

Abstract
Obesity is a neurological disorder which operates by favoring energy storage within adipose depots and increased caloric intake. Most cases of human obesity are acquired without any underlying genetic basis. We suggest that obesity can impair the function of some hypothalamic neurons critical to body weight regulation. Genetic ablation of the retinoblastoma gene within pro-opiomelanocortin neurons leads to death of the neurons and subsequent obesity. The retinoblastoma protein (pRb), a key inhibitor of the cell cycle, can also be inactivated by CDK-mediated phosphorylation. Extensive development led to the production of FDA-approved CDK4/6 inhibitors. Based on our own results, we propose that maintaining or re-instating pRb function using CDK4/6 inhibitors are potentially effective treatments of diet-induced obesity
Keywords: Cell cycle, Diet-induced Obesity, Hypothalamus
Diet-induced obesity: an incomplete picture
Obesity is one of the most prevalent threats to human health: the current adult obesity rate in developed countries is 19.5%, with a staggering 38.2% obesity rate in the United States alone [1]. New insights into the etiology of this pandemic are desperately required in order to introduce successful therapeutic options in the near future. In the twenty-five years since the discovery of the “satiety hormone” leptin [2, 3], work in the field has progressively identified key cellular and molecular determinants of energy balance regulation. However, early studies in obese humans [4] found no evidence to support any obvious genetic defects in the leptin pathway, a finding which still holds true in recent high throughput genetic screens [5, 6] With the molecular etiology of obesity remaining unclear in the context of these genetic studies, it has been hypothesized that the rising prevalence of obesity in the developed world can be largely attributed to environmental factors such as decreased energy expenditure and increased availability of calorie-dense foods. The most widely used laboratory rodent model of leptin-replete obesity utilized the administration of a high-fat diet (HFD, in which 60% kilocalorie content was derived from fat), and was subsequently characterized as diet-induced obesity (DIO) [7]. However, early work to determine the etiology of DIO reached contradictory conclusions. Several studies reported a phenomenon of HFD-induced hyperphagia, in which HFD was shown to either increase overall caloric intake, directly contributing to obesity due to an increase in positive energy balance [8, 9]. However, a near equivalent number of studies reported a complete absence of this hyperphagic phenotype during HFD feeding [10, 11]. Molecular genetic studies have reached similarly contradictory conclusions—early studies of HFD effects on the hypothalamus showed a modest HFD-induced decrease of LEPR-B (B isoform of leptin receptor) expression in the mediobasal hypothalamus (MBH) of C57BL/6J mice, proposing a “decreased available receptor quantity” model of leptin resistance [12]. However, more recent studies of the effects of DIO on differential gene expression in global hypothalamic neurons [13] and specifically anorexigenic POMC neurons [14] reached the opposite conclusion, demonstrating either no gene expression changes [13], or a modest HFD-mediated increase in both Lepr-b and Socs3 expression [14]—both suggesting the HFD-mediated “leptin resistance” in MBH neurons cannot be attributed to lower leptin receptor expression level. One of the most striking conclusions of these studies was that “leptin sensitivity” could be functionally restored in DIO mice by simply switching from HFD to standard chow diet, with returning levels of leptin sensitivity directly correlated to loss of fat mass [13].
