

Abstract
The role of even the strongest genetic risk factor for Alzheimer’s disease (AD), the APOE ε4 allele, in its etiology remains poorly understood. We examined molecular signatures of AD defined as differences in linkage disequilibrium patterns between AD-affected and unaffected Whites (2,673/16,246), Hispanics (392/867), and African Americans (285/1,789), separately. We focused on 29 polymorphisms from five genes in the APOE region emphasizing beneficial and adverse effects of the APOE ε2-and ε4-coding SNPs, respectively, and the differences in the linkage disequilibrium structures involving these alleles between AD-affected and unaffected subjects. Susceptibility to AD is likely the result of complex interactions of the ε2 and ε4 alleles with other polymorphisms in the APOE region, and these interactions differ across races/ethnicities corroborating differences in the adverse and beneficial effects of the ε4 and ε2 alleles. Our findings support complex race/ethnicity-specific haplotypes promoting and protecting against AD in this region. They contribute to better understanding of polygenic and resilient mechanisms, which can explain why even homozygous ε4 carriers may not develop AD.
Keywords: ApoE polymorphism, neurodegenerative disorders, Alzheimer’s disease, health span, life span, aging
1. INTRODUCTION
Alzheimer’s disease (AD), an age-related neurodegenerative disorder characterized by accumulation of amyloid-β plaques and neurofibrillary tangles, causes a progressive decline in cognitive function and eventual dementia (Selkoe, 2001). AD affects 5.4 million Americans and its prevalence sharply increases from about 13% at age 65+ years (one in eight people aged 65+) to 45% at age 85+ years (Alzheimer’s, 2012). AD-caused deaths in the U.S. recently increased by 66%. This population trend indicates that mechanisms triggering the development of AD remain largely unknown (Alzheimer’s, 2012), except rare forms of familial (early-onset) AD, which are believed to be primarily caused by mutations in the APP, PSEN1, and PSEN2 genes (Levy-Lahad, et al., 1995,Rogaev, et al., 1995,Sherrington, et al., 1995).
Although the discovery of these genes raised enthusiasm for identifying the genetic basis of late-onset AD (Chouraki and Seshadri, 2014,Reitz and Mayeux, 2014), no causative variants have been reported to date. The APOE ε4 allele (19q13.3 region) remains the main single genetic risk factor for AD development (Raichlen and Alexander, 2014). However, this allele has heterogeneous effects and ε4 carriers in some populations (e.g., Nigerians (Gureje, et al., 2006)) may not be strongly exposed to AD risks. The association of the ε4 allele with AD is also weaker in African Americans (AAs) and Hispanics compared to Whites (Farrer, et al., 1997). The APOE ε2 allele is associated with decreased risk of AD development (Corder, et al., 1994,Strittmatter, et al., 1993,Suri, et al., 2013) and it appears to have the same protective effects across Whites, Hispanics, and AAs (Farrer, et al., 1997).
Despite nearly two decades of APOE research, even the pathogenic role of the ε4 allele in AD remains poorly understood, consistent with competing hypotheses for AD development including the amyloid (Bu, 2009) and mitochondrial (Roses, et al., 2010,Swerdlow, et al., 2014) cascade hypotheses, the infection hypothesis (Fulop, et al., 2018), and others. Understanding the protective role of the ε2 allele has lagged behind that of ε4 due, in part, to its seemingly smaller effects on AD (Corder, et al., 1994,Suri, et al., 2013).
Understanding genetic predisposition to complex traits such as AD is complicated by the inherent heterogeneity of their genetic architecture, caused by polygenicity and etiologic complexity, and the uncertain role of evolution in establishing molecular mechanisms of traits manifested in post-reproductive life (Nesse, et al., 2012). This problem is further augmented by increased life expectancy (Oeppen and Vaupel, 2002), the expanding number of elderly people, and changes in the environment (Corella and Ordovas, 2014,Crespi, et al., 2010,Kulminski, 2013,Vijg and Suh, 2005). Because of these complications, genetic predisposition to AD is likely affected by both evolutionarily established differences between populations of different ancestries and natural-selection-free origins of complex diseases shaped by modern environments.
Following the framework of evolutionary biology, we examined associations of single nucleotide polymorphisms (SNPs) from the APOE region (BCAM, NECTIN2, TOMM40, APOE, and APOC1 genes) with AD and the differences in linkage disequilibrium (LD) structures in AD-affected and unaffected subjects of White, Hispanic, and AA ancestries. We emphasized the beneficial and adverse relationships of APOE ε2- and ε4-coding rs7412 and rs429358 SNPs, respectively, and differences in the LD structures involving these SNPs between AD-affected and unaffected subjects. We show that AD susceptibility is likely the result of complex interactions of the ε2- and ε4 alleles with alleles from other genes in the APOE region, which differ among race/ethnic groups. These findings support complex race/ethnicity-specific haplotypes promoting and protecting against AD in the APOE region.
2. METHODS
2.1. Data availability
This manuscript was prepared using limited access datasets obtained though dbGaP and the University of Michigan. Phenotypic HRS data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data.
2.2. Experimental design
We used data from five cohorts from four studies (described below) to examine the LD structure of the APOE region using 29 SNPs from five genes, including BCAM, NECTIN2, TOMM40, APOE, and APOC1. This set included rs429358 and rs7412 SNPs coding the APOE ε2/ε3/ε4 polymorphism. All selected SNPs were not in perfect LD (r2<0.8). We focused on analysis of genetic associations and the LD structures in Whites, Hispanics, and AAs; men and women combined; affected and unaffected by AD.
