

Abstract
Tertiary lymphoid structures (TLS) accumulate at sites of chronic injury where they function as an ectopic germinal center, fostering local autoimmune responses. Vascular injury leads to the release of endothelial‐derived apoptotic exosome‐like vesicles (ApoExo) that contribute to rejection in transplanted organs. The purpose of the study was to evaluate the impact of ApoExo on TLS formation in a model of vascular allograft rejection. Mice transplanted with an allogeneic aortic transplant were injected with ApoExo. The formation of TLS was significantly increased by ApoExo injection along with vascular remodeling and increased levels of antinuclear antibodies and anti‐perlecan/LG3 autoantibodies. ApoExo also enhanced allograft infiltration by γδT17 cells. Recipients deficient in γδT cells showed reduced TLS formation and lower autoantibodies levels following ApoExo injection. ApoExo are characterized by proteasome activity, which can be blocked by bortezomib. Bortezomib treated ApoExo reduced the recruitment of γδT17 cells to the allograft, lowered TLS formation, and reduced autoantibody production. This study identifies vascular injury‐derived extracellular vesicles (ApoExo), as initiators of TLS formation and demonstrates the pivotal role of γδT17 in coordinating TLS formation and autoantibody production. Finally, our results suggest proteasome inhibition with bortezomib as a potential option for controlling TLS formation in rejected allografts.
Keywords: antigen presentation/ recognition, autoantibody, basic (laboratory) research/science, cell death: apoptosis, immunobiology, rejection: vascular, vasculopathy
Short abstract
In this study, Dieudé et al identify vascular injury–derived extracellular vesicles as initiators of tertiary lymphoid structure formation and autoantibody production, demonstrate the pivotal role of γδT17 in coordinating formation of these structures, and suggest proteasome inhibition with bortezomib to control tertiary structure formation in rejected allografts.
Abbreviations
- AID
activation‐induced cytidine deaminase
- ANA
antinuclear antibodies
- ApoExo
apoptotic exosome‐like vesicles
- AT1R
angiotensin II type 1 receptors
- BOS
bronchiolitis obliterans syndrome
- DMEM
Dulbecco's modified eagle medium
- DMSO
dimethyl sulfoxide
- DSA
donor‐specific antibodies
- dsDNA
double stranded DNA
- EC
endothelial cell
- ECGS
endothelial cell growth supplements
- FBS
fetal bovine serum
- H&E
hematoxylin and eosin
- H2SO4
sulfuric acid
- mEC
murine endothelial cells
- PE
phycoerythrin
- RORγ
transcription factor retinoic acid receptor‐related orphan receptor gamma
- TCRβ
TCR beta
- TCRγ
TCR gamma
- TCRγδ
TCR gamma delta
- Th17
T helper 17
- TLS
tertiary lymphoid structures
- γδKO
gamma delta T cell–knockout
- γδT
gamma delta T
- γδTh17
gamma delta T helper 17
1. INTRODUCTION
Tertiary lymphoid structures (TLS) are associated with chronic infections, chronically rejected organ transplants and autoimmune conditions.1 TLS are nodular structures comprised of segregated T and B cells reminiscent of secondary lymphoid organs. They function as an ectopic germinal center, allowing local antigen presentation and maturation of B cells into memory B cells and antibody‐secreting plasma cells. The production of antibodies within TLS, whether autoantibodies in autoimmune conditions or allo‐ and autoantibodies in the case of transplanted organs, fuels local inflammation, complement activation, and further tissue destruction. The presence of TLS in heart, kidney, and lung allografts has been associated with severe and/or chronic forms of rejection.2 TLS formation has also been reported in the adventitia of epicardial coronary arteries with coronary artery vasculopathy following heart transplantation.3 Yet, the specific pathways and immune cells that trigger the formation of TLS in organ transplants and control their organization at sites of chronic inflammation remain ill defined.
