

Abstract
Background
Long‐term population‐representative data on motor fluctuations and levodopa‐induced dyskinesias in Parkinson's disease is lacking.
Methods
The Cambridgeshire Parkinson's Incidence from GP to Neurologist (CamPaIGN) cohort comprises incident PD cases followed for up to 13 years (n = 141). Cumulative incidence of motor fluctuations and levodopa‐induced dyskinesias and risk factors were assessed using Kaplan‐Meyer and Cox regression analyses.
Results
Cumulative incidence of motor fluctuations and levodopa‐induced dyskinesias was 54.3% and 14.5%, respectively, at 5 years and 100% and 55.7%, respectively, at 10 years. Higher baseline UPDRS‐total and SNCA rs356219(A) predicted motor fluctuations, whereas higher baseline Mini‐mental State Examination and GBA mutations predicted levodopa‐induced dyskinesias. Early levodopa use did not predict motor complications. Both early motor fluctuations and levodopa‐induced dyskinesias predicted reduced mortality in older patients (age at diagnosis >70 years).
Conclusions
Our data support the hypothesis that motor complications are related to the severity of nigrostriatal pathology rather than early levodopa use and indicate that early motor complications do not necessarily confer a negative prognosis. © 2019 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: levodopa‐induced dyskinesias, motor complications, motor fluctuations, Parkinson's disease
After a variable period of dopaminergic treatment, Parkinson's disease (PD) patients develop levodopa‐induced motor complications including motor fluctuations (MFs) and levodopa‐induced dyskinesias (LIDs), with important implications for quality of life. Understanding which patients are at high risk for these complications would be of benefit in terms of prognostication and planning management.
The natural history and risk factors for MF and LID are still incompletely understood. Current knowledge is mostly derived from cross‐sectional studies or clinical trials on highly selected patient groups,1, 2, 3, 4, 5, 6 not representative of PD in the population. To overcome this, some studies have employed population‐based PD cohorts,7, 8, 9, 10, 11 but only 2 have described the incidence and risk factors for MF and LID in a prospective manner, and follow‐up was limited to 5 years.9, 10
The Cambridgeshire Parkinson's Incidence from GP to Neurologist (CamPaIGN) cohort is an unbiased incident population‐based PD cohort comprising newly diagnosed cases recruited within the county of Cambridgeshire, UK, between December 2000 and December 2002.12 This study was established to provide comprehensive and true‐to‐life data on the natural history of idiopathic PD in terms of both motor symptoms and nonmotor symptoms. Baseline data and follow‐up data 3.5, 5, 7, and 10 years from diagnosis have been published previously12, 13, 14, 15, 16 and shown that there is not only motor but also cognitive heterogeneity in PD and that baseline clinical and genetic variables are predictive of prognosis. Now, follow‐up data to 13 years are available.
Using data from this well‐characterized cohort, we analyzed incidence of MF and LID and their baseline clinical and genetic risk factors, which, to our knowledge, have not been explored in population‐based cohorts followed for more than 10 years. In addition, we examined for the first time the association of MF and LID with later PD outcomes including dementia and death.
Methods
Subjects
Subjects were from the CamPaIGN cohort.12 Idiopathic PD was diagnosed using UK Parkinson's Disease Society Brain Bank (UKPDSBB) criteria. All cases were followed up at approximately 2‐year intervals. At the 3‐ to 4‐year point, diagnostic reevaluation was undertaken with repeated application of UKPDSBB criteria to maximize diagnostic accuracy.14 Of 142 patients included in the last published analysis at 10 year,16 1 was excluded because of a change in diagnosis (essential tremor and osteoarthritis); thus, 141 were included in this study. The study protocol was approved by the local ethics committee, and written informed consent was obtained from all subjects.
