been reported, the underlying mechanisms are unknown. For example, our data suggest that a novel HCN4-associated factor may be responsible for the age-dependent changes in If in SAMs (66, 67), but this factor has yet to be identified.
The lack of data on age-dependent ionic remodeling in single cells is compounded at the tissue level. Little is known about innervation and cell–cell coupling in general, much less how they may change with age. And while studies suggest that gap junction expression is altered with aging (46), cell–cell coupling between SAMs remains understudied.
The dearth of mechanistic data at the cellular and tissue levels in turn hinders the generation of validated mathematical models of APs in aged SAMs and precludes scaling of cellular data to the tissue level at this time. Modeling is a particularly important tool for understanding the complex and interrelated mechanisms that drive pacemaking in part because experimental perturbations often cause the entire dynamical system to fail. Acquisition of robust data sets under standardized conditions is a priority for future work in order to refine and improve mathematical models of the SAN.
Another important area for future work is a description of gender-dependent differences in the aging SAN. Such differences seem likely to exist—and to have profound implications for treatments—based on gender differences in aging of other regions of the heart (148). Indeed, while the rate of decline of mHR and iHR is indistinguishable between men and women (20), some studies suggest that women have higher rHRs and iHRs than men (149). Women are also more likely to have an electronic pacemaker implanted for SND or atrial fibrillation with bradycardia (150), and numerous cardiovascular drugs have different effects in men and women (151).
SUMMARY POINTS.
Aging reduces mHR by reducing iHR and thus by reducing the pacemaker activity of the sinoatrial node of the heart.
The progressive decrease in iHR with age necessitates a corresponding increase in sympathetic tone in order to maintain rHR.
The age-dependent reduction in mHR is crucial for individuals and society because it causes an inevitable reduction in VO2max, which not only limits athletic performance but can also limit functional independence and quality of life for the growing population of elderly people.
The mechanisms by which aging slows the heart include electrical and structural remodeling as well as a reduced response to βAR stimulation. Of these factors, the reductions in cellular mechanisms that are responsible for iHR predominate and represent the best potential for development of drugs that can selectively increase HR and VO2max in elderly individuals.
There is an urgent need for additional functional data to identify molecular mechanisms that contribute to decreased SAN function with age. Important areas for future work include acquisition of expanded data sets to improve mathematical models, description of gender differences in SAN aging, and additional information about electrical coupling in the SAN to improve understanding of tissue-level properties.
Given that aging affects all individuals, the clinical implications and benefits of future work deciphering the underlying mechanisms of cardiac pacemaker decline are clear.
ACKNOWLEDGMENTS
This work was supported by grants from the US National Institutes of Health (R01-HL088427, T32-AG000279, F31-HL132408) and the American Heart Association (19POST34380777). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health or the American Heart Association.
Glossary
- mHR
maximum heart rate
- VO2max
maximum rate of oxygen consumption
- iHR
intrinsic heart rate
- SAN
sinoatrial node
- rHR
resting heart rate
- SAM
sinoatrial node myocyte
- AP
action potential
- DDR
diastolic depolarization rate
- MDP
maximum diastolic potential
- If
funny current
- SR
sarcoplasmic reticulum
- NCX
Na+-Ca2+ exchanger
- RYR
ryanodine receptor
- SERCA
sarco-/endoplasmic reticulum Ca2+ ATPase
- SND
sinoatrial node dysfunction
- βAR
β-adrenergic receptor
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Homer. 1990. Iliad. London: Penguin [Google Scholar]
- 2.Robinson S. 1938. Experimental studies of physical fitness in relation to age. Eur. J. Appl. Physiol 10(3):251–323 [Google Scholar]; Seminal study showing age-dependent declines in mHR.