Pathophysiology of HFD-induced damage to energy balance neurons
The mediobasal hypothalamus is a key central regulator of energy balance and comprises the arcuate nucleus (ARC) and the ventromedial nucleus (VMN). The ARC contains two antagonistic neuron populations, the anorexigenic pro-opiomelanocortin (POMC) and the orexigenic agouti-related peptide/Neuropeptide Y (AgRP/NPY) neurons [15]. Elevated leptin levels during a positive energy state simultaneously activates POMC neurons and inhibits AGRP neurons though the leptin receptor (LEPR). A physiologically appropriate response in these two cell types to leptin signaling is critical for maintaining energy balance [16, 17], and thus, there has been much speculation around their sensitivity to changes in dietary intake. Moreover, ARC neurons are anatomically adjacent to the Median Eminence (ME), the inferior boundary of the hypothalamus and site of hormone release into the hypophyseal portal system [18], making it one of areas of the brain devoid of a blood-brain barrier with ME-adjacent neurons being accesssible to circulating factors. Indeed, it has been shown that parenteral administration of agents such as monosodium glutamate can result in the selective destruction of ARC neurons due to direct exposure [19]. Additional relevant studies have shown that HFD leads to increased serum free fatty acids (FFA), which can be directly sensed by hypothalamic neurons [20]. Integrating these studies suggests that ARC-ME neurons may be selectively vulnerable to changes in serum factors in response to HFD intake. Indeed, it has been shown that HFD intake leads to an acute rise in both inflammatory mediators and gliosis markers in the rodent ARC in response to only several days exposure to HFD—well before the onset of obesity; the same study demonstrated an increased level of gliosis in the MBH of live obese humans [21]. Further studies in the role of neural inflammation in DIO demonstrate that activation of hypothalamic inflammatory pathways such as inhibitor of nuclear factor-κβ kinase-(IKKβ) or nuclear factor-κβ (NF-κβ) promotes a rapid hyperphagic response and weight gain, and effectively desensitizes the leptin responsiveness of MBH neurons [22]. Additional studies confirmed this effect was mediated by high fat diet intake (specifically elevated fat macronutrient composition and not caloric content) and reversing the hypothalamic inflammation subsequently restored central leptin and insulin responses—again indicating the existence of a direct and reversible HFD-neural injury pathway [23]. An alternate line of studies has shown hypothalamic neurons experience an endoplasmic reticulum (ER) mediated stress response during obesity, and that the reversal of ER stress can restore leptin sensitivity in DIO mice [24]. The implications of the ER-stress model are in part concordant with the inflammatory/gliosis model, in that both demonstrate leptin-independent pathways of MBH neuron damage brought on by environmental models of obesity.
Cell Cycle Events as a Biomarker of Neuronal Damage
Postulating that DIO can be the result of direct MBH neuronal injury underlies two key unknowns to the translational significance of this hypothesis: what the molecular basis of neuronal injury is, and how it can be targeted for reversal in obese patients. Recent work proposes cell cycle regulation (Box 1) as both a biomarker and druggable target for the neuronal damage causal to obesity, demonstrating unexpected sites of de novo neurogenesis, aberrant neuronal cell-cycle reentry and DNA replication, and dysregulation in neurons in response to a variety of toxic environmental stimuli.
An overview of the cell cycle.
The eukaryotic cell cycle is divided into two major phases: the mitotic state of cell division, and interphase. Interphase is further subdivided in three stages: G1, when the proteins responsible for DNA replication are synthesized, S phase, when nuclear DNA is replicated, and G2, when the proteins responsible for cell division are synthesized. Progression through interphase is tightly regulated by a class of proteins known as cyclins and partner cyclin-dependent kinases (CDKs): Cyclin D is synthesized at the beginning of G1, where it activates CDK4/6 to phosphorylate retinoblastoma protein (pRb) and activate the transcription factor E2F1, which induces the synthesis of the proteins necessary for DNA replication[42]. Full progression to S phase and DNA replication is completed via the successive associations of cyclin E and cyclin A to CDK2, which phosphorylate additional residues of pRb[43]. A cyclin A/CDK1 complex activates late replication origins during G2 phase, inducing thecondensation of chromosomes prior to mitotic entry [44]. An additional class of regulatory proteins counteracts cyclin/complexes to negatively regulate cell cycle and are accordingly called Cyclin Kinase Inhibitors (CKIs); there are two major classes of CKIs, namely the Ink and Cip/Kip families. The Ink family proteins p15Ink4b, p16Ink4a, p18Ink4c, and p19Ink4d are the major regulators of entry to G1 state: they actively bind CDK4/6 and block cyclin D association, preventing pRb phospho-inactivation and E2F target gene expression [45]. The Cip/Kip family of p21Cip1, p27Kip1, and p57Kip2proteins function on a more global scale throughout interphase, and are able to inhibit multiple CDK/cyclin complexes during interphase [46]. Interestingly, a population of this progenitor population is found in the MBH, denoted hypothalamic neural stem cells (htNSCs), suggesting that there may be de novo neurogenesis in the MBH in response to environmental stimuli or in order to accommodate neuronal turnover [47].