Assessments
At baseline and follow‐up, patients underwent detailed clinical and neuropsychological tests as previously reported.15 Disease severity was evaluated using the Unified Parkinson's Disease Rating Scale (UPDRS) and Hoehn and Yahr scale. The presence of MFs and LIDs was assessed using UPDRS section 4. Dementia was diagnosed according to Dianostic and Statistical Manual fourth edition criteria in patients with Mini‐Mental State Examination (MMSE) ≤ 24.
Genotyping was performed for candidate genes with potential prognostic significance including: MAPT H1 versus H2 haplotype, SNCA rs356219, APOE ε‐2/3/4 alleles, COMT val(158)met, BDNF val(66)met, and GBA sequencing for pathogenic variants.16, 17, 18, 19
Statistical Analysis
Time of PD diagnosis was defined as t = 0. Cumulative incidence of MFs and LIDs was calculated using Kaplan‐Meier survival analysis. Time of onset of MFs, LIDs, and dementia was calculated as the midpoint of the interval between the assessment at which the outcome was first recorded and the preceding assessment.
Cox regression analysis was used to investigate covariates that might influence development of MFs and LIDs. All demographic, clinical, and genetic covariates putatively relevant to outcome were evaluated using univariate analyses (Table 1). Multivariate analysis was then performed using covariates with an unadjusted P ≤ 0.1 and a backward stepwise approach. Influence of MFs and LIDs on mortality and risk of dementia was analyzed using similar univariate and multivariate Cox regression analyses with MFs and LIDs as time‐dependent covariates. Statistical analyses were performed using IBM SPSS version 22 or SAS.
Table 1.
Cox regression analysis of predictor variables for development of motor fluctuations and levodopa‐induced dyskinesias
| Motor fluctuations | Levodopa‐induced dyskinesias | |||||||
|---|---|---|---|---|---|---|---|---|
| Baseline variables | Unadjustedd HR (95% CI) | P | Adjustedd HR (95% CI) | P | Unadjustedd HR (95% CI) | P | Adjustedd HR (95% CI) | P |
| Sex | 0.955 | 0.149 | ||||||
| Smoking history | 0.609 (0.393–0.945) | 0.027 | 0.818 | |||||
| Years of education | 0.397 | 0.167 | ||||||
| Age at diagnosis | 0.548 | 0.109 | ||||||
| UPDRS3 | 1.020 (1.001–1.040) | 0.043 | 0.398 | |||||
| UPDRS total | 1.014 (1.000–1.028) | 0.054 | 1.020 (1.005–1.036) | 0.010 | 0.420 | |||
| UPDRS dopa‐resistanta | 0.327 | 0.755 | ||||||
| HY | 0.405 | 0.939 | ||||||
| MMSE | 1.155 (0.986–1.353) | 0.074 | 1.395 (1.095–1.776) | 0.007 | 1.509 (1.170–1.947) | 0.002 | ||
| Tower of London | 0.352 | 0.124 | ||||||
| Pentagon copying | 0.308 | 0.201 | ||||||
| Letter fluency (FAS) | 0.254 | 0.274 | ||||||
| Animal fluency | 0.928 | 1.034 (0.999–1.071) | 0.058 | |||||
| BDIb | 0.709 | 0.516 | ||||||
| Motor phenotype (PIGD/intermediate vs tremor‐dominant) | 0.586 (0.372–0.924) | 0.021 | 0.691 | |||||
| LEDDc at baseline | 1.001 (1.000–1.003) | 0.022 | 1.002 (1.000–1.004) | 0.018 | ||||
| Levodopa use at baseline | 0.113 | 0.218 | ||||||
| MAPT (H1/H1 vs H2 carrier), n = 130 | 0.192 | 1.000 | ||||||
| COMT val158met (Met carrier vs Val/Val), n = 128 | 0.230 | 0.166 | ||||||
| BDNF val66met (Met carrier vs Val/Val), n = 128 | 0.300 | 0.177 | ||||||
| SNCA rs356219 (A carrier vs GG), n = 124 | 1.808 (0.982–3.328) | 0.057 | 1.902 (1.034–3.499) | 0.039 | 0.907 | |||
| APOE (ε4 noncarrier vs ε4 carrier), n = 124 | 0.795 | 0.143 | ||||||
| GBA (mutation carrier vs wild type), n = 113 | 0.406 | 2.753 (0.942–8.04) | 0.064 | 4.497 (1.454–13.906) | 0.009 | |||
Scores for speech, posture, gait, postural stability, and rising from sitting.
