

Abstract
Neurodevelopmental programming — the implementation of the genetic and epigenetic blueprints that guide and coordinate normal brain development — requires tight regulation of transcriptional processes. During prenatal and postnatal time periods, epigenetic processes fine-tune neurodevelopment towards an end product that determines how an organism interacts with and responds to exposures and experiences throughout life. Epigenetic processes also have the ability to reprogramme the epigenome in response to environmental challenges, such as maternal stress, making the organism more or less adaptive depending on the future challenges presented. Epigenetic marks generated within germ cells as a result of environmental influences throughout life can also shape future generations long before conception occurs.
Environmental changes that perturb an animal’s homeostasis — including stress, nutritional challenges and infection — can influence the developing and maturing brain and thereby alter the original blueprint and establish a new one in a process known as reprogramming1. There is a great history of studies that have examined this process, and it has recently become clear that epigenetic mechanisms, including DNA methylation, histone modification and the actions of small non-coding RNAs, have an important role in such neurodevelopmental reprogramming2–5. However, the precise mechanisms by which exposure to environmental perturbations can influence long-term outcomes are highly complex and depend on the type and the severity of the exposure, whether it occurs during a window of vulnerability in the target circuit or system, the organismal sex and whether there are epigenetic changes in germ cells (FIG. 1).
Figure 1 |. Complex interactions between the maternal milieu, placenta and fetal compartments during gestation.
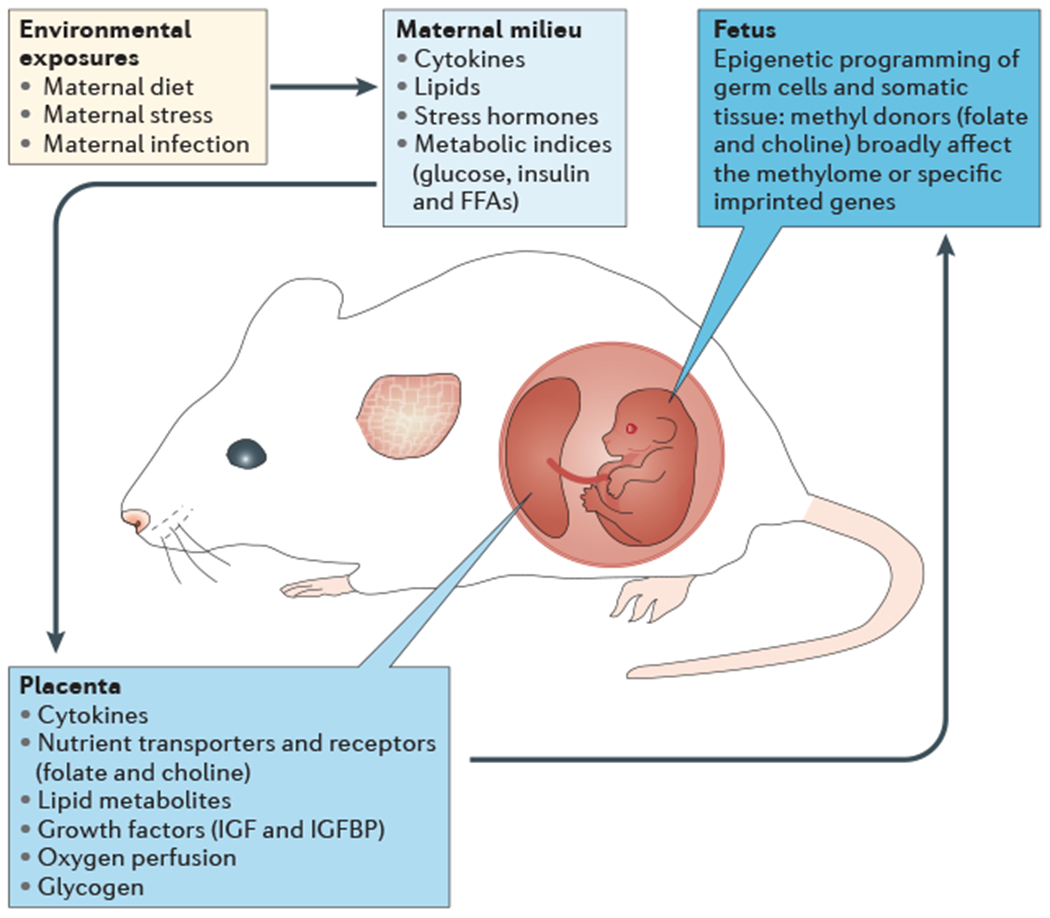
Fetal antecedents such as maternal dietary deficiencies, chronic stress or infection can promote endocrine disruptions in the maternal milieu, including increases in pro-inflammatory cytokines and stress hormones, and shifts in metabolic indices. In addition, these environmental exposures can indirectly alter placental development and function by changing local energy metabolism and lipid storage and metabolism. This can alter the transmission of key nutrients that are important for fetal growth and brain development, including growth factors and methyl donor nutrients such as folate and choline, which can affect fetal somatic and germ cell epigenetic programming. FFAs, free fatty acids; IGF, insulin-like growth factor; IGFBP, IGF-binding protein.
The timing of an environmental exposure or experience may determine whether the event has limited and specific epigenetic effects or whether it more broadly affects the epigenome (BOX 1) as well as how long the effect persists (acutely or across generations). As epigenetic processes are integrally involved in neurodevelopment and maturation, insults that are experienced during these key periods (which roughly correspond to early or late gestation) would be predicted to reprogramme the epigenome more extensively and, if also incorporated into the germ cells, their effects may become transgenerational1. By contrast, events that occur outside a sensitive period of brain development (that is, as an adult) may be more limited in their effect and duration, as they may produce specific epigenetic changes that do not manifest in epigenomic or germ cell reprogramming.
Box 1 |. Defining epigenetics and epigenomics.
The definition of the term ‘epigenetic’ has been subject to continuous evolution. Waddington172 coined the term to describe the premise that genes can respond to a dynamic environment and thereby shape phenotypes such that they are acutely advantageous to the individual (as opposed to advantageous on a much longer evolutionary scale). A more recent operational definition narrowed the usage to include the necessity of heritability, either across generations or to daughter cells, which involves the erasure and subsequent replacement of epigenetic marks173. However, over the past decade, developmental and behavioural neuroscientists have used the term ‘epigenetic modifications’ to refer to functionally relevant chromatin modifications that alter gene expression but that do not involve a change in DNA sequence. Recent studies have produced mechanistic insight both at the level of epigenetics, which examines changes in the expression of specific genes or loci that result from the addition of epigenetic marks, such as DNA methylation, or from changes in the levels of a given microRNA, and at the level of epigenomics, which examines genome-wide changes in gene expression that can be attributed to broad chromatin modifications. Such outcomes, which might be directed by a given histone modification (such as histone H3 lysine 4 (H3K4) acetylation) or by changes in the methylome, can influence large sets of genes that are involved in specific functions and that generate changes in developmental programming in response to an insult or a perturbation9.
According to the Barker hypothesis6, neurodevelopmental reprogramming induces adaptations to the early environment in anticipation of a similar postnatal environment. When there is a mismatch between these two environments, such reprogramming may increase disease risk. For example, if a mother is exposed to increased stress during pregnancy but the postpartum environment is not stressful, the offspring’s brain may continue to respond inappropriately to stressors that are experienced throughout life6.
Recent reviews in this area have focused on the specific developmental periods (including prenatal, postnatal and pubertal periods) during which such reprogramming occurs, the types of stimuli that are involved (including specific diets and exposure to infection or to stress) and specific epigenetic outcomes2–5,7–9. The goal of this Review is to integrate these points and highlight potential shared mechanisms that are important in reprogramming the developing brain. I examine results from human and animal studies that shed light on the factors that direct and shape brain development, and I discuss how differing parental contributions may affect offspring disease risk and resilience. In particular, I examine transgenerational epigenetic effects on neurodevelopment and discuss the interactions between specific perturbations, developmental windows of susceptibility and the sex-specificity of these effects.
Developmental windows of susceptibility
Studies evaluating susceptibility factors in neurodevelopmental disorders have indicated that certain outcomes are commonly linked to exposure to different types of gestational and early-life environments. For example, exposure to stress or infection during either of these developmental periods is linked to an increased risk of depression, schizophrenia and autism spectrum disorders (ASDs)1,10–15. This suggests that these insults may promote common mechanisms irrespective of the time at which they occurred in development. However, epidemiological evidence also supports the idea that the time of exposure during development may be important for some types of environmental challenge, as similar perturbations occurring at earlier or later stages have been associated with different long-term outcomes1,16–18. Prepubertal and pubertal brain maturation has been less well examined; however, these periods also seem to be susceptible to environmental disturbances. This is especially true for the prefrontal cortex and its crucial connections within the limbic system, in which disruptions have been linked to an increased risk of attention deficit hyperactivity disorder (ADHD) and schizophrenia19–21.
The importance of the prenatal period.
The best evidence that the timing of insults is a factor in disease risk comes from birth cohort studies examining the effects of maternal (prepartum) exposure to specific stress or immune challenges. These studies compile birth registries that enable prospective studies to be carried out and thus may have access to more accurate records of the exposure under investigation than do retrospective studies. They also allow the collection of plasma and tissue samples, which enables potential biomarkers and genetic and/or epigenetic factors associated with susceptibility to be assessed. For example, birth cohort studies have revealed strong associations between in utero exposure to infection, hypoxia or starvation and an increased risk of schizophrenia22,23, as well as an association with impairments in executive function in patients with schizophrenia15, which indicates that prenatal brain development, not surprisingly, is a uniquely susceptible period. Furthermore, recent studies showed marked changes in cortical layer patterning, which is a prenatal process, in children with ASD, suggesting that a disturbance occurred before birth24, However, in-depth neurodevelopmental assessments that compare similar perturbations across these prenatal and postnatal periods will be necessary to identify a specific period of development as a crucial factor in disease risk.