- 3.Robinson S, Dill DB, Tzankoff SP, Wagner JA, Robinson RD. 1975. Longitudinal studies of aging in 37 men. J. Appl. Physiol 38(2):263–67 [DOI] [PubMed] [Google Scholar]
- 4.Heath GW, Hagberg JM, Ehsani AA, Holloszy JO. 1981. A physiological comparison of young and older endurance athletes. J. Appl. Physiol 51(3):634–40 [DOI] [PubMed] [Google Scholar]
- 5.Tanaka H, Monahan KD, Seals DR. 2001. Age-predicted maximal heart rate revisited. J. Am. Coll. Cardiol 37(1):153–56 [DOI] [PubMed] [Google Scholar]
- 6.Tanaka H, Desouza CA, Jones PP, Stevenson ET, Davy KP, Seals DR. 1997. Greater rate of decline in maximal aerobic capacity with age in physically active versus sedentary healthy women. J. Appl. Physiol 83(6):1947–53 [DOI] [PubMed] [Google Scholar]
- 7.Fox SM, Naughton JP, Haskell WL. 1971. Physical activity and the prevention of coronary heart disease. Ann. Clin. Res 3(6):404–32 [PubMed] [Google Scholar]
- 8.Nes BM, Janszky I, Wisløff U, Støylen A, Karlsen T. 2013. Age-predicted maximal heart rate in healthy subjects: the HUNT fitness study. Scand. J. Med. Sci. Sports 23(6):697–704 [DOI] [PubMed] [Google Scholar]
- 9.Di Gennaro M, Bernabei R, Sgadari A, Carosella L, Carbonin PU. 1987. Age-related differences in isolated rat sinus node function. Basic Res. Cardiol 82(6):530–36 [DOI] [PubMed] [Google Scholar]
- 10.Alings AM, Bouman LN. 1993. Electrophysiology of the ageing rabbit and cat sinoatrial node—a comparative study. Eur. Heart J 14(9):1278–88 [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Sirenko S, Juhaszova M, Sollott SJ, Shukla S, et al. 2014. Age-associated abnormalities of intrinsic automaticity of sinoatrial nodal cells are linked to deficient cAMP-PKA-Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol 306(10):H1385–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yaniv Y, Ahmet I, Tsutsui K, Behar J, Moen JM, et al. 2016. Deterioration of autonomic neuronal receptor signaling and mechanisms intrinsic to heart pacemaker cells contribute to age-associated alterations in heart rate variability in vivo. Aging Cell 15(4):716–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larson ED, St Clair JR, Sumner WA, Bannister RA, Proenza C. 2013. Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. PNAS 110(44):18011–16 [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of age-dependent declines in pacemaking and membrane currents of isolated sinoatrial myocytes.
- 14.Yin FC, Spurgeon HA, Greene HL, Lakatta EG, Weisfeldt ML. 1979. Age-associated decrease in heart rate response to isoproterenol in dogs. Mech. Ageing Dev 10(1–2):17–25 [DOI] [PubMed] [Google Scholar]
- 15.Hagberg JM, Allen WK, Seals DR, Hurley BF, Ehsani AA, Holloszy JO. 1985. A hemodynamic comparison of young and older endurance athletes during exercise. J. Appl. Physiol 58(6):2041–46 [DOI] [PubMed] [Google Scholar]
- 16.Tanaka H, Seals DR. 2008. Endurance exercise performance in Masters athletes: age-associated changes and underlying physiological mechanisms. J. Physiol 586(1):55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paterson DH, Govindasamy D, Vidmar M, Cunningham DA, Koval JJ. 2004. Longitudinal study of determinants of dependence in an elderly population. J. Am. Geriatr. Soc 52(10):1632–38 [DOI] [PubMed] [Google Scholar]
- 18.Shephard RJ. 2009. Maximal oxygen intake and independence in old age. Br. J. Sports Med 43(5):342–46 [DOI] [PubMed] [Google Scholar]
- 19.Hawkins S, Wiswell R. 2003. Rate and mechanism of maximal oxygen consumption decline with aging: implications for exercise training. Sports Med. 33(12):877–88 [DOI] [PubMed] [Google Scholar]
- 20.Jose AD, Collison D. 1970. The normal range and determinants of the intrinsic heart rate in man. Cardiovasc. Res 4(2):160–67 [DOI] [PubMed] [Google Scholar]; Seminal study on age-dependent declines in iHR.
- 21.Jose AD, Stitt F, Collison D. 1970. The effects of exercise and changes in body temperature on the intrinsic heart rate in man. Am. Heart J 79(4):488–98 [DOI] [PubMed] [Google Scholar]
- 22.Christou DD, Seals DR. 2008. Decreased maximal heart rate with aging is related to reduced β-adrenergic responsiveness but is largely explained by a reduction in intrinsic heart rate. J. Appl. Physiol 105(1):24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]; Study measuring the contribution of decreased iHR and βAR agonist sensitivity to declines in mHR.
- 23.Silverman ME, Hollman A. 2007. Discovery of the sinus node by Keith and Flack: on the centennial of their 1907 publication. Heart 93(10):1184–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fye WB. 1987. The origin of the heart beat: a tale of frogs, jellyfish, and turtles. Circulation 76(3):493–500 [DOI] [PubMed] [Google Scholar]
- 25.Keith A, Flack M. 1907. The form and nature of the muscular connections between the primary divisions of the vertebrate heart. J. Anat. Physiol 41(Part 3):172–89 [PMC free article] [PubMed] [Google Scholar]; Description of SAN anatomy in multiple species.
