

Abstract
A substantial proportion of risk for Parkinson disease (PD) is driven by genetics. Progress in understanding the genetic basis of PD has been significant. So far, highly-penetrant rare genetic alterations in SNCA, LRRK2, VPS35, PRKN, PINK1, DJ-1 and GBA have been linked with typical familial PD and common genetic variability at 90 loci have been linked to risk for PD. In this review, we outline the journey thus far of PD genetics, highlighting how significant advances have improved our knowledge of the genetic basis of PD risk, onset and progression. Despite remarkable progress, our field has yet to unravel how genetic risk variants disrupt biological pathways and molecular networks underlying the pathobiology of the disease. We highlight that currently identified genetic risk factors only represent a fraction of the likely genetic risk for PD. Identifying the remaining genetic risk will require us to diversify our efforts, performing genetic studies across different ancestral groups. This work will inform us on the varied genetic basis of disease across populations and also aid in fine mapping discovered loci. If we are able to take this course, we foresee that genetic discoveries in PD will directly influence our ability to predict disease and aid in defining etiological subtypes, critical steps for the implementation of precision medicine for PD.
Keywords: Genetics, Parkinson disease, risk, Post-GWAS era
Introduction
Parkinson Disease (PD) is a neurodegenerative disease characterized by the presence of Lewy bodies in the midbrain and the loss of activity of dopaminergic neurons, particularly in the substantia nigra. First clinically described by James Parkinson in 1817, PD exhibits symptoms that include muscle tremors and rigidity, bradykinesia and loss of balance, among others (Poewe et al. 2017). Years before distinct neuromotor symptoms are manifest, PD initially begins in a long prodromal period. Patients with prodromal PD present non-motor symptoms such as constipation and REM sleep disorder (Heinzel et al. 2019). PD currently has no cure and treatments are only symptomatic.
PD exerts a significant burden on the global economy and society, and this is expected to worsen; as of 2016, there are estimated to be more than 6 million PD cases. As the fastest growing neurological disorder in disability-adjusted life years, deaths, and prevalence (GBD 2016 Neurology Collaborators 2019), the number of PD cases is expected to grow to over 12 million by 2040 (GBD 2016 Neurology Collaborators 2019; Dorsey et al. 2018).
Age is the biggest risk factor to PD, and sex is a contributing factor with men being disproportionately affected (Wright Willis et al. 2010; Van Den Eeden 2003). A wide number of studies have shown that known and unknown environmental factors can contribute to PD risk. Environmental factors such as pesticide exposure, well water consumption, and head injury, as well as premotor symptoms including constipation and depression among others (Noyce et al. 2012), have been associated with increased risk for PD (van der Mark et al. 2012), while other factors such as tobacco, coffee, and alcohol usage have controversially shown possible protective associations with PD (Li et al. 2015; R. Liu et al. 2012; D. Zhang, Jiang, and Xie 2014).
The genetics of Parkinson disease in the pre-GWAS and GWAS era
Monogenic Parkinson disease
Historically, PD was considered a sporadic disorder in which environmental factors and age were the main risk factors. Indeed, before the 1990s, there was significant doubt that PD had any heritability (Duvoisin 1984). However, family history of PD (first degree relative definition) is found in approximately 15% of the patients and 5–10% of PD patients follow a classical Mendelian inheritance pattern (Lesage and Brice 2009).
Since the discovery of disease causing mutations in SNCA in a large Italian kindred and three unrelated Greek families in the late nineties (Polymeropoulos 1997), several genes have been linked to both autosomal dominant and recessive familial PD. However, only SNCA, LRRK2, VPS35, PRKN, PINK1, GBA and DJ-1 have been convincingly associated with typical PD (Figure 1). It is noteworthy that additional genes harbor rare mutations that lack replication and functional validation, and thus should be considered putative. Despite the high disease penetrance of familial cases, known monogenic loci only explain a small fraction of the observed familial aggregation of PD, suggesting that much remains to be found, likely in both genetic and non-genetic domains.
Figure 1.
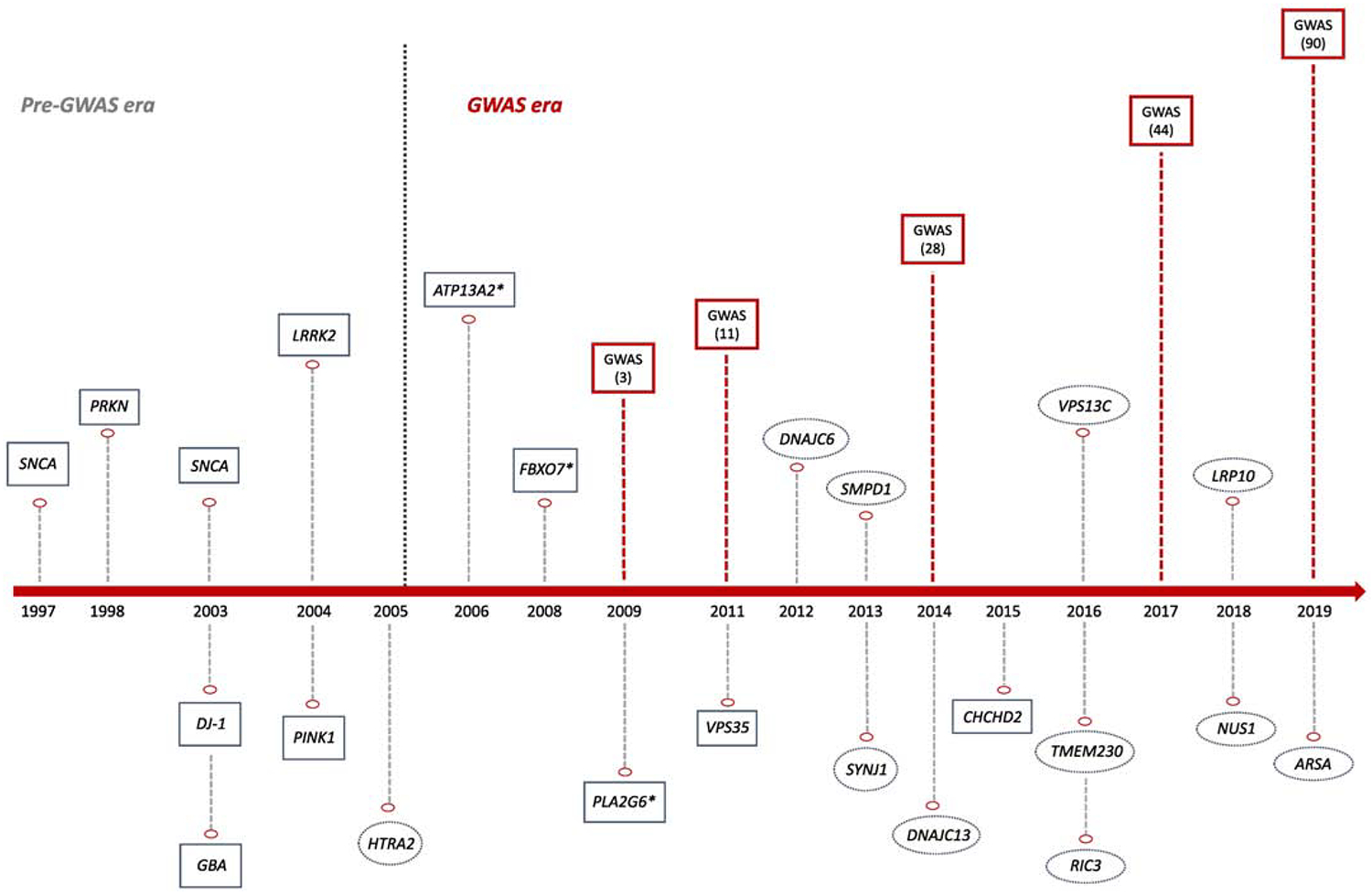
The genetics of Parkinson disease over time.
Red squares represent genome-wide association studies and number of discovered risk loci in brackets. * indicate genes associated with atypical parkinsonism related syndromes. Green squares represent controversial or not widely validated genes linked to typical Parkinson disease
Apart from the widely reviewed genes involved in Mendelian PD, recent findings have nominated TMEM230, LRP10, NUS1 and ARSA as putative disease-causing candidates. In 2016, Deng and colleagues identified TMEM230 as the disease-causing gene in the same Canadian family where mutations in DNAJC13 had been previously found to be causative (Deng et al. 2016; Vilariño-Güell et al. 2014). Three other PD-associated TMEM230 variants were detected in 7 additional Chinese families (Deng et al. 2016). However, many other studies, including large series of PD patients, have failed to replicate this association (Yan et al. 2017; Giri et al. 2017; He et al. 2017; Baumann et al. 2017; Wu et al. 2017; Quadri et al. 2017; Buongarzone et al. 2017; X. Yang et al. 2017; Shi et al. 2017; Wei et al. 2018; Ibanez et al. 2017; Ma et al. 2017; Fan et al. 2017; Conedera et al. 2018; Tejera-Parrado et al. 2018); thus, casting doubt on its implication in PD etiology (Iqbal and Toft 2019; Deng, Pericak-Vance, and Siddique 2019; Farrer 2019).
Genome-wide linkage analysis of a large Italian family with 13 members affected by autosomal-dominant PD and one affected by dementia with Lewy bodies (DLB) led to the identification of heterozygous variants in LRP10 associated with PD, PD dementia (PDD), and DLB (Quadri et al. 2018). Further screening of LRP10 in an international cohort of 660 probands revealed 8 additional rare and potentially-pathogenic LRP10 variants (Quadri et al. 2018). Therefore, LRP10 together with SNCA, LRRK2, and GBA, would support the idea that PD, PDD and DLB are parts of a continuum of Lewy body disease (Langston et al. 2015; Jellinger and Korczyn 2018). However, replication of this association in different populations remains inconsistent (Kia, Sabir, et al. 2018; Guerreiro et al. 2018; Pihlstrøm et al. 2018; Tesson et al. 2018; Shi et al. 2018; Y. Chen et al. 2019; Vergouw et al. 2019), and further studies are needed to determine the involvement of LRP10 in PD pathogenesis.