There is growing evidence that there are specific developmental windows of increased vulnerability throughout pregnancy itself10,25–27. Indeed, a recent epidemiological study reported an association between maternal stress that is experienced specifically during the first trimester of pregnancy and an increased risk of schizophrenia in male offspring28. Exposure to maternal infection and high levels of pro-inflammatory cytokines during early or mid-gestation was also associated with an increased risk of neurodevelopmental disorders18. For example, children of women who were exposed to influenza during early or mid-pregnancy, but not during late gestation, had a threefold higher risk of schizophrenia than those of women who had not been exposed13. Similarly, studies using the Danish medical birth register reported that maternal viral infection resulting in hospitalization in the first trimester, but not later in pregnancy, was associated with a threefold higher risk of ASD in the child29,30. Furthermore, the Early Markers for Autism study found increased levels of 17 cytokines in early and mid-gestation in the mothers of children that were later diagnosed with ASD31. These studies broadly suggest that early pregnancy is a particularly sensitive period in development, during which time insults such as stress and inflammation can promote programming changes that will increase the risk of disease. This increased vulnerability may lie in the sensitivity of the brain, the reactivity of the mother in early gestation and/or the ability of the fetus to recover from such insults.
In addition to stress or infection during pregnancy, reduced maternal nutrition can also have a negative influence on fetal growth and can induce epigenetic changes, thereby influencing fetal brain development. Long-term epidemiological studies investigating famine exposures during specified periods of pregnancy or postnatal life are beginning to discern associated risks for neuropsychiatric disease in first- and second-generation offspring. Therefore, these studies have the potential to uncover transgenerational epigenetic mechanisms as well as direct effects on neurodevelopment. Initial studies have reported an increased risk of neurodevelopmental disorders in children of women who were exposed to severe caloric restriction or famine during pregnancy compared with other periods of postnatal development32,33. For instance, children of women who were pregnant during the Dutch ‘hunger winter’ of 1944–1945 were more likely to have altered stress responsiveness and schizophrenia as adults32,34. These outcomes were also associated with changes in circulating growth factors and altered DNA methylation of several genes, including the maternally imprinted gene insulin-like growth factor 2 (IGF2), which were measured 60 years after the famine exposure35. In addition, their second-generation offspring had increased adiposity and overall worse health as adults32. These transgenerational outcomes suggest that the initial maternal dietary restriction induced epigenetic marks in fetal germ cells and thus the first-generation offspring were able to transmit a phenotype without re-exposure to the insult. Of course, epidemiological studies are limited by an inability to control for the effects of additional maternal or paternal lifetime experiences and/or interactions with the maternal milieu in subsequent generations. In addition, it is important to note that famine experience is also an enormous physiological and psychological stressor, and stress insults are also metabolic challenges. Therefore, it is not surprising that both maternal stress and maternal famine early in pregnancy produce similar neurodevelopmental disorder risk13–15,32.
The current obesity epidemic in Western society has shifted the focus from understanding the effect of undernutrition to examining the effects of maternal overnutrition on neurodevelopment. Indeed, it has been proposed that transgenerational epigenetic mechanisms may contribute to the rapid rise in obesity rates1. Maternal diet and body composition are less dynamic factors than stress or infection and are therefore more difficult to designate to a specific period of gestation or even to be limited to the prenatal environment. However, maternal body mass and obesity preceding and during pregnancy have been linked to severity in childhood ADHD symptoms and other executive function deficits36,37. Exposure to maternal obesity during gestation has also been associated with a twofold–threefold increased risk of schizophrenia in offspring in two separate birth cohorts32,33.
As is clear from the findings described above, gestation is one of the most studied developmental windows of vulnerability38,39 and is a critical time in neurodevelopmental programming13–15,24,32. In this context, it is important to consider that early pregnancy involves processes such as embryo implantation and placental differentiation, whereas mid- and late gestation are characterized by somatic growth, including considerable brain development40. Therefore, exposure to environmental challenges will influence different developmental processes depending on when in pregnancy the exposure occurred. In addition, the primordial germ cells, which will give rise to the next generation, undergo remethylation of imprinted genes before birth in males, whereas this occurs postnatally in females40. Therefore, if an insult occurs during a period in which the germ cells are undergoing this normal epigenetic process, changes could be incorporated that would give rise to a phenotype in future generations.
Mechanisms of reprogramming
Animal models have been used in an attempt to mimic the epidemiological findings described above by examining offspring outcomes that result from specific types of insults during specific periods of development. These studies have provided an insight into the mechanisms that are involved in reprogramming the brain. Rodent brain development begins during gestation but mostly continues postnatally. Therefore, using these animal models, researchers have been able to examine offspring phenotypes that are relevant to neurodevelopmental disorders by studying maternal exposure to stress, dietary challenges and infection both during pregnancy as well as in the early postnatal period.
Prenatal maternal stress.
Studies in which the pregnant dam is exposed to perturbations that provoke activation of her neuroendocrine stress system have shown that the effect of maternal stress during pregnancy on offspring outcomes, including on stress sensitivity, reward and cognition, depends on the gestational timing of the exposure (as well as on the sex of the fetus)41–47. In many species, including mice, rats, guinea pigs and non-human primates, maternal stress increases offspring stress sensitivity such that the overall production of glucocorticoids following hypothalamic–pituitary–adrenal stress axis (HPA stress axis) activation is increased, behavioural stress responses are heightened and cognitive deficits are present, all of which are endophenotypes that are associated with neuropsychiatric disease42,46,48–56.
To compare the effects of maternal stress experience across early, mid- or late pregnancy on the programming of offspring stress outcomes, studies in mice and guinea pigs have used a chronic stress paradigm42,46,49,50. In mice, exposure to stress very early in pregnancy (embryonic days 1–7) resulted in male, but not female, offspring that showed increased immobility in both tail suspension and forced swim tests as well as cognitive deficits in spatial learning and memory tasks as adults42,50. Early stress exposure also affected the programming of stress neurocircuitry: male offspring showed increased corticosterone production in response to acute restraint stress, and this was associated with increased levels of corticotropin-releasing factor (CRF) in the amygdala and reduced hippocampal glucocorticoid receptor expression42. These changes were associated with corresponding alterations in promoter methylation, which suggests that the prenatal stress experience resulted in long-term epigenetic reprogramming of genes involved in the development of stress neurocircuitry. The timing specificity of the effects of prenatal stress was similarly explored using short periods of daily flashing light as a maternal stressor in guinea pigs at mid- or late gestation49. Adult offspring that were exposed to stress in mid-gestation showed increased anxiety-like behaviours, increased basal corticosterone levels and lower hippocampal glucocorticoid receptor expression; however, late-gestation stress did not produce these outcomes. This evidence for specific effects of maternal stress during mid-gestation complements the evidence arising from the epidemiological studies described above28,47,49.
Interestingly, in many studies, male offspring are the most severely affected sex. They often present with a ‘dysmasculinized’ phenotype; that is, the prenatally stressed males seem to be more similar to control females than to control males in terms of their behaviours, physiology and gene expression patterns. For instance, male mice exposed to maternal stress very early in gestation showed significant changes in spatial learning and memory performance in a Barnes maze and used ‘female-like’ strategies to solve the task50. In addition, these mice showed dysmasculinized patterns of stress physiology, behaviour and cognitive performance, which indicates a potential disruption in normal perinatal brain masculinization44,52. Similar dysmasculinizing results have been reported in prenatally stressed rats and guinea pigs43,46,47,49,57,58. Interestingly, several recent studies have reported an association between steroidogenic activity and testosterone levels in fetuses and autism or autistic-like traits in young children59, and low serum testosterone levels have been associated with certain schizophrenia symptoms such as reduced verbal and working memory in males60. Fetal testosterone levels also predict the area of the sexually dimorphic grey matter in the human brain61, and male patients with schizophrenia have orbitofrontal cortex-to-amygdala volume ratios that are more similar to those of control women than to those of control men62. These findings support the idea that brain masculinization is an important part of normal brain development in males and suggest that this process may be a point of vulnerability to perturbations in the environment.
Postnatal maternal stress.
Complex interactions between the neonate and its mother occur in the postnatal period, and maternal behaviours such as nursing and grooming can strongly influence neurodevelopment63–77. Rodent studies have shown that both enhanced and fragmented maternal care in the early postnatal period can result in marked differences in the behaviours of the adult offspring. It is well established that the quality of maternal care influences epigenetic processes in the developing brain8,73,78. This was first shown in a rat model in which dams showed consistent high or low levels of maternal care — identified as increased or decreased licking and grooming — and this has since been demonstrated in many additional species, including mice, hamsters and non-human primates65–67,70,73,76,79–82. These studies have shown that the mother’s interactions with her offspring predict their behavioural and hormonal responses to stress in adulthood. Specifically, in rats, high rates of licking and grooming by the mother were associated with more rapid negative feedback to the HPA stress axis as a result of enhanced glucocorticoid receptor sensitivity as well as reduced levels of anxiety-like behaviours72. At an epigenetic level, these outcomes were correlated with changes in DNA methylation of the glucocorticoid receptor promoter and in glucocorticoid receptor expression in the hippocampus74. Although other genes are probably also affected, the glucocorticoid receptor gene, Nr3c1, seems to have been the most consistently examined across models and studies. NR3C1 expression and promoter methylation has also been examined in humans following early-life trauma, showing a potentially translational outcome for an epigenetic finding from animal models83,84. However, to adequately address the question of how maternal stress during pregnancy ultimately reprogrammes the developing brain, it is necessary to consider the epigenome and the broader epigenetic mechanisms that would be capable of establishing changes that are marked enough to alter organismal behaviour decades later.
Chronic stress during the postnatal period — affecting the dam and/or the pups — can also affect the cognitive performance of adult offspring through functional and structural changes in specific brain regions, including the hippocampus and the hypothalamus. For example, in a rat model of altered early postpartum maternal care resulting from the stress of repeated maternal separation, the offspring showed lifelong epigenetic changes in the hippocampus68. Specifically, the ability of a transcriptional repressor to bind to Crf ‘was decreased because of increased DNA methylation, and this was associated with a reduced ability of the offspring to appropriately respond to and to cope during stressful experiences as adults. This shows an interesting epigenetic reprogramming effect in response to postnatal stress that opposes changes that have been observed following prenatal stress: that is, the Crf promoter was hypomethylated following postnatal stress42. This again highlights the importance of the timing of environmental exposures in producing their long-term consequences. Interestingly, similar long-term changes in the expression of CRF in the hypothalamus were also found in offspring in a model of fragmented maternal care, in which the rat dam is given limited and insufficient material with which to build her nest, suggesting that alterations in maternal behaviour during early postnatal life can drive similar epigenetic events in her offspring65,70.