- 26.Bexton RS, Nathan AW, Hellestrand KJ, Cory-Pearce R, Spurrell RA, et al. 1984. Sinoatrial function after cardiac transplantation. J. Am. Coll. Cardiol 3(3):712–23 [DOI] [PubMed] [Google Scholar]
- 27.Strobel JS, Epstein AE, Bourge RC, Kirklin JK, Kay GN. 1999. Nonpharmacologic validation of the intrinsic heart rate in cardiac transplant recipients. J. Interv. Card. Electrophysiol. Int. J. Arrhythm. Pacing 3(1):15–18 [DOI] [PubMed] [Google Scholar]
- 28.Robinson BF, Epstein SE, Beiser GD, Braunwald E. 1966. Control of heart rate by the autonomic nervous system. Studies in man on the interrelation between baroreceptor mechanisms and exercise. Circ. Res 19(2):400–11 [DOI] [PubMed] [Google Scholar]
- 29.Masson-Pévet M, Bleeker WK, Gros D. 1979. The plasma membrane of leading pacemaker cells in the rabbit sinus node. A qualitative and quantitative ultrastructural analysis. Circ. Res 45(5):621–29 [DOI] [PubMed] [Google Scholar]
- 30.James TN, Sherf L, Fine G, Morales AR. 1966. Comparative ultrastructure of the sinus node in man and dog. Circulation 34(1):139–63 [DOI] [PubMed] [Google Scholar]
- 31.Opthof T, de Jonge B, Jongsma HJ, Bouman LN. 1987. Functional morphology of the mammalian sinuatrial node. Eur. Heart J 8(11):1249–59 [DOI] [PubMed] [Google Scholar]
- 32.Boyett MR, Honjo H, Kodama I. 2000. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res 47(4):658–87 [DOI] [PubMed] [Google Scholar]
- 33.Csepe TA, Zhao J, Hansen BJ, Li N, Sul LV, et al. 2016. Human sinoatrial node structure: 3D microanatomy of sinoatrial conduction pathways. Prog. Biophys. Mol. Biol 120(1–3):164–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwong KF, Schuessler RB, Green KG, Laing JG, Beyer EC, et al. 1998. Differential expression of gap junction proteins in the canine sinus node. Circ. Res 82(5):604–12 [DOI] [PubMed] [Google Scholar]
- 35.Coppen SR, Kodama I, Boyett MR, Dobrzynski H, Takagishi Y, et al. 1999. Connexin45, a major connexin of the rabbit sinoatrial node, is co-expressed with connexin43 in a restricted zone at the nodal-crista terminalis border. J. Histochem. Cytochem 47(7):907–18 [DOI] [PubMed] [Google Scholar]
- 36.Kreuzberg MM, Söhl G, Kim J-S, Verselis VK, Willecke K, Bukauskas FF. 2005. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res 96(11):1169–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haubrich S, Schwarz HJ, Bukauskas F, Lichtenberg-Fraté H, Traub O, et al. 1996. Incompatibility of connexin 40 and 43 Hemichannels in gap junctions between mammalian cells is determined by intracellular domains. Mol. Biol. Cell 7(12):1995–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreuzberg MM, Willecke K, Bukauskas FF. 2006. Connexin-mediated cardiac impulse propagation: connexin 30.2 slows atrioventricular conduction in mouse heart. Trends Cardiovasc. Med 16(8):266–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anumonwo JM, Wang HZ, Trabka-Janik E, Dunham B, Veenstra RD, et al. 1992. Gap junctional channels in adult mammalian sinus nodal cells. Immunolocalization and electrophysiology. Circ. Res 71(2):229–39 [DOI] [PubMed] [Google Scholar]
- 40.Joyner RW, van Capelle FJ. 1986. Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys. J 50(6):1157–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto M, Honjo H, Niwa R, Kodama I. 1998. Low-frequency extracellular potentials recorded from the sinoatrial node. Cardiovasc. Res 39(2):360–72 [DOI] [PubMed] [Google Scholar]
- 42.Fedorov VV, Glukhov AV, Chang R, Kostecki G, Aferol H, et al. 2010. Optical mapping of the isolated coronary-perfused human sinus node. J. Am. Coll. Cardiol 56(17):1386–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J, Fedorov VV. 2015. Fibrosis: a structural modulator of sinoatrial node physiology and dysfunction. Front. Physiol 6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]; Review of fibrosis in the dysfunction of aging SAN.
- 44.Valiunas V, Weingart R, Brink PR. 2000. Formation of heterotypic gap junction channels by connexins 40 and 43. Circ. Res 86(2):E42–49 [DOI] [PubMed] [Google Scholar]
- 45.Bukauskas FF, Angele AB, Verselis VK, Bennett MVL. 2002. Coupling asymmetry of heterotypic connexin 45/connexin 43-EGFP gap junctions: properties of fast and slow gating mechanisms. PNAS 99(10):7113–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones SA, Lancaster MK, Boyett MR. 2004. Ageing-related changes of connexins and conduction within the sinoatrial node. J. Physiol 560(Part 2):429–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moghtadaei M, Jansen HJ, Mackasey M, Rafferty SA, Bogachev O, et al. 2016. The impacts of age and frailty on heart rate and sinoatrial node function. J. Physiol 594(23):7105–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tellez JO, Mączewski M, Yanni J, Sutyagin P, Mackiewicz U, et al. 2011. Ageing-dependent remodelling of ion channel and Ca2+ clock genes underlying sino-atrial node pacemaking. Exp. Physiol 96(11):1163–78 [DOI] [PubMed] [Google Scholar]; Review of fibrosis in the dysfunction of aging SAN.