2.3. Study cohorts and phenotypes
Data on individuals of European ancestry were drawn from the Framingham Heart Study (FHS) original (FHS_C1) and offspring (FHS_C2) cohorts (Cupples, et al., 2009), Cardiovascular Health Study (CHS) (Fried, et al., 1991), Health and Retirement Study (HRS) (Juster and Suzman, 1995), and NIA Late-Onset Alzheimer Disease Family Study (LOADFS) (Lee, et al., 2008). Data on Hispanics were from HRS and LOADFS and data on AAs were from HRS, LOADFS, and CHS (Table 1). The LOADFS was designed to ascertain dementias of Alzheimer’s type in the elderly and recruited cases and controls. The FHS and CHS collected information on AD in population samples during follow up. In LOADFS, FHS, and CHS, AD was defined based on diagnoses made according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association. The HRS is linked with Medicare service use files and includes enrollment information as well as the diagnoses made (International Classification of Disease [ICD]-revision 9, Clinical Modification) during episodes of care paid for by the Medicare system. A diagnosis of AD was defined based on ICD-9:331.0x in claims paid for by either Medicare Part A (facility-based services [e.g., hospitals]) or Medicare Part B (professional services [e.g., physician practices]). Individuals with AD constituted the case group of n=2,673 Whites, n=392 Hispanics, and n=285 AAs. Individuals without AD constituted the non-case group of n=16,246 Whites, n=867 Hispanics, and n=1,789 AAs.
Table 1.
Basic demographic information for AD-affected and unaffected subjects of different ancestries in the examined cohorts.
| Study | Status | Whites | Hispanics | AAs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N (%) | Men (%) | Age (SD), yrs | N (%) | Men (%) | Age (SD), yrs | N (%) | Men (%) | Age (SD), yrs | ||
| LOADFS | Non-cases | 1865 (50.2) | 751 (40.3) | 76.6 (8.3) | 184 (33.5) | 66 (35.9) | 73.7 (11.0) | 61 (41.5) | 20 (32.8) | 75.9 (7.1) |
| Cases | 1850 (49.8) | 644 (34.8) | 76.6 (7.1) | 366 (66.5) | 122 (33.3) | 78.5 (8.7) | 86 (58.5) | 30 (34.9) | 78.0 (8.8) | |
| HRS | Non-cases | 6963 (96.4) | 3030 (43.5) | 79.0 (8.0) | 683 (96.3) | 283 (41.4) | 76.3 (7.7) | 963 (86.0) | 351 (36.4) | 76.8 (8.3) |
| Cases | 263 (3.6) | 100 (38.0) | 81.5 (7.1) | 26 (3.7) | 8 (30.8) | 82.9 (6.3) | 157 (14.0) | 66 (42.0) | 77.2 (8.1) | |
| CHS | Non-cases | 4074 (94.2) | 1790 (43.9) | 83.4 (5.4) | 765 (94.8) | 288 (37.6) | 81.6 (5.8) | |||
| Cases | 252 (5.8) | 94 (37.3) | 83.2 (5.3) | 42 (5.2) | 14 (33.3) | 82.9 (6.1) | ||||
| FHS_C1 | Non-cases | 426 (67.5) | 149 (35.0) | 88.4 (5.3) | ||||||
| Cases | 205 (32.5) | 61 (29.8) | 87.9 (5.0) | |||||||
| FHS_C2 | Non-cases | 2918 (96.6) | 1332 (45.6) | 69.5 (9.2) | ||||||
| Cases | 103 (3.4) | 51 (49.5) | 77.2 (7.5) | |||||||
| Mega | Non-cases | 16246 (85.9) | 7052 (43.4) | 77.5 (9.9) | 867 (68.9) | 370 (40.3) | 76.4 (7.8) | 1789 (86.3) | 659 (36.8) | 78.9 (7.7) |
| Cases | 2673 (14.1) | 950 (35.5) | 79.4 (7.7) | 392 (31.1) | 131 (33.2) | 78.6 (8.6) | 285 (13.7) | 110 (38.6) | 78.3 (8.3) | |
AD denotes Alzheimer’s disease.
N denotes genotyped sample after excluding individuals with missingness for SNPs greater than 5% and missing information on AD.
SD denotes standard deviation.
LOADFS = the NIA Late Onset Alzheimer’s disease Family Study; HRS = the Health and Retirement Study; CHS = the Cardiovascular Health Study; FHS_C1 = the Framingham Heart Study (FHS) original cohort; FHS_C2 = the FHS Offspring cohort.
Age is given at onset of AD or the end of follow up.
Large proportion of AD cases in LOADFS is due to case-control design.
Large proportion of AD cases in FHS is due to older age of participants of this cohort at the end of follow up and small proportion of men who are at smaller risk of AD.
2.4. Genotypes
We used genotyping data available from the same customized Illumina iSelect array (the IBC-chip, ~50K SNPs) in the FHS and CHS cohorts, Affymetrix 500K in the FHS, Illumina HumanCNV370v1 chip (370K SNPs) in the CHS, Illumina HumanOmni 2.5 Quad chip (~2.5 M SNPs) in the HRS, and Illumina Human 610Quadv1_B Beadchip (~610K SNPs) in the LOADFS.
The analyses focused on 29 SNPs from the BCAM, NECTIN2, TOMM40, APOE, and APOC1 genes in the 19q13.3 region (Table S1). These SNPs were not in perfect LD (r2<0.8). We excluded individuals with >5% missingness. To facilitate cross-platform comparisons, we selected directly genotyped target SNPs using all available arrays for each study. We also selected a proxy for rs1871047 available for Whites (rs1871046, r2=0.82 in the 1000 Genomes Project, CEU population) in the FHS and CHS on the IBC-chip. Non-genotyped SNPs were imputed (IMPUTE2 (Howie, et al., 2009)) according to the 1000 Genomes Project Phase 3 integrated variant set release (SHAPEIT2) in the NCBI build 37 (hg19) coordinate. Most imputed SNPs included in the analyses had high imputation quality (info>0.8) in each study. We also included five SNPs (rs4803763, rs17561351, rs11673139, rs283813, rs12721046) with moderate imputation quality (info>0.3) in LOADFS for Hispanics and AAs (Table S2).