Tissue injury, including ischemia reperfusion at time of transplantation, can trigger autoantibody production. In turn, various types of autoantibodies have been shown to induce or accelerate rejection of allogeneic organs, either on their own or in association with allogeneic donor‐specific antibodies. Autoantibodies against vimentin, angiotensin II type 1 receptors (AT1R), collagen V, fibronectin and perlecan/LG3 as well as natural autoantibodies targeting constituents of apoptotic cells have been associated with severe rejection episodes and reduced survival of heart, lung, and kidney allografts.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 Most of these autoantibodies increase vascular inflammation and complement deposition within the allograft. Recently, our group identified a novel type of membrane vesicles released at the time of ischemia reperfusion by dying endothelial cells as a trigger for autoantibody production and acceleration of rejection. We showed that vascular injury resulting in caspase‐3‐dependent death of endothelial cells fuels the release of apoptotic exosome‐like vesicles (ApoExo) that in turn can foster the production of autoantibodies such as anti‐perlecan/LG3.15 ApoExo differ from classical apoptotic bodies in size, protein content, and function. They are characterized by the presence of a functionally active 20S proteasome core complex and perlecan/LG3 autoantigen and aggravate vascular allograft rejection.15 Bearing in mind the association between TLS, autoimmunity, and vascular inflammation we evaluated whether ApoExo regulate the formation of TLS in rejecting allografts.
Here, we identify extracellular vesicles derived from vascular injury, ApoExo, as novel inducers of tertiary lymphoid structure formation. We also identify their proteasome activity as a pivotal mediator of TLS formation, attracting IL‐17‐producing gamma delta T cells to the allograft that in turn control TLS formation.
2. MATERIALS AND METHODS
2.1. Endothelial cell (EC) culture and preparation of conditioned media
Murine ECs (mEC) were isolated from the aorta of C57BL/6 or BALB/c mice grown in Dulbecco's modified eagle medium (DMEM) low‐glucose culture media supplemented with endothelial cell growth supplements (ECGS; Alfa Aesar, Haverhill, MA), 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), 10% newborn calf serum (Invitrogen), heparin (12.6 U/mL, Sandoz, Holzkirchen, Germany), 1% penicillin‐streptomycin, and 1% amphotericin B. To generate conditioned media, cells were exposed for 9 hours (mEC) to serum‐free media RPMI‐1640 (Invitrogen) alone or in the presence of either the proteasome inhibitor bortezomib (100 µg/mL; Stressmarq, Victoria, BC, Canada) or its vehicle, dimethyl sulfoxide (DMSO). In previous work, we demonstrated that this system leads to the release of active mediators by apoptotic ECs downstream of caspase‐3 activation without cell membrane permeabilization (Figure S1).
2.2. Animal studies
Adult C57BL/6, BALB/c (20‐22 g; Charles River, Kingston, NY), and T cell receptor (TCR) delta−/− (20‐22 g; B6.129P2‐Tcrdtm1Mom/J; JAX stock #002120; Jackson Laboratory, Bar Harbor, ME) mice were housed in sterilized, ventilated cages in a specific pathogen‐free animal facility under a standard 12‐hour light/12‐hour dark cycle and fed a normal diet ad libitum. All animal experiments and methods were performed in accordance with the relevant guidelines and regulations approved by the CRCHUM’s Comité Institutionnel de Protection des Animaux (CIPA).
2.3. Aortic transplantation procedures
Mice were anesthetized with isoflurane (2%) by inhalation. Aortic transplantation was performed as described previously.16 Briefly, 1 mL of heparinized saline (50 μL/mL) was injected into the vena cava to flush the aorta. A 6‐mm segment of abdominal aorta measured from below the renal arteries to just above the aortic bifurcation was excised and soaked in ice‐cold 0.9% normal saline. When mentioned, warm ischemia was induced by clamping the aorta for 15 minutes before excision from the donor. The grafts were then excised and sutured orthotopically with end‐to‐end anastomoses using 11‐0 nylon interrupted sutures.
2.4. Injection of murine apoptotic endothelial membrane vesicles
Serum‐free media conditioned by 1 × 104 mECs from C57 C57BL/6 or BALB/c mice were fractionated using sequential centrifugation: a first centrifugation 1200g for 15 minutes at 4°C to pellet cell debris; a second centrifugation at 50 000g for 15 minutes at 4°C to pellet apoptotic bodies; and a final ultracentrifugation at 200 000g for 18 hours at 4°C to pellet exosome‐like vesicles. Pellets containing either apoptotic bodies or exosome‐like vesicles were resuspended in half of the initial volume of conditioned medium. Transplanted mice received tail vein (150 µL) intravenous injections of resuspended apoptotic bodies or ApoExo preparations every other day during 3 weeks, for a total of eight doses.
