

To the Editor:
Immunodeficiency, centromeric instability, and facial anomalies syndrome (ICF) is a rare autosomal recessive disorder characterized by DNA hypomethylation at the pericentromeric regions of several chromosomes.1 Variants in DNMT3B (ICF-1) account for over half of cases, and variants in ZBTB24 (ICF-2),2 CDCA7 (ICF-3), and HELLS (ICF-4)3 comprise the other half of cases. The majority of patients have hypo- or agammaglobulinemia with normal B cells, though additional defects have been reported.4–6 We present the case of a female patient with impaired NK cell function, decreased NK cell numbers, and abnormal NK cell phenotype who was found to have a novel homozygous ZBTB24 pathogenic variant. She subsequently developed EBV-driven lymphoproliferative disease and was successfully treated with chemotherapy followed by hematopoietic stem cell transplantation (HSCT).
The patient presented at 3 months of age with fever and respiratory distress. After brief hospitalization for presumed bronchiolitis, she returned with new fever, worsening of respiratory symptoms, hepatosplenomegaly, leukopenia, and diffuse hazy infiltrates on chest radiograph (Figure 1A). Further evaluation revealed a whole-blood CMV PCR of 125,257 copies/mL and she was diagnosed with CMV pneumonitis.
Figure 1. Imaging during hospitalizations.



A) CMV pneumonitis. EBV-driven lymphoproliferative disease B) radiograph, C) PET.
She was born full-term and Pennsylvania newborn screen was normal, including T cell receptor excision circles. She had received her 2-month vaccinations—including live oral rotavirus vaccine—without issue, was formula-fed, and had no allergies. Her parents are first cousins from Saudi Arabia, and she has a healthy older brother. Her family history was significant for “blood cancers” in two female cousins and a maternal aunt.
Initial workup (summarized in Table E1 in the Online Repository) was significant for undetectable IgA, low IgG, low CD8+ cells, low NK cells, and low B cells. She received IVIG and valganciclovir. She was discharged on hospital day 32 after CMV PCR was undetectable and continued valganciclovir prophylaxis as an outpatient.
Immunoglobulins and T and B cells normalized over the 9 months after admission. CMV remained undetectable (Table E1). NK cell number remained low, and NK cell function was decreased when repeated three times as a clinical send-out lab to the Cancer & Blood Diseases Institute Clinical Laboratories at Cincinnati Children’s Hospital. Further NK cell analysis at 17 months of age on a research basis (laboratory of EMM and JSO, then at Texas Children’s Hospital) confirmed this functional abnormality and showed an increased percentage of CD56bright cells and a decreased percentage of CD16+ cells (Figure 2). These findings suggest either impaired transition from immature to mature NK cells or impaired survival of mature NK cells. NK phenotype was further defined by 5 flow cytometry panels designed to interrogate human NK cell phenotype and function7 that confirmed that the over-represented CD56bright subset were bona fide immature NK cells (Figure E1 in the Online Repository).
Fig. 2. Impaired NK cell phenotype and function.