2.5. Association analysis
Univariate associations between SNPs and AD were evaluated using an additive genetic model with the minor allele as an effect allele in each study separately and in a mega-sample of all studies combined, and in Whites, Hispanics, and AAs separately. Given limited information on AD age at onset in the LOADFS, the associations were characterized using a logistic model with AD as a binary outcome and random effects to adjust for potential familial clustering (gee package in R). All statistical tests were adjusted for age (defined as age at onset of AD or the end of follow up), sex, and a variable with information on the sample composition, i.e., field center for CHS, enrollment cohorts in FHS and HRS, and, in addition, study cohorts in the mega-sample.
2.6. LD analysis
We have used methods detailed in Kulminski et al. (Kulminski, et al., 2018). In brief, LD was characterized by the correlation coefficient r using a haplotype-based method (Weir, 1979) because all SNPs were in Hardy-Weinberg Equilibrium (HWE) in each study for each race/ethnicity (Table S1). This method is useful because it leverages biologically plausible deviation from HWE, which can occur in subsamples and/or at the haplotype level (Nielsen, et al., 1998).
Significance of the LD estimates was characterized using chi-square statistics, defined as χ2=r2N, where N=2n is the number of gametes and n is the sample size (Lewontin, 1988). Given potential loss of power because of inferring haplotypes from genotypes (Wellek and Ziegler, 2009), we used a more conservative estimate, with n instead of N. We employed a LD contrast test (Zaykin, et al., 2006) to compare the LD estimates between the AD-affected and unaffected groups. This test was used to characterize the significance of the differences in pair-wise estimates of LD between these two groups.
The significance of the r2 estimates and the differences in the pair-wise estimates of LD were corrected for multiple testing. For the 29 SNPs examined, this represented 406 (=29×28/2) tests. We adopted a conservative Bonferroni correction for significance, p≤1.2×10−4, despite some correlation between these SNPs.
Asymptotically valid confidence intervals were constructed using asymptotic variance adapted from Wellek and Ziegler (Wellek and Ziegler, 2009). This asymptotic variance closely coincided with the exact variance in a sample of n≥60 individuals.
3. RESULTS
3.1. Study overview
Genetic associations with AD and differences in LD between AD-affected and unaffected subjects were examined in mega-samples of Whites (2,673 cases and 16,246 non-cases), Hispanics (392 cases and 867 non-cases), and AAs (285 cases and 1,789 non-cases), separately. The data are from the LOADFS, HRS, CHS, FHS_C1, and FHS_C2 cohorts (Table 1). Mega-samples included data on individuals from all available studies for each race/ethnic group separately. LD patterns were characterized by 29 non-proxy SNPs (defined as LD with r2<0.8) representing the BCAM (4 SNPs), NECTIN2 (16 SNPs), TOMM40 (3 SNPs), APOE (4 SNPs), and APOC1 (1 SNPs) genes in the 19q13.3 region (Table S1), including two SNPs, rs429358 and rs7412, whose minor alleles coded the APOE ε4 and ε2 alleles, respectively (see Section 2. Methods).
3.2. Associations of the rs429358 and rs7412 minor alleles with AD
The minor allele frequency (MAF) of rs429358 was larger in cases than non-cases in all race/ethnic groups. The smallest and the largest MAFs were in White non-cases and cases, respectively, followed by Hispanics and AAs (Fig. 1A). The MAF changed linearly, decreasing in cases and increasing in non-cases across these race/ethnic groups. The minor allele rs429358 was associated with a significantly higher risk of AD development in Whites (β=1.28, p=3.87×10−177) than in Hispanics (β=0.47, p=1.07×10−2) or AAs (β=0.74, p=2.09×10−7), as evidenced by well separated 95% confidence intervals (CIs).
Figure 1. Minor allele frequencies (MAF) and the estimates of genetic effects.

MAF (right y-axis) and statistics for the effects (left y-axis) are given for (A) rs429358 coding the APOE ε4 allele and (B) rs7412 coding the APOE ε2 allele for Whites (W), Hispanics (H), and African Americans (AA), separately. MAF is given for AD-affected (cases, crosses) and unaffected (non-cases, diamonds) subjects. Bars show 95% confidence intervals.
The MAF of rs7412 was the same in Hispanics with and without AD, but it was larger in non-cases than cases of Whites and AAs (Fig. 1B). The beneficial association of the rs7412 minor allele attained significance in Whites (β=−0.78, p=3.35×10−14), but not in Hispanics (β=−0.29, p=3.70×10−1) or AAs (β=−0.36, p=1.14×10−1). Although the association of rs7412 with AD was the most favorable in Whites, no significant differences in these effects were observed across race/ethnic groups, as evidenced by overlapping 95% CIs. The effect directions for rs429358 and rs7412 were consistent in all studies, except for rs7412 in the FHS_C1 study (Table S1).
3.3. Differences in LD between AD-affected and unaffected Whites, Hispanics, and AAs
We contrasted pair-wise LD estimates for the selected 29 SNPs between AD-affected and unaffected subjects in each of three race/ethnic groups separately (Table S3) because LD structures in these groups are significantly different (Fig. S1 Table S4). In Whites and Hispanics, this analysis identified 162 and 19 SNP pairs, respectively, with a Δr=rcases-rnon-cases significant at the Bonferroni-adjusted level, p≤pBonf=1.2×10−4 (for 406 SNP pairs), and 53 and 29 SNP pairs, respectively, at suggestive significances, i.e., pBonf<p<5×10−3. In AAs, no significant differences at p≤pBonf were identified, whereas suggestive significances pBonf<p<5×10−3 were observed for 11 SNP pairs. These molecular signatures of AD, characterized by the differences in LD, are illustrated by heat maps for Δr (Fig. 2).