Beck Depression Inventory.
Levodopa‐equivalent daily dose.
”Unadjusted” indicates univariate analysis, and “adjusted” indicates multivariate analysis.
Results
Patients were followed up for a maximum of 13 years. Mean follow‐up duration was 7.8 ± 3.5 years. Mean age at diagnosis was 70.2 ± 9.6 years. All patients received dopaminergic therapy during the course of follow‐up. Sixty‐nine were receiving treatment at baseline, of whom 42 were receiving levodopa. No patients had undergone deep brain stimulation or were receiving continuous infusion therapies during the follow‐up period. At 13 years, 61 of 141 patients were alive.
Risk Factors for MF and LID
Eighty‐three patients developed MF, with a cumulative incidence of 54.3% and 100% at 5 and 10 years, respectively. Median time to MF was 4.7 years (Fig. 1). The multivariate model revealed that higher baseline UPDRS‐total and the SNCA rs356219 A allele were independently associated with increased risk of MF (Table 1).
Figure 1.
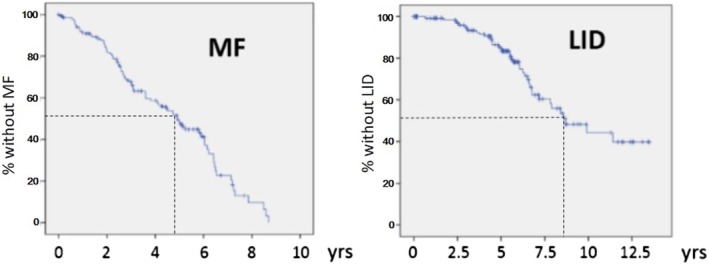
Kaplan‐Meier plots of survival to onset of motor fluctuations (MFs) and levodopa‐induced dyskinesias (LIDs). Dotted line denotes median time to outcome. [Color figure can be viewed at http://wileyonlinelibrary.com]
LID developed in 39 patients, with a cumulative incidence of 14.5% and 55.7% at 5 and 10 years, respectively. Median time to LID was 8.7 years (Fig. 1). Multivariate analysis showed that higher MMSE at baseline and carrying a GBA mutation were independently associated with increased risk of LID (Table 1).
MF and LID as Prognostic Factors
In univariate analyses, both MF and LID were independently associated with reduced mortality (unadjusted HR, 0.239; 95% CI, 0.133–0.430; P < 0.001 for MF; unadjusted HR, 0.355, 95% CI, 0.186–0.676; P = 0.002 for LID). Multivariate analysis adjusting for covariates independently associated with mortality (diagnosis age, UPDRS‐total, baseline levodopa use, smoking) showed similar results (adjusted HR, 0.259; 95% CI, 0.136–0.493; P < 0.001 for MF; adjusted HR, 0.426; 95% CI, 0.210–0.865; P = 0.018 for LID). Stratification by age at diagnosis revealed associations with lower mortality reached significance only in patients over the cohort median of 70 (unadjusted HR, 0.225; 95% CI, 0.114–0.442; P < 0.001 for MF; unadjusted HR, 0.296; 95% CI, 0.121–0.724; P = 0.008 for LID). Neither MF nor LID was associated with risk of dementia.
Discussion
This long‐term study in an incident population‐based cohort provides “real‐world” data for the frequency of motor complications in a typical PD population throughout the course of the disease and indicates that at 10 years virtually all surviving patients with PD have developed MF, whereas only 56% experience LID.