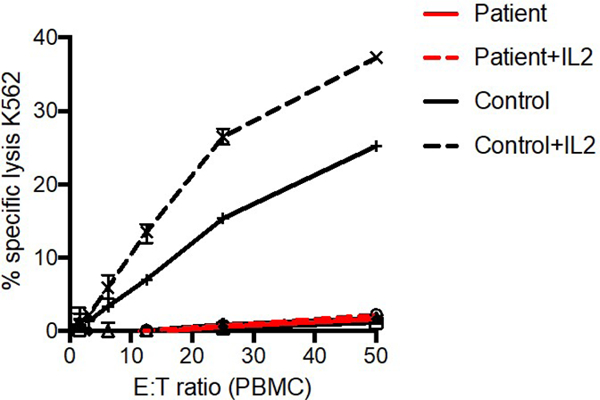
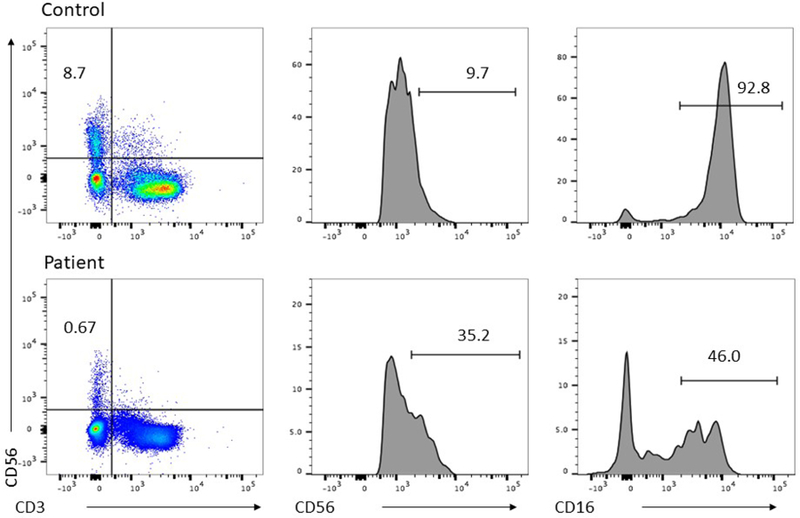
PBMC isolated by standard Ficoll density centrifugation. A) 51Cr cytotoxicity assay against K562 target cells in the presence (dashed line) or absence (solid line) of IL-2. B) FACS analysis of PBMC demonstrating NK cell frequency within the lymphocyte gate (left) and frequency of CD56bright (center) and CD16+ NK cells (right).
Medical Genetics evaluation noted hypotonia, arched eyebrows with blepharophimosis, telecanthus, ptosis, saddle-shaped nose, recessed chin, and gross motor and speech delays at age 17 months. Whole exome sequencing revealed a novel homozygous frameshift variant in ZBTB24 (c.1492_1493del, p.Q498Vfs) consistent with a diagnosis of ICF-2 (MIM phenotype number 614069), which was confirmed with Sanger sequencing. Karyotyping, C-banding, and centromeric instability studies were normal.
Continued evaluation at 32 months of age was significant for low NK cells, low memory B cells, and low pneumococcal titers (Table E1). PPSV23 was administered shortly thereafter.
At 34 months of age, she developed fever and respiratory distress. Initial evaluation revealed bilateral infiltrates on chest radiograph (Figure 1B), respiratory viral panel positive for rhinovirus/enterovirus, and elevated whole-blood EBV PCR of 6,500 copies/mL. CMV PCR was negative. Immunological evaluation showed loss of previously protective tetanus and diphtheria titers, no response to PPSV23, low IgG, and additional abnormalities in lymphocyte subsets (Table E1). Due to persistent high fevers and lack of clinical improvement, chest CT and bronchoscopy with bronchoalveolar lavage (BAL) were pursued. EBV PCR from the BAL specimen was elevated at 83,000 copies/mL, and CT was concerning for lymphoproliferative disease. Due to her tenuous clinical status, one dose of rituximab was administered for presumptive EBV-driven lymphoproliferative disease. She subsequently underwent thoracoscopic lung wedge resection biopsy which was consistent with EBV-driven lymphoproliferative disorder with features of a CD20-negative large B cell lymphoma.
At the time of diagnosis, both HSCT and medical therapy for lymphoma were considered and discussed. Due to the previous success of HSCT for ICF, 8 the evolution of the patient’s immunodeficiency to include persistent hypogammaglobulinemia and a memory B-cell defect in addition to NK abnormalities, and the occurrence of a second severe, life-threatening infection at a young age, HSCT to correct the underlying immunodeficiency was considered an clinically-relevant therapeutic option for this patient. Chemotherapy was initiated, however, due to the patient’s severe illness, the large tumor burden, and the time necessary to orchestrate HSCT.