While entry from interphase into mitosis is required for the propagation of stem/progenitor cells such as hypothalamic neural stem cells (htNSCs), most fully-differentiated cells are not destined for any further self-replication and are termed “post-mitotic”. Adult neurons are the archetypical example of this post-mitotic cell fate, which is an interphase state of cell cycle withdrawal known as G0 [25]. As such, a widely held belief is that adult neurons do not have the ability to reenter cell cycle, and thus cannot self-replicate. However, a substantial body of recent work has demonstrated nuances in the permanence of the neuronal G0 state, particularly during states of cellular stress or injury [26]. A prevailing hypothesis in this area is that neuronal quiescence is not a terminal fate per se, but rather a state of constant vigilance that is actively regulated by cyclin kinase inhibitors (CKIs) and other intracellular factors; “relaxation” of this vigilance leads to cell-cycle reentry, which can manifest as neuronal dysfunction along a continuum of severity, and can eventually result in cell death if not reversed [27]. Factors which have been shown to induce neuronal cell cycle reentry include DNA damage, which leads to induction of CDK4/cyclin D and eventual neuronal death, but can be rescued with constitutively active CKIs p16, p21, and p27 [28]. Oxidative stress has been additionally implicated as an environmental factor which can dysregulate neuronal cell cycle, and in fact has been repeatedly shown to be a pathology which precedes neuronal apoptosis in several human neurodegenerative diseases [29]. These cell cycle reentry events are usually catastrophic and irreversible, and lead to activation of an E2F1 mediated apoptosis pathway directly after G1 entry, termed “abortive cell cycle reentry” [30, 31].
Interestingly, emerging research indicates that not all neuronal cell-cycle reentry events are unproductive and fated for apoptosis: evidence shows neurons can successfully undergo S phase and exist in a tetraploid state of stable pre-mitotic G2, although it is unclear what degree pre-replication neuronal function remains. On one end of the spectrum, tetraploidy in a terminally-differentiated neuron forecasts inescapable cell death; for example, hypoxic-ischemic stress leads to cell-cycle reentry and completion of S phase in rodent hippocampal neurons, but these neurons rapidly undergo apoptosis soon after [32]. On the other end of the spectrum, there is evidence of stable somatic tetraploidy in fully functional adult neurons, such as retinal ganglion cells induced to re-enter cell cycle using nerve growth factor. In this scenario tetraploidy creates no measurable detriment on cell function, and these cells exist in their functional tetraploid state throughout adulthood [33]. For the most part, the majority of identified tetraploid adult neurons seem to have some degree of either functional deficit or increased vulnerability.
Using cell cycle reentry as a disease hallmark in neurodegenerative disease not only underscores the importance of cell cycle vigilance in neuron function, but also uncovers possible therapeutic options towards restoring neuron function. Similar to AD patients, a certain degree of cell cycle reentry is seen in the dysfunctional neurons of PD patients as well, as evidenced by DNA content, pRb phospho-inactivation, and E2F1 target gene expression [34]. Remarkably, small-molecule inhibition of CDK4 by the pan-CDK inhibitor flavopiridol has been shown to have neuroprotective effects in PD models, and even attenuate apoptosis in re-entrant neurons [35]. This provides compelling evidence that while cell cycle reentry is catastrophic for neuron function, this process can be targeted by drug therapy and may be reversible.
As cell cycle reentry has been shown to be a cause of neuron functional failure, in the context of DIO, the question again arises if elevated dietary fat intake can be a trigger for this damaged neuronal state. As discussed earlier, elevated dietary fat intake can certainly trigger neuronal stress states through inflammatory and other mitogenic pathways. Thus, whether elevated dietary fat intake can generate enough neuronal stress to result in a cell cycle reentry event is by no means a far-reaching speculation. However, there is scant evidence that high fat feeding leads to active proliferation of neurons. Rather, a more molecularly focused aspect of our hypothesis is the induction of E2F target gene expression that could generate aberrant neuronal functions. It is likely that there may be a small suite of genes, which remain to be identified, that fit this description.