NUS1 has been nominated as a possible candidate gene for PD in the Han Chinese population (Guo et al. 2018). Guo et al. performed whole-exome sequencing of 39 EOPD patients, their parents and 20 unaffected siblings in order to investigate the effects of de novo mutations in PD. They identified 12 genes with plausible functional relevance to PD pathogenesis. Further analyses in two independent case-control cohorts found a significant association between NUS1 and PD (p-value combined = 1.01 × 10−5). Subsequent functional studies on Drosophila demonstrated that the loss of NUS1 affects climbing ability, dopamine level and the number of dopaminergic neurons; thus, supporting a potential link between NUS1 and PD pathogenesis (Guo et al. 2018). NUS1 encodes the Nogo-B receptor (NgBR) that is a type I single transmembrane domain specific receptor for the neural and cardiovascular regulator Nogo-B. Mutations in NUS1 have been previously associated with a congenital disorder of glycosylation (E. J. Park et al. 2014) and epileptic encephalopathies (Hamdan et al. 2017). It is also involved in intracellular cholesterol trafficking by interacting and stabilizing Niemann-Pick type C2 protein (NPC2) (Harrison et al. 2009). Although a recent study does not support a role for NPC1/2 mutations in the pathogenesis of PD (Zech et al. 2013), the association between cholesterol and PD risk has been suggested in multiple studies (Klemann et al. 2017; Arenas, Garcia-Ruiz, and Fernandez-Checa 2017; García-Sanz et al. 2017; Huang et al. 2019).
More recently, pathogenic and protective mutations in the arylsulfatase A gene (ARSA) have been linked to PD (J. S. Lee et al. 2019). The authors analyzed ARSA mutations in a family with a history of PD and identified two compound heterozygous missense mutations. ARSA encodes a lysosomal hydrolase and its complete deficiency causes metachromatic leukodystrophy, an autosomal recessive lysosomal storage disease. Therefore, these data provide additional support for the lysosomal system in PD pathogenesis (Klein and Mazzulli 2018; Robak et al. 2017). Further screening in 92 familial and 92 sporadic PD cases did not detect additional pathogenic ARSA variants, but identified a non-synonymous variant that was proposed to be protective (J. S. Lee et al. 2019). Interestingly, the authors found that ARSA acts as a cytosolic molecular chaperone regulating α-synuclein accumulation and propagation. However, large international consortiums have failed to replicate the association between ARSA and PD (Makarious et al. 2019).
The number of familial and early onset cases with no known genetic cause remains high, and as demonstrated above, the validation of novel genes associated with PD remains extremely challenging. Families harbouring rare genetic variants are uncommon and globally dispersed, thus making replication of segregating mutations or mutations in the same gene difficult. Further, modern genetic approaches such as whole genome sequencing remain expensive and are not always readily available. A logical path to solving this challenge is the creation of a global resource for mutation discovery, which democratizes the analysis of rare, but highly valuable families.
Sporadic Parkinson disease
Only a small percentage of sporadic PD cases harbor mutations in known PD genes. Over the past decade, many studies have pursued the ‘common disease-common variant’ (CDCV) hypothesis, which postulates that the genetic component of most common and complex disorders, such as PD, is due to a large number of common low-risk alleles (Lohmueller et al. 2003). Genome-wide association (GWA) studies are the gold-standard tool for testing the CDCV hypothesis. The first GWAS in PD to identify genome wide significant associations were reported in 2009. The first consisted of 5,074 cases and 8,551 controls of European origin (Simón-Sánchez et al. 2009). The authors identified two strong association signals, one in SNCA and the other in MAPT locus, as major risk factors for PD (Simón-Sánchez et al. 2009). The second, a GWA in an ancestral Asian population, replicated association at SNCA and identified 3 additional PD susceptibility loci, including PARK16, LRRK2 and BST1 (Satake et al. 2009). Since then, numerous GWA studies with increasing number of participants have been performed across populations (Kara et al. 2014; Siitonen et al. 2017; Bandres-Ciga, Ahmed, et al. 2019). The last and largest PD GWAS to date including around 37,700 cases, 18,600 ‘proxy’ cases and 1.4 million controls has robustly identified 90 independent risk signals associated with sporadic PD (Nalls et al. 2019).
In spite of the success of GWA at expanding our understanding of the genetic basis of human complex diseases, for most traits, including PD, the associated loci do not account for all the genetic variance underlying the disease (Manolio et al. 2009). Analysis of common genetic variability suggests that only ~22% of the liability for PD is driven by these variants and the known associated GWAS SNPs only represent a fraction of this detected heritability (Nalls et al., 2019). Different explanations for the ‘missing heritability problem’ have been proposed (Young 2019; Génin 2019). First, GWA analyses are not currently powered to capture all of the common genetic variability contributing to disease susceptibility, particularly those that may be rare and / or have small effect sizes. Increasing sample sizes of GWA allows researchers to identify new susceptibility loci in situations where statistical power is low and therefore may improve the proportion of explained heritability. It is also worth noting that rare variants of both large and small effect, and some structural variation, are not well-captured by GWA chips and imputation. There is increasing evidence that these may indeed play a major role in common diseases (Gibson 2012; Bomba, Walter, and Soranzo 2017; J. Yang et al. 2015; Weissbrod et al. 2019). In addition, gene-gene and gene-environment interactions could also be contributing to common disease susceptibility (Zuk et al. 2012; Cordell 2009; Ritchie and Van Steen 2018; Cannon and Greenamyre 2013). In PD, there is scant evidence for epistatic interactions, although this is a common theme in disease genetics because of the extreme sample sizes required to reliably detect such associations (Fernández-Santiago et al. 2019; Pecanka et al. 2017; Kuwahara et al. 2016; Beilina et al. 2014; Göbel et al. 2012). It is noteworthy however, that the explanation of epistasis as a piece of the missing heritability puzzle is controversial since dominance variance (due to genetic interactions) appears to have a minimal effect on heritability estimates and does not explain the missing heritability for some complex diseases (Zhu et al. 2015; Guerreiro et al. 2019; Diez-Fairen et al. 2019). Lastly, epigenetic factors can be transmitted through generations and thus may also contribute to disease risk (Bourrat, Lu, and Jablonka 2017; Trerotola et al. 2015; Cortijo et al. 2014; Bell and Spector 2011).
Linking familial and sporadic forms of the disease
Besides identifying several risk loci for sporadic PD, large population-based GWAS and meta-analyses have also established that disease linked common variability exists at loci known to contain rare causal mutations. These include SNCA, LRRK2, GBA and VPS13C, previously associated with Mendelian forms of PD (Satake et al. 2009; Chang et al. 2017; Nalls et al. 2014; Simón-Sánchez et al. 2009); thus, suggesting a link between familial and sporadic forms of disease. These loci harboring both rare large effect and common smaller effect variants are known as pleomorphic risk loci (A. Singleton and Hardy 2011). Therefore, both the CDCV hypothesis and the multiple rare variant hypothesis are not mutually exclusive. Instead, various disease-related genetic mechanisms may coexist at the same locus, each influencing disease through different biological effects on a single gene. For example, SNCA locus has been linked to PD etiology through: (i) duplications and triplications with a clear gene-dose effect (A. B. Singleton et al. 2003; Ibáñez et al. 2004; Chartier-Harlin et al. 2004); (ii) coding mutations causing early-onset PD familial cases (Krüger et al. 1998; Zarranz et al. 2004; Polymeropoulos 1997; Lesage et al. 2013; Kiely et al. 2013; Pasanen et al. 2014); (iii) different association signals in GWAS illustrating the presence of low-effect common variability in this locus (Satake et al. 2009; Chang et al. 2017; Nalls et al. 2014; Simón-Sánchez et al. 2009); and (iv) non-coding risk/protective variants (Trotta et al. 2012).
Understanding the biological consequences of genetic risk
Many of the PD-related genes are involved in common biological pathways, which suggests that several critical cellular routes contribute to the disease risk. Mitochondrial dysfunction was one of the first biological processes to be associated with PD pathogenesis through the analysis of the recessive Mendelian genes. PINK1, a mitochondrial kinase, and PRKN, an E3 ubiquitin ligase, converge in the mitophagy pathway where PINK1 phosphorylates PRKN to eliminate damaged mitochondria (Kane et al. 2014; J. Park et al. 2006; I. E. Clark et al. 2006). DJ-1, another ubiquitin ligase, promotes PINK1 transcriptional activity and is also involved in mitochondrial physiology (Requejo-Aguilar et al. 2015; Cookson 2010). Other genes previously linked to classical and atypical PD risk such as FBXO7, PLA2G6, VPS13C and CHCHD2, have also been linked to the mitochondrial quality control system (Burchell et al. 2013; Lesage et al. 2016; Funayama et al. 2015; R. G. Lee et al. 2018; Cieri, Brini, and Calì 2017). A recent study estimated that common variation within the mitochondria function associated genes can contribute around 7% of the overall heritability (22%) of PD and that mitochondrial processes are involved in both monogenic and sporadic PD pathogenesis and contribute to later age at onset (Kimberley J. Billingsley et al. 2019).
Pathway-based analyses have started to be implemented in PD research in order to identify biological processes that contribute to the disease. Recent studies have pointed out a high genetic risk burden for PD in the lysosomal and endocytic membrane trafficking pathways (Robak et al. 2017; Bandres-Ciga, Saez-Atienzar, et al. 2019). The immune response is another pathway that has been strongly associated with PD susceptibility (M. Zhang et al. 2017; Holmans et al. 2013), through both inflammation and autoimmune response. In fact, major histocompatibility complex (MHC) proteins are displayed on dopamine neurons of the substantia nigra, suggesting that antigenic epitopes could activate T cells involved in autoimmune responses and cell death (Garretti et al. 2019). Previous PD GWAS have identified risk loci spanning key immune associated genes such as BST1 (bone marrow stromal cell antigen 1) and HLA (human leukocyte antigen) (Satake et al. 2009; Nalls et al. 2014). BST1 has been proposed to play a role in neutrophil adhesion and migration, and it has been reported that it could be a cause of selective vulnerability of dopaminergic neurons in PD (K. J. Billingsley et al. 2018). Additionally, the HLA locus which encodes the MHC-II has been associated with PD (Nalls et al., 2019). GWAS have identified an association of PD with two MHC genes, including HLA-DRB1 and HLA-DRB5 (Hamza et al. 2010; Nalls et al. 2019). Indeed, it has been reported that α-synuclein-derived fragments act as antigenic epitopes displayed by HLA receptors, where both helper and cytotoxic T-cell responses are present in a high percentage of patients when tested (Bandres-Ciga and Cookson 2017; Sulzer et al. 2017). Further investigation of these pathways can provide novel insights into PD pathogenesis and uncover novel therapeutic targets.
Predicting risk, progression and defining etiological subtypes of disease
Individually GWA identified loci confer relatively small amounts of disease risk; however, the use of polygenic risk scores (PRS) affords the ability to attribute a total known genetic risk score to an individual by summing their collective genetic risk. To date, the PRS reveals that, collectively, the 90 susceptibility loci confer considerable risk for disease, with those in the top decile of genetic risk being 6-fold more likely to have PD than those in the lowest decile of genetic risk (Nalls et al. 2019). Additionally, by creating a composite risk score for PD diagnosis that combines the cumulative effect of genetic risk variants as well as the presence or absence of anosmia, age, sex, and family history, the ability to predict individuals at high risk for PD is remarkable, showing an AUC sensitivity of ~ 83.4% and specificity of ~ 90% (Nalls, McLean, et al. 2015).