Maternal nutrition and obesity.
Rodent studies have shown a link between prenatal availability of nutrients and offspring brain development85. In mice, females receiving a protein-deficient diet throughout pregnancy and lactation produced offspring with intrauterine growth restriction and marked changes in dopamine circuitry and reward-related behaviours as adults86. Interestingly, the dopamine reward neurocircuitry was also affected when pregnant dams were fed a high-fat diet throughout pregnancy and lactation, which suggests that the developing reward system is particularly sensitive to gestational nutrition (probably stemming from an evolutionary benefit of linking motivational behaviours with caloric need)87–92. Reduced reward sensitivity (anhedonia) is a symptom of depression in humans; thus, there may be a connection between prenatal dietary perturbations and an increased risk of affective disorders or addictions. As described above, maternal undernutrition also increases stress responsivity in pregnant females and thus contributes to offspring phenotypes that are similar to those produced by maternal stress93. For example, rats that were exposed to 50% food restriction during pregnancy had increased plasma corticosterone levels and their adult offspring had higher adrenal weights94. These maternally food-restricted offspring also showed changes in the expression of key stress genes, including reduced levels of hippocampal Nr3c1 expression. Furthermore, exposure of female guinea pigs to nutrient restriction for 2 days in the last third of pregnancy increased maternal and fetal cortisol levels and reduced the expression of glucocorticoid receptor in the offspring hypothalamus and hippocampus in adulthood95. These outcomes are similar to those reported in studies investigating the effects of prenatal stress and maternal dexamethasone treatment1,22,42,55,56,94,96–98, which suggests that common signals are produced in the maternal milieu during maternal stress and undernutrition. These common signals are probably related to the similar catabolic metabolism that occurs during these experiences93.
Maternal dietary challenges also affect the maternal and fetal immune systems. For instance, increases in the number of hypothalamic microglia and pro-inflammatory cytokine levels were found in fetuses of macaque mothers who received a high-fat diet before and during pregnancy99. As juveniles, the female, but not the male, offspring showed increased anxiety-like behaviours100. Similarly, increased numbers of activated microglia in the hippocampus were reported for the adult offspring of rat dams fed a diet high in saturated fat throughout pregnancy and lactation and these animals also showed increased anxiety-like behaviours and deficits in spatial learning101. Together, these studies indicate that there are sex-specific interactions between the maternal diet during early life and long-term neuroimmune programming100,101.
The placenta may function as an important intermediary between an unhealthy maternal milieu and fetal markers of neuroinflammation by producing its own pro-inflammatory cytokines. Placentas from pregnant macaques receiving a high-fat diet throughout pregnancy showed increased levels of the pro-inflammatory cytokines tumour necrosis factor (TNF) and interleukin-6 (IL-6), and of Toll-like receptor 4 (REF. 102). Surprisingly, in pregnant rats with restricted access to low-protein food, levels of TNF and IL-6 were also increased in placental tissue103. Timing also seems to be important for the interaction between the immune system and the placenta, as maternal immune activation during early gestation in mice can result in implantation failure104,105, and mid-gestation inflammation in the placenta has been linked to schizophrenia-like and ASD-like phenotypes106.
Maternal infection and immune activation.
Animal studies examining the effects of maternal infection during pregnancy have reported many behavioural outcomes in the adult offspring that are similar to those detected in offspring exposed to prenatal stress, including increased stress reactivity and impaired cognitive performance11,12,18,107. This suggests that the two manipulations may affect the fetus and the developing brain through common mediators, including glucocorticoids and/or pro-inflammatory cytokines96,108–112. In fact, inhibition of inflammatory processes in a mouse model of maternal chronic stress ameliorated the effects of stress on the offspring108. The pro-inflammatory cytokines produced by the mother and/or in the placenta during maternal infection or chronic stress can elicit their long-term effects on fetal somatic cells through interactions with epigenetic machinery. For example, IL-6 increases the expression and the activity of DNA (cytosine 5)-methyltransferase 1 by regulating its gene promoter113. In another example, in a mouse model of early pregnancy stress, changes in the expression of the chromatin-remodelling enzyme O-GlcNAc transferase (OGT) were detected in male but not in female placental tissue and were found to regulate a broad pattern of gene expression that is associated with the active histone mark H3K4me3 (histone H3 lysine 4 trimethylation)38,114.
Transgenerational programming
Although some environmental factors only ‘programme’ the individual that is directly exposed to them, others are transmitted across one or more generations115. The transmission of epigenetic marks to future generations through germ cell reprogramming adds complexity to our attempts to establish causal links between environmental risk factors and disease. True transgenerational reprogramming requires an epigenetic mark to be erased and then re-established in the germ cell in the absence of re-exposure or continuation of the exposure115 (FIG. 2). Tight control of epigenetic regulation is crucial during the early phase of gestation, which is characterized by a wave of genome demethylation followed by de novo remethylation, establishment of imprints and sex determination, and is therefore particularly sensitive to maternal influences that could result in embryonic and germ cell reprogramming40. The different ways in which germ cell epigenetic marks are transmitted and inherited can also be examined; these include whether the mother and/or the father pass on the trait and whether male or female offspring (or both) inherit it. Epidemiological data from Swedish famine periods suggest that there may be specific developmental windows during which germ cells are more vulnerable to this type of environmental influence116; boys exposed to famine before puberty were more likely to have children and grandchildren with an increased risk of cardiovascular disease and diabetes than males exposed outside this period117. However, studies have also shown that the type of perturbation or exposure may be an important determinant in how or if the germ cells are affected118.
Figure 2 |. Modes of maternal and paternal transgenerational epigenetic transmission.
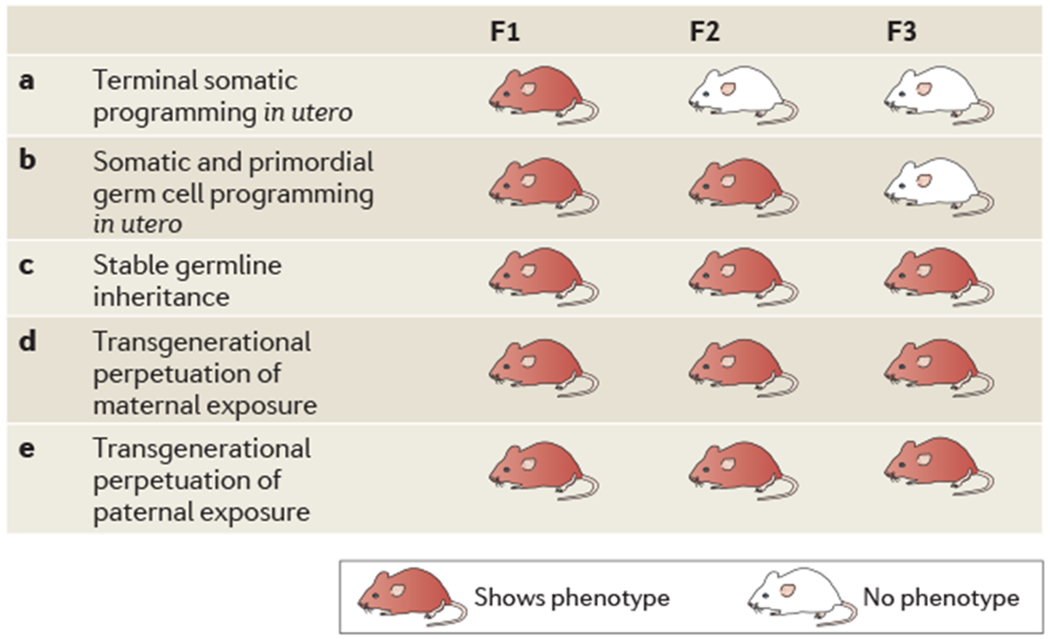
Several mechanisms for transgenerational epigenetic transmission are possible, asshown in the table, in which a red mouse indicates that the phenotype is shown in a particular generation, a | In utero somatic programming may affect tissue and brain development inthe fetus, initiating a developmental trajectory that results in an adult phenotype inthe first (F1) generation. However, this phenotype is not transmitted to the next generation, as there is no germ cell involvement. b | If in utero somatic programming is accompanied by changes in primordial germ cells, the phenotype will be transmitted to a second (F2) generation. In order for this to occur, germ cells must be exposed to a maternal insult during gestation. c | If the in utero exposure is able to alter epigenetic marks in the germ line of the F1 offspring and this reprogramming is able to withstand erasure and re-establishment during subseguent generations’ germ cell development, the phenotype will be perpetuated across generations withoutthe need for re-exposure to the original insult. This is defined as a truly transgenerational epigenetic programming. d | Transgenerational phenotypes may also occurthrough a repeated maternal effect by which programming during gestation or through exposure to altered maternal behaviours results in an adult phenotype that is only found in the maternal lineage. In order for this to occur, the offspring must be re-exposed to the insult in each generation to promote the continuation of the phenotype from the dam. This trait would not be passed through a paternal lineage. e | Transgenerational programming through a paternal lineage originating from an initial exposure inthe male reguires establishment or reprogramming of epigenetic marks in the sperm. Adapted with permission from REF 115, Elsevier.
Animal models have been used to examine the transgenerational outcomes of germ cell reprogramming that are induced by maternal and/or paternal exposure to an environmental factor. For example, male mice that are exposed to stress in utero (that is, their mothers are stressed during pregnancy), which results in increased stress sensitivity and an increased corticosterone response to acute restraint stress as adults, pass on this phenotype to their male, but not their female, offspring119. Similarly, when mice were exposed to early postnatal stress (chronic and unpredictable maternal separation from postnatal days 1 to 14), their stress-sensitive behavioural phenotype in adulthood continued through to the third generation120. Changes were detected in DNA methylation in the germ line of this third generation, which suggests that germ cell epigenetic marks can be maintained across generations without re-exposure to the insult120. These studies support the idea that the germ line can be manipulated, and they show the potential ability of external influences to increase susceptibility or resilience to disease in subsequent generations. It is important to note that an environmental signal that can influence the germ line does not necessarily produce a notable phenotype in the first generation if there is no effect on somatic tissues (including the brain). Whether such an effect occurs probably depends on the timing, the type or the severity of the exposure121. However, in such a scenario, if the germ cell were reprogrammed this would give rise to a phenotype in the second generation; thus, phenotypes can skip a generation, making it more difficult to form clear risk analyses for pregnant women or for other at-risk groups (FIG. 3). As this premise has not yet been mechanistically examined, the specific signals or insults remain to be determined.