Previous population‐based prospective cohort studies have only provided data to 5 years.9, 10 The cumulative incidence of MF at 5 years was higher, at 54.3%, in our study compared with those studies showing 43% and 23%, which may reflect differences in the population, treatment approaches, and data collection methods. Our high incidence of MF at 5 years appears at odds with the classical view that MF is associated with advanced PD. However, studies have shown that MF can occur as early as 6 months from levodopa initiation,2, 20 and 1 study showed that 25% of patients initially treated with dopaminergic agonists and 43% of patients initially treated with levodopa developed MF in 2 years.21 At 10 years, the cumulative incidence of MF was 100% in our cohort. The Sydney Multicentre Study22 similarly reported that all surviving patients had MF at 20 years, supporting our finding that virtually all patients eventually develop MF. An important clinical implication of this is that almost all patients still benefit from dopaminergic treatment in the long term, as evidenced by the presence of MF, and this should be considered when adjusting medications. However, it remains possible that there is a subgroup that does not benefit from levodopa but is censored from analysis at an earlier stage because of death or withdrawal.
It has previously been reported that up to 80% of patients develop LID within 5 years of diagnosis, and 80%–90% of patients experience LID after 10 years,1, 3, 23 but these estimates come from studies of selected cohorts. The older average age of our cohort may be relevant, as patients with a younger age at onset are more likely to develop LID.10, 24, 25 Our 10‐year data are comparable to data from previous retrospective community‐based studies with a similar age at onset,7, 8 reporting that 53%–59% had LID at 10 or more years. However, the reason for the low cumulative incidence (14.5%) of LID at 5 years, when compared with previously mentioned population‐based prospective cohort studies with a similar age at onset (cumulative incidence at 5 years 24% and 30%9, 10), is not clear.
In agreement with other studies,5, 9 our data show MFs are associated with increased baseline disease severity, but in contrast with some previous reports,2, 5, 7 we did not find an association with levodopa use at baseline or levodopa‐equivalent daily dose26 in the multivariate analysis. This indicates that early levodopa is not a major factor in driving earlier motor complications, in keeping with studies comparing motor complications in subgroups of patients with differing baseline treatment approaches (levodopa versus dopamine agonists versus no treatment).27 This supports the general shift in opinion favoring earlier rather than delayed onset of levodopa treatment.28
Higher MMSE at baseline was associated with earlier LID in multivariate analysis, which is a novel finding, but previous studies have not specifically examined the relationship between cognition and LID, to our knowledge. A plausible explanation for this finding is that patients developing early LID represent a subgroup of highly levodopa‐responsive patients in whom pathology is relatively confined to the nigrostriatal system, with minimal extrastriatal and cortical involvement. The observation that LID and MF are not associated with earlier dementia (despite association with disease severity) provides further support for this hypothesis. However, it also is possible that this observation could be influenced by a tendency to use lower medication doses in individuals with cognitive impairment.
Association of the SNCA rs356219 A allele and MF is an unexpected finding because this variant has been associated with reduced PD susceptibility and older age at onset,29, 30, 31 as well as reduced plasma α‐synuclein levels in PD.32 Our observed association may reflect less severe diffuse α‐synuclein pathology in levodopa‐responsive patients who develop early MF, but it is a finding that requires further replication in an independent cohort.
We observed an increased LID risk in GBA mutation carriers, in line with reports that GBA mutations are associated with more rapid motor progression,19, 33 earlier age at onset,33, 34, 35 and possibly earlier DBS surgery.36 GBA‐PD is also generally associated with rapid cognitive decline and earlier dementia,19, 33, 34 whereas we found higher baseline MMSE also increased LID risk. However, longitudinal studies have shown that GBA status does not determine cognitive status at baseline,19, 34, 35 and we propose that the GBA association with LID is driven by the more aggressive pathology that tends to characterize this condition.