Studies which have attempted to look at the role of HFD on the MBH have shown a direct suppression of htNSC neurogenesis in response to HFD, which was phenocopied in ob/ob mice, demonstrating a htNSC replication deficiency may in part be causal to obesity [36]. A similar study which further integrates the HFD-mediated neuron inflammatory response with HFD effects on htNSCs replicated the finding that chronic HFD administration leads to both the depletion and neurogenic impairment of htNSCs, but went on to directly link this phenomenon to selective IKKβ/NF-κB activation mediated apoptosis in the htNSCs population [37]. However, not all studies in this area have had concordant results: another line of work looking into the role of htNSCs in the ARC-ME found that while neurogenesis was in fact sensitive to dietary intake, HFD feeding actually increased neurogenesis in this area; furthermore, selectively inhibiting neurogenesis in the ventrobasal hypothalamus prevents HFD-induced weight gain [38]. This may suggest that elevated MBH neurogenesis alters the ratio of positive to negative energy balance neurons in an obesity-promoting manner. In short, it remains unclear whether HFD induces activation or prevention of neurogenesis, as does the role of hypothalamic neurogenesis in DIO.
Together, these studies suggest HFD-mediated cell cycle injury in the MBH may be a “two-prong” effect: (A) adult post-mitotic MBH neurons experience HFD-mediated stress, initiating a transcriptional response triggered by activated E2Fs, compromising neuron function, and possibly slowly undergoing apoptosis, while the (B) htNSC population, which would otherwise replace this depleted pool of adult MBH neurons, experiences impaired neuronal differentiation due to HFD. Although it is unclear whether HFD-related neurogenesis protects against or accelerates DIO, it is clear that altered dietary intake can certainly perturb the baseline htNSC replication dynamic. Combined, these events alter the functional population of MBH neurons leading to a deficiency in melanocortinergic transmission and DIO.
The retinoblastoma protein pathway as a druggable target for DIO
The effect of HFD on adult ARC-ME neuron cell cycle remains unclear. One of the earliest reports of the effects of HFD on adult ARC-MRE demonstrated that chronic HFD feeding leads to phosphorylation of pRb (Box 2) in the MBH (including in POMC neurons), and that these neurons re-entered cell cycle as measured by E2F1 target gene expression; pRb’s role as an “obesity suppressor” made it critical in POMC neurons but dispensable in AgRP neurons [39]. Interestingly, pRb is not the only cell cycle regulator affected by HFD in the MBH. A very recent study demonstrated that the tumor suppressor p53 also plays a major role in the prevention of DIO—the loss of p53 function specifically in AgRP neurons increases vulnerability to DIO by increasing food intake during HFD feeding, suggesting that p53 is required for feeding adaptations against DIO [40]. It is interesting to note that while these antagonistic neuron populations can both demonstrate compromised function in response to HFD-induced cell cycle stress, they have a differential preference between p53 and pRb as their respective master regulators.
The retinoblastoma protein.
Whether cell cycle progression is occurring in adult or progenitor neuron populations, the master regulator of this process is both cell types is the retinoblastoma protein (pRb). The core function of pRb is to bind and repress the E2F family of transcription factors, preventing cell cycle progression into S phase [48]. The bi-allelic loss of pRb inappropriately accelerates cell proliferation, and as expected, leads to tumorigenesis in several tissue types, including the eponymous retinoblastoma [49]. pRb is additionally often cited as the classical examples of a “two-hit tumorigenesis” model: while pRb is haplosufficent to prevent tumorigenesis in heterozygous individuals, a second genetic insult to the functional copy of RB1 (or loss of heterozygosity), statistically pre-disposes heterozygotes to developing early retinoblastoma, as well as other cancers in later life [50]. Finally, pRb function can be blocked at the protein level to drive tumorigenesis. Several oncovirus gene products directly target pRb, most often through direct binding and degradation of pRb; one of the most well studied clinical examples of this phenomenon is the human papillomavirus (HPV) protein E7 [50]. The second method by which “genetically intact” pRb can be functionally inactivated is through phosphorylation: pRb is an extensively phospho-regulated protein, as discussed below.
There are 15 known consensus phospho-acceptor sites for CDK-Cyclin mediated phosphorylation (S/TPxK/R motif) in pRb [51-53]. A recent study used 2-D isoelectric focusing electrophoresis to reveal the relationship between extent of pRb phosphorylation and its functional inactivation. When quiescent fibroblasts were stimulated to re-enter cell cycle to early and mid G1 pRb became mono-phosphorylated on any one of the 15 consensus S/Ts by cyclin D/CDK4. Mono-phosphorylated pRb remained bound to E2F but allows cyclin E/CDK2 to phosphorylate a total of 12 or more of the 15 consensus S/Ts. This degree of hyper-phosphorylation inactivates pRb to activate E2F for target gene expression and completes entry into S phase [54]. Thus CDK4-cyclin D can initiate pRb phosphorylation, but functional inactivation requires hyper-phosphorylation of at least 12 CDK consensus S/Ts. Consequently, this hyper-phosphorylation cascade can be prevented by CDK4 blockade, establishing a “druggable” target for aberrant pRb-inactivated cell-cycle reentry.