Research focused on age at onset disease modifiers is one area where consistent effort has been seen. The largest PD age at onset GWAS to date (Blauwendraat et al. 2019) included data from > 25K cases and identified two GWAS significant signals; one at SNCA and the other was a protein-coding variant in TMEM175, both of which are known PD risk loci. Notably, these results showed that not all PD risk loci influence age at onset and therefore suggest the idea that risk and onset might operate through mechanisms that do not completely overlap.
Despite the progress made in dissecting risk and understanding onset due to the availability of large sets of samples for genome-wide assessment, for the most part, these cohorts lack substantial clinical data, making it difficult to investigate progression in a well-powered manner. Collecting comprehensive longitudinal data is costly, and harmonizing and combining individually small sample sets is time consuming and challenges. Despite these difficulties, the latest PD progression GWAS to date (Iwaki et al. 2019), managed to identify two GWAS signals in loci not previously linked to PD risk (SLC44A1 associated with a faster rate of progression via Hohn and Yar score, and an intergenic variant related to ADRA2A expression associated with insomnia) (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/).
Although laborious, accurate models of disease progression will enable the next generation of clinical trials. In recent years, several initiatives such as the Parkinson’s Progression Markers Initiative (PPMI) (https://www.ppmi-info.org/), the Parkinson’s Disease Biomarkers Program (PDBP) (https://pdbp-demo.cit.nih.gov/), Predict PD (Noyce, R’Bibo, et al. 2017), and the Accelerating Medicines Partnership for Parkinson Disease (AMP-PD) (https://www.nih.gov/research-training/accelerating-medicines-partnership-amp) have emerged as valuable resources to aggregate and harmonize efforts in PD.
Predicting the precise clinical, molecular, and pathological subtypes in such a complex and heterogeneous disorder, will prove invaluable and will likely have an impact on drug development, therapeutic testing, and ultimately treatment (Leonard et al. 2019).
Inferring causal relationships across phenotypic traits and exploring shared polygenic risk
A corollary of abundant GWA data is the ability to test other traits that can predispose or protect individuals to disease. Access to large amounts of GWA data affords the opportunity to explore causal inferences between myriad traits and PD through Mendelian Randomization (MR), the gold standard approach for causality in genetic studies that sits at the interface between observational epidemiology and interventional trials (Lawlor et al., n.d.). MR has emerged as a statistical approach that uses genetic data in the form of SNPs to study whether an exposure exerts a causal effect in an outcome. This addresses the question of whether an observational association between a risk or protective factor and a disease of interest is consistent with a causal effect by focusing usually only on genome-wide significant SNPs. Put simply, genetic variants that explain variation in a certain exposure can be used as proxies to determine how a change in that exposure might influence a disease outcome. One of the key strengths of this method is that it relies on genetic variants that remain constant over the lifespan of an individual, are randomized during gametogenesis and fixed at conception, which means that genetic variants are not associated with all the confounder factors that affect an observational study.
In the PD field, two-sample MR has been applied considering both targeted (hypothesis-driven) exploration of causal associations, as well as hypothesis-generating approaches in a large-scale, high-throughput, unbiased manner.
For the former, an inverse causal association between body mass index (BMI) and risk of PD has been reported. A genetically-estimated 5kg/m2 higher BMI was associated with a reduced risk of PD (Noyce, Kia, et al. 2017). Negative causal associations have also been necessary to refute spurious associations. For instance, observational studies have reported that modulating plasma urate could be a potential preventive avenue for PD (Noyce et al. 2012; Bellou et al. 2016). However, MR did not find evidence for a causal protective effect of urate levels on PD risk (Kia, Noyce, et al. 2018), corroborating the results from the Phase III of the SURE-PD3 clinical trial. The randomized, placebo-controlled, double-blinded trial conducted nationwide at 57 clinical sites of the Parkinson Study Group (PSG), failed at demonstrating that a treatment with Inosine raised blood levels of the natural antioxidant urate and therefore these molecular changes could slow the rate of progression in PD.
For the latter, a useful interactive resource for the PD community has been made publicly available (https://pdgenetics.shinyapps.io/pdgenetics) (Noyce et al. 2019). By using the PD MR Research Portal, users can search for evidence of causality on a broad range of exposures from 5,839 different GWASes. This resource can be used to provide evidence to support, and over time, evidence against, causality when undertaking observational studies or pursuing interventions aimed at reducing the risk of PD.
To date, the main limitations of applying MR, is that many studies are underpowered, either as a consequence of relatively low samples sizes in the exposure GWAS data or the small amount of variation (heritability) in an exposure trait that is explained by the reported common genetic variation.
Another approach that has recently taken importance is applying Linkage disequilibrium score regression (LDSC) to investigate the extent to which genetic etiologies are shared across different diseases. In the PD genetics field, it has been successfully implemented to test whether polygenic risk contributing to a phenotype of interest might also contribute to the risk of PD. Cross-trait genetic correlations between the latest PD GWAS (Nalls et al. 2019) summary statistics and 757 other GWAS available datasets revealed four significant genetic correlations, including positive correlations with intracranial volume and putamen volume (suggesting that both may prove to be valuable PD biomarkers) and negative correlations with current tobacco use and “academic qualifications: National Vocational Qualifications (NVQ) or Higher National Diploma (HND) or Higher National Certificate (HNC) or equivalent”. Interestingly, the negative association with one’s academic qualifications indicates that those individuals without a college education may have a lower likelihood of developing PD than individuals with higher levels of education, but again, correlation does not imply causation. Although there remains a lot to be done, the current work represents a novel approach towards the foundation to pursue PD research in the Post-GWAS era.
Translating Genetics into Precision Medicine
Pharmacogenetics
The decrease in genetic testing cost has positioned precision medicine, medical practice specifically attuned to genetics of the individual patient, as the focus of clinical genetics research and the US health care policy (Collins and Varmus 2015). Genetics-informed personalized therapeutics, also called pharmacogenetics, are the future of healthcare, and already an indication for drug usage and dosing. Currently, there are clinical trials that take into account crucial genes associated with PD such as GBA (Silveira et al. 2019) and LRRK2 (J. Chen, Chen, and Pu 2018; Whiffin et al., n.d.).
As one of the most well understood PD risk genes, GBA is frequently seen as a treatment target. In general, GBA carrier PD cases present worse disease symptoms and mutation severity has been associated with differences in age-at-onset (Brockmann et al. 2015; Gan-Or, Liong, and Alcalay 2018). For instance, GBA p.N370S variant is known to increase PD risk and when homozygous cause Gaucher Disease (GD) (L. N. Clark et al. 2005). In the largest PD age at onset GWAS to date, significant hits within the GBA region were variants p.N370S, p.E326K, and p.T369M with effect sizes between 2.6 to 0.9 year reduction in ageat-onset (Blauwendraat et al. 2019). In a longitudinal study, PD patients with GBA variants that cause neuropathic GD have shown to have accelerated cognitive decline over time compared to other PD patients (G. Liu et al. 2016). Despite these findings, the role and the mechanism of GBA and its product Glucocerebrosidase (GCase) remains unclear. In addition, many GBA carriers never get PD. GBA modifiers may help answer this conundrum. PD PRS is higher in GBA carrier cases compared to carrier controls, with variants near CTSB and SNCA showing potential gene-gene interaction with GBA (Blauwendraat et al. 2020). Nevertheless, clinical trials have started for GBA targeted therapies, all trying increase GCase production or activity. One uses glucosylceramide synthase inhibitor GZ/SAR402671 (ClinicalTrials.gov identifier NCT02906020), while others are using ambroxol hydrochloride (ClinicalTrials.gov identifier NCT02941822 and NCT02914366).
Patient-specific dosage and management of side effects is an important aspect of personalized medicine. Genes that influence the patient response often are involved in pathways that interact with the treatment in question. Many of the current PD treatments focus on supplying the brain with additional dopamine, either by providing the body with precursor molecules (levodopa), metabolic enzyme inhibitors (rasagiline), or dopamine receptor agonists (ropinirole). As such, genes associated with treatment effectiveness are involved in pathways that interact with dopamine and metabolism of dopamine precursors. For instance, DDC and COMT encode enzymes that metabolize levodopa to dopamine, and variations in both are associated with dosage and side effects of levodopa (Bialecka et al. 2008; de Lau et al. 2012; Cheshire et al. 2014). Dopamine transporter gene DAT has been associated with psychosis with levodopa treatment (Kaiser et al. 2003) and dopamine receptor genes, including DRD2 and DRD3, have been associated with side effects in levodopa and MAO-B inhibitors (Masellis et al. 2016; Rieck et al. 2012; Krishnamoorthy et al. 2016). In addition, there is evidence that monogenic PD patients have varied response to deep brain stimulation therapy (DBS) depending on the gene and variant in question; the LRRK2 p.G2019S variant has shown positive outcomes to DBS, while LRRK2 p.R1441G has shown poor outcomes (Kuusimäki et al. 2019).
Future Directions
Genomics Need Diversity
Most work in genomics has been done in subjects of Northern European Ancestry (Popejoy and Fullerton 2016) and PD genetic research is no exception (Foo et al. 2017). This lack of diversity reveals three limitations of our current approach: We don’t understand the applicability of genetic risk and PRS across populations; the LD structure of a single ancestral group means that it is difficult to fine map risk loci, and there is missing novel ancestry underlying specific genetic risk.
Expanding our investigation to more diverse populations is the right thing to do. Given our discussion of the potential impact of individualized genetic risk on treatment, it is critical that we understand genetic risk in individuals of varied genetic backgrounds, so that we can treat everyone effectively. A clear benefit of the expansion of this effort is the potential to identify rare but informative ancestry specific risk alleles and the genes these alleles influence, providing new therapeutic targets; however, this work also affords the ability to perform trans ethnic fine mapping, an approach that facilitates the reduction of risk regions to smaller credible intervals, thus simplifying their functional characterization.
Merely adding additional non-European participants will not be enough to remove the current bias. We will have to produce reference data and develop genetic tools specifically tailored to underserved populations. The current PD GWAS includes large amount of data from genotyping arrays designed around European populations (Nalls, Bras, et al. 2015; Blauwendraat et al. 2017; Bycroft et al. 2018). Designing arrays based on more global population or use of whole-genome sequencing in conjunction with diverse participant population will be crucial to reducing systematic European bias. Outside of PD, there has been at least one study on an underrepresented population that found a rare SNP with an effect on both the underrepresented and European populations (Fumagalli et al. 2015). In the same manner, PD may have “missing heritability” ready to be discovered through the study of non-European populations.