Figure 3 |. Programming of phenotypes and disease risk can skip generations.
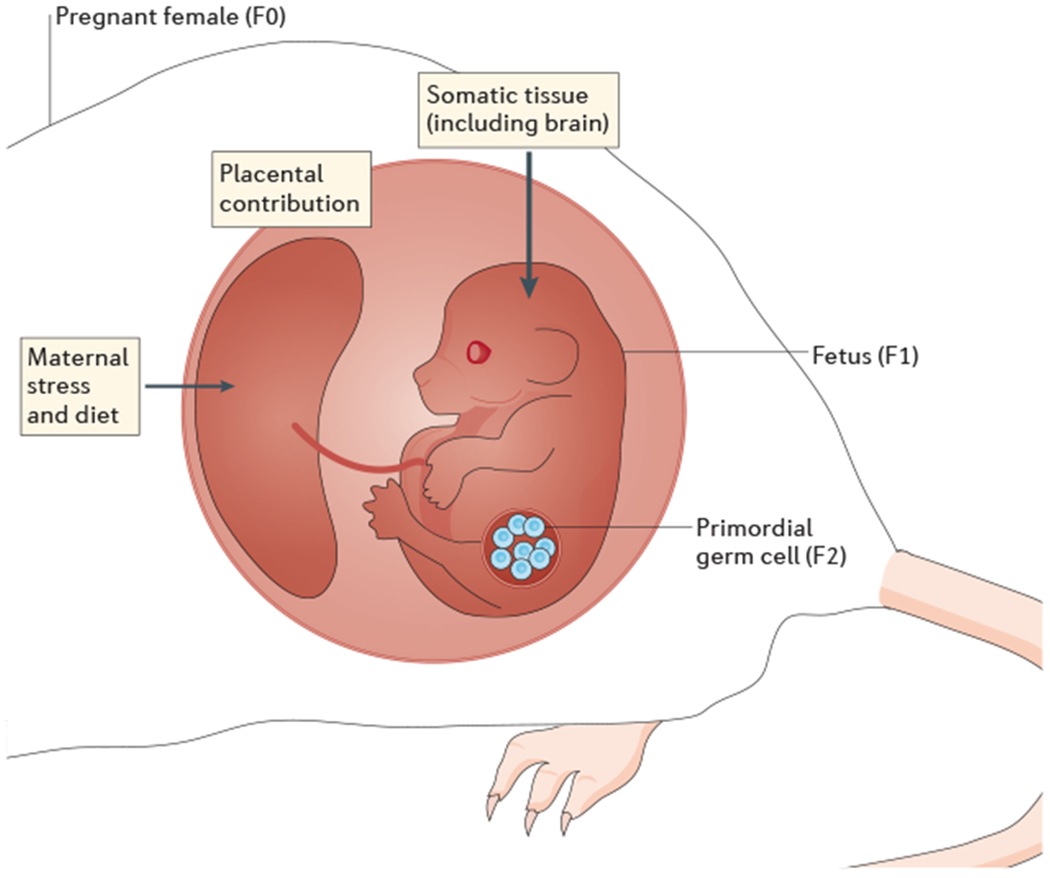
Maternal stress or dietary insults can generate signals, in the form of changes in the hormonal milieu, that are transmitted from the maternal to the fetal compartment via the placenta. The resulting maternal programming of fetal somatic tissues can lead to changes in long-term health outcomes in the first generation (F1). Furthermore, in utero exposure can also occur for the primordial germ cells that will give rise to the second generation (F2), as these germ cells are also present and undergo reprogramming during embryonic development. If the exposure does not produce a detectable effect on somatict issues but is still able to reprogramme epigenetic marks in the germ cell, a phenotype could effectively arise in the F2 generation that was not present in the F1 generation, thus having the appearance of ‘skipping a generation’. Adapted with permission from REF 115, Elsevier.
Maternal transmission.
Studies in which female mice were exposed to nutritional challenges have provided some of the clearest examples of how phenotypes can be lost or retained across generations121–126. These studies have monitored body length, adiposity, glucose regulation, metabolic rate and behaviour through at least three generations and have reported epigenetic changes, including effects on imprinted genes. Maternal obesity or exposure to a high-fat diet during pregnancy is associated with reduced insulin and glucose sensitivity and increased body size and adiposity in the first-generation offspring of humans, non-human primates and rodents122,127–131. However, not all of these traits will arise in the second generation, which supports the idea that many phenotypes are the result of direct somatic cell programming and not of altered epigenetic marks in the germ cell. For example, one study in mice found that an increased offspring body size was the only phenotype of many from the first generation that was retained into the second and the third generations following the initial exposure to a maternal high-fat diet during pregnancy123. A closer examination of the transmission pattern of this phenotype revealed that only second-generation fathers could pass on the trait and only to their third-generation daughters, indicating an epigenetic mechanism (as male mice were not permitted any interaction with the litter, any passage of traits to their offspring must have been programmed in their germ cells) involving a paternally expressed gene (as only the second-generation fathers were able to pass on the trait) that may be on the X chromosome (as only their daughters inherited the trait)123. Indeed, the expression pattern of imprinted genes from these females supported this hypothesis: paternally expressed genes showed increased overall expression in the livers of third-generation female offspring born to the high-fat diet lineage second-generation males but not in those born to the females.
Paternal transmission.
Although transgenerational effects of various changes in the maternal environment are well characterized, fewer studies have examined possible paternal modes of transmission. In such studies, male mice or rats exposed to social defeat, chronic stress, cocaine, a high-fat diet, endocrine disrupters or olfactory cues before breeding have been examined132–139. These models provide unique opportunities to examine mechanisms of transgenerational transmission. In a research laboratory environment, male rodents do not normally participate in the rearing of their offspring and thus have no postpartum behavioural influence on their development. Therefore, the ability of the sire to transmit a trait to his offspring is limited to the genetic and the epigenetic material in his sperm, which is an accessible and plentiful tissue source for epigenetic analyses. Therefore, paternal models provide a more transparent method by which to study mechanisms of epigenetic transmission than models of maternal transmission, which are complicated by the potentially confounding effects of maternal behaviours, parturition and lactation. As stated above, the term ‘transgenerational’ requires that an epigenetic mark giving rise to a phenotype is found in the germ cell and is passed on to the next generation in the absence of re-exposure. Therefore, to be considered a transgenerational epigenetic mechanism in a male model of transmission, a phenotype need be detected only into the second generation. Perturbations that occur during pregnancy, such as maternal stress or infection, must affect the third generation for their influence to be truly transgenerational, because the gestating first-generation offspring and the germ cells that will give rise to the second-generation offspring are simultaneously exposed to the initial perturbation115 (FIG. 2).
Epigenetic marks normally found in sperm include changes in the expression of non-coding RNAs (micro-RNAs (miRNAs) and PlWI-interacting RNAs (piRNAs; which are important in silencing retrotransposons)), histone modifications (for the few retained histones in sperm) and DNA methylation140. Perturbations such as stress can alter these marks, resulting in offspring with detectable phenotypes. For instance, when male mice were exposed to chronic social defeat or chronic variable stress before breeding, their offspring showed behavioural and physiological phenotypes of stress dysregulation, including altered HPA axis responses to stress132,134. Furthermore, one study showed that, in sperm from fathers who were chronically stressed either during puberty or as adults, the levels of a subset of specific miRNAs were increased134. Interestingly, rewarding or reinforcing environmental factors, such as drugs of abuse, pleasant odours or high-fat diets, can also produce germline epigenetic marks that are transmitted to the offspring. For example, male rats exposed to a specific odour paired with a shock passed this conditioned experience to their offspring through an epigenetic germ cell mechanism133. Similarly, male offspring of male rats that were exposed to chronic cocaine self-administration showed reward neurocircuitry hypofunction136. This phenotype was associated with a change in histone H3 acetylation at the brain-derived neurotrophic factor (Bdnf) promoter in the sperm of the sires that were initially exposed, and the same change was also detected in the brains of their male offspring. In another study, female but not male offspring of male rats that were exposed to a high-fat diet for 10 weeks had reduced glucose sensitivity and β-cell dysfunction135. Such sex-specific outcomes suggest that the epigenetic mark or machinery involved is found on the X or the Y chromosome and/or that the offspring phenotype is dependent on programming or activational effects of oestradiol or testosterone. The mechanism by which paternal life experiences such as stress, diets or drugs can reprogramme epigenetic marks in the germ cell are not currently understood; however, normal processes and development across spermatogenesis and the stages at which epigenetic or transcriptional machinery is present in the cell must be considered (FIG. 4).
Figure 4 |. Windows of vulnerability to environmental reprogramming in spermatogenesis.
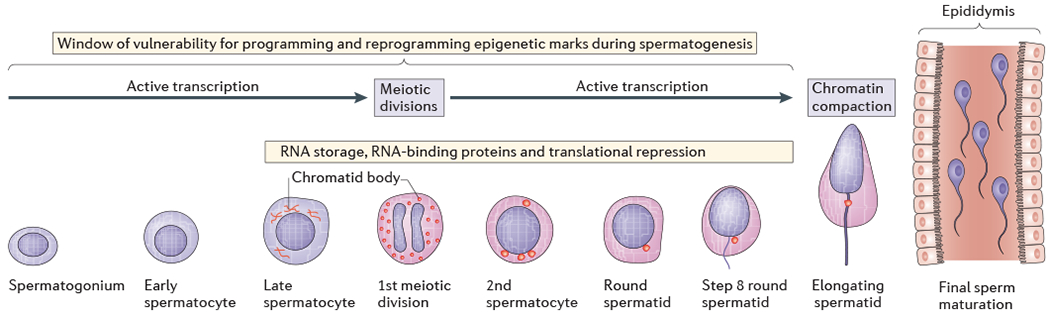
The figure shows the stages of spermatogenesis and sperm maturation, highlighting the periods in which environmental exposures can reprogramme epigenetic marks. Active transcription and storage of RNA is ongoing through the spermatogonium, spermatocyte and spermatid stages. However, once compaction of paternal DNA has occurred, which involves replacing the majority of histones with protamines, there is not a clear mechanism by which epigenetic marks can be further altered in the mature sperm. This suggest sthat an environmental exposure must occur within a sensitive time period of spermatogenesis in order to pass on a new or reprogrammed epigenetic mark to future offspring. In most mammals, complete spermatogenesis occurs in 6–8 weeks, by which point sperm are present in the epididymis for final maturation processes. The time between chromatin compaction and the transit of mature sperm to the epididymis is at least 10 days, during which time epigenetic and transcriptional machineries are no longer active. However, the secretion of exosomes and other factors that interact with the maturing sperm in the epididymis may impart lasting epigenetic marks, such as microRNAs. Figure is reproduced from BaleT. Lifetime stress experience: transgenerational epigenetics and germ cell programming. Dialogues Clin. Neurosci. 2014;16:297–305 (REF. 179), with the permission of © AICH-Servier Research Group, Suresnes, France, and adapted from REF. 180, Nature Publishing Group.