Chemotherapy was administered per protocol ANHL1131, group B (pre-phase with cyclophosphamide, vincristine, prednisone, intrathecal methotrexate/hydrocortisone; courses 1 and 2 with cyclophosphamide, vincristine, prednisone, doxorubicin, high-dose methotrexate, intrathecal methotrexate/hydrocortisone; and courses 3 and 4 with cytarabine, high-dose methotrexate, intrathecal methotrexate/hydrocortisone, intrathecal cytarabine/hydrocortisone). She ultimately underwent reduced-intensity conditioning per institutional protocol with hydroxyurea, alemtuzumab, fludarabine, melphalan, and thiotepa followed by CD34-selected, 12/12 HLA-matched, unrelated-donor peripheral blood HSCT. Her early post-transplant course was complicated by adeno-, EBV, and CMV viremia which responded quickly to antiviral medications, donor lymphocyte infusion, and rituximab.
She is now greater than 300 days post-transplant, off immunosuppression with 98–100% donor engraftment, without evidence of organ toxicity or graft-versus-host disease, and with excellent immune reconstitution. Recent CMV and EBV whole-blood PCRs were undetectable, and she is receiving IVIG for mild hypogammaglobulinemia thought to be secondary to rituximab given several times throughout her course.
Previously described immune defects in patients with ICF other than hypogammaglobulinemia include decreased CD4+ T cells5 and defective lymphocyte mitogen response.6 This is the first report of impaired NK cell number and function with likely related EBV-driven lymphoma. Little is known about NK cells in ICF, with one patient with ICF-2 who gradually developed decreased NK cells,6 and an additional patient with ICF-2 with low NK cell number and hemophagocytic lymphohistiocytosis.9 Further investigations may include NK phenotyping of other patients with ICF.
This case adds an unexpected disorder to the differential diagnosis of an infant with low NK cell number and/or function and also adds a new presenting phenotype to the known clinical spectrum of ICF-2. Of interest, though this patient had characteristic facies and a novel pathogenic variant in a gene associated with ICF, she had neither centromeric instability nor the typical hypogammaglobulinemia associated with the disorder. The absence of centromeric instability has important implications for diagnosis – a negative centromeric instability test may not rule out ICF. This case also demonstrates a novel requirement for ZBTB24 in human NK cell maturation and function.
Regarding the relationship between this patient’s immunodeficiency, her susceptibility to EBV, and her ultimate diagnosis of EBV-driven malignancy, we suggest that her NK dysfunction was most clinically significant. It has been described that surveillance by NK and antigen-specific CD8+ T-cells are the main protectors against primary EBV infection, and that lymphoproliferative disorders (LPDs) can emerge when EBV infection escapes the cellular immune response. It has recently been reported that two-thirds of EBV-associated LPDs occur in patients with B- and T-cell deficiency, highlighting the importance of cellular immunity in protection against EBV-driven processes.10 In a recent review by Tangye et al, the importance of NK cells in human anti-EBV immunity is reviewed, in addition to several recent studies in mice which suggest that NK dysfunction leads to higher incidence and increased severity of EBV driven lymphoproliferation compared to NK-sufficient mice. 11 Regarding the patient’s hypogammaglobulinemia, EBV-specific antibodies have been shown to possibly modulate disease severity rather than to prevent disease, and cellular-based mechanisms appear to play the largest role in EBV immunity.11
It has been suggested that HSCT should be considered for patients with ICF with evidence of T cell dysfunction.8 This case suggests that patients with ICF-2 syndrome should have their NK cell function thoroughly investigated and transplant should be considered as a potential therapeutic option if NK cell defects are found.
Supplementary Material
Clinical implications:
This is the first reported case of both 1) ICF syndrome presenting as NK dysfunction and 2) EBV-driven malignancy in ICF syndrome. Hematopoietic stem cell transplantation treated the immunodeficiency and should be considered early in ICF-2 with NK cell defects.