The good news is that there is a growing push to include diverse ancestral populations in genetics work. Pan-continental programs such as H3 Africa (H3Africa Consortium et al. 2014) have already begun establishing networks of international labs in genetically understudied regions. In the United States, the All of Us initiative is building a diverse genetic database that encompasses the population of the United States, including historically underrepresented populations (Investigators and The All of Us Research Program Investigators 2019). In the PD genetics field, pilot genomic studies have started to arise; the Michael J Fox Foundation (MJFF) has recently committed pilot support across non-European populations (Williams, Bandmann, and Walker 2018) including initiatives to extend large scale genotyping efforts to Latin Americans (LARGE-PD) (Zabetian, Mata, and Latin American Research Consortium on the Genetics of PD (LARGE-PD) 2017), Africans, African-Americans, the Luxembourg-German-Indian Alliance on Neurodegenerative diseases and Therapeutics (Lux GIANT) and East-Asians under the wing of the International Parkinson Disease Genomics Consortium (IPDGC; https://pdgenetics/org). We envisage that funding agencies will incentivize studying diverse populations and PD genetics research will make in-roads with non-European academia to establish collaborative large-scale genetic studies.
Conclusion
Within two decades, PD genetics has come a long way, from the field doubting the role of genetics in PD to an increased understanding its impact on PD risk, onset, progression, and treatment response, as well as phenotypic etiologies and causal comorbidities. Yet only a fraction of the heritability is known and the relationship between genetics and PD pathology is poorly understood. The nascent movement to expand this work at scale and to encourage diverse participation in PD genetics research will not only reduce systematic health disparities but also promote discoveries of the missing pieces of PD genetics.
Figure 2.
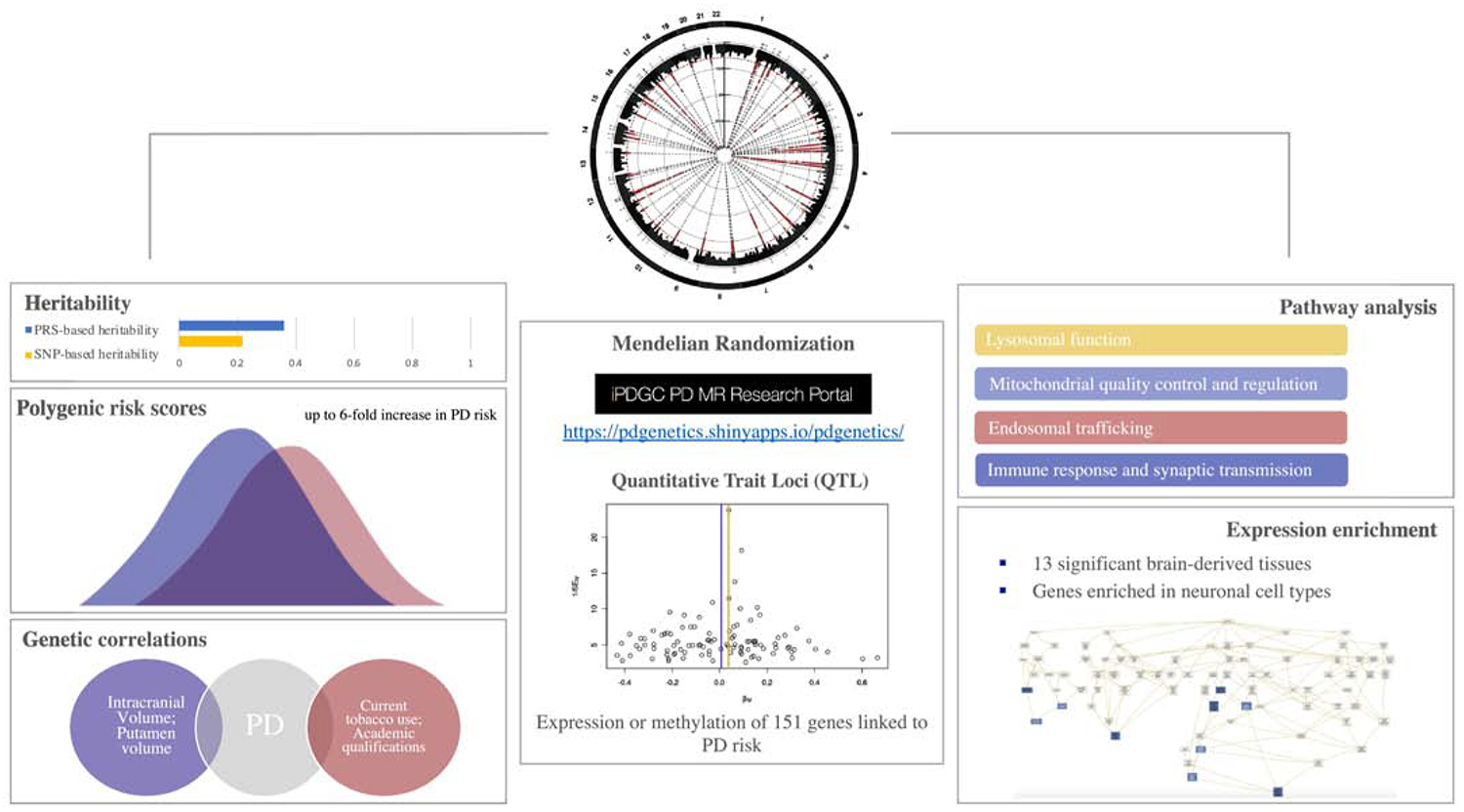
Characterizing the genetic architecture of Parkinson disease.
Highlights:
A total of 90 risk loci have been associated with PD to date representing 16–36% of the heritable component of the disease.
The number of familial and early onset cases with no known genetic cause remains high and the validation of novel genes remains extremely challenging.
PD genetics is disproportionately based on studying populations of European ancestry. Diversity will not only reduce systematic health disparities but also promote discoveries of the missing pieces of PD genetics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have nothing to disclose
REFERENCES
- Arenas Fabian, Garcia-Ruiz Carmen, and Fernandez-Checa Jose C.. 2017. “Intracellular Cholesterol Trafficking and Impact in Neurodegeneration.” Frontiers in Molecular Neuroscience 10 (November): 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres-Ciga Sara, Ahmed Sarah, Sabir Marya S., Blauwendraat Cornelis, Adarmes-Gómez Astrid D., Bernal-Bernal Inmaculada, Bonilla-Toribio Marta, et al. 2019. “The Genetic Architecture of Parkinson Disease in Spain: Characterizing Population-Specific Risk, Differential Haplotype Structures, and Providing Etiologic Insight.” 10.1101/609016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandres-Ciga Sara, and Cookson Mark R.. 2017. “Alpha-Synuclein Triggers T-Cell Response. Is Parkinson’s Disease an Autoimmune Disorder?” Movement Disorders. 10.1002/mds.27116. [DOI] [PubMed] [Google Scholar]
- Bandres-Ciga Sara, Sara Saez-Atienzar Luis Bonet-Ponce, Billingsley Kimberley, Vitale Dan, Blauwendraat Cornelis, Gibbs Jesse Raphael, et al. 2019. “The Endocytic Membrane Trafficking Pathway Plays a Major Role in the Risk of Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 34 (4): 460–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann Hauke, Wolff Simone, Alexander Münchau Johann M. Hagenah, Lohmann Katja, and Klein Christine. 2017. “Evaluating the Role of TMEM230 Variants in Parkinson’s Disease.” Parkinsonism & Related Disorders. [DOI] [PubMed] [Google Scholar]
- Beilina Alexandria, Rudenko Iakov N., Kaganovich Alice, Civiero Laura, Chau Hien, Kalia Suneil K., Kalia Lorraine V., et al. 2014. “Unbiased Screen for Interactors of Leucine-Rich Repeat Kinase 2 Supports a Common Pathway for Sporadic and Familial Parkinson Disease.” Proceedings of the National Academy of Sciences of the United States of America 111 (7): 2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell Jordana T., and Spector Tim D.. 2011. “A Twin Approach to Unraveling Epigenetics.” Trends in Genetics: TIG 27 (3): 116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellou Vanesa, Belbasis Lazaros, Tzoulaki Ioanna, Evangelou Evangelos, and Ioannidis John P. A.. 2016. “Environmental Risk Factors and Parkinson’s Disease: An Umbrella Review of Meta-Analyses.” Parkinsonism & Related Disorders. 10.1016/j.parkreldis.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Bialecka Monika, Kurzawski Mateusz, Gabriela Klodowska-Duda Grzegorz Opala, Tan Eng-King, and Drozdzik Marek. 2008. “The Association of Functional Catechol-O-Methyltransferase Haplotypes with Risk of Parkinson’s Disease, Levodopa Treatment Response, and Complications.” Pharmacogenetics and Genomics 18 (9): 815–21. [DOI] [PubMed] [Google Scholar]
- Billingsley Kimberley J., International Parkinson’s Disease Genomics Consortium (IPDGC), Barbosa Ines A., Bandrés-Ciga Sara, Quinn John P., Bubb Vivien J., Deshpande Charu, et al. 2019. “Mitochondria Function Associated Genes Contribute to Parkinson’s Disease Risk and Later Age at Onset.” Npj Parkinson’s Disease. 10.1038/s41531-019-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S, and Singleton AB. 2018. “Genetic Risk Factors in Parkinson’s Disease.” Cell and Tissue Research 373 (1): 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat Cornelis, Faghri Faraz, Pihlstrom Lasse, Geiger Joshua T., Elbaz Alexis, Lesage Suzanne, Corvol Jean-Christophe, et al. 2017. “NeuroChip, an Updated Version of the NeuroX Genotyping Platform to Rapidly Screen for Variants Associated with Neurological Diseases.” Neurobiology of Aging 57 (September): 247.e9–247.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat Cornelis, Heilbron Karl, Vallerga Costanza L., Bandres-Ciga Sara, von Coelln Rainer, Pihlstrøm Lasse, Simón-Sánchez Javier, et al. 2019. “Parkinson’s Disease Age at Onset Genome-Wide Association Study: Defining Heritability, Genetic Loci, and α-Synuclein Mechanisms.” Movement Disorders: Official Journal of the Movement Disorder Society 34 (6): 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat Cornelis, Reed Xylena, Krohn Lynne, Heilbron Karl, Bandres-Ciga Sara, Tan Manuela, Gibbs J. Raphael, et al. 2020. “Genetic Modifiers of Risk and Age at Onset in GBA Associated Parkinson’s Disease and Lewy Body Dementia.” Brain: A Journal of Neurology 143 (1): 234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomba Lorenzo, Walter Klaudia, and Soranzo Nicole. 2017. “The Impact of Rare and Low-Frequency Genetic Variants in Common Disease.” Genome Biology 18 (1): 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourrat Pierrick, Lu Qiaoying, and Jablonka Eva. 2017. “Why the Missing Heritability Might Not Be in the DNA.” BioEssays. 10.1002/bies.201700067. [DOI] [PubMed] [Google Scholar]
- Brockmann Kathrin, Srulijes Karin, Pflederer Sylvia, Ann-kathrin Hauser Claudia Schulte, Maetzler Walter, Gasser Thomas, and Berg Daniela. 2015. “GBA - associated Parkinson’s Disease: Reduced Survival and More Rapid Progression in a Prospective Longitudinal Study.” Movement Disorders. 10.1002/mds.26071. [DOI] [PubMed] [Google Scholar]
- Buongarzone Gabriele, Monfrini Edoardo, Franco Giulia, Trezzi Ilaria, Borellini Linda, Frattini Emanuele, Melzi Valentina, et al. 2017. “Mutations in TMEM230 Are Rare in Autosomal Dominant Parkinson’s Disease.” Parkinsonism & Related Disorders 39 (June): 87–88. [DOI] [PubMed] [Google Scholar]
- Burchell Victoria S., Nelson David E., Alvaro Sanchez-Martinez Marta Delgado-Camprubi, Ivatt Rachael M., Pogson Joe H., Randle Suzanne J., et al. 2013. “The Parkinson’s Disease–linked Proteins Fbxo7 and Parkin Interact to Mediate Mitophagy.” Nature Neuroscience. 10.1038/nn.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft Clare, Freeman Colin, Petkova Desislava, Band Gavin, Elliott Lloyd T., Sharp Kevin, Motyer Allan, et al. 2018. “The UK Biobank Resource with Deep Phenotyping and Genomic Data.” Nature 562 (7726): 203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon Jason R., and Greenamyre J. Timothy. 2013. “Gene-Environment Interactions in Parkinson’s Disease: Specific Evidence in Humans and Mammalian Models.” Neurobiology of Disease 57 (September): 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Diana, Nalls Mike A., Hallgrímsdóttir Ingileif B., Hunkapiller Julie, van der Brug Marcel, Cai Fang, International Parkinson’s Disease Genomics Consortium, et al. 2017. “A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci.” Nature Genetics 49 (10): 1511–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin Marie-Christine, Kachergus Jennifer, Roumier Christophe, Mouroux Vincent, Douay Xavier, Lincoln Sarah, Levecque Clotilde, et al. 2004. “Alpha-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease.” The Lancet 364 (9440): 1167–69. [DOI] [PubMed] [Google Scholar]