Another novel finding is that MF and LID were associated with reduced mortality in patients >70 at diagnosis, suggesting that levodopa responsiveness predicts better prognosis in older patients. Levodopa‐responsive symptoms are associated with presynaptic nigrostriatal dopaminergic loss,37, 38 whereas mortality in advanced disease likely results from pathology outside the nigrostriatal system.16 This suggests that severity of motor dysfunction at baseline is not necessarily a poor prognostic feature, if the motor deficits are treatment responsive.
In conclusion, our findings are in keeping with the idea that the timing of motor complications reflects the severity of pathology in the dopaminergic nigrostriatal system rather than levodopa use at baseline.39 We propose that idiopathic PD patients are on a spectrum from those with disease predominantly confined to the nigrostriatal system, with more prominent motor symptoms at baseline and earlier motor complications, but generally preserved cognition and prolonged survival to those with diffuse α‐synuclein pathology with less treatment responsiveness and less motor complications but more cognitive impairment and reduced survival. Future work is required to develop prognostic models to predict motor complications at the individual patient level.
Authors’ Roles
Dr. H. J. Kim: drafting the manuscript, study concept and design, analysis and interpretation of data, statistical analysis.
Dr. Sarah Mason: acquisition of data, critical review and revising the manuscript.
Dr. Thomas Foltynie: acquisition of data, critical review and revising the manuscript.
Dr. Sophie Winder‐Rhodes: acquisition of data, critical review and revising the manuscript.
Dr. Roger A. Barker: acquisition of data, critical review and revising the manuscript, study set up and supervision.
Dr. Caroline H. Williams‐Gray: study concept and design, acquisition of data, analysis and interpretation of data, critical review and revising the manuscript, study supervision.
Financial Disclosures of all authors (for the preceding 12 months)
Dr. H. J. Kim: Employment at Seoul National University Hospital; travel grants from International Parkinson and Movement Disorder Society, Korean Movement Disorder Society; research grants from Seoul National University Hospital, New York University.
Dr. Sarah Mason: employment at Huntington's disease Association and the NIHR Cambridge Biomedical Research Centre awarded to Addenbrooke's Hospital.
Dr. Thomas Foltynie: grant funding from Michael J. Fox Foundation, National Institute of Health Research, John Black Charitable Foundation, Cure Parkinson's Trust and Innovate UK; honoraria for speaking at meetings sponsored by Boston Scientific, Bial, Profile Pharma.
Dr. Sophie Winder‐Rhodes: employment at NHS clinical.
Dr. Roger A Barker: grants from the MRC, Wellcome Trust, Cure Parkinson's Trust, Parkinson's UK, Birax, Huntington's Disease Association and Rosetrees Trust; royalties from Springer Nature and Wiley; consulting fees from UCB, LCT, Oxford Biomedica, Cellular Dynamics International, Cellino, Nova Nordisk, and F‐prime.
Dr. Caroline H. Williams‐Gray: salary funded by an MRC Clinician Scientist Fellowship; additional grant funding from the Rosetrees Trust, the Evelyn Trust, the Michael J. Fox Foundation, Addenbrooke's Charitable Trust, and Parkinson's UK; consultancy work for Modus Outcomes.
Acknowledgments
The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. We appreciated the statistical advice from the Medical Research Collaborating Center at the Seoul National University Hospital and the Seoul National University College of Medicine.
Relevant conflicts of interest/financial disclosures: C.W.G. is supported by a Medical Research Council Clinician Scientist Fellowship. R.A.B. is an NIHR Senior Investigator.
Funding agencies: This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013‐R1A1A2010499). The CamPaIGN study was supported by the Wellcome Trust, the Medical Research Council, the Patrick Berthoud Trust, Parkinson's UK, and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre Dementia and Neurodegeneration Theme (Grant Reference Number 146281).
Contributor Information
Han‐Joon Kim, Email: movement@snu.ac.kr.
Caroline H. Williams‐Gray, Email: chm27@cam.ac.uk.