Sex specificity in epigenetic mechanisms
As described above, epigenetic responses to environmental changes can be sex specific at several levels. The perception or the response to the exposure itself can differ between sexes, so that one sex produces a higher or a lower change in hormone levels or greater neural activation, leading to a cascade of differing outcomes. The placenta is an example of a tissue that shows sex-specific effects; for example, maternal stress or dietary challenge during pregnancy produces lasting gene expression changes in the placentas of male fetuses but not in the placentas of female fetuses38,39,42. This provides a mechanism by which the effects of stress can be transmitted in a sex-specific manner to the developing brain. Epidemiological studies have shown that gender is an important determinant in the severity and the onset of diseases that are associated with certain fetal antecedents. For example, male, but not female, children of women who were in their second trimester of pregnancy during the 1940 invasion of the Netherlands (and therefore who presumably suffered high stress levels at that time) had an increased risk of schizophrenia141. A recent report found that maternal depression during pregnancy correlated with increased postnatal anxiety development in children and that males were significantly more likely to be affected142. Many neurodevelopmental disorders, such as ASD, have a strong sex bias143. Exposure to maternal stress before week 32 of gestation has been suggested to be a contributing factor to ASD144. Therefore, studies examining the role of fetal sex in determining the specific outcomes of maternal stress during pregnancy may provide a unique insight into neurodevelopmental disease aetiologies145.
There is also evidence that the effects of activation of the maternal immune system on the offspring are sex dependent. For example, male offspring of pregnant rats exposed to lipopolysaccharides late in gestation showed deficits in pie-pulse inhibition in adulthood, whereas female offspring were unaffected146. Similarly, male, but not female, mice exposed during late gestation to maternal injection of the viral mimic polyinosinic-polycytidylic acid (poly(I:C)) showed behavioural and cognitive inflexibility in adulthood, resembling some of the negative symptoms of schizophrenia18. These effects were also associated with reduced levels of glutamate in the prefrontal cortex in males. Such studies support the widely held view that males are more vulnerable to prenatal insults than females, who are more susceptible to insults that occur in the postnatal environment147.
Animal studies have also begun to examine the developmental timing of such sex-specific outcomes more closely1. For example, as described above, gestation has been established as a period in which important masculinization of the male brain can be disrupted by maternal stress22,42,50,57. Disruptions in the organizational programming of the sexually dimorphic brain may also have a role in the aetiology of neurodevelopmental disorders. For example, structural brain volume analyses showed a reduction of the normal sexual dimorphism of the brain in male patients with schizophrenia62,148. These studies are exciting because they suggest a specific time window in development (probably prenatal) in which an exposure or insult disrupted this normal masculinizing process. As the female brain is not exposed to gonadal hormones during this early developmental period, these findings indicate a sex-specific process that is a point of potential disruption in males. This may ultimately lead to a mismatch for these males later in life, as they would be genetically XY but would not have a fully masculinized brain.
Gonadal hormones and their receptors also interact with epigenetic mechanisms to promote normal brain maturation and development149. For instance, oestradiol can affect epigenetic machinery, including DNA methyl-transferases and histone deacetylases (HDACs), in the neonate as a part of sexually dimorphic brain development, and can alter DNA methylation patterns3,150–152. Therefore, alterations in the levels of oestrogen, or its precursor testosterone, that result from perturbations experienced during early life can manifest as long-term reprogramming of brain function149. Oestradiol can also alter histone modification through actions on HDACs149. A single postnatal administration of an HDAC inhibitor in male mice was shown to disrupt the sexual dimorphic development of the bed nucleus of the stria terminalis (BNST), which is a crucial part of the neurocircuitry involved in regulating stress responses153. Small non-coding miRNAs also seem to be responsive to oestradiol levels in the early postnatal mouse brain; more than 250 of the most abundantly expressed miRNAs in the brain significantly differ in their expression between males and females119. These sex differences are dependent on the conversion of gonadal testosterone to oestradiol in males154, as a single administration of an aromatase inhibitor at birth was able to completely shift the entire male miRNA expression pattern to that of a female119.
These studies show that gonadal hormones can, during important periods, promote epigenomic effects and thereby broadly influence neurodevelopment, effectively producing the sexually dimorphic brain. Importantly, these regulatory mechanisms can be sensitive to perturbations in the environment. As described above, maternal stress and infection are both associated with dysmasculinized phenotypes, which suggests that they cause a disruption in male sexual differentiation or gonad development that in turn alters testosterone production or its actions in the brain during this critical neonatal organizational period1,155,156. Finally, it is also interesting to note that although the Y chromosome contains no miRNA genes, the mammalian X chromosome is highly enriched for them and their density is approximately twofold higher than it is on autosomes in mice and humans147. In circumstances in which these genes escape X inactivation, females would have twice the expression level of males, thus producing a sex-specific post-transcriptional mode of regulation that provides females with the select ability to buffer or to respond differently to insults.
Conclusions and future directions
It is clear that the outcomes and the long-term sustainability of dynamic epigenetic marks are dependent on factors such as the timing and the type of exposure, the sex of the offspring and, in the case of transgenerational effects, the sex of the parent and the offspring. According to the Barker hypothesis, whether such programming ultimately results in disease risk or resilience mainly depends on the concordance between the expected and actual environments. As discussed above, there are several common signalling mechanisms through which perturbations during early development are likely to function, including inflammation and energy metabolism in the maternal milieu and in the placenta (BOX 2). Studies examining the early postnatal environment have also identified common epigenetic gene targets in the brain. In addition, recent studies have provided insight into the novel contributions of the gut microbiome to early brain development; thus, metabolic and immune processes may interact to ultimately affect neurodevelopment157–159. Taken together with other important gene targets, these findings will provide the clues necessary to discover the upstream transcriptional regulatory mechanisms, which will increase our understanding of the links between developmental experience and ultimate brain function. Studies in mice focusing on brain region-specific epigenetic processes and important behaviours related to cognitive performance, learning and memory, and appetitive behaviours have repeatedly shown how dynamic the epigenome continues to be into adulthood1,9,69,160–170. This reminds us that insults that occur early in life may be just a ‘first hit,’ which primes the brain to be more or less responsive to future insults, with disease presentation occurring following multiple insults in life. Support for this hypothesis can be seen in neuropsychiatric diseases in which adult stress-induced onset occurs, such as depression or post-traumatic stress disorder. Perturbations experienced during neurodevelopment that promote dysregulation of stress pathways would certainly be considered an important first hit12,22,171.
Box 2 |. Common cellular mechanisms?
In thinking about the mechanisms by which prenatal perturbations promote transgenerational outcomes, it is interesting to note that several seemingly different insults, such as maternal stress, diet and infection, can produce similar neurodevelopmental and phenotypic outcomes. Mechanistically, this suggests that there are a limited number of physiological signals from the maternal milieu that are responsible for transmitting information from the in utero environment to the developing fetus and that physiological disturbances produced by maternal stress, infection, famine or obesity share some common end points.
The most evolutionarily important signals are likely to be related to energy availability, which is important in determining future fitness, and are therefore likely to include metabolic signals such as insulin, glucose, free fatty acids and glucocorticoids, as well as inflammatory cytokines (FIG. 1). Glucocorticoids are certainly an important downstream indicator of stress, and some aspects of prenatal stress models can be recapitulated by simply administering high levels of glucocorticoids to the pregnant animal55,56,96,174,175. However, the stress response encompasses other stress hormones, such as catecholamines, as well as numerous downstream metabolic factors. The involvement of these additional metabolic processes in developmental programming at the level of the placenta and the fetus is probable and has been shown to be important in rodent models of prenatal stress38,93,97,114,176.
There can only be a limited number of biochemical inputs that a given cell can to respond to and, as such, it is likely that different environmental challenges impinge on the same signalling cascades and downstream effectors to promote changes in existing epigenetic marks or to apply new ones — effectively reprogramming similar offspring phenotypes from different starting points. Therefore, there must be a cellular barometer by which the quantitative and the qualitative changes in the hormonal milieu are surveyed. For instance, during maternal or paternal stress exposure, for a signal to be gauged as important enough to warrant activation of the epigenetic machinery to facilitate the creation of new or changed epigenetic marks, does a stressor need to be of a certain duration, to reach a certain level (perceived as mild versus severe or chronic) or to induce a certain level of stress hormones? How does a germ cell know whether a stressor warrants passing on the information to future generations? Coincident detection of key signals occurring over precise periods is probably required.
The field of transgenerational epigenetics has been on a fast-paced trajectory over the past decade, with the development of animal models that reliably and predictively mimic epidemiological studies in order to determine the mechanisms by which perturbations in the environment are associated with risk of neurodevelopmental disorders (BOX 3). Future studies will need to use ‘omic’ technologies to better understand the broader reprogramming abilities of the environment on the epigenome. For instance, metabolomic analyses of the maternal and the fetal milieu would be useful in models of maternal stress or infection to determine the metabolic signals that are involved in neurodevelopment. There remain several considerable gaps in our knowledge: what are the physiological and the cellular signals by which experience is able to reprogramme an epigenetic mark (BOX 1), and when and how are the germ cells involved? Identifying these mechanisms in a stepwise manner will facilitate the discovery of better disease predictive biomarkers and will perhaps lead to effective interventions for disease prevention and earlier treatments (BOX 3).