- Chen Jinhua, Chen Ying, and Pu Jiali. 2018. “Leucine-Rich Repeat Kinase 2 in Parkinson’s Disease: Updated from Pathogenesis to Potential Therapeutic Target.” European Neurology. 10.1159/000488938. [DOI] [PubMed] [Google Scholar]
- Chen You, Cen Zhidong, Zheng Xiaosheng, Pan Qinqing, Chen Xinhui, Zhu Lili, Chen Si, et al. 2019. “LRP10 in Autosomal-Dominant Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 34 (6): 912–16. [DOI] [PubMed] [Google Scholar]
- Cheshire Perdita, Bertram Kelly, Ling Helen, O’Sullivan Sean S., Halliday Glenda, McLean Catriona, Bras Jose, Foltynie Tom, Storey Elsdon, and Williams David R.. 2014. “Influence of Single Nucleotide Polymorphisms in COMT, MAO-A and BDNF Genes on Dyskinesias and Levodopa Use in Parkinson’s Disease.” Neuro-Degenerative Diseases 13 (1): 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri Domenico, Brini Marisa, and Calì Tito. 2017. “Emerging (and Converging) Pathways in Parkinson’s Disease: Keeping Mitochondrial Wellness.” Biochemical and Biophysical Research Communications 483 (4): 1020–30. [DOI] [PubMed] [Google Scholar]
- Clark Ira E., Dodson Mark W., Jiang Changan, Cao Joseph H., Huh Jun R., Jae Hong Seol Soon Ji Yoo, Hay Bruce A., and Guo Ming. 2006. “Drosophila pink1 Is Required for Mitochondrial Function and Interacts Genetically with Parkin.” Nature 441 (7097): 1162–66. [DOI] [PubMed] [Google Scholar]
- Clark Lorraine N., Nicolai Angelique, Afridi Shehla, Harris Juliette, Helen Mejia-Santana Lisa Strug, Cote Lucien J., et al. 2005. “Pilot Association Study of the Beta-Glucocerebrosidase N370S Allele and Parkinson’s Disease in Subjects of Jewish Ethnicity.” Movement Disorders: Official Journal of the Movement Disorder Society 20 (1): 100–103. [DOI] [PubMed] [Google Scholar]
- Collins Francis S., and Varmus Harold. 2015. “A New Initiative on Precision Medicine.” New England Journal of Medicine. 10.1056/nejmp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conedera Silvio A., Li Yuanzhe, Funayama Manabu, Yoshino Hiroyo, Nishioka Kenya, and Hattori Nobutaka. 2018. “Genetic Analysis of TMEM230 in Japanese Patients with Familial Parkinson’s Disease.” Parkinsonism & Related Disorders 48 (March): 107–8. [DOI] [PubMed] [Google Scholar]
- Cookson Mark R. 2010. “DJ-1, PINK1, and Their Effects on Mitochondrial Pathways.” Movement Disorders: Official Journal of the Movement Disorder Society 25 Suppl 1: S44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell Heather J. 2009. “Detecting Gene-Gene Interactions That Underlie Human Diseases.” Nature Reviews. Genetics 10 (6): 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo Sandra, Wardenaar René, Maria Colomé-Tatché Arthur Gilly, Etcheverry Mathilde, Labadie Karine, Caillieux Erwann, et al. 2014. “Mapping the Epigenetic Basis of Complex Traits.” Science 343 (6175): 1145–48. [DOI] [PubMed] [Google Scholar]
- Deng Han-Xiang, Pericak-Vance Margaret A., and Siddique Teepu. 2019. “Reply to ‘TMEM230 Variants in Parkinson’s Disease’ and ‘Doubts about TMEM230 as a Gene for Parkinsonism.’” Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Deng Han-Xiang, Shi Yong, Yang Yi, Ahmeti Kreshnik B., Miller Nimrod, Huang Cao, Cheng Lijun, et al. 2016. “Identification of TMEM230 Mutations in Familial Parkinson’s Disease.” Nature Genetics 48 (7): 733–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Fairen Monica, Sara Bandres-Ciga Gabrielle Houle, Nalls Mike A., Girard Simon L., Dion Patrick A., Blauwendraat Cornelis, Singleton Andrew B., Rouleau Guy A., and Pastor Pau. 2019. “Genome-Wide Estimates of Heritability and Genetic Correlations in Essential Tremor.” Parkinsonism & Related Disorders, May 10.1016/j.parkreldis.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey E. Ray, Sherer Todd, Okun Michael S., and Bloem Bastiaan R.. 2018. “The Emerging Evidence of the Parkinson Pandemic.” Journal of Parkinson’s Disease 8 (s1): S3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoisin Roger C. 1984. “Is Parkinson’s Disease Acquired or Inherited?” The Canadian Journal of Neurological Sciences. Le Journal Canadien Des Sciences Neurologiques 11 (S1): 151–55. [DOI] [PubMed] [Google Scholar]
- Fan Tian-Sin, Lin Chin-Hsien, Lin Hang-I, Chen Meng-Ling, and Wu Ruey-Meei. 2017. “Lack of TMEM230 Mutations in Patients with Familial and Sporadic Parkinson’s Disease in a Taiwanese Population.” American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: The Official Publication of the International Society of Psychiatric Genetics 174 (7): 751–56. [DOI] [PubMed] [Google Scholar]
- Farrer Matt J. 2019. “Doubts about TMEM230 as a Gene for Parkinsonism.” Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Fernández-Santiago Rubén, Núria Martín-Flores Francesca Antonelli, Cerquera Catalina, Moreno Verónica, Sara Bandres-Ciga Elisabetta Manduchi, et al. 2019. “SNCA and mTOR Pathway Single Nucleotide Polymorphisms Interact to Modulate the Age at Onset of Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society, June 10.1002/mds.27770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo Jia Nee, Tan Louis C., Irwan Ishak D., Au Wing-Lok, Low Hui Qi, Prakash Kumar-M, Ahmad-Annuar Azlina, et al. 2017. “Genome-Wide Association Study of Parkinson’s Disease in East Asians.” Human Molecular Genetics 26 (1): 226–32. [DOI] [PubMed] [Google Scholar]
- Fumagalli Matteo, Moltke Ida, Grarup Niels, Racimo Fernando, Bjerregaard Peter, Jørgensen Marit E., Korneliussen Thorfinn S., et al. 2015. “Greenlandic Inuit Show Genetic Signatures of Diet and Climate Adaptation.” Science 349 (6254): 1343–47. [DOI] [PubMed] [Google Scholar]
- Funayama Manabu, Ohe Kenji, Amo Taku, Furuya Norihiko, Yamaguchi Junji, Saiki Shinji, Li Yuanzhe, et al. 2015. “CHCHD2 Mutations in Autosomal Dominant Late-Onset Parkinson’s Disease: A Genome-Wide Linkage and Sequencing Study.” Lancet Neurology 14 (3): 274–82. [DOI] [PubMed] [Google Scholar]
- Gan-Or Ziv, Liong Christopher, and Alcalay Roy N.. 2018. “GBA-Associated Parkinson’s Disease and Other Synucleinopathies.” Current Neurology and Neuroscience Reports. 10.1007/s11910-018-0860-4. [DOI] [PubMed] [Google Scholar]
- García-Sanz Patricia, Orgaz Lorena, Guillermo Bueno-Gil Isabel Espadas, Eva Rodríguez-Traver Jaime Kulisevsky, Gutierrez Antonia, et al. 2017. “N370S-GBA1 Mutation Causes Lysosomal Cholesterol Accumulation in Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (10): 1409–22. [DOI] [PubMed] [Google Scholar]
- Garretti Francesca, Agalliu Dritan, Arlehamn Cecilia S. Lindestam, Sette Alessandro, and Sulzer David. 2019. “Autoimmunity in Parkinson’s Disease: The Role of α-Synuclein-Specific T Cells.” Frontiers in Immunology 10 (February): 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2016 Neurology Collaborators. 2019. “Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016.” Lancet Neurology 18 (5): 459–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Génin Emmanuelle. 2019. “Missing Heritability of Complex Diseases: Case Solved?” Human Genetics, June 10.1007/s00439-019-02034-4. [DOI] [PubMed] [Google Scholar]
- Gibson Greg. 2012. “Rare and Common Variants: Twenty Arguments.” Nature Reviews. Genetics 13 (2): 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri Anamika, Mok Kin Y., Jansen Iris, Sharma Manu, Tesson Christelle, Mangone Graziella, Lesage Suzanne, et al. 2017. “Lack of Evidence for a Role of Genetic Variation in TMEM230 in the Risk for Parkinson’s Disease in the Caucasian Population.” Neurobiology of Aging 50 (February): 167.e11–167.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbel Anna, Macklin Eric A., Winkler Susen, Betensky Rebecca A., Klein Christine, Lohmann Katja, and Simon David K.. 2012. “Genetic Risk Factors in Parkinson’s Disease: Single Gene Effects and Interactions of Genotypes.” Journal of Neurology 259 (11): 2503–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro Rita, Valentina Escott-Price Dena G. Hernandez, Celia Kun-Rodrigues Owen A. Ross, Orme Tatiana, Neto Joao Luis, et al. 2019. “Heritability and Genetic Variance of Dementia with Lewy Bodies.” Neurobiology of Disease 127 (July): 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro Rita, Orme Tatiana, Neto João Luís, Bras Jose, and International DLB Genetics Consortium. 2018. “LRP10 in α-Synucleinopathies.” Lancet Neurology. [Google Scholar]