2.5. Assessment of circulating levels of antinuclear antibodies (ANA), total IgGs, anti‐double stranded DNA (dsDNA), anti‐AT1R, anti‐perlecan/LG3, anti‐vimentin, and anti‐fibronectin
ANA, total IgG, anti‐dsDNA, and anti‐AT1R levels were assessed using ANA mouse bioassay kits (US Biologicals, Salem, MA), Mouse IgG Total Ready‐SET‐Go kits (Affymetrix, Santa Clara, CA), Anti‐dsDNA mouse ELISA kits (BioVendor, Asheville, NC), and Angiotensin 1 Receptor Antibody (Anti AT1R) BioAssay™ ELISA Kit (Mouse; US Biological), respectively, in accordance with the manufacturers’ instructions. Anti‐LG3, anti‐vimentin, and anti‐fibronectin titers were measured with locally developed ELISAs. Recombinant perlecan fragment LG3 was produced and purified as previously described.17 The purity of the recovered LG3 protein was assessed by reducing SDS‐PAGE and Coomassie Blue R‐250 staining. Recombinant mouse LG3 (5 ng/µL), vimentin (5 ng/µL, Cloud‐Clone Corp., Katy, TX) or fibronectin (5 ng/µL, MyBioSource, San Diego, CA) was first coated onto 96‐well Immulon II HB plates (Thermo Electron, Waltham, MA), for a total of 0.5 µg per well. Notably, mouse and human LG3 fragments are highly homologous at the amino acid level (87%). The sera were diluted (1:100), and 100 µL were added to each well. The plates were washed, and bound IgG was detected using horseradish peroxidase coupled with anti‐mouse IgG (Amersham, Piscataway, NJ). Reactions were revealed with 100 µL of tetramethylbenzidine substrate (BD Biosciences, San Jose, CA) and stopped with 50 µL of sulfuric acid (1 mol/L H2SO4). Spectrophotometric analysis was taken at 450 nm, and the results were expressed as optical density × 1000.
2.6. Measurement of murine antidonor IgG
Sera were diluted 1:100 in FACS buffer and incubated with 1 × 106 BALB/c splenocyte targets for 30 minutes at 4°C. The samples were then washed three times and stained with phycoerythrin (PE) goat anti‐mouse IgG and Alexa 488 anti‐mouse CD3e (BD Biosciences) in FACS buffer for 30 minutes in the dark at 4°C. Samples were run on a flow cytometer (FACScan, BD) and analyzed using the computer software FACS DIVA (Becton Dickinson, Franklin Lakes, NJ). A CD3+ parent gate was used to avoid nonspecific background signals from Fc receptor–expressing cells.
2.7. Immunohistochemistry
Transplanted aortas were harvested 3 weeks posttransplantation. Tissues were fixed with 10% neutral‐buffered formalin and paraffin‐embedded according to established methods. Samples were cut into 4‐μm slices. Immunohistochemical staining against CD20 epitope was carried out using the automated Discovery XT staining platform from Ventana Medical Systems (Roche Group, Tucson, AZ) and with the automated Bond RX staining platform (Leica Biosystems, Wetzlar, Germany) for CD3, IL‐17, and activation‐induced cytidine deaminase (AID) stainings. Sections were deparaffinized inside immunostainer. For the CD20, staining antigen recovery was conducted using heat‐induced epitope retrieval with citrate buffer. For CD3 staining, antigen recovery was conducted using protease‐induced epitope retrieval with Enzyme 1 (Leica Biosystems) and with heat‐induced epitope retrieval with ER1 (Leica Biosystems) for IL‐17 and AID stainings. Sections were then incubated with anti‐CD20 antibody (Acris, Rockville, MD, 1/50 dilution), anti‐CD3 (BIO‐RAD, Hercules, CA, 1/100), anti‐IL‐17 (Abcam, Cambridge, UK, 1/400), or AID antibody (LSBio, Seattle, WA, 1/100). Detection of specific signal for CD20 staining was acquired by using DABmap detection kit (#760‐124, Ventana Medical Systems ‐ Roche, Oro Valley, AZ) followed by Biotin‐SP‐conjugated Affinipure Donkey Anti‐Rabbit IgG (Jackson ImmunoResearch, West Grove, PA, 1/100) and slides were counterstained manually with hematoxylin and eosin (H&E). Detection of specific signal was acquired by using Bond Intense R Detection System (#DS9263, Leica Biosystems) for CD3 staining and with Bond Polymer DAB Refine kit (#DS9800, Leica Biosystems) for Il‐17 and AID stainings. Slides were counterstained automatically with H&E included in the Polymer DAB kit.