Acknowledgements/Funding Statement:
This work was supported by NIH-NIAID R01 AI120989 to JSO and NIH-NHGRI UM1 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure Statement:
C.M. Burk reports a travel grant from the American Academy of Allergy, Asthma, and Immunology outside the submitted work. B.L. Bostwick reports personal fees from Baylor Genetics and Alnylam Pharmaceuticals, and also support from Biomarin Pharmaceuticals outside of the submitted work. I.K. Chinn reports grant support from the Jeffrey Modell Foundation and from the NIH during the conduct of the study, and also personal fees from Wolters Kluwer/UpToDate (royalties), the American Academy of Pediatrics (author stipend), and the Clinical Immunology Society (travel grant) outside the submitted work. J. R. Lupski reports personal fees from Regeneron Genetics Center (Scientific Advisory Board), stock options from 23andMe, and grant support from NIH/NHGRI, NIH/NIGMS, NIH/NINDS, and the Muscular Dystrophy Association. D. Ortiz reports personal fees from Sanofi-Genzyme (honoraria for consult work) outside the submitted work. J.S. Orange reports personal fees from ADMA Biologist (Scientific Advisory Board), Takeda (consult work), Grifols (consult work), CSL Behring (consult work), Wolters Kluwer/UpToDate (editor/author stipend) outside the submitted work. H.J. Chong reports personal fees from Horizon Pharma (Actimmune Advisory Board) during the conduct of the study. The rest of the authors declare that they have no conflict of interest to report (KEC, EMM, ZHC, SNJ, DO, JLB, SWA, LR).
REFERENCES
- 1.Jeanpierre M, Turleau C, Aurias A, Prieur M, Ledeist F, Fischer A, Viegas-Pequignot E. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet 1993;2(6):731. [DOI] [PubMed] [Google Scholar]
- 2.de Greef JC, Wang J, Balog J1, den Dunnen JT, Frants RR, Straasheijm KR, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet 2011. June 10;88(6):796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thijssen PE, Ito Y, Grillo G, Wang J, Velasco G, Nitta H, et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat Commun 2015;6:7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hagleitner MM, Lankester A, Maraschio P, Hultén M, Fryns JP, Schuetz C, et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J Med Genet 2008;45:93–99. [DOI] [PubMed] [Google Scholar]
- 5.Rechavi E, Lev A, Eyal E, Barel O, Kol N, Barhom SF, et al. A Novel Mutation in a Critical Region for the Methyl Donor Binding in DNMT3B Causes Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome (ICF). J Clin Immunol 2016. November;36(8):801–809. [DOI] [PubMed] [Google Scholar]
- 6.von Bernuth H, Ravindran E, Du H, Fröhler S, Strehl K, Krämer N, et al. Combined immunodeficiency develops with age in Immunodeficiency-centromeric instability-facial anomalies syndrome 2 (ICF2). Orphanet J Rare Dis 2014. October 21;9:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahapatra S, Mace EM, Minard CG, Forbes LR, Vargas-Hernandez A, Duryea TK, et al. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS One 2017. August 2;12(8):e0181134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gennery AR, Slatter MA, Bredius RG, Hagleitner MM, Weemaes C, Cant AJ, et al. Hematopoietic stem cell transplantation corrects the immunologic abnormalities associated with immunodeficiency-centromeric instability-facial dysmorphism syndrome. Pediatrics 2007. November;120(5):e1341–4. [DOI] [PubMed] [Google Scholar]
- 9.Harnisch E, Buddingh EP, Thijssen PE, Brooks AS, Driessen GJ, Kersseboom R. Hematopoietic Stem Cell Transplantation in a Patient With ICF2 Syndrome Presenting With EBV-Induced Hemophagocytic Lymphohystiocytosis. Transplantation 2016. July;100(7):e35–6. [DOI] [PubMed] [Google Scholar]
- 10.Riaz IB, Faridi W, Patnaik MM, Abraham RS. A Systematic Review on Predisposition to Lymphoid (B and T cell) Neoplasias in Patients With Primary Immunodeficiencies and Immune Dysregulatory Disorders (Inborn Errors of Immunity). Front Immunol 2019. April 16;10:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tangye SG, Palendira U, Edwards ESJ. Human immunity against EBV—lessons from the clinic. J Exp Med 2017. February; 214(2): 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.