References
- 1. Ahlskog JE, Muenter MD. Frequency of levodopa‐related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001;16:448–458. [DOI] [PubMed] [Google Scholar]
- 2. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson's disease. N Engl J Med 2004;351(24):2498–2508. [DOI] [PubMed] [Google Scholar]
- 3. Hauser RA, Rascol O, Korczyn AD, et al. Ten‐year follow‐up of Parkinson's disease patients randomized to initial therapy with ropinirole or levodopa. Mov Disord 2007;22:2409–2417. [DOI] [PubMed] [Google Scholar]
- 4. Nicoletti A, Mostile G, Nicoletti G, et al. Clinical phenotype and risk of levodopa‐induced dyskinesia in Parkinson's disease. J Neurol 2016;263:888–894. [DOI] [PubMed] [Google Scholar]
- 5. Olanow CW, Kieburtz K, Rascol O, et al. Factors predictive of the development of Levodopa‐induced dyskinesia and wearing‐off in Parkinson's disease. Mov Disord 2013;28:1064–1071. [DOI] [PubMed] [Google Scholar]
- 6. Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE‐PD study. Ann Neurol 2010;68:18–27. [DOI] [PubMed] [Google Scholar]
- 7. Schrag A, Quinn N. Dyskinesias and motor fluctuations in Parkinson's disease: A community‐based study. Brain 2000;123:2297–2305. [DOI] [PubMed] [Google Scholar]
- 8. Van Gerpen JA, Kumar N, Bower JH, Weigand S, Ahlskog JE. Levodopa‐associated dyskinesia risk among Parkinson disease patients in Olmsted County, Minnesota, 1976‐1990. Arch Neurol 2006;63:205–209. [DOI] [PubMed] [Google Scholar]
- 9. Bjornestad A, Forsaa EB, Pedersen KF, Tysnes O‐B, Larsen JP, Alves G. Risk and course of motor complications in a population‐based incident Parkinson's disease cohort. Parkinsonism Relad Disord 2016;22:48–53. [DOI] [PubMed] [Google Scholar]
- 10. Scott N, Macleod A, Counsell C. Motor complications in an incident Parkinson's disease cohort. Eur J Neurol 2016;23:304–312. [DOI] [PubMed] [Google Scholar]
- 11. Turcano P, Mielke MM, Bower JH, et al. Levodopa‐induced dyskinesia in Parkinson disease: A population‐based cohort study. Neurology 2018;91:e2238–e2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foltynie T, Brayne CE, Robbins TW, Barker RA. The cognitive ability of an incident cohort of Parkinson's patients in the UK. The CamPaIGN study. Brain 2004;127:550–560. [DOI] [PubMed] [Google Scholar]
- 13. Evans JR, Mason SL, Williams‐Gray CH, et al. The natural history of treated Parkinson's disease in an incident, community based cohort. J Neurol Neurosurg Psychiatry 2011;82:1112–1118. [DOI] [PubMed] [Google Scholar]
- 14. Williams‐Gray C, Foltynie T, Brayne C, Robbins T, Barker R. Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain 2007;130:1787–1798. [DOI] [PubMed] [Google Scholar]
- 15. Williams‐Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow‐up of the CamPaIGN cohort. Brain 2009;132:2958–2969. [DOI] [PubMed] [Google Scholar]
- 16. Williams‐Gray CH, Mason SL, Evans JR, et al. The CamPaIGN study of Parkinson's disease: 10‐year outlook in an incident population‐based cohort. J Neurol Neurosurg Psychiatry 2013;84:1258–1264. [DOI] [PubMed] [Google Scholar]
- 17. de Lau LM, Verbaan D, Marinus J, Heutink P, van Hilten JJ. Catechol‐O‐methyltransferase Val158Met and the risk of dyskinesias in Parkinson's disease. Mov Disord 2012;27:132–135. [DOI] [PubMed] [Google Scholar]
- 18. Foltynie T, Cheeran B, Williams‐Gray CH, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson's disease. J Neurol Neurosurg Psychiatry 2009;80:141–144. [DOI] [PubMed] [Google Scholar]
- 19. Winder‐Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain 2013;136:392–399. [DOI] [PubMed] [Google Scholar]