- 49.Cavoto FV, Kelliher GJ, Roberts J. 1974. Electrophysiological changes in the rat atrium with age. Am. J. Physiol 226(6):1293–97 [DOI] [PubMed] [Google Scholar]
- 50.Rickert C, Proenza C. 2017. ParamAP: standardized parameterization of sinoatrial node myocyte action potentials. Biophys. J 113(4):765–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mangoni ME, Nargeot J. 2008. Genesis and regulation of the heart automaticity. Physiol. Rev 88(3):919–82 [DOI] [PubMed] [Google Scholar]
- 52.Lakatta EG, DiFrancesco D. 2009. JMCC point-counterpoint. J. Mol. Cell. Cardiol 47(2):157–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monfredi O, Dobrzynski H, Mondal T, Boyett MR, Morris GM. 2010. The anatomy and physiology of the sinoatrial node—a contemporary review. PACE Pacing Clin. Electrophysiol 33(11):1392–406 [DOI] [PubMed] [Google Scholar]
- 54.Brown H, DiFrancesco D, Noble S. 1979. Cardiac pacemaker oscillation and its modulation by autonomic transmitters. J. Exp. Biol 81:175–204 [DOI] [PubMed] [Google Scholar]
- 55.Moosmang S, Biel M, Hofmann F, Ludwig A. 1999. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol. Chem 380(7–8):975–80 [DOI] [PubMed] [Google Scholar]
- 56.Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. 2001. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur. J. Biochem 268(6):1646–52 [DOI] [PubMed] [Google Scholar]
- 57.Li N, Csepe TA, Hansen BJ, Dobrzynski H, Higgins RSD, et al. 2015. Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ. Arrhythm. Electrophysiol 8(5):1219–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang X, Yang P, Yang Z, Zhang H, Ma A. 2016. Age-associated expression of HCN channel isoforms in rat sinoatrial node. Exp. Biol. Med 241(3):331–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrmann S, Layh B, Ludwig A. 2011. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J. Mol. Cell. Cardiol 51(6):997–1006 [DOI] [PubMed] [Google Scholar]
- 60.Moroni A, Gorza L, Beltrame M, Gravante B, Vaccari T, et al. 2001. Hyperpolarization-activated cyclic nucleotide-gated channel 1 is a molecular determinant of the cardiac pacemaker current If. J. Biol. Chem 276(31):29233–41 [DOI] [PubMed] [Google Scholar]
- 61.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, et al. 2003. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 22(2):216–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flynn GE, Zagotta WN. 2018. Insights into the molecular mechanism for hyperpolarization-dependent activation of HCN channels. PNAS 115(34):E8086–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bucchi A, Barbuti A, Difrancesco D, Baruscotti M. 2012. Funny current and cardiac rhythm: insights from HCN knockout and transgenic mouse models. Front. Physiol 3:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verkerk AO, Wilders R. 2013. Hyperpolarization-activated current, If, in mathematical models of rabbit sinoatrial node pacemaker cells. BioMed. Res. Int 2013:872454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharpe EJ, Gantz SC, Liu P, Bean BP, Proenza C. 2017. Characteristics of ivabradine-sensitive currents in mouse sinoatrial node myocytes. Biophys. J 112(3):35a–36a [Google Scholar]
- 66.Sharpe EJ, Larson ED, Proenza C. 2017. Cyclic AMP reverses the effects of aging on pacemaker activity and If in sinoatrial node myocytes. J. Gen. Physiol 149(2):237–47 [DOI] [PMC free article] [PubMed] [Google Scholar]; Study showing cAMP can restore If and the firing rate of aged SAMs to youthful levels.