Box 3 |. Caution in translation of epigenetic outcomes.
We currently lack the ability to accurately predict who will present with a neuropsychiatric disease. However, the collection of biological samples as part of prospective studies may enable us to identify predictive biomarkers, which may include epigenetic marks, and to distinguish between disease cause and effect if changes in a given outcome precede disease onset rather than being a response to it. Of course, this raises the question of whether epigenetic and epigenomic changes in peripheral cells and tissues are informative about what goes on in the brain, as most epigenetic changes are cell and tissue specific177. A great deal more evidence is still needed before we can draw firm conclusions as to how accurate the epigenome will be in relaying the story of an individual’s life exposures and experiences and our ability to translate that into predictions for disease risk and resilience.
Using established epidemiological and clinical measures to design better animal models that reproduce specific endophenotypes rather than attempting to model mental health disorders in totality is an important goal that may facilitate study of the translational potential of the epigenome. Such models may enable us, for example, to better understand how an environmental factor may programme susceptibility or resilience to disease in humans. However, if the field of transgenerational epigenetics has taught us anything, it is that experience is everything. Results from studies showing transgenerational effects of stress, odorant exposure, diet and infection have opened our eyes to the dynamic (that is, programmable and reprogrammable) epigenome, including — and importantly — the germline epigenome5,133–135,178. The seemingly standard experiences that our experimental animals encounter, including shipping, dietary changes, parasitic infections and treatments, construction noise and different environments (such as germ-free versus conventional facilities), can produce substantial epigenetic differences within otherwise genetically identical inbred strains. If present in the germ cells, these marks can last for generations, promoting phenotypic variability within and between research laboratories. Controlling for such variables is difficult but important.
Acknowledgements
This work was supported by grants from the US National Institutes of Health MH099910, MH104184, MH091258, MH087597 and MH073030.
Glossary
- Birth cohort studies
Longitudinal prospective studies in which pregnancies are followed through birth and into childhood to correlate the risk forgiven outcomes with specified experiences or exposures during gestation. These cohorts of individuals differ from other epidemiological studies in which the outcome measures are mainly retrospective.
- Biomarkers
Specific biochemical, molecular, anatomical or physiological characteristics that are used to predict, measure or indicate the presence or progress of disease or the effects of treatment.
- Executive function
A set of cognitive abilities that include inhibition (resisting habits, temptations or distractions), switching (adjusting to change) and working memory (mentally holding and using information).
- Stress responsiveness
Behavioural or physiological measures in response to a stress provocation. In all mammals, the physiological hypothalamic—pituitary—adrenal stress axis is the standard measure of stress experience. Behavioural changes in response to stress or threat can also be examined as an indication of stress state.
- Imprinted gene
A gene expressed from only one allele in a manner that depends on the parent of origin and that is regulated by DNA methylation.
- Hypothalamic—pituitary—adrenal stress axis
(HPA stress axis). The neuroendocrine core of the stress system. Its activation results in the release of corticotropin-releasing factor from the hypothalamus, adrenocorticotropic hormone from the pituitary and cortisol (corticosterone in rats and mice) from the adrenal glands.
- Endophenotypes
Quantifiable phenotypes with an assumed intermediate role in the pathway from genes to complex phenotypes. It is thought that the action of an endophenotype is easier to understand biologically and genetically than the action of the complex phenotype of primary interest. They enable the examination of specific aspects of complex human diseases in animal models.
- Barnes maze
A spatial learning and memory task in which the animal is placed on a large, open table that has holes around the circumference. Only one of the holes has an escape tunnel, and the animal must learn to use distal cues to identify this hole and escape in the shortest possible time. Learning in this task involves the hippocampus.
- Microglia
Glia of mesodermal origin and the resident macrophages of the CNS.
- Social defeat
A behavioural model in which rodents (predominantly males) are repeatedly exposed to a more aggressive conspecific. The outcomes of these confrontations are repeated losses fertile subject. This exposure is then followed by a period of housing the defeated animal in close proximity to the aggressor to establish the conditioning.
- Pre-pulse inhibition
A reduction in the magnitude of the startle reflex that Occurs when an Organism is presented with a non-startling stimulus (a pre-pulse) before being presented with the startling stimulus. Deficits in pre-pulse inhibition have been Observed in patients with schizophrenia.
Footnotes
Competing interests statement
The author declares no competing interests.
References
- 1.Bale TL et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68, 314–319 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curley JP, Jensen CL, Mashoodh R & Champagne FA Social influences on neurobiology and behavior: epigenetic effects during development. Psychoneuroendocrinology 36, 352–371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy MM & Nugent BM Epigenetic contributions to hormonally mediated sexual differentiation of the brain. J. Neuroendocrinol 25,1133–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodes GE Sex, stress, and epigenetics: regulation of behavior in animal models of mood disorders. Biol. Sex Differ 4, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohacek J, Gapp K, Saab BJ & Mansuy IM Transgenerational epigenetic effects on brain functions. Biol. Psychiatry 73, 313–320 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Hales CN & Barker DJ The thrifty phenotype hypothesis. Br. Med. Bull 60, 5–20 (2001). [DOI] [PubMed] [Google Scholar]; In this paper, the thrifty phenotype hypothesis describes how a mismatch between fetal programming and the postnatal environment can result in disease risk.
- 7.Binder EB & Nemeroff CB The CRF system, stress, depression and anxiety-insights from human genetic studies. Mol. Psychiatry 15, 574–588 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackman DA, Farah MJ & Meaney MJ Socioeconomic status and the brain: mechanistic insights from human and animal research. Nature Rev. Neurosci 11, 651–659 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sweatt JD The emerging field of neuroepigenetics. Neuron 80, 624–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glynn LM, Wadhwa PD, Dunkel-Schetter C, Chicz-Demet A & Sandman CA When stress happens matters: effects of earthquake timing on stress responsivity in pregnancy. Am. J. Obstet. Gynecol 184, 637–642 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Deverman BE & Patterson PH Cytokines and CNS development. Neuron 64, 61–78 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Howerton CL & Bale TL Prenatal programing: at the intersection of maternal stress and immune activation. Horm. Behav 62, 237–242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AS Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol 72, 1272–1276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AS et al. Prenatal infection and cavum septum pellucidum in adult schizophrenia. Schizophr. Res 108, 285–287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown AS et al. Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am. J. Psychiatry 166, 683–690 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selevan SG, Kimmel CA & Mendola P Identifying critical windows of exposure for children’s health. Environ. Health Perspect 108 (Suppl. 3), 451–455 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapoport JL, Addington AM, Frangou S & Psych MR The neurodevelopmental model of schizophrenia: update 2005. Mol. Psychiatry 10, 434–449 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Meyer U, Feldon J & Dammann O Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr. Res 69, 26R–33R (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examines the shared mechanistic link between neurodevelopmental disorders that are related to prenatal inflammation.
- 19.Morrison KE, Rodgers AB, Morgan CP & Bale TL Epigenetic mechanisms in pubertal brain maturation. Neuroscience 264, 7–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis EP, Sandman CA, Buss C, Wing DA & Head K Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol. Psychiatry 74, 647–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malter Cohen M et al. Early-life stress has persistent effects on amygdala function and development in mice and humans. Proc. Natl Acad. Sci. USA 110, 18274–18278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bale TL Sex differences in prenatal epigenetic programming of stress pathways. Stress 14, 348–356 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Susser E, St Clair D & He L Latent effects of prenatal malnutrition on adult health: the example of schizophrenia. Ann. NY Acad. Sci 1136, 185–192 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Stoner R et al. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med 370, 1209–1219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avishai-Eliner S, Brunson KL, Sandman CA & Baram TZ Stressed-out, or in (utero)? Trends Neurosci. 25, 518–524 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wadhwa PD, Sandman CA & Garite TJ The neurobiology of stress in human pregnancy: implications for prematurity and development of the fetal central nervous system. Prog. Brain Res 133, 131–142 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Wadhwa PD, Sandman CA, Porto M, Dunkel-Schetter C & Garite TJ The association between prenatal stress and infant birth weight and gestational age at birth: a prospective investigation. Am. J. Obstet. Gynecol 169, 858–865 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Khashan AS et al. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch. Gen. Psychiatry 65, 146–152 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Atladottir HO et al. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch. Pediatr. Adolesc. Med 164, 470–477 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Atladottir HO et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord 40, 1423–1430 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Goines PE et al. Increased midgestational IFNγ, IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol. Autism 2, 13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AS & Susser ES Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull 34, 1054–1063 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu MQ et al. Prenatal malnutrition and adult schizophrenia: further evidence from the 1959–1961 Chinese famine. Schizophr. Bull 35, 568–576 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravelli GP, Stein ZA & Susser MW Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med 295, 349–353 (1976). [DOI] [PubMed] [Google Scholar]
- 35.Heijmans BT et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl Acad. Sci. USA 105, 17046–17049 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Lieshout RJ, Taylor VH & Boyle MH Pre-pregnancy and pregnancy obesity and neurodevelopmental outcomes in offspring: a systematic review. Obes. Rev 12, e548–e559 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Buss C et al. Impaired executive function mediates the association between maternal pre-pregnancy body mass index and child ADHD symptoms. PLoS ONE 7, e37758 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howerton CL, Morgan CP, Fischer DB & Bale TL O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl Acad. Sci. USA 110, 5169–5174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao J et al. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl Acad. Sci. USA 107, 5557–5562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weaver JR, Susiarjo M & Bartolomei MS Imprinting and epigenetic changes in the early embryo. Mamm. Genome 20, 532–543 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Mueller BR & Bale TL Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol. Behav 88, 605–614 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Mueller BR & Bale TL Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci 28, 9055–9065 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies sex-specific changes in the placenta in response to early gestational stress that are associated with changes in offspring neurodevelopment.