- 20. Fahn S, Group PS . Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol 2005;252:iv37–iv42. [DOI] [PubMed] [Google Scholar]
- 21. Group PS . Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. JAMA 2000;284:1931–1938. [DOI] [PubMed] [Google Scholar]
- 22. Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord 2008;23(6):837–844. [DOI] [PubMed] [Google Scholar]
- 23. Bastide MF, Meissner WG, Picconi B, et al. Pathophysiology of L‐dopa‐induced motor and non‐motor complications in Parkinson's disease. Prog Neurobiol 2015;132:96–168. [DOI] [PubMed] [Google Scholar]
- 24. Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five‐year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. New Engl J Med 2000;342:1484–1491. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y‐h, Tang B‐s, Song C‐y, et al. The relationship between the phenotype of Parkinson's disease and levodopa‐induced dyskinesia. Neurosci Lett 2013;556:109–112. [DOI] [PubMed] [Google Scholar]
- 26. Brodsky MA, Godbold J, Roth T, Olanow CW. Sleepiness in Parkinson's disease: a controlled study. Mov Disord 2003;18:668–672. [DOI] [PubMed] [Google Scholar]
- 27. Katzenschlager R, Head J, Schrag A, Ben‐Shlomo Y, Evans A, Lees A. Fourteen‐year final report of the randomized PDRG‐UK trial comparing three initial treatments in PD. Neurology 2008;71:474–480. [DOI] [PubMed] [Google Scholar]
- 28. Fox SH, Lang AE. ‘Don't delay, start today’: delaying levodopa does not delay motor complications. Brain 2014;137:2628–2630. [DOI] [PubMed] [Google Scholar]
- 29. Campêlo CL, Cagni FC, de Siqueira Figueredo D, et al. Variants in SNCA Gene Are Associated with Parkinson's Disease Risk and Cognitive Symptoms in a Brazilian Sample. Front Aging Neurosci 2017;9:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Han W, Liu Y, Mi Y, Zhao J, Liu D, Tian QB. Alpha‐synuclein (SNCA) polymorphisms and susceptibility to Parkinson's disease: A meta‐analysis. Am J Med Genet Part B 2015;168B:123–134. [DOI] [PubMed] [Google Scholar]
- 31. Mata IF, Yearout D, Alvarez V, et al. Replication of MAPT and SNCA, but not PARK16‐18, as susceptibility genes for Parkinson's disease. Mov Disord 2011;26:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mata IF, Shi M, Agarwal P, et al. SNCA variant associated with Parkinson disease and plasma α‐synuclein level. Arch Neurol 2010;67:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol 2016;80:674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lunde KA, Chung J, Dalen I, et al. Association of glucocerebrosidase polymorphisms and mutations with dementia in incident Parkinson's disease. Alzheimers Dement 2018;14:1293–1301. [DOI] [PubMed] [Google Scholar]
- 36. Pal GD, Hall D, Ouyang B, et al. Genetic and Clinical Predictors of Deep Brain Stimulation in Young‐Onset Parkinson's Disease. Mov Disord Clin Pract 2016;3:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chung SJ, Lee Y, Oh JS, Kim JS, Lee PH, Sohn YH. Putaminal dopamine depletion in de novo Parkinson's disease predicts future development of wearing‐off. Parkinsonism Relat Disord 2018;53:96–100. [DOI] [PubMed] [Google Scholar]
- 38. Hong JY, Oh JS, Lee I, et al. Presynaptic dopamine depletion predicts levodopa‐induced dyskinesia in de novo Parkinson disease. Neurology 2014;82:1597v1604. [DOI] [PubMed] [Google Scholar]
- 39. Cilia R, Akpalu A, Sarfo FS, et al. The modern pre‐levodopa era of Parkinson's disease: insights into motor complications from sub‐Saharan Africa. Brain 2014;137:2731–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]