A report [41] directly tested the possibility of reversing/preventing diet induced obesity from HFD using two methods: A) expression of a non-phosphorylable pRb in which 18 potential phosphorylable Ser/Thr residues had been altered to alanine and B) treatment with a CDK4/6 inhibitor (Box 3) to prevent pRb inactivation. Virally delivered expression of the nonphosphorylable pRb, within the mediobasal hypothalamus or within ARC POMC neurons only, prevented excess fat mass gain in C57BL/6J mice on HFD. Abemaciclib, a CDK4/6 inhibitor, was effective in both preventing and reversing HFD-induced obesity with a significant reduction of fat content without affecting fat-free mass. Significantly, control animals on the same high fat diet that underwent a similar degree of weight reduction by food restriction exhibited loss of lean mass and fat mass, suggesting that inhibition of CDK4/6 promoted lipolysis as the principal mode for weight loss. Another significant finding is that DIO is reversible with abemaciclib treatment, suggesting that the energy homeostatic system is not permanently damaged and could be rendered functional with appropriate treatment. A final finding was that mice on normal chow were not affected by abemaciclib, supporting the notion that weight loss due to CDK4/6 inhibition was due to normalization of the function of the weight-regulatory system rather than being a pharmacological effect of drug treatment. These findings are in contrast to the mouse model wherein Rb deletion in POMC neurons led to irreversible loss of POMC neurons. The differences between the models could be due to influences of Rb deletion during neuronal development or incomplete inactivation of pRb by CDK4/6. Further studies with long term feeding of the HFD will be needed to determine if POMC neurons are lost in significant numbers and whether pRb is involved in the process.
Cyclin-dependent kinase inhibitors.
In tumorigenesis, pRb can be genetically unaltered but functionally inactivated by hyper-phosphorylation by CDKs. This aberrant CDK activity is usually secondary to a genetic loss of a CKI such as p16 or p21 in cancer cells [55].pRb phosphorylation is initiated by cyclin D/Cdk4 and completed by cyclin E or A/CDK2 or 1 [51-53]. As such, a modern cancer therapy rationale has been to directly block the activity of CDKs, to “re-activate” pRb in tumor cells and prevent tumorigenic cell-cycle activity [56]. While early pan-CDK inhibitors showed serious side effects, CDK4/6 selective inhibitors have shown remarkable efficacy in treating cancers with WT RB1 genetic background [56, 57], indicating pRb can be functionally reactivated by CDK4/6 inhibition. The FDA approved CDK4/6 inhibitor Palbociclib for treatment of ER+/HER2−RB1wt breast cancer in 2015, and new inhibitors Ribociclib and Abemaciclib in 2017 [57, 58].
Concluding Remarks
We have provided a framework for approaching obesity as an environmentally induced disorder of cell cycle control in hypothalamic neurons. The identification of phospho-inactivated retinoblastoma protein in these neurons implicates cyclin dependent kinases as instigators of the process leading to neuronal dysfunction. As pRb is turned over, inhibition of CDKs leads to the reinstatement of pRb function and potential reversal of hyperphagia and obesity. The further exploration of fundamental cellular mechanisms in regulating neuron health will provide insights to novel therapeutic modalities for obesity and other metabolic disorders.
Figure 1. High Fat Feeding Triggers An E2F Transcriptional Program Leading to POMC Neuronal Dysfunction.