Figure 2. Molecular signatures of Alzheimer’s Disease (AD) for Whites (A), Hispanics (B), and African Americans (C).
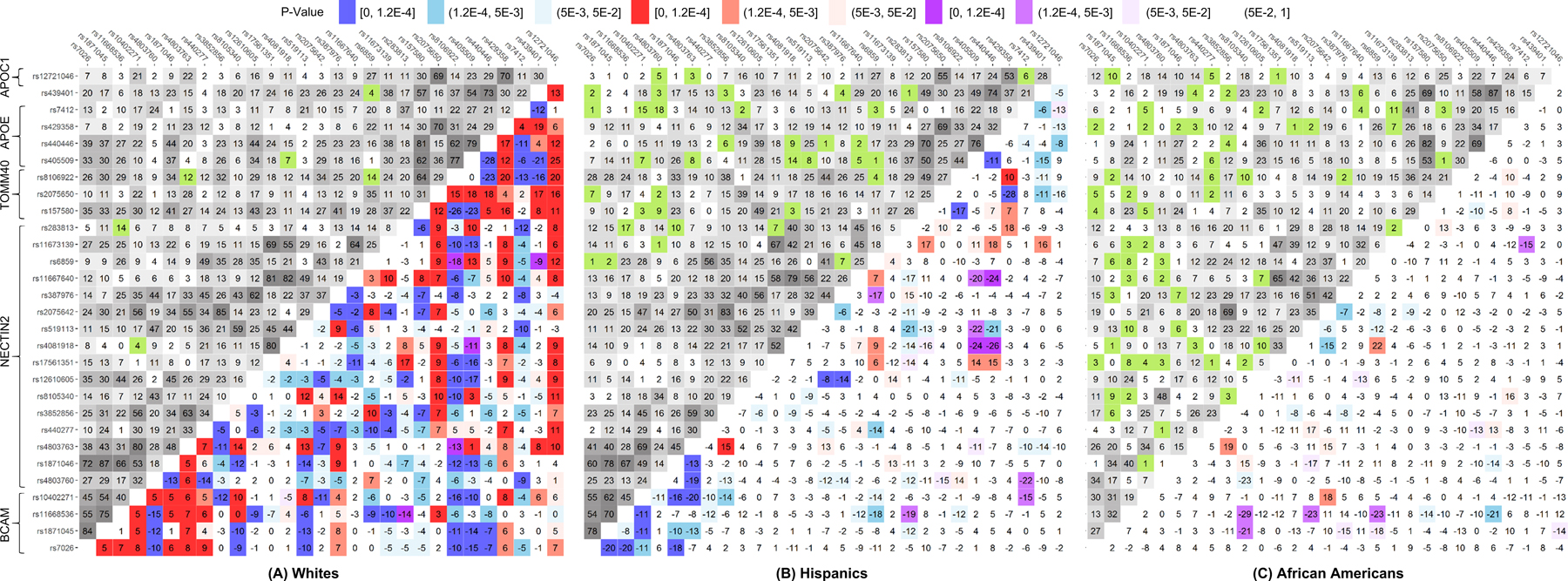
Upper-left triangle: Linkage disequilibrium (LD) pattern (r, %) in the mega-samples, non-cases, for 29 single nucleotide polymorphisms (SNPs). Lower-right triangle: heat map for Δr=rcases−rnon-cases representing the molecular signatures of AD. Red denotes rcases>rnon-cases and blue denotes rcases<rnon-cases. Purple and green indicate the estimates of LD characterized by opposite signs of rcases and rnon-cases. For convenience, positive sign of rnon-cases has been selected. Legend on the top shows color-coded p-values and grey coded LD. Numerical estimates are shown in Table S3.
We observed 15 SNP pairs with significant differences at p≤pBonf=1.2×10−4 both in Whites and Hispanics. For five of them, mostly in the TOMM40-APOE locus, LD changed in the same directions in Whites and Hispanics (i.e., it decreased in AD cases for 4 of 5 SNP pairs), whereas for 10 SNP pairs in the BCAM-NECTIN2 locus LD changed in the opposite directions (i.e., it decreased in AD cases in Hispanics for 9 of 10 SNP pairs) (Fig. 2). LD for one of these 15 pairs (rs4803763 and rs3852856) attained pBonf<p<5×10−3 in AAs and the effect direction aligned with that in Hispanics. Of the remaining 147 (=162–15) SNP pairs with a significant Δr at p≤pBonf in Whites, we observed 14 SNP pairs at suggestive significances (pBonf<p<5×10−3) in Hispanics and 6 in AAs. Likewise, of the remaining four (=19–15) SNP pairs with a significant Δr at p≤pBonf in Hispanics, two SNP pairs (rs12610605 and rs387976, rs2075650 and rs429358) were at suggestive significances (pBonf<p<5×10−3) in Whites, but none in AAs (Table S3).
3.4. The APOE ε2- and ε4-coding rs7412 and rs429358 SNPs, respectively, and the molecular signatures of AD
In non-cases, rs7412 and rs429358 SNPs were in LD with r=11.6%, 9.5%, and 16% in Whites, Hispanics, and AAs, respectively, and that LD was highly significant. In non-cases of these race/ethnic groups, rs429358 (rs7412) was in significant LD (at the Bonferroni-adjusted level for estimates of r using 28 SNPs, i.e., p≤1.8×10−3) with 25 (25), 24 (14), and 17 (18) of 28 SNPs (Table S3) in Whites, Hispanics, and AAs, respectively. For rs429358, the strongest LD was observed with rs2075650 (r~70%, TOMM40) in Whites and Hispanics, with rs12721046 (r=70%, APOC1) in Whites, and with rs405509 (r=34%, APOE) in AAs. For rs7412, the strongest LD was observed with the same SNP, rs283813 (NECTIN2), in Whites (r=37%), Hispanics (r=24%), and AAs (r=42%).