- 67.Robertson GA. 2017. It’s not funny: how changes in If limit maximum heart rate with aging. J. Gen. Physiol 149(2):177–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yanni J, Tellez JO, Sutyagin PV, Boyett MR, Dobrzynski H. 2010. Structural remodelling of the sinoatrial node in obese old rats. J. Mol. Cell. Cardiol 48(4):653–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang X, Yang P, Du Y, Zhang J, Ma A. 2007. Age-related down-regulation of HCN channels in rat sinoatrial node. Basic Res. Cardiol 102(5):429–35 [DOI] [PubMed] [Google Scholar]
- 70.Li Y-D, Hong Y-F, Yusufuaji Y, Tang B-P, Zhou X-H, et al. 2015. Altered expression of hyperpolarization-activated cyclic nucleotide-gated channels and microRNA-1 and -133 in patients with age-associated atrial fibrillation. Mol. Med. Rep 12(3):3243–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ragueneau I, Laveille C, Jochemsen R, Resplandy G, Funck-Brentano C, Jaillon P. 1998. Pharmacokinetic-pharmacodynamic modeling of the effects of ivabradine, a direct sinus node inhibitor, on heart rate in healthy volunteers. Clin. Pharmacol. Ther 64(2):192–203 [DOI] [PubMed] [Google Scholar]
- 72.Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, et al. 2011. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. PNAS 108(4):1705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Milano A, Vermeer AMC, Lodder EM, Barc J, Verkerk AO, et al. 2014. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol 64(8):745–56 [DOI] [PubMed] [Google Scholar]
- 74.Pilcher JD. 1912. The action of caffein on the mammalian heart. J. Pharmacol. Exp. Ther 3(6):609–24 [Google Scholar]
- 75.Tsien RW, Kass RS, Weingart R. 1979. Cellular and subcellular mechanisms of cardiac pacemaker oscillations. J. Exp. Biol 81:205–15 [DOI] [PubMed] [Google Scholar]
- 76.Rapp PE, Berridge MJ. 1977. Oscillations in calcium-cyclic AMP control loops form the basis of pacemaker activity and other high frequency biological rhythms. J. Theor. Biol 66(3):497–525 [DOI] [PubMed] [Google Scholar]
- 77.Rubenstein DS, Lipsius SL. 1989. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ. Res 64(4):648–57 [DOI] [PubMed] [Google Scholar]
- 78.Rigg L, Terrar DA. 1996. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol 81(5):877–80 [DOI] [PubMed] [Google Scholar]
- 79.Chen B, Wu Y, Mohler PJ, Anderson ME, Song LS. 2009. Local control of Ca2+-induced Ca2+ release in mouse sinoatrial node cells. J. Mol. Cell. Cardiol 47(5):706–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bogdanov KY, Vinogradova TM, Lakatta EG. 2001. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger. Circ. Res 88(12):1254–58 [DOI] [PubMed] [Google Scholar]
- 81.Li J, Qu J, Nathan RD. 1997. Ionic basis of ryanodine’s negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am. J. Physiol. Heart Circ. Physiol 273(5):H2481–89 [DOI] [PubMed] [Google Scholar]
- 82.Vinogradova TM, Brochet D, Sirenko S, Li Y, Spurgeon H, Lakatta EG. 2010. Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ. Res 107(6):767–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dun W, Yagi T, Rosen MR, Boyden PA. 2003. Calcium and potassium currents in cells from adult and aged canine right atria. Cardiovasc. Res 58(3):526–34 [DOI] [PubMed] [Google Scholar]
- 84.Xu G-J, Gan T-Y, Tang B-P, Chen Z-H, Jiang T, et al. 2013. Age-related changes in cellular electrophysiology and calcium handling for atrial fibrillation. J. Cell. Mol. Med 17(9):1109–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Herraiz-Martínez A, Álvarez-García J, Llach A, Molina CE, Fernandes J, et al. 2015. Ageing is associated with deterioration of calcium homeostasis in isolated human right atrial myocytes. Cardiovasc. Res 106(1):76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jones SA, Boyett MR, Lancaster MK. 2007. Declining into failure the age-dependent loss of the L-type calcium channel within the sinoatrial node. Circulation 115(10):1183–90 [DOI] [PubMed] [Google Scholar]
- 87.Lipsius SL, Vassalle M. 1978. Dual excitatory channels in the sinus node. J. Mol. Cell. Cardiol 10(8):753–67 [DOI] [PubMed] [Google Scholar]
- 88.Muramatsu H, Zou AR, Berkowitz GA, Nathan RD. 1996. Characterization of a TTX-sensitive Na+ current in pacemaker cells isolated from rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol 270(6):H2108–19 [DOI] [PubMed] [Google Scholar]