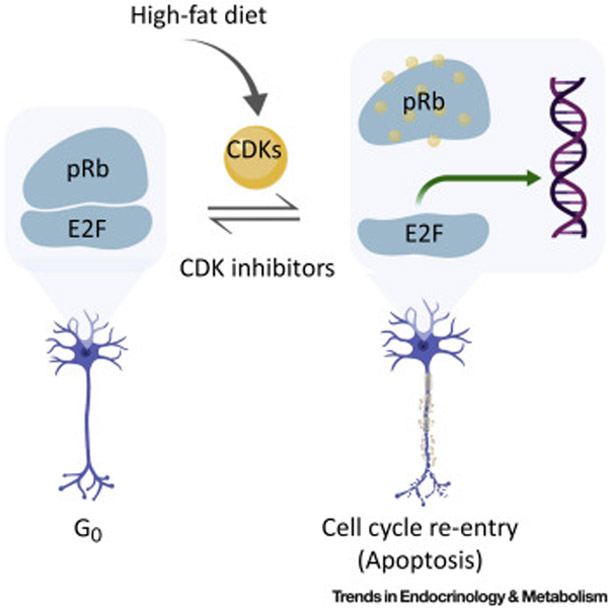
Mature neurons are mitotically quiescent, due to active repression of E2Fs by pRb. Phospho-inactivation of pRb by cyclin dependent kinases (CDK2/CDK4/CDK6) releases E2Fs to find their target genes and initiates cell cycle re-entry. Neurons in most cases appear to be intolerant of expression of some E2F target genes and become dysfunctional. The pRb in hypothalamic POMC neurons is susceptible to CDK-mediated phospho-inactivation, leading to hyperphagia and obesity. Inhibition of CDKs by CDK inhibitors, such as abemaciclib, can reverse this process. In the case of complete pRb absence, POMC neurons (as well as many other neuron types) undergo cell death. (This figure was created using BioRender (https://biorender.com/).
Outstanding Questions.
1. Is there a unique neuronal signature of E2F1 target genes that are induced by high fat feeding?
As pRb mainly acts via inhibition of E2F1, the disinhibition of E2F1 by pRb inactivation would lead to a set of genes whose expression would be induced. A set of E2F1 regulated genes that is unique to neurons could provide a potential set of drug targets with CNS specificity.
2. What are the mechanisms by which pRb inactivation leads to neuronal dysfunction?
Specifically, POMC neurons are dysfunctional after pRb inactivation. However, the molecular and specific cellular deficits caused by an inactive pRb remain unknown. Specific tests for sensitivity to hormones/nutrients could address the ability of POMC neurons to sense environmental cues as well as specific signal transduction pathways while testing of neurons downstream of POMC neurons could address connectivity issues.
3. Is the reversibility of high fat diet induced obesity by CDK4/6 inhibition constrained within a critical period?
It is possible that the mechanism for causing dysfunction in POMC neurons can cause irreversible cellular damage after a certain period of time. However, even in the most severe case of genetic obesity, complete leptin deficiency, leptin is able to reverse the obese phenotype, suggesting that irreversible damage to the brain's weight-regulating system has not occurred even after lifelong obesity.
Highlights:
Genetic studies have identified numerous genes and neural circuits that cause hyperphagia and obesity although the majority of obese humans have no identifiable mutations in these genes. Diet-induced obesity (DIO) with a calorically dense diet indicates environmental factors directly perturb the functional status of the energy balance neuronal circuitry.
Adult neurons, terminally differentiated and mitotically quiescent, can be triggered to re-enter the cell cycle due to elevated cellular stress. This process leads to impaired neuronal function and neurodegeneration.
Studies approaching obesity as a neuronal cell cycle disorder have revealed the critical role of the retinoblastoma protein (pRb) in maintaining neuronal health. Novel therapeutic options are available with repurposing of drugs developed to treat cancers dependent on inactivation of pRb.
Acknowledgements
Funding: This work was funded by NIH grant 5 R01 DK111043 to SC and LZ, and F30 DK116532 to NJI. Imaging analysis was supported by the Einstein Analytical Imaging Facility, NCI Cancer Center Support grant P30CA013330. Additional support and resources were provided by Einstein Sinai Diabetes Research Center Grant P60DK020541, the New York Obesity Research Center Grant 1P01DK026687, and Liver Pathobiology and Gene Therapy Research Core Center Grant 5P30-DK041296.