To contrast the LD for rs429358 between AD cases and non-cases, we selected all SNPs for which Δr attained Bonferroni-adjusted level p<pBonf=1.2×10−4 in Whites (13 SNPs) or Hispanics (rs2075650), or marginal significance p=2×10−4 (2 SNPs) in Whites. We observed an increase in the LD of rs429358 with these 16 SNPs in Whites with AD (Fig. 3A), as well as a consistent increase in the LD of rs429358 with rs10402271 (BCAM) and two TOMM40 SNPs (rs157580 and rs8106922) in Hispanics and AAs. For TOMM40 SNPs, the increase was observed despite modest LD between rs157580 and rs8106922 in Whites (r=64%), Hispanics (r=49%), and AAs (r=24%) (Fig. 3). Interestingly, a small but consistent increase in LD in AD cases was also observed between rs429358 and rs7412 SNPs in Whites (p<pBonf), Hispanics (p=8.9×10−2), and AAs (p=1.5×10−1). The estimates of rcases, rnon-cases, and Δr=rcases-rnon-cases for rs4803763 and rs11667640 (NECTIN2) were not significantly (p>0.05) different from zero in AAs (Fig. 3A, rectangles; Table S3). Unlike Whites, LD between rs429358 and several other SNPs virtually did not change in Hispanics and AAs (Fig. 3A). LD in unaffected Hispanics tend to be smaller than in Whites, although locus-wide significance (p<pBonf=1.2×10−4) was attained for three SNPs only. LD in unaffected AAs was significantly smaller than in Whites at p<pBonf=1.2×10−4 for eight SNPs.
Figure 3. The ε4- and ε2-related molecular signatures of Alzheimer’s Disease (AD).


Differences in linkage disequilibrium (LD) between cases and non-cases (Δr=rcases−rnon-cases) are given for (A) the APOE ε4-coding rs429358 SNP and (B) the APOE ε2-coding rs7412 SNP for Whites (W), Hispanics (H), and African Americans (AA). Figure A shows Δr significant at the Bonferroni-adjusted level p<pBonf=1.2×10−4 in at least one race/ethnicity and at the marginal significances p=2×10−4 for 2 SNPs in Whites. Figure B shows Δr significant at the Bonferroni-adjusted level p<pBonf=1.2×10−4 (attained only in Whites) and the suggestive significances (pBonf<p<5×10−3). Cone vertex represent LD in cases, whereas cone base corresponds to LD in non-cases. LD difference Δr for non-significant estimates of rcases and rnon-cases is shown as rectangles. Color of contour of these cones and rectangles codes race/ethnicity as shown on the top. Legends in the insets show color-coded p-values filling cones. Purple shade for SNPs shows the same SNPs in Figures A and B. Boxes show groups of SNPs within genes shown beneath them. Numerical estimates are shown in Table S3. Asterisks above x-axis indicate significant differences in LD between AD unaffected Whites and Hispanics (pink) and Whites and AAs (brown): *0.005<p≤0.05, **1.2×10−4<p≤0.005, and ***p≤1.2×10−4. Boxes around asterisks in figure (A) indicate SNPs with larger LD in Hispanics (pink) and AAs (brawn), respectively, than in Whites. For all other significant estimates, LD is larger in Whites than in non-Whites. Numerical estimates are shown in Table S4.
In contrast to the change in LD for rs429358, we found that the LD of rs7412 with 15 SNPs (excluding rs429358) decreased in Whites, which attained either p<pBonf (8 SNPs) or the suggestive significances pBonf<p<5×10−3 (7 SNPs) (Fig. 3B). The decrease in LD with these SNPs attained suggestive significance for 3 SNPs in Hispanics (rs10402271, rs4803760, and rs439401) and for rs11673139 (NECTIN2) in AAs. The decrease in LD was also observed at nominal (i.e., 5×10−3≤p<5×10−2) significance for rs440446 (APOE) (p=8.2×10−3) in Hispanics. LD of rs7412 consistently decreased in AD cases in Whites, Hispanics and AAs with six SNPs, five of them adjacent to the APOE gene, although these decreases were smaller than in Whites. For five SNPs in the BCAM-NECTIN2 locus, the estimates of rcases, rnon-cases, and Δr=rcases-rnon-cases were not significantly (p>0.05) different from zero in Hispanics or AAs (Fig. 3B, rectangles; Table S3). LD in unaffected Hispanics and AAs tend to be smaller than in Whites attaining significance at p<0.05 for five SNPs in Hispanics and ten SNPs in AAs.
Fig. 3 shows that the observed rearrangements of LD for rs429358 and rs7412 SNPs affected the same 8 SNPs in different genes.
4. DISCUSSION
Our analyses using mega-samples of each of the three race/ethnic groups (Whites, Hispanics, and AAs) from different studies confirmed that the risk of AD for the APOE ε4 allele carriers is significantly higher in Whites than in Hispanics or AAs (Farrer, et al., 1997). We also show that, although the beneficial association of the APOE ε2 allele was more pronounced in Whites, no significant differences were identified across these racial groups, which resembles the results in (Farrer, et al., 1997).
We identified that pair-wise LD between the same SNPs can be different in AD-affected and unaffected individuals in each of these race/ethnic groups. The sets of SNP pairs with significant differences in LD constitute molecular signatures of AD. The most extensive molecular signature, comprising 162 SNP pairs, was observed in Whites, followed by the signature comprising 19 SNP pairs in Hispanics. Although no significant differences in LD at the Bonferroni-adjusted level were identified in AAs, 11 SNP pairs with suggestive significances may contribute to the molecular signatures in AAs. The molecular signatures of AD support the hypothesis that AD is associated with complex haplotypes rather than single alleles in the APOE region (Jazwinski, et al., 2010,Lescai, et al., 2011). Then, the significant changes in LD between AD-affected and unaffected subjects highlight interacting SNPs (or their proxies), which can be involved in AD pathogenesis in a beneficial or adverse manner. Accordingly, analysis of molecular signatures provides an alternative opportunity to identify interacting SNPs, which otherwise could be difficult to do, especially in the case of multi-SNP interactions when the number of tests sharply increases.