- 89.Maier SKG, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, et al. 2003. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. PNAS 100(6):3507–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ruan Y, Liu N, Priori SG. 2009. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol 6(5):337–48 [DOI] [PubMed] [Google Scholar]
- 91.Wu J, Zhang Y, Zhang X, Cheng L, Lammers WJ, et al. 2012. Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/ΔKPQ hearts suggest an overlap syndrome. Am. J. Physiol. Heart Circ. Physiol 302(7):H1510–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Protas L, Oren RV, Clancy CE, Robinson RB. 2010. Age-dependent changes in Na current magnitude and TTX-sensitivity in the canine sinoatrial node. J. Mol. Cell. Cardiol 48(1):172–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bettahi I, Marker CL, Roman MI, Wickman K. 2002. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J. Biol. Chem 277(50):48282–88 [DOI] [PubMed] [Google Scholar]
- 94.Weisbrod D, Peretz A, Ziskind A, Menaker N, Oz S, et al. 2013. SK4 Ca2+ activated K+ channel is a critical player in cardiac pacemaker derived from human embryonic stem cells. PNAS 110(18):E1685–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lai MH, Wu Y, Gao Z, Anderson ME, Dalziel JE, Meredith AL. 2014. BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am. J. Physiol. Heart Circ. Physiol 307(9):H1327–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weisbrod D, Khun SH, Bueno H, Peretz A, Attali B. 2016. Mechanisms underlying the cardiac pacemaker: the role of SK4 calcium-activated potassium channels. Acta Pharmacol. Sin 37(1):82–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aziz Q, Li Y, Tinker A. 2018. Potassium channels in the sinoatrial node and their role in heart rate control. Channels 12(1):356–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomas D, Wimmer A-B, Karle CA, Licka M, Alter M, et al. 2005. Dominant-negative IKs suppression by KCNQ1-ΔF339 potassium channels linked to Romano-Ward syndrome. Cardiovasc. Res 67(3):487–97 [DOI] [PubMed] [Google Scholar]
- 99.Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, et al. 2010. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ. Arrhythm. Electrophysiol 3(1):10–17 [DOI] [PubMed] [Google Scholar]
- 100.Wilders R, Verkerk AO. 2018. Long QT syndrome and sinus bradycardia—a mini review. Front. Cardiovasc. Med 5:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang L, Swirp S, Duff H. 2000. Age-dependent response of the electrocardiogram to K+ channel blockers in mice. Am. J. Physiol. Cell Physiol 278(1):C73–80 [DOI] [PubMed] [Google Scholar]
- 102.Furukawa Y, Miyashita Y, Nakajima K, Hirose M, Kurogouchi F, Chiba S. 1999. Effects of verapamil, zatebradine, and E-4031 on the pacemaker location and rate in response to sympathetic stimulation in dog hearts. J. Pharmacol. Exp. Ther 289(3):1334–42 [PubMed] [Google Scholar]
- 103.Kuß J, Stallmeyer B, Goldstein M, Rinné S, Pees C, et al. 2019. Familial sinus node disease caused by a gain of GIRK (G-protein activated inwardly rectifying K+ channel) channel function. Circ. Genomic Precis. Med 12(1):e002238. [DOI] [PubMed] [Google Scholar]
- 104.Wickman K, Nemec J, Gendler SJ, Clapham DE. 1998. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20(1):103–14 [DOI] [PubMed] [Google Scholar]
- 105.Liu Y, Beyer A, Aebersold R. 2016. On the dependency of cellular protein levels on mRNA abundance. Cell 165(3):535–50 [DOI] [PubMed] [Google Scholar]
- 106.Shiraishi I, Takamatsu T, Minamikawa T, Onouchi Z, Fujita S. 1992. Quantitative histological analysis of the human sinoatrial node during growth and aging. Circulation 85(6):2176–84 [DOI] [PubMed] [Google Scholar]
- 107.Thery C, Gosselin B, Lekieffre J, Warembourg H. 1977. Pathology of sinoatrial node. Correlations with electrocardiographic findings in 111 patients. Am. Heart J 93(6):735–40 [DOI] [PubMed] [Google Scholar]
- 108.Inoue S, Shinohara F, Niitani H, Gotoh K. 1986. A new method for the histological study of aging changes in the sinoatrial node. Jpn. Heart J 27(5):653–60 [DOI] [PubMed] [Google Scholar]
- 109.Kistler PM, Sanders P, Fynn SP, Stevenson IH, Spence SJ, et al. 2004. Electrophysiologic and electroanatomic changes in the human atrium associated with age. J. Am. Coll. Cardiol 44(1):109–16 [DOI] [PubMed] [Google Scholar]
- 110.Sanders P, Morton JB, Kistler PM, Spence SJ, Davidson NC, et al. 2004. Electrophysiological and electroanatomic characterization of the atria in sinus node disease. Circulation 109(12):1514–22 [DOI] [PubMed] [Google Scholar]