Digital images of tissues were captured using a Leica DMLS microscope and Leica DFC420C camera (Leica Microsystems) for H/E, CD20 and CD3 stainings and an Olympus VS110 slide scanner microscope (Model BX61VSF) was used to capture IL‐7 and AID stainings. Grafts intimal‐ and medial‐area were outlined and quantified using a digital image analysis program (ImageJ 1.42q, NIH). The numbers of T cells and B cells in the tissue were measured by quantification of the number of CD3+ and CD20+ cells, respectively, per 6 high‐power fields per allograft IL‐17, and AID deposition was quantified using VisiomorphTM VIS Histoinformatics Software (Olympus). For each graft, the number of TLS was recorded by counting the number of TLS on 3 standardized unconnected sections set X µmol/L apart.
2.8. Flow cytometry analyses of aortic graft infiltrates
Transplanted aortas were harvested 3 weeks posttransplantation. Leukocytes were retrieved from the aorta as previously described.18 Briefly, harvested aortas were digested in a phosphate‐buffered saline solution with 450 U/mL collagenase type I, 125 U/mL collagenase type XI, 60 U/mL hyaluronidase type I‐S, and 60 U/mL deoxyribonuclease I for an hour at 37°C. After passing through a 70 µm cell strainer, cell suspension was stimulated O/N with Phorbol 12‐myristate 13‐acetate (Sigma, St. Louis, MO) 10 ng/mL and Ionomycine (EMD Millipore, Burlington, MA) 1 µg/mL O/N at 37°C followed by Brefeldine A (Sigma) 5 µg/mL treatment for 3 hours at 37°C. Infiltrating T cells subsets were characterized using CD3 APC/Fire750 (Biolegend, San Diego, CA), anti‐TCR gamma delta (TCRγδ) (clone GL3), BV421 (BD Biosciences), TCR beta (TCRβ) chain (clone: H57‐597), Alexa Fluor 488 (BD Biosciences), transcription factor retinoic acid receptor‐related orphan receptor gamma (RORγ) PerCP‐efluor 710 (eBioscience, San Diego, CA), IL‐17A BV711 (Biolegend), and IL‐17F Alexa fluor 647 (Biolegend) antibody staining. Samples were run on a flow cytometer (FACScan, Becton Dickinson) and analyzed on the computer software FlowJo.
2.9. Statistical analyses
All data are expressed as means ± SEM derived from at least three independent experiments, unless otherwise specified. Biological and histological data were compared using a Student's t test. Statistical analyses were performed using Prism 4 (Prism‐GraphPad Software, Inc). P values of <.05 were considered significant.
2.10. Study approval
All experiments were performed according to the animal experimental guidelines upon the approval CRCHUM’s Comité Institutionnel de Protection des Animaux .