Our results show that the significant changes in LD between AD-affected and unaffected Hispanics for vast majority of SNP pairs (15 of 19, 79%) were also significant in Whites, although the directions of change in these race/ethnic groups varied. The opposite-direction changes of LD in Whites and Hispanics were observed for SNPs from the BCAM-NECTIN2 locus, whereas same-direction changes were primarily observed for SNPs from the TOMM40-APOE locus. These findings indicate that the BCAM-NECTIN2 locus can highlight race/ethnic-specific difference in the genetic architecture of AD between Whites and Hispanics, whereas the TOMM40-APOE locus likely indicates common genetic architecture of AD in these race/ethnic groups.
We found that AD in Whites was associated with increases in the LD of rs429358 (SNP coding the strongest genetic risk factor for AD, the APOE ε4 allele) with 15 SNPs from all five genes at the Bonferroni-adjusted and marginal significances (Fig. 3A, all SNPs except rs2075650). In Hispanics and AAs, the changes in LD of rs429358 with these SNPs (except rs2075650 in Hispanics) were of different directions and non-significant. This finding indicates that the adverse effect of the ε4 allele in Whites is likely enhanced by interactions with alleles from other SNPs in this locus. This result is consistent with significantly larger effect size for the ε4 allele in Whites compared with Hispanics and AAs.
We identified consistent increases in LD between the rs429358 and rs8106922 TOMM40 SNP in AD-affected individuals, which attained Bonferroni-adjusted (Whites and Hispanics) and nominal (AAs) significance levels. The LD increases were also observed between rs429358 and rs157580 (TOMM40) in the AD-affected Whites, Hispanics, and AAs that cannot likely be explained by LD between rs157580 and rs8106922 TOMM40 SNPs. These findings suggest these TOMM40 SNPs as potential contributors to common genetic architecture of AD in Whites, Hispanics, and AAs, involving the APOE ε4 allele. Significant changes in LD between rs429358 and the other SNPs in Whites, the lack of such changes in Hispanics and AAs, and smaller LD in unaffected Hispanics and AAs compared to Whites (Fig. 3A) likely indicate genetic mechanisms underlying differences in the APOE-ε4-specific risks of AD across different race/ethnic groups (Fig. 1A).
Contrary to rs429358, AD in Whites was associated with decreases in LD of rs7412 (SNP coding beneficial APOE ε2 allele) with 15 SNPs in the APOE region at the Bonferroni-adjusted (8 SNPs) and suggestive (7 SNPs) (Fig. 3B) significances. This finding suggests that the larger LD strengthens the beneficial effect of the ε2 allele because the large LD was observed in AD unaffected Whites. Accordingly, the smaller LD of rs7412 with the other SNPs in unaffected Hispanics and AAs and smaller and mixed-direction changes in LD between affected and unaffected subjects (Figs. 1 and 2B) may explain, at least in part, smaller magnitudes of the protective effects of the ε2 allele in these race/ethnic groups in our study. These results supports previous findings suggesting that AD can be tailored to the ancestral-specific genetic profiles within different sites at the APOE locus (Babenko, et al., 2018).
Despite the rigor of this study, we acknowledge its limitations. The first is the relatively small samples of Hispanics and AAs compared to Whites in the available data that can limit the power to identify significant differences in LD for some SNP pairs in these race/ethnic groups. The second is that the observed LD differences in AD-affected and unaffected Hispanics and AAs require replication in independent studies, as was done for Whites (Kulminski, et al., 2018). The third is moderate imputation quality for five SNPs in LOADFS for Hispanics and AAs that, however, does not affect the main conclusions of the paper. The fourth is that despite the study researchers followed the same diagnostic criteria for AD in different race/ethnic groups, the AD ascertainment may be affected by various factors driven by racial and ethnic differences such as those related to disease detection, measurement issues, methods, perceptions of the normality of cognition, subjective concerns, etc. (Babulal, et al., 2019).
Thus, our findings indicate that the roles of the ε4- and ε2-coding SNPs in AD were dependent on the other SNPs in this locus. Differences between White and non-White populations in LD structure and changes in LD between the AD-affected and unaffected subjects may explain differences in risks of AD for these alleles in these populations. Our results contribute to a better understanding of resilient mechanisms, which can explain why even homozygous ε4 carriers may not develop AD (Corder, et al., 1993) and live long and healthy lives (Freudenberg-Hua, et al., 2014). Thus, further research is needed to gain insights into the ε2-based protective and ε4-based resilient genetic profiles and their driving forces.
Supplementary Material
Upper-left triangle: LD structures (|r|, %) in the mega-samples of AD unaffected Whites (A), African Americans (B), and Hispanics (C) for 29 single nucleotide polymorphisms (SNPs). Lower-right triangle: heat map for Δr defined as differences in |r| between Whites and Hispanics (A), Whites and African Americans (B), and Hispanics and African Americans (C). Red denotes larger LD in Whites (A and B) or Hispanics (C) compared to the contrasted races/ethnicities in these figures. Blue denotes the opposite. Legend on the top shows color-coded p-values and grey coded LD. Numerical estimates are shown in Table S4.
Table S1. Basic information on SNPs and the associations of these SNPs with AD in mega-samples of different races/ethnicities.
Table S2. Quality of imputation for SNPs used in the analyses.