- 43.Brunton PJ & Russell JA Prenatal social stress in the rat programmes neuroendocrine and behavioural responses to stress in the adult offspring: sex-specific effects. J. Neuroendocrinol 22, 258–271 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Brunton PJ & Russell JA Allopregnanolone and suppressed hypothalamo—pituitary—adrenal axis stress responses in late pregnancy in the rat. Stress 14, 6–12 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Weinstock M Gender differences in the effects of prenatal stress on brain development and behaviour. Neurochem. Res 32, 1730–1740 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Kapoor A, Kostaki A, Janus C & Matthews SG The effects of prenatal stress on learning in adult offspring is dependent on the timing of the stressor. Behav. Brain Res 197, 144–149 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Kapoor A, Leen J & Matthews SG Molecular regulation of the hypothalamic–pituitary–adrenal axis in adult male guinea pigs after prenatal stress at different stages of gestation. J. Physiol 586, 4317–4326 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider ML, Moore CF, Kraemer GW, Roberts AD & DeJesus OT The impact of prenatal stress, fetal alcohol exposure, or both on development: perspectives from a primate model. Psychoneuroendocrinology 27, 285–298 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Kapoor A & Matthews SG Short periods of prenatal stress affect growth, behaviour and hypothalamo–pituitary–adrenal axis activity in male guinea pig offspring. J. Physiol 566, 967–977 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller BR & Bale TL Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol. Behav 91, 55–65 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Lemaire V, Koehl M, Le Moal M & Abrous DN Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc. Natl Acad. Sci. USA 97, 11032–11037 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darnaudery M & Maccari S Epigenetic programming of the stress response in male and female rats by prenatal restraint stress. Brain Res. Rev 57, 571–585 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Weinstock M Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Prog. Neurobiol 65, 427–451 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Coe CL et al. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biol. Psychiatry 54, 1025–1034 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Welberg LA, Seckl JR & Holmes MC Prenatal glucocorticoid programming of brain corticosteroid receptors and corticotrophin-releasing hormone: possible implications for behaviour. Neuroscience 104, 71–79 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Welberg LA & Seckl JR Prenatal stress, glucocorticoids and the programming of the brain. J. Neuroendocrinal 13, 113–128 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Ward IL Prenatal stress feminizes and demasculinizes the behavior of males. Science 175, 82–84 (1972). [DOI] [PubMed] [Google Scholar]; This is one of the first studies to determine the importance of sex-specific neurodevelopmental programming that can be disrupted by maternal stress.
- 58.Ward IL & Stehm KE Prenatal stress feminizes juvenile play patterns in male rats. Physiol. Behav 50, 601–605 (1991). [DOI] [PubMed] [Google Scholar]
- 59.Baron-Cohen S et al. Elevated fetal steroidogenic activity in autism. Mol. Psychiatry 20, 369–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moore L et al. Serum testosterone levels are related to cognitive function in men with schizophrenia. Psychoneuroendocrinology 38, 1717–1728 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Lombardo MV et al. Fetal testosterone influences sexually dimorphic gray matter in the human brain. J. Neurosci 32, 674–680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gur RE et al. A sexually dimorphic ratio of orbitofrontal to amygdala volume is altered in schizophrenia. Biol. Psychiatry 55, 512–517 (2004). [DOI] [PubMed] [Google Scholar]; This paper reports the dysmasculinization of limbic brain regions in men with schizophrenia.
- 63.Fenoglio KA et al. Enduring, handling-evoked enhancement of hippocampal memory function and glucocorticoid receptor expression involves activation of the corticotropin-releasing factor type 1 receptor. Endocrinology 146, 4090–4096 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fenoglio KA, Brunson KL & Baram TZ Hippocampal neuroplasticity induced by early-life stress: functional and molecular aspects. Front. Neuroendocrinol 27, 180–192 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ivy AS, Brunson KL, Sandman C & Baram TZ Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience 154, 1132–1142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivy AS et al. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci 30, 13005–13015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korosi A & Baram TZ Plasticity of the stress response early in life: mechanisms and significance. Dev. Psychobiol 52, 661–670 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Korosi A et al. Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J. Neurosci 30, 703–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClelland S, Korosi A, Cope J, Ivy A & Baram TZ Emerging roles of epigenetic mechanisms in the enduring effects of early-life stress and experience on learning and memory. Neurobiol. Learn. Mem 96, 79–88 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rice CJ, Sandman CA, Lenjavi MR & Baram TZ A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology 149, 4892–4900 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This novel and reproducible model shows how disruptions in maternal care can manifest in reprogramming of offspring stress pathways in the brain.
- 71.Ladd CO et al. Long-term behavioral and neuroendocrine adaptations to adverse early experience. Prog. Brain Res 122, 81–103 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Liu D et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic–pituitary–adrenal responses to stress. Science 277, 1659–1662 (1997). [DOI] [PubMed] [Google Scholar]
- 73.Meaney MJ Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci 24, 1161–1192 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Weaver IC et al. Epigenetic programming by maternal behavior. Nature Neurosci. 7, 847–854 (2004). [DOI] [PubMed] [Google Scholar]; This important paper was the first to establish epigenetic mechanisms in the developing brain that were altered by the quality of maternal care.
- 75.Barha CK, Pawluski JL & Galea LA Maternal care affects male and female offspring working memory and stress reactivity. Physiol. Behav 92, 939–950 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Sanchez MM, Ladd CO & Plotsky PM Early adverse experience as a developmental risk factor for later psychopathology: evidence from rodent and primate models. Dev. Psychopathol 13, 419–449 (2001). [DOI] [PubMed] [Google Scholar]
- 77.Caldji C et al. Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proc. Natl Acad. Sci. USA 95, 5335–5340 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weaver IC, Diorio J, Seckl JR, Szyf M & Meaney MJ Early environmental regulation of hippocampal glucocorticoid receptor gene expression: characterization of intracellular mediators and potential genomic target sites. Ann. NY Acad. Sci 1024, 182–212 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Korosi A & Baram TZ The pathways from mother’s love to baby’s future. Front. Behav. Neurosci 3, 27 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Graham YP, Heim C, Goodman SH, Miller AH & Nemeroff CB The effects of neonatal stress on brain development: implications for psychopathology. Dev. Psychopathol 11, 545–565 (1999). [DOI] [PubMed] [Google Scholar]
- 81.Lyons DM, Martel FL, Levine S, Risch NJ. & Schatzberg AF. Postnatal experiences and genetic effects on squirrel monkey social affinities and emotional distress. Horm. Behav 36, 266–275 (1999). [DOI] [PubMed] [Google Scholar]
- 82.Barr CS et al. Sexual dichotomy of an interaction between early adversity and the serotonin transporter gene promoter variant in rhesus macaques. Proc. Natl Acad. Sci. USA 101, 12358–12363 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGowan PO et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neurosci. 12, 342–348 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vukojevic V et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J. Neurosci 34, 10274–10284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koshy TS, Sara VR, King TL & Lazarus L The influence of protein restriction imposed at various stages of pregnancy on fetal and placental development. Growth 39, 497–506 (1975). [PubMed] [Google Scholar]
- 86.Vucetic Z et al. Early life protein restriction alters dopamine circuitry. Neuroscience 168, 359–370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pankevich DE, Teegarden SL, Hedin AD, Jensen CL & Bale TL Caloric restriction experience reprograms stress and orexigenic pathways and promotes binge eating. J. Neurosci 30, 16399–16407 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teegarden SL & Bale TL Decreases in dietary preference produce increased emotionality and risk for dietary relapse. Biol. Psychiatry 61, 1021–1029 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Teegarden SL & Bale TL Effects of stress on dietary preference and intake are dependent on access and stress sensitivity. Physiol. Behav 93, 713–723 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teegarden SL, Scott AN & Bale TL Early life exposure to a high fat diet promotes long-term changes in dietary preferences and central reward signaling. Neuroscience 162, 924–932 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Will MJ, Franzblau EB & Kelley AE Nucleus accumbens μ-opioids regulate intake of a high-fat diet via activation of a distributed brain network. J. Neurosci 23, 2882–2888 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pecina S, Cagniard B, Berridge KC, Aldridge JW & Zhuang X Hyperdopaminergic mutant mice have higher “wanting” but not “liking” for sweet rewards. J. Neurosci 23, 9395–9402 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cottrell EC, Holmes MC, Livingstone DE, Kenyon CJ & Seckl JR Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J. 26, 1866–1874 (2012). [DOI] [PubMed] [Google Scholar]
- 94.Lesage J, Blondeau B, Grino M, Breant B & Dupouy JP Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo–pituitary–adrenal axis in the newborn rat. Endocrinology 142, 1692–1702 (2001). [DOI] [PubMed] [Google Scholar]
- 95.Lingas R, Dean F & Matthews SG Maternal nutrient restriction (48 h) modifies brain corticosteroid receptor expression and endocrine function in the fetal guinea pig. Brain Res. 846, 236–242 (1999). [DOI] [PubMed] [Google Scholar]
- 96.Cottrell EC & Seckl JR Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci 3, 19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seckl JR & Holmes MC Mechanisms of disease: glucocorticoids, their placental metabolism and fetal ‘programming’ of adult pathophysiology. Nature Clin. Pract. Endocrinol. Metab 3, 479–488 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Welberg LA, Thrivikraman KV & Plotsky PM Chronic maternal stress inhibits the capacity to up-regulate placental 11 β-hydroxysteroid dehydrogenase type 2 activity. J. Endocrinol 186, R7–R12 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Grayson BE et al. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 151, 1622–1632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sullivan EL et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci 30, 3826–3830 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bilbo SD & Tsang V Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 24, 2104–2115 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Frias AE et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152, 2456–2464 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shen Q et al. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: implications for schizophrenia. Schizophr. Res 99, 48–55 (2008). [DOI] [PubMed] [Google Scholar]
- 104.Ilievski V, Lu SJ & Hirsch E Activation of toll-like receptors 2 or 3 and preterm delivery in the mouse. Reprod. Sci 14, 315–320 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Hsiao EY & Patterson PH Placental regulation of maternal-fetal interactions and brain development. Dev. Neurobiol 72, 1317–1326 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Malkova NV, Yu CZ, Hsiao EY, Moore MJ & Patterson PH Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun 26, 607–616 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bilbo SD & Schwarz JM Early-life programming of later-life brain and behavior: a critical role for the immune system. Front. Behav. Neurosci 3, 14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bronson SL & Bale TL Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology 155, 2635–2646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hsiao EY & Patterson PH Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun 25, 604–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lucassen PJ et al. Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: possible key role of placental 11 β-hydroxysteroid dehydrogenase type 2. Eur. J. Neurosci 29, 97–103 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Mairesse J et al. Maternal stress alters endocrine function of the feto-placental unit in rats. Am. J. Physiol. Endocrinol. Metab 292, E1526.- (2007). [DOI] [PubMed] [Google Scholar]
- 112.Garbett KA, Hsiao EY, Kalman S, Patterson PH & Mirnics K Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl. Psychiatry 2, e98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hodge DR et al. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J. Biol. Chem 276, 39508–39511 (2001). [DOI] [PubMed] [Google Scholar]
- 114.Howerton CL & Bale TL Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl Acad. Sci. USA 111, 9639–9644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dunn GA, Morgan CP & Bale TL Sex-specificity in transgenerational epigenetic programming. Horm. Behav 59, 290–295 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Pembrey ME et al. Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet 14, 159–166 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Kaati G, Bygren LO & Edvinsson S Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet 10, 682–688 (2002). [DOI] [PubMed] [Google Scholar]
- 118.Pembrey M, Saffery R & Bygren LO Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J. Med. Genet 51, 563–572 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a comprehensive review that covers the field of transgenerational epigenetics in relation to human health and disease.