3. RESULTS
3.1. Apoptotic exosome‐like vesicles trigger the formation of lymphoid‐like structures within vascular allografts in association with the production of autoantibodies
We previously showed that ApoExo, extracellular vesicles reminiscent of exosomes produced by apoptotic endothelial cells, but not classical apoptotic bodies, foster the production of autoantibodies and enhance vascular inflammation.15 This prompted us to consider the possibility that ApoExo could trigger the formation of TLS, which in turn favors the development of autoimmune responses and allograft inflammation. C57Bl/6 mice transplanted with a major histocompatibility complex (MHC) mismatched allogeneic aortic transplant from BALB/c donor mice were injected intravenously every other day for 3 weeks posttransplantation with ApoExo produced by serum starved apoptotic ECs in vitro as previously described.15 In this system, ApoExo are released through caspase‐3–dependent pathways in the early phases of endothelial apoptosis (Figure S1).15 ApoExo injection increased allograft vascular remodelling with enhanced neointima formation (Figure 1A,B) as reported previously.15 It also led to a significant increase in the number of infiltrating CD20+ B cells and CD3+ T cells. Increased CD20+ B cell infiltration was found throughout the vessel wall whereas CD3+ T cell infiltration was significantly enhanced only in the adventitial perivascular area. Perivascular infiltration was highly organized resulting in the formation of nodular structures (Figure 1A,D,E) reminiscent of TLS. These structures were characterized by polarized clusters of CD20+ B cell and CD3+ T cell aggregates (Figure 1A,C). The expression of AID, a key enzyme that controls class switch recombination and somatic hypermutation, was detected in the B cell region of the TLS, indicating germinal centre activity (Figure 1A,F). Staining for IL‐17, a cytokine associated with autoimmunity and TLS formation, was enhanced throughout the vessel wall in allograft recipients injected with ApoExo (Figure 1A,G). Of note, vascular inflammation, TLS formation, and neointima formation were specific to the allogeneic graft, as the native aorta adjacent to the allograft did not show leukocyte infiltration, TLS formation, or neointima formation (Figure S2).
Figure 1.
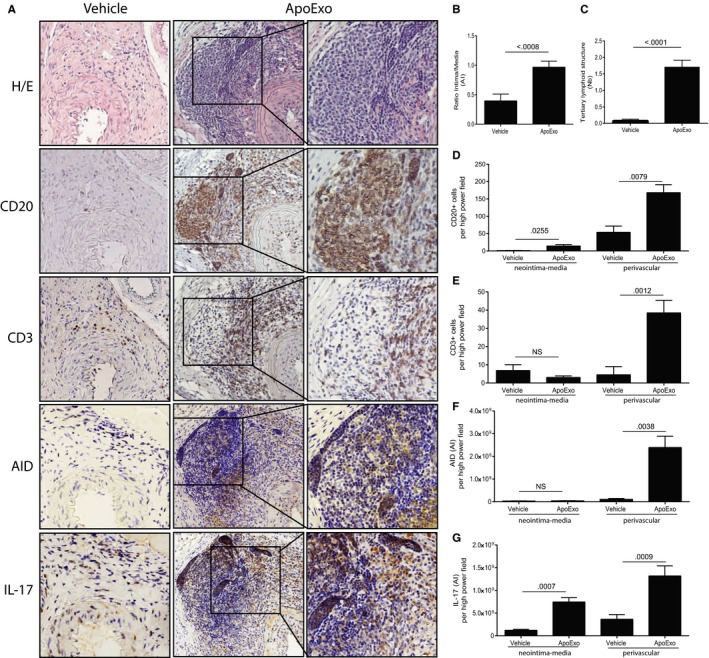
Apoptotic exosome‐like vesicles (ApoExo) are novel triggers of IL‐17 production and tertiary lymphoid structure (TLS) formation in vascular allografts. Mice were injected with the vehicle (n = 13) or ApoExo (n = 16) for 3 weeks posttransplantation: (A) Aortic allograft sections stained with H&E, CD20, CD3, AID, and IL‐17 (magnification: 5×; magnification of right inset panels: 20×). (B) Ratio intima/media in the allografts. (C) Mean number of TLS per allograft. Neointima‐media and perivascular quantification of CD20+ B cells (D), CD3+ T cells (E), AID (F), and IL‐17 (G) staining in each high‐power field of the allografts. Data were pooled from 3 independent experiments and expressed as means ± SEM. Comparison with the vehicle was done with a Student's t test
We also evaluated the autoimmune and alloimmune antibody responses developing in allograft recipients injected in ApoExo. Injection of ApoExo significantly increased the production of anti‐perlecan/LG3 IgG antibodies (Figure 2A) and ANA (Figure 2B), compared to mice injected with apoptotic bodies or the vehicle. However, endothelial ApoExo did not induce a generalized autoimmune response as levels of anti‐dsDNA, anti‐fibronectin, anti‐vimentin, and anti‐AT1R, were not affected by the injection of ApoExo, apoptotic bodies, or the vehicle (Figure 2C‐F, respectively). Injection of ApoExo did not affect levels of donor‐specific antibodies (DSA), a finding that was expected since injections were performed with autologous ApoExo (Figure 2G,H).