Table S3. Linkage disequilibrium estimates using haplotype-based method in the mega samples of whites, Hispanics, and African-Americans, separately.
Table S4. Differences in magnitudes of linkage disequilibrium between unaffected Whites, Hispanics and African Americans (AAs) evaluated using haplotype-based method.
Acknowledgements:
This manuscript was prepared using limited access datasets obtained though dbGaP (accession numbers phs000007.v28.p10, phs000287.v5.p1, phs000428.v1.p1, and phs000168.v2.p2) and the University of Michigan. Phenotypic HRS data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data. The authors thank Arseniy P. Yashkin for help in preparation of phenotypes in HRS.
The Framingham Heart Study (FHS) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195 and HHSN268201500001I). This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02-HL-64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding for CARe genotyping was provided by NHLBI Contract N01-HC-65226. Funding support for the Framingham Dementia dataset was provided by NIH/NIA grant R01 AG08122.
The Cardiovascular Health Study (CHS) was supported by contracts HHSN268201200036C, HHSN268200800007C, N01-HC-85079, N01-HC-85080, N01-HC-85081, N01-HC-85082, N01-HC-85083, N01-HC-85084, N01-HC-85085, N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, and N01-HC-85239; grant numbers U01 HL080295 and U01 HL130014 from the National Heart, Lung, and Blood Institute (NHLBI), and R01 AG-023629 from the National Institute on Aging, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at https://chs-nhlbi.org/pi. This manuscript was not prepared in collaboration with CHS investigators and does not necessarily reflect the opinions or views of CHS, or the NHLBI. Additional support for infrastructure was provided by HL105756 and additional genotyping among the African-American cohort was supported in part by HL085251. DNA handling and genotyping at Cedars-Sinai Medical Center was supported in part by National Center for Research Resources grant UL1RR033176, now at the National Center for Advancing Translational Technologies CTSI grant UL1TR000124; in addition to the National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The Health and Retirement Study (HRS) genetic data is sponsored by the Genetics Resource with HRS April 21, 2010, version G Page 5 of 7 National Institute on Aging (grant numbers U01AG009740, RC2AG036495, and RC4AG039029) and was conducted by the University of Michigan. This manuscript was not prepared in collaboration with HRS investigators and does not necessarily reflect the opinions or views of HRS.
Funding support for the Late Onset Alzheimer’s Disease Family Study (LOADFS) was provided through the Division of Neuroscience, NIA. The LOADFS includes a genome-wide association study funded as part of the Division of Neuroscience, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Genetic Consortium for Late Onset Alzheimer’s Disease. This manuscript was not prepared in collaboration with LOADFS investigators and does not necessarily reflect the opinions or views of LOADFS.
Funding: This research was supported by the National Institute on Aging [Grant numbers P01 AG043352, R01 AG047310, and R01 AG061853]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: none.
References:
- Alzheimer’s A. 2012. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement 8(2), 131–68. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Babenko VN, Afonnikov DA, Ignatieva EV, Klimov AV, Gusev FE, Rogaev EI. 2018. Haplotype analysis of APOE intragenic SNPs. Bmc Neurosci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babulal GM, Quiroz YT, Albensi BC, Arenaza-Urquijo E, Astell AJ, Babiloni C, Bahar-Fuchs A, Bell J, Bowman GL, Brickman AM, Chetelat G, Ciro C, Cohen AD, Dilworth-Anderson P, Dodge HH, Dreux S, Edland S, Esbensen A, Evered L, Ewers M, Fargo KN, Fortea J, Gonzalez H, Gustafson DR, Head E, Hendrix JA, Hofer SM, Johnson LA, Jutten R, Kilborn K, Lanctot KL, Manly JJ, Martins RN, Mielke MM, Morris MC, Murray ME, Oh ES, Parra MA, Rissman RA, Roe CM, Santos OA, Scarmeas N, Schneider LS, Schupf N, Sikkes S, Snyder HM, Sohrabi HR, Stern Y, Strydom A, Tang Y, Terrera GM, Teunissen C, Melo van Lent D, Weinborn M, Wesselman L, Wilcock DM, Zetterberg H, O’Bryant SE, International Society to Advance Alzheimer’s, R., Treatment, A.s.A. 2019. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: Update and areas of immediate need. Alzheimers Dement 15(2), 292–312. doi: 10.1016/j.jalz.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. 2009. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10(5), 333–44. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouraki V, Seshadri S. 2014. Genetics of Alzheimer’s disease. Adv Genet 87, 245–94. doi: 10.1016/B978-0-12-800149-3.00005-6. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL, Pericakvance MA. 1994. Protective Effect of Apolipoprotein-E Type-2 Allele for Late-Onset Alzheimer-Disease. Nature Genetics 7(2), 180–4. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123), 921–3. [DOI] [PubMed] [Google Scholar]
- Corella D, Ordovas JM. 2014. Aging and cardiovascular diseases: the role of gene-diet interactions. Ageing research reviews 18, 53–73. doi: 10.1016/j.arr.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Crespi B, Stead P, Elliot M. 2010. Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A 107 Suppl 1, 1736–41. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupples LA, Heard-Costa N, Lee M, Atwood LD. 2009. Genetics Analysis Workshop 16 Problem 2: the Framingham Heart Study data. BMC Proc 3 Suppl 7, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278(16), 1349–56. [PubMed] [Google Scholar]
- Freudenberg-Hua Y, Freudenberg J, Vacic V, Abhyankar A, Emde AK, Ben-Avraham D, Barzilai N, Oschwald D, Christen E, Koppel J, Greenwald B, Darnell RB, Germer S, Atzmon G, Davies P. 2014. Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med 2(5), 438–50. doi: 10.1002/mgg3.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. 1991. The Cardiovascular Health Study: design and rationale. Annals of epidemiology 1(3), 263–76. [DOI] [PubMed] [Google Scholar]