- 111.Monfredi O, Boyett MR. 2015. Sick sinus syndrome and atrial fibrillation in older persons—a view from the sinoatrial nodal myocyte. J. Mol. Cell. Cardiol 83:88–100 [DOI] [PubMed] [Google Scholar]
- 112.Hao X, Zhang Y, Zhang X, Nirmalan M, Davies L, et al. 2011. TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythm. Electrophysiol 4(3):397–406 [DOI] [PubMed] [Google Scholar]
- 113.Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, et al. 2015. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur. Heart J 36(11):686–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.de Melo SR, de Souza RR, Mandarim-de-Lacerda CA. 2002. Stereologic study of the sinoatrial node of rats—age related changes. Biogerontology 3(6):383–90 [DOI] [PubMed] [Google Scholar]
- 115.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, et al. 2011. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Investig 121(8):3277–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kharche S, Beling J, Biktasheva IV, Zhang H, Biktashev VN. 2013. Simulating cell apoptosis induced sinus node dysfunction. Paper presented at the 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Osaka, Japan, July 3–7 [DOI] [PubMed] [Google Scholar]
- 117.Lakatta EG, Gerstenblith G, Angell CS, Shock NW, Weisfeldt ML. 1975. Diminished inotropic response of aged myocardium to catecholamines. Circ. Res 36(2):262–69 [DOI] [PubMed] [Google Scholar]
- 118.Stratton JR, Cerqueira MD, Schwartz RS, Levy WC, Veith RC, et al. 1992. Differences in cardiovascular responses to isoproterenol in relation to age and exercise training in healthy men. Circulation 86(2):504–12 [DOI] [PubMed] [Google Scholar]
- 119.Fleg JL, Schulman S, O’Connor F, Becker LC, Gerstenblith G, et al. 1994. Effects of acute beta-adrenergic receptor blockade on age-associated changes in cardiovascular performance during dynamic exercise. Circulation 90(5):2333–41 [DOI] [PubMed] [Google Scholar]
- 120.Abhishekh HA, Nisarga P, Kisan R, Meghana A, Chandran S, et al. 2013. Influence of age and gender on autonomic regulation of heart. J. Clin. Monit. Comput 27(3):259–64 [DOI] [PubMed] [Google Scholar]
- 121.Freeling JL, Li Y. 2015. Age-related attenuation of parasympathetic control of the heart in mice. Int. J. Physiol. Pathophysiol. Pharmacol 7(3):126–35 [PMC free article] [PubMed] [Google Scholar]
- 122.Baker SE, Limberg JK, Dillon GA, Curry TB, Joyner MJ, Nicholson WT. 2018. Aging alters the relative contributions of the sympathetic and parasympathetic nervous system to blood pressure control in women. Hypertension 72(5):1236–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vestal RE, Wood AJJ, Shand DG. 1979. Reduced β-adrenoceptor sensitivity in the elderly. Clin. Pharmacol. Ther 26(2):181–86 [DOI] [PubMed] [Google Scholar]
- 124.Alboni P, Malcarne C, Pedroni P, Masoni A, Narula OS. 1982. Electrophysiology of normal sinus node with and without autonomic blockade. Circulation 65(6):1236–42 [DOI] [PubMed] [Google Scholar]
- 125.St. Clair JR, Liao Z, Larson ED, Proenza C. 2013. PKA-independent activation of If by cAMP in mouse sinoatrial myocytes. Channels 7(4):318–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. 2014. L-type Ca2+ channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal 3(2):15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vinogradova TM, Bogdanov KY, Lakatta EG. 2002. β-Adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res 90(1):73–79 [DOI] [PubMed] [Google Scholar]
- 128.Shen J. 2006. Isoprenaline enhances local Ca2+ release in cardiac myocytes. Acta Pharmacol. Sin 27(7):927–32 [DOI] [PubMed] [Google Scholar]
- 129.Yoshida A, Takahashi M, Imagawa T, Shigekawa M, Takisawa H, Nakamura T. 1992. Phosphorylation of ryanodine receptors in rat myocytes during beta-adrenergic stimulation. J. Biochem 111(2):186–90 [DOI] [PubMed] [Google Scholar]
- 130.Uehara A, Yasukochi M, Mejía-Alvarez R, Fill M, Imanaga I. 2002. Gating kinetics and ligand sensitivity modified by phosphorylation of cardiac ryanodine receptors. Pflügers Arch. 444(1–2):202–12 [DOI] [PubMed] [Google Scholar]
- 131.Kaese S, Bögeholz N, Pauls P, Dechering D, Olligs J, et al. 2017. Increased sodium/calcium exchanger activity enhances beta-adrenergic-mediated increase in heart rate: whole-heart study in a homozygous sodium/calcium exchanger overexpressor mouse model. Heart Rhythm 14(8):1247–53 [DOI] [PubMed] [Google Scholar]