Glossary
- ARC
arcuate nucleus of the hypothalamus
- AgRP
agouti gene related peptide
- CDK
cyclin dependent kinase
- CKI
cyclin dependent kinase inhibitor
- DIO
diet induced obesity
- htNSC
nypothalamic neural stem cells
- HFD
high fat diet
- LEPR
leptin receptor
- MBH
medio-basal hypothalamus
- ME
median eminence
- NFkappaB
nuclear factor kappa light chain enhancer of B cells
- NPY
Neuropeptide Y
- POMC
pro-opiomelanocortin
- Rb
retinoblastoma
- VMH
ventromedial hypothalamus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.OECD. (2017) OECD Obesity Update 2017, OECD Publishing. [Google Scholar]
- 2.Chua SC et al. (1996) Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271, 994–996. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y et al. (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372 (6505), 425–32. [DOI] [PubMed] [Google Scholar]
- 4.Considine RV et al. (1995) Evidence against either a premature stop codon or the absence of obese gene mRNA in human obesity. J Clin Invest 95 (6), 2986–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huvenne H et al. (2016) Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes Facts 9 (3), 158–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nunziata A et al. (2017) Estimated prevalence of potentially damaging variants in the leptin gene. Molecular and cellular pediatrics 4 (1), 10–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin S et al. (2000) Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord 24 (5), 639–46. [DOI] [PubMed] [Google Scholar]
- 8.Ziotopoulou M et al. (2000) Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiol Endocrinol Metab 279 (4), E838–45. [DOI] [PubMed] [Google Scholar]
- 9.Farley C et al. (2003) Meal pattern analysis of diet-induced obesity in susceptible and resistant rats. Obes Res 11 (7), 845–51. [DOI] [PubMed] [Google Scholar]
- 10.West DB and York B (1998) Dietary fat, genetic predisposition, and obesity: lessons from animal models. Am J Clin Nutr 67 (3 Suppl), 505s–512s. [DOI] [PubMed] [Google Scholar]
- 11.Alexander J et al. (2006) Distinct phenotypes of obesity-prone AKR/J, DBA2J and C57BL/6J mice compared to control strains. Int J Obes (Lond) 30 (1), 50–9. [DOI] [PubMed] [Google Scholar]
- 12.Townsend KL et al. (2008) High-fat diet-induced changes in body mass and hypothalamic gene expression in wild-type and leptin-deficient mice. Endocrine 33 (2), 176–88. [DOI] [PubMed] [Google Scholar]
- 13.Enriori PJ et al. (2007) Diet-Induced Obesity Causes Severe but Reversible Leptin Resistance in Arcuate Melanocortin Neurons. Cell Metabolism 5 (3), 181–194. [DOI] [PubMed] [Google Scholar]
- 14.Gamber KM et al. (2012) Over-expression of leptin receptors in hypothalamic POMC neurons increases susceptibility to diet-induced obesity. PloS one 7 (1), e30485–e30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nogueiras R et al. (2008) Central nervous system regulation of adipocyte metabolism. Regulatory Peptides 149 (1–3), 26–31. [DOI] [PubMed] [Google Scholar]
- 16.Myers MG et al. (2010) Obesity and leptin resistance: distinguishing cause from effect. Trends in Endocrinology & Metabolism 21 (11), 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers MG et al. (2008) Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70, 537–56. [DOI] [PubMed] [Google Scholar]
- 18.Palkovits M (1984) Neuropeptides in the hypothalamo-hypophyseal system: lateral retrochiasmatic area as a common gate for neuronal fibers towards the median eminence. Peptides 5 Suppl 1, 35–9. [DOI] [PubMed] [Google Scholar]
- 19.Peruzzo B et al. (2000) A second look at the barriers of the medial basal hypothalamus. Experimental Brain Research 132 (1), 10–26. [DOI] [PubMed] [Google Scholar]
- 20.Myers MG (2008) Metabolic sensing and regulation by the hypothalamus. American Journal of Physiology-Endocrinology and Metabolism 294 (5), E809–E809. [DOI] [PubMed] [Google Scholar]
- 21.Thaler JP et al. (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122 (1), 153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X et al. (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135 (1), 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posey KA et al. (2009) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296 (5), E1003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozcan L et al. (2009) Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metabolism 9 (1), 35–51. [DOI] [PubMed] [Google Scholar]
- 25.Fisher RP (2012) The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes & cancer 3 (11-12), 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frade JM and Ovejero-Benito MC (2015) Neuronal cell cycle: the neuron itself and its circumstances. Cell cycle (Georgetown, Tex.) 14 (5), 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrup K and Yang Y (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nature Reviews Neuroscience 8, 368. [DOI] [PubMed] [Google Scholar]