- 119.Morgan CP & Bale TL Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci 31, 11748–11755 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Franklin TB et al. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 68, 408–415 (2010). [DOI] [PubMed] [Google Scholar]
- 121.King V et al. Maternal obesity has little effect on the immediate offspring but impacts on the next generation. Endocrinology 154, 2514–2524 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Dunn GA & Bale TL Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 150, 4999–5009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dunn GA & Bale TL Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152, 2228–2236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jimenez-Chillaron JC et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 58, 460–468 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thamotharan M et al. Transgenerational inheritance of the insulin-resistant phenotype in embryo-transferred intrauterine growth-restricted adult female rat offspring. Am. J. Physiol. Endocrinol. Metab 292, E1270–E1279 (2007). [DOI] [PubMed] [Google Scholar]
- 126.Burdge GC et al. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr 97, 435–439 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Boney CM, Verma A, Tucker R & Vohr BR Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 115, e290–e296 (2005). [DOI] [PubMed] [Google Scholar]
- 128.McCurdy CE et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest 119, 323–335 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sullivan EL, Smith MS & Grove KL Perinatal exposure to high-fat diet programs energy balance, metabolism and behavior in adulthood. Neuroendocrinology 93, 1–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper examines the sex-specific neurodevelopmental programming and resulting increased anxiety in the non-human primate that follows exposure to a maternal high-fat diet during pregnancy.
- 130.Tamashiro KL, Terrillion CE, Hyun J, Koenig JI & Moran TH Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes 58, 1116–1125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shiell AW, Campbell DM, Hall MH & Barker DJ Diet in late pregnancy and glucose-insulin metabolism of the offspring 40 years later. BJOG 107, 890–895 (2000). [DOI] [PubMed] [Google Scholar]
- 132.Dietz DM & Nestler EJ From father to offspring: paternal transmission of depressive-like behaviors. Neuropsychopharmacology 37, 311–312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dias BG & Ressler KJ Parental olfactory experience influences behavior and neural structure in subsequent generations. Naure Neurosci. 17, 89–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a comprehensive examination, in adult male mice, of epigenetic changes in germ cells that occur in response to exposure to an odour cue in fear conditioning that is related to reprogramming of offspring brain and behaviour.
- 134.Rodgers AB, Morgan CP, Bronson SL, Revello S & Bale TL Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J. Neurosci 33, 9003–9012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identifies important changes in specific sperm miRNAs produced by chronic stress in adult mice that correlate with programming of offspring stress reactivity.
- 135.Ng SF et al. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966 (2010). [DOI] [PubMed] [Google Scholar]
- 136.Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G & Pierce RC. Epigenetic inheritance of a cocaine-resistance phenotype. Nature Neurosci. 16, 42–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Carone BR et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143, 1084–1096 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Anway MD, Cupp AS, Uzumcu M & Skinner MK Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Anderson LM et al. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrition 22, 327–331 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Jenkins TG & Carrell DT The sperm epigenome and potential implications for the developing embryo. Reproduction 143, 727–734 (2012). [DOI] [PubMed] [Google Scholar]
- 141.van Os J & Selten JP Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 invasion of The Netherlands. Br. J. Psychiatry 172, 324–326 (1998). [DOI] [PubMed] [Google Scholar]
- 142.Gerardin P et al. Depression during pregnancy: is the developmental impact earlier in boys? A prospective case-control study. J. Clin. Psychiatry 72, 378–387 (2010). [DOI] [PubMed] [Google Scholar]
- 143.Newschaffer CJ et al. The epidemiology of autism spectrum disorders. Annu. Rev. Publ. Health 28, 235–258 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Beversdorf DQ et al. Timing of prenatal stressors and autism. J. Autism Dev. Disord 35, 471–478 (2005). [DOI] [PubMed] [Google Scholar]
- 145.Sandman CA, Glynn LM & Davis EP Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J. Psychosomat. Res 75, 327–335 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smith SE, Li J, Garbett K, Mirnics K & Patterson P H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci 27, 10695–10702 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Morgan CP & Bale TL Sex differences in microRNA regulation of gene expression: no smoke, just miRs. Biol. Sex Differ 3, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cowell PE, Kostianovsky DJ, Gur RC, Turetsky BI & Gur RE Sex differences in neuroanatomical and clinical correlations in schizophrenia. Am. J. Psychiatry 153, 799–805 (1996). [DOI] [PubMed] [Google Scholar]
- 149.McCarthy MM et al. The epigenetics of sex differences in the brain. J. Neurosci. 29, 12815–12823 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nugent BM & McCarthy MM Epigenetic underpinnings of developmental sex differences in the brain. Neuroendocrinology 93, 150–158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Westberry JM, Trout AL & Wilson ME Epigenetic regulation of estrogen receptor-α gene expression in the mouse cortex during early postnatal development. Endocrinology 151, 731–740 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kurian JR, Olesen KM & Auger A P Sex differences in epigenetic regulation of the estrogen receptor-α promoter within the developing preoptic area. Endocrinology 151, 2297–2305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Murray EK, Hien A, de Vries GJ & Forger NG Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 150, 4241–4247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.McCarthy MM Molecular aspects of sexual differentiation of the rodent brain. Psychoneuroendocrinology 19, 415–427 (1994). [DOI] [PubMed] [Google Scholar]
- 155.Bale TL Stress sensitivity and the development of affective disorders. Horm. Behav 50, 529–533 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Bale TL Neuroendocrine and immune influences on the CNS: it’s a matter of sex. Neuron 64, 13–16 (2009). [DOI] [PubMed] [Google Scholar]
- 157.Cryan JF & Dinan TG Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nature Rev. Neurosci 13, 701–712 (2012). [DOI] [PubMed] [Google Scholar]
- 158.Dinan TG & Cryan JF Regulation of the stress response by the gut microbiota: implications for psychoneuroendocrinology. Psychoneuroendocrinology 37, 1369–1378 (2012). [DOI] [PubMed] [Google Scholar]
- 159.Clarke G et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 18, 666–673 (2013). [DOI] [PubMed] [Google Scholar]
- 160.Borrelli E, Nestler EJ, Allis CD & Sassone-Corsi P Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Colvis CM et al. Epigenetic mechanisms and gene networks in the nervous system. J. Neurosci 25, 10379–10389 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tsankova N, Renthal W, Kumar A & Nestler EJ Epigenetic regulation in psychiatric disorders. Nature Rev. Neurosci 8, 355–367 (2007). [DOI] [PubMed] [Google Scholar]
- 163.Day JJ et al. DNA methylation regulates associative reward learning. Nature Neurosci. 16, 1445–1452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roth TL & Sweatt JD Regulation of chromatin structure in memory formation. Curr. Opin. Neurobiol 19, 336–342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sweatt JD Experience-dependent epigenetic modifications in the central nervous system. Biol. Psychiatry 65, 191–197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zovkic IB, Guzman-Karlsson MC & Sweatt JD Epigenetic regulation of memory formation and maintenance. Learn. Mem 20, 61–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zovkic IB & Sweatt JD Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology 38, 77–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mahgoub M & Monteggia LM A role for histone deacetylases in the cellular and behavioral mechanisms underlying learning and memory. Learn. Mem 21, 564–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Adachi M & Monteggia LM Decoding transcriptional repressor complexes in the adult central nervous system. Neuropharmacology 80, 45–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lester BM et al. Behavioral epigenetics. Ann. NY Acad. Sci 1226, 14–33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Morrison KE, Rodgers AB, Morgan CP & Bale TL Epigenetic mechanisms in pubertal brain maturation. Neuroscience 264, 17–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Waddington CH Canalization of development and genetic assimilation of acquired characters. Nature 183, 1654–1655 (1959). [DOI] [PubMed] [Google Scholar]; This paper laid the foundation for the field of epigenetic programming by defining the ‘epigenetic landscape.
- 173.Berger SL, Kouzarides T, Shiekhattar R & Shilatifard A An operational definition of epigenetics. Genes Dev. 23, 781–783 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Drake AJ, Walker BR & Seckl JR Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 288, R34–R38 (2005). [DOI] [PubMed] [Google Scholar]
- 175.O’Regan D, Welberg LL, Holmes MC & Seckl JR Glucocorticoid programming of pituitary-adrenal function: mechanisms and physiological consequences. Semin. Neonatol 6, 319–329 (2001). [DOI] [PubMed] [Google Scholar]
- 176.Nyirenda MJ, Welberg LA & Seckl JR Programming hyperglycaemia in the rat through prenatal exposure to glucocorticoids — fetal effect or maternal influence? J. Endocrinol 170, 653–660 (2001). [DOI] [PubMed] [Google Scholar]
- 177.Heller EA et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nature Neurosci. 17, 1720–1727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Brunner AM, Nanni P & Mansuy IM Epigenetic marking of sperm by post-translational modification of histones and protamines. Epigenetics Chromatin 7, 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bale TL Lifetime stress experience: transgenerational epigenetics and germ cell programming. Dialogues Clin. Neurosci 16, 297–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kotaja N & Sassone-Corsi P The chromatid body: a germ-cell-specific RNA-processing centre. Nature Rev. Mol. Cell Biol 8, 85–90 (2007). [DOI] [PubMed] [Google Scholar]