- 132.Sanders L, Rakovic S, Lowe M, Mattick PAD, Terrar DA. 2006. Fundamental importance of Na+−Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol 571(Part 3):639–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liao Z, Lockhead D, Larson E, Proenza C. 2010. Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J. Gen. Physiol 136(3):247–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Simmerman HKB, Jones LR. 1998. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev 78(4):921–47 [DOI] [PubMed] [Google Scholar]
- 135.O’Connor SW, Scarpace PJ, Abrass IB. 1981. Age-associated decrease of adenylate cyclase activity in rat myocardium. Mech. Ageing Dev 16(1):91–95 [DOI] [PubMed] [Google Scholar]
- 136.Hardouin S, Bourgeois F, Toraasson M, Oubenaissa A, Elalouf JM, et al. 1998. β-Adrenergic and muscarinic receptor mRNA accumulation in the sinoatrial node area of adult and senescent rat hearts. Mech. Ageing Dev 100(3):277–97 [DOI] [PubMed] [Google Scholar]
- 137.Yaniv Y, Ahmet I, Liu J, Lyashkov AE, Guiriba T-R, et al. 2014. Synchronization of sinoatrial node pacemaker cell clocks and its autonomic modulation impart complexity to heart beating intervals. Heart Rhythm 11(7):1210–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Behar J, Yaniv Y. 2017. Age-related pacemaker deterioration is due to impaired intracellular and membrane mechanisms: insights from numerical modeling. J. Gen. Physiol 149(10):935–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ortman JM, Velkoff VA, Hogan H. 2014. An aging nation: the older population in the United States Curr. Pop. Rep P25–1140, US Dep. Commer., Washington, DC: https://www.census.gov/prod/2014pubs/p25-1140.pdf [Google Scholar]
- 140.Semelka M, Gera J, Usman S. 2013. Sick sinus syndrome: a review. Am. Fam. Physician 87(10):691–96 [PubMed] [Google Scholar]
- 141.Sideris S, Archontakis S, Dilaveris P, Gatzoulis KA, Trachanas K, et al. 2017. Leadless cardiac pacemakers: current status of a modern approach in pacing. Hellenic J. Cardiol 58(6):403–10 [DOI] [PubMed] [Google Scholar]
- 142.Stieber J, Wieland K, Stöckl G, Ludwig A, Hofmann F. 2006. Bradycardic and proarrhythmic properties of sinus node inhibitors. Mol. Pharmacol 69(4):1328–37 [DOI] [PubMed] [Google Scholar]
- 143.Lees-Miller JP, Guo J, Wang Y, Perissinotti LL, Noskov SY, Duff HJ. 2015. Ivabradine prolongs phase 3 of cardiac repolarization and blocks the HERG1 (KCNH2) current over a concentration-range overlapping with that required to block HCN4. J. Mol. Cell. Cardiol 85:71–78 [DOI] [PubMed] [Google Scholar]
- 144.Melgari D, Brack KE, Zhang C, Zhang Y, El Harchi A, et al. 2015. hERG potassium channel blockade by the HCN channel inhibitor bradycardic agent ivabradine. J. Am. Heart Assoc 4(4):e001813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kragie L, Sekovski B. 1992. Theophylline—an alternative therapy for bradyarrhythmia in the elderly. Pharmacotherapy 12(4):324–30 [PubMed] [Google Scholar]
- 146.Pasnoori VR, Leesar MA. 2004. Use of aminophylline in the treatment of severe symptomatic bradycardia resistant to atropine. Cardiol. Rev 12(2):65–68 [DOI] [PubMed] [Google Scholar]
- 147.Proenza C. 2018. Ion channels: exploiting natural regulation. eLife 7:e39664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kane AE, Howlett SE. 2018. Differences in cardiovascular aging in men and women. Adv. Exp. Med. Biol 1065:389–411 [DOI] [PubMed] [Google Scholar]
- 149.Burke JH, Goldberger JJ, Ehlert FA, Kruse JT, Parker MA, Kadish AH. 1996. Gender differences in heart rate before and after autonomic blockade: evidence against an intrinsic gender effect. Am. J. Med 100(5):537–43 [DOI] [PubMed] [Google Scholar]
- 150.Nowak B, Misselwitz B, Erdogan A, Funck R, et al. 2010. Do gender differences exist in pacemaker implantation?—results of an obligatory external quality control program. Europace 12(2):210–15 [DOI] [PubMed] [Google Scholar]
- 151.Tamargo J, Rosano G, Walther T, Duarte J, Niessner A, et al. 2017. Gender differences in the effects of cardiovascular drugs. Eur. Heart J. Cardiovasc. Pharmacother 3(3):163–82 [DOI] [PubMed] [Google Scholar]
- 152.Park DS, Fishman GI. 2017. Development and function of the cardiac conduction system in health and disease. J. Cardiovasc. Dev. Dis 4(2):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kharche SR, Vigmond E, Efimov IR, Dobrzynski H. 2017. Computational assessment of the functional role of sinoatrial node exit pathways in the human heart. PLOS ONE 12(9):e0183727. [DOI] [PMC free article] [PubMed] [Google Scholar]