- Guo Ji-Feng, Zhang Lu, Li Kai, Mei Jun-Pu, Xue Jin, Chen Jia, Tang Xia, et al. 2018. “Coding Mutations in Contribute to Parkinson’s Disease.” Proceedings of the National Academy of Sciences of the United States of America 115 (45): 11567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- H3Africa Consortium, Rotimi Charles, Abayomi Akin, ‘le Abimiku Alash, Adabayeri Victoria May, Adebamowo Clement, Adebiyi Ezekiel, et al. 2014. “Research Capacity. Enabling the Genomic Revolution in Africa.” Science 344 (6190): 1346–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan Fadi F., Myers Candace T., Cossette Patrick, Lemay Philippe, Spiegelman Dan, Alexandre Dionne Laporte Christina Nassif, et al. 2017. “High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies.” American Journal of Human Genetics 101 (5): 664–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza Taye H., Zabetian Cyrus P., Tenesa Albert, Laederach Alain, Montimurro Jennifer, Yearout Dora, Kay Denise M., et al. 2010. “Common Genetic Variation in the HLA Region Is Associated with Late-Onset Sporadic Parkinson’s Disease.” Nature Genetics. 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison Kenneth D., Robert Qing Miao Carlos Fernandez-Hernándo, Yajaira Suárez Alberto Dávalos, and Sessa William C.. 2009. “Nogo-B Receptor Stabilizes Niemann-Pick Type C2 Protein and Regulates Intracellular Cholesterol Trafficking.” Cell Metabolism 10 (3): 208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel Sebastian, Berg Daniela, Gasser Thomas, Chen Honglei, Yao Chun, Postuma Ronald B., and MDS Task Force on the Definition of Parkinson’s Disease. 2019. “Update of the MDS Research Criteria for Prodromal Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society, August 10.1002/mds.27802. [DOI] [PubMed] [Google Scholar]
- He Ya-Chao, Huang Pei, Li Qiong-Qiong, Sun Qian, Li Dun-Hui, Wang Tian, Shen Jun-Yi, and Chen Sheng-Di. 2017. “TMEM230 Stop Codon Mutation Is Rare in Parkinson’s Disease and Essential Tremor in Eastern China.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (2): 301–2. [DOI] [PubMed] [Google Scholar]
- Holmans Peter, Moskvina Valentina, Jones Lesley, Sharma Manu, International Parkinson’s Disease Genomics Consortium, Vedernikov Alexey, Buchel Finja, et al. 2013. “A Pathway-Based Analysis Provides Additional Support for an Immune-Related Genetic Susceptibility to Parkinson’s Disease.” Human Molecular Genetics 22 (5): 1039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Xuemei, Sterling Nicholas W., Du Guangwei, Sun Dongxiao, Stetter Christina, Kong Lan, Zhu Yusheng, et al. 2019. “Brain Cholesterol Metabolism and Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 34 (3): 386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez Laura, Dube Umber, Budde John, Black Kathleen, Medvedeva Alexandra, Davis Albert A., Perlmutter Joel S., Benitez Bruno A., and Cruchaga Carlos. 2017. “TMEM230 in Parkinson’s Disease.” Neurobiology of Aging 56 (August): 212.e1–212.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet A-M, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, and Brice A. 2004. “Causal Relation between Alpha-Synuclein Gene Duplication and Familial Parkinson’s Disease.” The Lancet 364 (9440): 1169–71. [DOI] [PubMed] [Google Scholar]
- Investigators, The All of Us Research Program, and The All of Us Research Program Investigators. 2019. “The ‘All of Us’ Research Program.” New England Journal of Medicine. 10.1056/nejmsr1809937. [DOI] [Google Scholar]
- Iqbal Zafar, and Toft Mathias. 2019. “TMEM230 Variants in Parkinson’s Disease.” Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Iwaki Hirotaka, Blauwendraat Cornelis, Leonard Hampton L., Kim Jonggeol J., Liu Ganqiang, Jodi Maple-Grødem Jean-Christophe Corvol, et al. 2019. “Genomewide Association Study of Parkinson’s Disease Clinical Biomarkers in 12 Longitudinal Patients’ Cohorts.” Movement Disorders: Official Journal of the Movement Disorder Society 34 (12): 1839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger Kurt A., and Korczyn Amos D.. 2018. “Are Dementia with Lewy Bodies and Parkinson’s Disease Dementia the Same Disease?” BMC Medicine. 10.1186/s12916-018-1016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser R, Hofer A, Grapengiesser A, Gasser T, Kupsch A, Roots I, and Brockmöller J. 2003. “L-Dopa-Induced Adverse Effects in PD and Dopamine Transporter Gene Polymorphism.” Neurology 60 (11): 1750–55. [DOI] [PubMed] [Google Scholar]
- Kane Lesley A., Lazarou Michael, Fogel Adam I., Li Yan, Yamano Koji, Sarraf Shireen A., Banerjee Soojay, and Youle Richard J.. 2014. “PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity.” The Journal of Cell Biology 205 (2): 143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara Eleanna, Xiromerisiou Georgia, Spanaki Cleanthe, Bozi Maria, Koutsis Georgios, Panas Marios, Dardiotis Efthimios, et al. 2014. “Assessment of Parkinson’s Disease Risk Loci in Greece.” Neurobiology of Aging 35 (2): 442.e9–442.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kia Demis A., Noyce Alastair J., White Jon, Speed Doug, Nicolas Aude, IPDGC collaborators Stephen Burgess, et al. 2018. “Mendelian Randomization Study Shows No Causal Relationship between Circulating Urate Levels and Parkinson’s Disease.” Annals of Neurology 84 (2): 191–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kia Demis A., Sabir Marya S., Ahmed Sarah, Trinh Joanne, Bandres-Ciga Sara, and International Parkinson’s Disease Genomics Consortium. 2018. “LRP10 in α-Synucleinopathies.” Lancet Neurology. [Google Scholar]
- Kiely Aoife P., Asi Yasmine T., Kara Eleanna, Limousin Patricia, Ling Helen, Lewis Patrick, Proukakis Christos, et al. 2013. “α-Synucleinopathy Associated with G51D SNCA Mutation: A Link between Parkinson’s Disease and Multiple System Atrophy?” Acta Neuropathologica 125 (5): 753–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Andrés D., and Mazzulli Joseph R.. 2018. “Is Parkinson’s Disease a Lysosomal Disorder?” Brain: A Journal of Neurology 141 (8): 2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemann CJH, Martens GJM, Sharma M, Martens MB, Isacson O, Gasser T, Visser JE, and Poelmans G. 2017. “Integrated Molecular Landscape of Parkinson’s Disease.” Npj Parkinson’s Disease 3 (1): 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy Soumya, Rajan Roopa, Banerjee Moinak, Kumar Hardeep, Sarma Gangadhara, Krishnan Syam, Sarma Sankara, and Kishore Asha. 2016. “Dopamine D3 Receptor Ser9Gly Variant Is Associated with Impulse Control Disorders in Parkinson’s Disease Patients.” Parkinsonism & Related Disorders. 10.1016/j.parkreldis.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, and Riess O. 1998. “Ala30Pro Mutation in the Gene Encoding Alpha-Synuclein in Parkinson’s Disease.” Nature Genetics 18 (2): 106–8. [DOI] [PubMed] [Google Scholar]
- Kuusimäki Tomi, Korpela Jaana, Pekkonen Eero, Martikainen Mika H., Antonini Angelo, and Kaasinen Valtteri. 2019. “Deep Brain Stimulation for Monogenic Parkinson’s Disease: A Systematic Review.” Journal of Neurology. 10.1007/s00415-019-09181-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara Tomoki, Inoue Keiichi, D’Agati Vivette D., Fujimoto Tetta, Eguchi Tomoya, Saha Shamol, Wolozin Benjamin, Iwatsubo Takeshi, and Abeliovich Asa. 2016. “LRRK2 and RAB7L1 Coordinately Regulate Axonal Morphology and Lysosome Integrity in Diverse Cellular Contexts.” Scientific Reports 6 (July): 29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston J. William, Schüle Birgitt, Rees Linda, Nichols R. Jeremy, and Barlow Carrolee. 2015. “Multisystem Lewy Body Disease and the Other Parkinsonian Disorders.” Nature Genetics 47 (12): 1378–84. [DOI] [PubMed] [Google Scholar]
- de Lau Lonneke M. L., Verbaan Dagmar, Marinus Johan, Heutink Peter, and van Hilten Jacobus J.. 2012. “Catechol-O-Methyltransferase Val158Met and the Risk of Dyskinesias in Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 27 (1): 132–35. [DOI] [PubMed] [Google Scholar]
- Lawlor Deborah A., Wade Kaitlin, Maria Carolina Borges Tom Palmer, Fernando Pires Hartwig Gibran Hemani, and Bowden Jack. n.d. “A Mendelian Randomization Dictionary: Useful Definitions and Descriptions for Undertaking, Understanding and Interpreting Mendelian Randomization Studies.” 10.31219/osf.io/6yzs7. [DOI] [Google Scholar]
- Lee Jun Sung, Kanai Kazuaki, Suzuki Mari, Kim Woojin S., Han Soo Yoo Yuhong Fu, Kim Dong-Kyu, et al. 2019. “Arylsulfatase A, a Genetic Modifier of Parkinson’s Disease, Is an α-Synuclein Chaperone.” Brain: A Journal of Neurology, July 10.1093/brain/awz205. [DOI] [PubMed] [Google Scholar]
- Lee Richard G., Sedghi Maryam, Salari Mehri, Shearwood Anne-Marie J., Stentenbach Maike, Kariminejad Ariana, Goullee Hayley, et al. 2018. “Early-Onset Parkinson Disease Caused by a Mutation in CHCHD2 and Mitochondrial Dysfunction.” Neurology. Genetics 4 (5): e276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard Hampton, Blauwendraat Cornelis, Krohn Lynne, Faghri Faraz, Iwaki Hirotaka, Furgeson Glen, Day-Williams Aaron G., et al. 2019. “Genetic Variability and Potential Effects on Clinical Trial Outcomes: Perspectives in Parkinson’s Disease.” 10.1101/427385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage Suzanne, Anheim Mathieu, Letournel Franck, Bousset Luc, Aurélie Honoré Nelly Rozas, Pieri Laura, et al. 2013. “G51D α-Synuclein Mutation Causes a Novel Parkinsonian-Pyramidal Syndrome.” Annals of Neurology 73 (4): 459–71. [DOI] [PubMed] [Google Scholar]
- Lesage Suzanne, and Brice Alexis. 2009. “Parkinson’s Disease: From Monogenic Forms to Genetic Susceptibility Factors.” Human Molecular Genetics 18 (R1): R48–59. [DOI] [PubMed] [Google Scholar]