- Fulop T, Witkowski JM, Bourgade K, Khalil A, Zerif E, Larbi A, Hirokawa K, Pawelec G, Bocti C, Lacombe G, Dupuis G, Frost EH. 2018. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front Aging Neurosci 10, 224. doi: 10.3389/fnagi.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureje O, Ogunniyi A, Baiyewu O, Price B, Unverzagt FW, Evans RM, Smith‐Gamble V, Lane KA, Gao S, Hall KS. 2006. APOE ε4 is not associated with Alzheimer’s disease in elderly Nigerians. Annals of neurology 59(1), 182–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. 2009. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6), e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM, Kim S, Dai J, Li L, Bi X, Jiang JC, Arnold J, Batzer MA, Walker JA, Welsh DA, Lefante CM, Volaufova J, Myers L, Su LJ, Hausman DB, Miceli MV, Ravussin E, Poon LW, Cherry KE, Welsch MA. 2010. HRAS1 and LASS1 with APOE are associated with human longevity and healthy aging. Aging Cell 9(5), 698–708. doi:ACE600 [pii] 10.1111/j.1474-9726.2010.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juster FT, Suzman R. 1995. An overview of the health and retirement study. Journal of Human Resources 30, S7–S56. [Google Scholar]
- Kulminski AM. 2013. Unraveling genetic origin of aging-related traits: evolving concepts. Rejuvenation research 16(4), 304–12. doi: 10.1089/rej.2013.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. 2018. Apolipoprotein E region molecular signatures of Alzheimer’s disease. Aging Cell, e12779. doi: 10.1111/acel.12779. [DOI] [PMC free article] [PubMed]
- Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study, G. 2008. Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol 65(11), 1518–26. doi: 10.1001/archneur.65.11.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescai F, Chiamenti AM, Codemo A, Pirazzini C, D’Agostino G, Ruaro C, Ghidoni R, Benussi L, Galimberti D, Esposito F, Marchegiani F, Cardelli M, Olivieri F, Nacmias B, Sorbi S, Tagliavini F, Albani D, Martinelli Boneschi F, Binetti G, Santoro A, Forloni G, Scarpini E, Crepaldi G, Gabelli C, Franceschi C. 2011. An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer’s disease. J Alzheimers Dis 24(2), 235–45. doi:M44461477M774757 [pii] 10.3233/JAD-2011-101764. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. 1995. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269(5226), 973–7. [DOI] [PubMed] [Google Scholar]
- Lewontin RC. 1988. On measures of gametic disequilibrium. Genetics 120(3), 849–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesse RM, Ganten D, Gregory TR, Omenn GS. 2012. Evolutionary molecular medicine. J Mol Med (Berl) 90(5), 509–22. doi: 10.1007/s00109-012-0889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DM, Ehm MG, Weir BS. 1998. Detecting marker-disease association by testing for Hardy-Weinberg disequilibrium at a marker locus. Am J Hum Genet 63(5), 1531–40. doi:Doi 10.1086/302114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeppen J, Vaupel JW. 2002. Demography. Broken limits to life expectancy. Science 296(5570), 1029–31. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- Raichlen DA, Alexander GE. 2014. Exercise, APOE genotype, and the evolution of the human lifespan. Trends Neurosci 37(5), 247–55. doi: 10.1016/j.tins.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Mayeux R. 2014. Genetics of Alzheimer’s disease in Caribbean Hispanic and African American populations. Biol Psychiatry 75(7), 534–41. doi: 10.1016/j.biopsych.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. 1995. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376(6543), 775–8. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. 2010. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10(5), 375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. 2001. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2), 741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375(6534), 754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. 1993. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90(17), 8098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri S, Heise V, Trachtenberg AJ, Mackay CE. 2013. The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE varepsilon2. Neurosci Biobehav Rev 37(10 Pt 2), 2878–86. doi: 10.1016/j.neubiorev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, Khan SM. 2014. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 1842(8), 1219–31. doi: 10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Suh Y. 2005. Genetics of longevity and aging. Annu Rev Med 56, 193–212. doi: 10.1146/annurev.med.56.082103.104617. [DOI] [PubMed] [Google Scholar]
- Weir BS. 1979. Inferences about linkage disequilibrium. Biometrics 35(1), 235–54. [PubMed] [Google Scholar]
- Wellek S, Ziegler A. 2009. A genotype-based approach to assessing the association between single nucleotide polymorphisms. Hum Hered 67(2), 128–39. doi: 10.1159/000179560. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Meng Z, Ehm MG. 2006. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am J Hum Genet 78(5), 737–46. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Upper-left triangle: LD structures (|r|, %) in the mega-samples of AD unaffected Whites (A), African Americans (B), and Hispanics (C) for 29 single nucleotide polymorphisms (SNPs). Lower-right triangle: heat map for Δr defined as differences in |r| between Whites and Hispanics (A), Whites and African Americans (B), and Hispanics and African Americans (C). Red denotes larger LD in Whites (A and B) or Hispanics (C) compared to the contrasted races/ethnicities in these figures. Blue denotes the opposite. Legend on the top shows color-coded p-values and grey coded LD. Numerical estimates are shown in Table S4.
Table S1. Basic information on SNPs and the associations of these SNPs with AD in mega-samples of different races/ethnicities.
Table S2. Quality of imputation for SNPs used in the analyses.
Table S3. Linkage disequilibrium estimates using haplotype-based method in the mega samples of whites, Hispanics, and African-Americans, separately.
Table S4. Differences in magnitudes of linkage disequilibrium between unaffected Whites, Hispanics and African Americans (AAs) evaluated using haplotype-based method.
Data Availability Statement
This manuscript was prepared using limited access datasets obtained though dbGaP and the University of Michigan. Phenotypic HRS data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p=data.