- 28.Park DS et al. (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 143 (2), 457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein JA and Ackerman SL (2003) Oxidative stress, cell cycle, and neurodegeneration. The Journal of Clinical Investigation 111 (6), 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park DS et al. (2000) Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci 20 (9), 3104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdaguer E et al. (2007) Implication of the transcription factor E2F-1 in the modulation of neuronal apoptosis. Biomed Pharmacother 61 (7), 390–9. [DOI] [PubMed] [Google Scholar]
- 32.Kuan CY et al. (2004) Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 24 (47), 10763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morillo SM et al. (2010) Somatic tetraploidy in specific chick retinal ganglion cells induced by nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America 107 (1), 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Höglinger GU et al. (2007) The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America 104 (9), 3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvira D et al. (2007) Inhibition of cyclin-dependent kinases is neuroprotective in 1-methyl-4-phenylpyridinium-induced apoptosis in neurons. Neuroscience 146 (1), 350–65. [DOI] [PubMed] [Google Scholar]
- 36.McNay DEG et al. (2012) Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. Journal of Clinical Investigation 122 (1), 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J et al. (2012) IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol 14 (10), 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee DA et al. (2012) Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nature Neuroscience 15 (5), 700–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Z et al. (2013) pRb is an obesity suppressor in hypothalamus and high-fat diet inhibits pRb in this location. The EMBO Journal 32 (6), 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quiñones M et al. (2018) p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nature Communications 9 (1), 3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iqbal NJ et al. (2018) Cyclin-dependent kinase 4 is a preclinical target for diet-induced obesity. JCI Insight 3 (17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu N et al. (1998) A new model of cell cycle-regulated transcription: repression of the cyclin A promoter by CDF-1 and anti-repression by E2F. Oncogene 16 (23), 2957–63. [DOI] [PubMed] [Google Scholar]
- 43.Sherr CJ and Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13 (12), 1501–12. [DOI] [PubMed] [Google Scholar]
- 44.Gong D and Ferrell JE Jr. (2010) The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol Biol Cell 21 (18), 3149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canepa ET et al. (2007) INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 59 (7), 419–26. [DOI] [PubMed] [Google Scholar]
- 46.Besson A et al. (2008) CDK inhibitors: cell cycle regulators and beyond. Dev Cell 14 (2), 159–69. [DOI] [PubMed] [Google Scholar]
- 47.Kokoeva MV et al. (2007) Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J Comp Neurol 505 (2), 209–20. [DOI] [PubMed] [Google Scholar]
- 48.Goodrich DW et al. (1991) The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 67 (2), 293–302. [DOI] [PubMed] [Google Scholar]
- 49.Murphree AL and Benedict WF (1984) Retinoblastoma: clues to human oncogenesis. Science 223 (4640), 1028–33. [DOI] [PubMed] [Google Scholar]
- 50.Kleinerman RA et al. (2005) Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol 23 (10), 2272–9. [DOI] [PubMed] [Google Scholar]
- 51.Brown VD et al. (1999) Cumulative Effect of Phosphorylation of pRB on Regulation of E2F Activity. Molecular and Cellular Biology 19 (5), 3246–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burke JR et al. (2012) Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes & Development 26 (11), 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harbour JW et al. (1999) Cdk Phosphorylation Triggers Sequential Intramolecular Interactions that Progressively Block Rb Functions as Cells Move through G1. Cell 98 (6), 859–869. [DOI] [PubMed] [Google Scholar]
- 54.Narasimha AM et al. (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinyol M et al. (1997) Deletions and Loss of Expression of P16<sup>INK4a</sup> and P21<sup>Waf1</sup> Genes Are Associated With Aggressive Variants of Mantle Cell Lymphomas. Blood 89 (1), 272. [PubMed] [Google Scholar]
- 56.Sherr CJ et al. (2016) Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 6 (4), 353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corona SP et al. (2017) CDK4/6 inhibitors in HER2-positive breast cancer. Crit Rev Oncol Hematol 112, 208–214. [DOI] [PubMed] [Google Scholar]
- 58.Patnaik A et al. (2016) Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 6 (7), 740–53. [DOI] [PubMed] [Google Scholar]