- Lesage Suzanne, Drouet Valérie, Majounie Elisa, Deramecourt Vincent, Jacoupy Maxime, Nicolas Aude, Cormier-Dequaire Florence, et al. 2016. “Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy.” American Journal of Human Genetics 98 (3): 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Ganqiang, Boot Brendon, Locascio Joseph J., Jansen Iris E., Sophie Winder-Rhodes Shirley Eberly, Elbaz Alexis, et al. 2016. “Specifically Neuropathic Gaucher’s Mutations Accelerate Cognitive Decline in Parkinson’s.” Annals of Neurology. 10.1002/ana.24781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Rui, Guo Xuguang, Park Yikyung, Huang Xuemei, Sinha Rashmi, Freedman Neal D., Hollenbeck Albert R., Blair Aaron, and Chen Honglei. 2012. “Caffeine Intake, Smoking, and Risk of Parkinson Disease in Men and Women.” American Journal of Epidemiology 175 (11): 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Xiao, Li Weihua, Liu Guixia, Shen Xu, and Tang Yun. 2015. “Association between Cigarette Smoking and Parkinson’s Disease: A Meta-Analysis.” Archives of Gerontology and Geriatrics. 10.1016/j.archger.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Lohmueller Kirk E., Pearce Celeste L., Pike Malcolm, Lander Eric S., and Hirschhorn Joel N.. 2003. “Meta-Analysis of Genetic Association Studies Supports a Contribution of Common Variants to Susceptibility to Common Disease.” Nature Genetics 33 (2): 177–82. [DOI] [PubMed] [Google Scholar]
- Ma Dongrui, Jia Nee Foo Ebonne Yulin Ng, Zhao Yi, Liu Jian-Jun, and Tan Eng King. 2017. “Screening for TMEM230 Mutations in Young-Onset Parkinson’s Disease.” Neurobiology of Aging 58 (October): 239.e9–239.e10. [DOI] [PubMed] [Google Scholar]
- Makarious Mary B., Monica Diez-Fairen Lynne Krohn, Blauwendraat Cornelis, Sara Bandres-Ciga Jinhui Ding, Lasse Pihlstrøm Henry Houlden, Scholz Sonja W., and Gan-Or Ziv. 2019. “ARSA Variants in α-Synucleinopathies.” Brain: A Journal of Neurology, October 10.1093/brain/awz340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio Teri A., Collins Francis S., Cox Nancy J., Goldstein David B., Hindorff Lucia A., Hunter David J., McCarthy Mark I., et al. 2009. “Finding the Missing Heritability of Complex Diseases.” Nature 461 (7265): 747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Mark Marianne, Brouwer Maartje, Kromhout Hans, Nijssen Peter, Huss Anke, and Vermeulen Roel. 2012. “Is Pesticide Use Related to Parkinson Disease? Some Clues to Heterogeneity in Study Results.” Environmental Health Perspectives 120 (3): 340–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masellis Mario, Collinson Shannon, Freeman Natalie, Tampakeras Maria, Levy Joseph, Tchelet Amir, Eyal Eli, et al. 2016. “Dopamine D2 Receptor Gene Variants and Response to Rasagiline in Early Parkinson’s Disease: A Pharmacogenetic Study.” Brain. 10.1093/brain/aww109. [DOI] [PubMed] [Google Scholar]
- Nalls Mike A., Blauwendraat Cornelis, Vallerga Costanza L., Heilbron Karl, Sara Bandres-Ciga Diana Chang, Tan Manuela, et al. 2019. “Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies.” Lancet Neurology 18 (12): 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls Mike A., Bras Jose, Hernandez Dena G., Keller Margaux F., Majounie Elisa, Renton Alan E., Saad Mohamad, et al. 2015. “NeuroX, a Fast and Efficient Genotyping Platform for Investigation of Neurodegenerative Diseases.” Neurobiology of Aging 36 (3): 1605.e7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls Mike A., McLean Cory Y., Rick Jacqueline, Eberly Shirley, Hutten Samantha J., Gwinn Katrina, Sutherland Margaret, et al. 2015. “Diagnosis of Parkinson’s Disease on the Basis of Clinical and Genetic Classification: A Population-Based Modelling Study.” Lancet Neurology 14 (10): 1002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls Mike A., Pankratz Nathan, Lill Christina M., Do Chuong B., Hernandez Dena G., Saad Mohamad, DeStefano Anita L., et al. 2014. “Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease.” Nature Genetics 46 (9): 989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce Alastair J., Sara Bandres-Ciga Jonggeol Kim, Heilbron Karl, Kia Demis, Hemani Gibran, Xue Angli, et al. 2019. “The Parkinson’s Disease Mendelian Randomization Research Portal.” Movement Disorders: Official Journal of the Movement Disorder Society, October 10.1002/mds.27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce Alastair J., Bestwick Jonathan P., Laura Silveira-Moriyama Christopher H. Hawkes, Giovannoni Gavin, Lees Andrew J., and Schrag Anette. 2012. “Meta-Analysis of Early Nonmotor Features and Risk Factors for Parkinson Disease.” Annals of Neurology. 10.1002/ana.23687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce Alastair J., Kia Demis A., Hemani Gibran, Nicolas Aude, Price T. Ryan, De Pablo-Fernandez Eduardo, Haycock Philip C., et al. 2017. “Estimating the Causal Influence of Body Mass Index on Risk of Parkinson Disease: A Mendelian Randomisation Study.” PLoS Medicine 14 (6): e1002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce Alastair J., Lea R’Bibo Luisa Peress, Bestwick Jonathan P., Adams-Carr Kerala L., Mencacci Niccolo E., Hawkes Christopher H., et al. 2017. “PREDICT-PD: An Online Approach to Prospectively Identify Risk Indicators of Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (2): 219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Eon Joo, Grabińska Kariona A., Guan Ziqiang, Stránecký Viktor, Hartmannová Hana, Hodaňová Kateřina, Barešová Veronika, et al. 2014. “Mutation of Nogo-B Receptor, a Subunit of Cis-Prenyltransferase, Causes a Congenital Disorder of Glycosylation.” Cell Metabolism 20 (3): 448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Jeehye, Sung Bae Lee Sungkyu Lee, Kim Yongsung, Song Saera, Kim Sunhong, Bae Eunkyung, et al. 2006. “Mitochondrial Dysfunction in Drosophila PINK1 Mutants Is Complemented by Parkin.” Nature 441 (7097): 1157–61. [DOI] [PubMed] [Google Scholar]
- Pasanen Petra, Myllykangas Liisa, Siitonen Maija, Raunio Anna, Kaakkola Seppo, Lyytinen Jukka, Tienari Pentti J., Pöyhönen Minna, and Paetau Anders. 2014. “Novel α-Synuclein Mutation A53E Associated with Atypical Multiple System Atrophy and Parkinson’s Disease-Type Pathology.” Neurobiology of Aging 35 (9): 2180.e1–5. [DOI] [PubMed] [Google Scholar]
- Pecanka Jakub, Jonker Marianne A., International Parkinson’S Disease Genomics Consortium (IPDGC), Bochdanovits Zoltan, and Van Der Vaart Aad W.. 2017. “A Powerful and Efficient Two-Stage Method for Detecting Gene-to-Gene Interactions in GWAS.” Biostatistics 18 (3): 477–94. [DOI] [PubMed] [Google Scholar]
- Pihlstrøm Lasse, Schottlaender Lucia, Chelban Viorica, Houlden Henry, and MSA Exome Consortium. 2018. “LRP10 in α-Synucleinopathies.” Lancet Neurology. [DOI] [PubMed] [Google Scholar]
- Poewe Werner, Seppi Klaus, Tanner Caroline M., Halliday Glenda M., Brundin Patrik, Volkmann Jens, Schrag Anette-Eleonore, and Lang Anthony E.. 2017. “Parkinson Disease.” Nature Reviews Disease Primers. 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH 1997. “Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease.” Science. 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Popejoy Alice B., and Fullerton Stephanie M.. 2016. “Genomics Is Failing on Diversity.” Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri Marialuisa, Breedveld Guido J., Chang Hsiu-Chen, Yeh Tu-Hsueh, Leonor Correia Guedes Vincenzo Toni, Fabrizio Edito, et al. 2017. “Mutations in TMEM230 Are Not a Common Cause of Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (2): 302–4. [DOI] [PubMed] [Google Scholar]
- Quadri Marialuisa, Mandemakers Wim, Grochowska Martyna M., Masius Roy, Geut Hanneke, Fabrizio Edito, Breedveld Guido J., et al. 2018. “LRP10 Genetic Variants in Familial Parkinson’s Disease and Dementia with Lewy Bodies: A Genome-Wide Linkage and Sequencing Study.” Lancet Neurology 17 (7): 597–608. [DOI] [PubMed] [Google Scholar]
- Requejo-Aguilar Raquel, Irene Lopez-Fabuel Daniel Jimenez-Blasco, Fernandez Emilio, Almeida Angeles, and Bolaños Juan P.. 2015. “DJ1 Represses Glycolysis and Cell Proliferation by Transcriptionally up-Regulating Pink1.” Biochemical Journal 467 (2): 303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck Mariana, Schumacher-Schuh Artur F., Altmann Vivian, Francisconi Carolina Lm, Fagundes Paulo Tb, Monte Thaís L., Callegari-Jacques Sidia M., Rieder Carlos Rm, and Hutz Mara H.. 2012. “DRD2 Haplotype Is Associated with Dyskinesia Induced by Levodopa Therapy in Parkinson’s Disease Patients.” Pharmacogenomics 13 (15): 1701–10. [DOI] [PubMed] [Google Scholar]
- Ritchie Marylyn D., and Van Steen Kristel. 2018. “The Search for Gene-Gene Interactions in Genome-Wide Association Studies: Challenges in Abundance of Methods, Practical Considerations, and Biological Interpretation.” Annals of Translational Medicine 6 (8): 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak Laurie A., Jansen Iris E., Jeroen van Rooij André G. Uitterlinden, Kraaij Robert, Jankovic Joseph, International Parkinson’s Disease Genomics Consortium (IPDGC), Heutink Peter, and Shulman Joshua M.. 2017. “Excessive Burden of Lysosomal Storage Disorder Gene Variants in Parkinson’s Disease.” Brain: A Journal of Neurology 140 (12): 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake Wataru, Nakabayashi Yuko, Mizuta Ikuko, Hirota Yushi, Ito Chiyomi, Kubo Michiaki, Kawaguchi Takahisa, et al. 2009. “Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease.” Nature Genetics 41 (12): 1303. [DOI] [PubMed] [Google Scholar]
- Shi Chang-He, Li Fang, Shi Meng-Meng, Yang Zhi-Hua, Mao Cheng-Yuan, Zhang Shu-Yu, Wang Hui, et al. 2017. “Genetic Analysis of the TMEM230 Gene in Chinese Han Patients with Parkinson’s Disease.” Scientific Reports 7 (1): 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Chang-He, Luo Hai-Yang, Fan Yu, Li Yu-Sheng, and Xu Yu-Ming. 2018. “LRP10 in α-Synucleinopathies.” Lancet Neurology. [DOI] [PubMed] [Google Scholar]
- Siitonen Ari, Nalls Michael A., Hernández Dena, Gibbs J. Raphael, Ding Jinhui, Ylikotila Pauli, Edsall Connor, Singleton Andrew, and Majamaa Kari. 2017. “Genetics of Early-Onset Parkinson’s Disease in Finland: Exome Sequencing and Genome-Wide Association Study.” Neurobiology of Aging 53 (May): 195.e7–195.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira CRA, MacKinley J, Coleman K, Li Z, Finger E, Bartha R, Morrow SA, et al. 2019. “Ambroxol as a Novel Disease-Modifying Treatment for Parkinson’s Disease Dementia: Protocol for a Single-Centre, Randomized, Double-Blind, Placebo-Controlled Trial.” BMC Neurology. 10.1186/s12883-019-1252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez Javier, Schulte Claudia, Bras Jose M., Sharma Manu, Gibbs J. Raphael, Berg Daniela, Paisan-Ruiz Coro, et al. 2009. “Genome-Wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease.” Nature Genetics 41 (12): 1308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, et al. 2003. “Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease.” Science 302 (5646): 841. [DOI] [PubMed] [Google Scholar]
- Singleton Andrew, and Hardy John. 2011. “A Generalizable Hypothesis for the Genetic Architecture of Disease: Pleomorphic Risk Loci.” Human Molecular Genetics 20 (R2): R158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer David, Alcalay Roy N., Garretti Francesca, Cote Lucien, Kanter Ellen, Julian Agin-Liebes Christopher Liong, et al. 2017. “T Cells from Patients with Parkinson’s Disease Recognize α-Synuclein Peptides.” Nature. 10.1038/nature22815. [DOI] [Google Scholar]
- Tejera-Parrado Cristina, Silvia Jesús Adrián López-Ruíz, Dolores Buiza-Rueda Marta Bonilla-Toribio, Inmaculada Bernal-Bernal María Teresa Periñán, Laura Vargas-González Pilar Gómez-Garre, and Mir Pablo. 2018. “TMEM230 in Parkinson’s Disease in a Southern Spanish Population.” PloS One 13 (5): e0197271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson Christelle, Christine Brefel-Courbon Jean-Christophe Corvol, Lesage Suzanne, Brice Alexis, and French Parkinson’s Disease Genetics Study Group. 2018. “LRP10 in α-Synucleinopathies.” Lancet Neurology. [DOI] [PubMed] [Google Scholar]
- Trerotola Marco, Relli Valeria, Simeone Pasquale, and Alberti Saverio. 2015. “Epigenetic Inheritance and the Missing Heritability.” Human Genomics 9 (July): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta Luca, Guella Ilaria, Giulia Soldà Francesca Sironi, Tesei Silvana, Canesi Margherita, Pezzoli Gianni, Goldwurm Stefano, Duga Stefano, and Asselta Rosanna. 2012. “SNCA and MAPT Genes: Independent and Joint Effects in Parkinson Disease in the Italian Population.” Parkinsonism & Related Disorders 18 (3): 257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Eeden SK 2003. “Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity.” American Journal of Epidemiology. 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- Vergouw Leonie J. M., Ruitenberg Annemieke, Tsz Hang Wong Shamiram Melhem, Breedveld Guido J., Criscuolo Chiara, De Michele Giuseppe, et al. 2019. “LRP10 Variants in Parkinson’s Disease and Dementia with Lewy Bodies in the South-West of the Netherlands.” Parkinsonism & Related Disorders, May 10.1016/j.parkreldis.2019.05.037. [DOI] [PubMed] [Google Scholar]
- Vilariño-Güell Carles, Rajput Alex, Milnerwood Austen J., Shah Brinda, Chelsea Szu-Tu Joanne Trinh, Yu Irene, et al. 2014. “DNAJC13 Mutations in Parkinson Disease.” Human Molecular Genetics 23 (7): 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Qianqian, Ou Ruwei, Zhou Qingqing, Chen Yongping, Cao Bei, Gu Xiaojing, Zhao Bi, Wu Ying, Song Wei, and Shang Hui-Fang. 2018. “TMEM230 Mutations Are Rare in Han Chinese Patients with Autosomal Dominant Parkinson’s Disease.” Molecular Neurobiology 55 (4): 2851–55. [DOI] [PubMed] [Google Scholar]
- Weissbrod Omer, Hormozdiari Farhad, Benner Christian, Cui Ran, Ulirsch Jacob, Gazal Steven, Schoech Armin P., et al. 2019. “Functionally-Informed Fine-Mapping and Polygenic Localization of Complex Trait Heritability.” bioRxiv. 10.1101/807792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin Nicola, Armean Irina M., Kleinman Aaron, Marshall Jamie L., Minikel Eric V., Goodrich Julia K., Quaife Nicholas M., et al. n.d. “Human Loss-of-Function Variants Suggest That Partial LRRK2 Inhibition Is a Safe Therapeutic Strategy for Parkinson’s Disease.” 10.1101/561472. [DOI] [Google Scholar]
- Williams Uduak, Bandmann Oliver, and Walker Richard. 2018. “Parkinson’s Disease in Sub-Saharan Africa: A Review of Epidemiology, Genetics and Access to Care.” Journal of Movement Disorders. 10.14802/jmd.17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis Wright, Allison Bradley A. Evanoff, Lian Min, Criswell Susan R., and Racette Brad A.. 2010. “Geographic and Ethnic Variation in Parkinson Disease: A Population-Based Study of US Medicare Beneficiaries.” Neuroepidemiology 34 (3): 143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Hongwei, Zheng Xiaosheng, Cen Zhidong, Xie Fei, Chen You, Lu Xingjiao, and Luo Wei. 2017. “Genetic Analysis of the TMEM230 Gene in Chinese Patients with Familial Parkinson Disease.” Parkinsonism & Related Disorders 36 (March): 105–6. [DOI] [PubMed] [Google Scholar]
- Yang Jian, Bakshi Andrew, Zhu Zhihong, Hemani Gibran, Vinkhuyzen Anna A. E., Sang Hong Lee Matthew R. Robinson, et al. 2015. “Genetic Variance Estimation with Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index.” Nature Genetics 47 (10): 1114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Xinglong, An Ran, Xi Jing, Zheng Jinhua, Chen Yalan, Huang Hongyan, Tian Sijia, Zhao Quanzhen, Ning Pingping, and Xu Yanming. 2017. “Sequencing TMEM230 in Chinese Patients with Sporadic or Familial Parkinson’s Disease.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (5): 800–802. [DOI] [PubMed] [Google Scholar]
- Yan Weiqian, Tang Beisha, Zhou Xiaoxia, Lei Lifang, Li Kai, Sun Qiying, Xu Qian, Yan Xinxiang, Guo Jifeng, and Liu Zhenhua. 2017. “TMEM230 Mutation Analysis in Parkinson’s Disease in a Chinese Population.” Neurobiology of Aging 49 (January): 219.e1–219.e3. [DOI] [PubMed] [Google Scholar]
- Young Alexander I. 2019. “Solving the Missing Heritability Problem.” PLoS Genetics 15 (6): e1008222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabetian Cyrus P., Mata Ignacio F., and Latin American Research Consortium on the Genetics of PD (LARGE-PD). 2017. “LARGE-PD: Examining the Genetics of Parkinson’s Disease in Latin America.” Movement Disorders: Official Journal of the Movement Disorder Society 32 (9): 1330–31. [DOI] [PubMed] [Google Scholar]
- Zarranz Juan J., Alegre Javier, Gómez-Esteban Juan C., Lezcano Elena, Ros Raquel, Ampuero Israel, Vidal Lídice, et al. 2004. “The New Mutation, E46K, of Alpha-Synuclein Causes Parkinson and Lewy Body Dementia.” Annals of Neurology 55 (2): 164–73. [DOI] [PubMed] [Google Scholar]
- Zech Michael, Georg Nübling Florian Castrop, Jochim Angela, Schulte Eva C., Mollenhauer Brit, Lichtner Peter, et al. 2013. “Niemann-Pick C Disease Gene Mutations and Age-Related Neurodegenerative Disorders.” PloS One 8 (12): e82879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Dongfeng, Jiang Hong, and Xie Junxia. 2014. “Alcohol Intake and Risk of Parkinson’s Disease: A Meta-Analysis of Observational Studies.” Movement Disorders: Official Journal of the Movement Disorder Society 29 (6): 819–22. [DOI] [PubMed] [Google Scholar]
- Zhang Mingming, Mu Hongbo, Shang Zhenwei, Kang Kai, Lv Hongchao, Duan Lian, Li Jin, et al. 2017. “Genome-Wide Pathway-Based Association Analysis Identifies Risk Pathways Associated with Parkinson’s Disease.” Neuroscience. 10.1016/j.neuroscience.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Zhu Zhihong, Bakshi Andrew, Vinkhuyzen Anna A. E., Hemani Gibran, Sang Hong Lee Ilja M. Nolte, van Vliet-Ostaptchouk Jana V., et al. 2015. “Dominance Genetic Variation Contributes Little to the Missing Heritability for Human Complex Traits.” American Journal of Human Genetics 96 (3): 377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk Or, Hechter Eliana, Sunyaev Shamil R., and Lander Eric S.. 2012. “The Mystery of Missing Heritability: Genetic Interactions Create Phantom Heritability.” Proceedings of the National Academy of Sciences of the United States of America 109 (4): 1193–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Others, 2019b. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. BioRxiv 388165. [Google Scholar]