Figure 2.
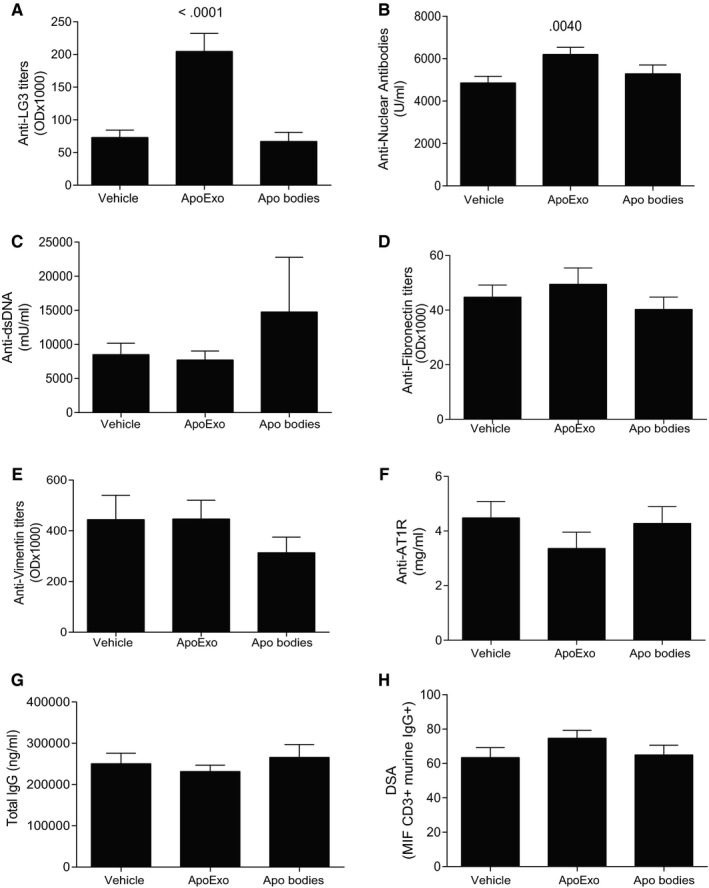
Apoptotic exosome‐like vesicles (ApoExo) trigger the production of anti‐LG3 IgG and ANA in allografted mice. Anti‐LG3 (A), ANA (B), anti‐dsDNA (C), anti‐fibronectin (D), anti‐vimentin (E), anti‐ AT1R (F), total IgG (G), and DSA (H) IgG levels in sera from allografted mice after 3 weeks of intravenous injections with vehicle (n = 13), ApoExo (n = 16), or apoptotic bodies (n = 17)
To evaluate the importance of the source of ApoExo (donor vs recipient), ApoExo from syngeneic (C56Bl/6) or from allogeneic (BALB/c) endothelial cells were injected to allografted mice using previously described protocol. Our results demonstrate that the syngeneic or allogeneic origin of ApoExo does not change their impact on vascular remodeling, TLS formation, and autoantibody production (Figure S3).
Additionally, to characterize the impact of ApoExo in a context exempt of alloimmune dependent vascular injury, we performed syngeneic aortic transplantations and injected the recipients with syngeneic ApoExo. ApoExo injection in the syngeneic graft model did not trigger DSAs but prompted the production of similar levels of anti‐LG3 autoantibodies and ANA to levels similar to as the ones observed in the allografted model (Figure S2). However, and contrary to allogeneic grafts, aortic graft from the syngeneic model injected with ApoExo did not show signs of enhanced vascular remodeling and TLS formation observed in the allografted model (Figure S2). These results demonstrate that ApoExo trigger autoantibody production independently of alloimmunity but that their pathophysiological impact is optimal develops only in the context of ongoing alloimmune‐dependent vascular injury.
Collectively, these results identify ApoExo as novel triggers of TLS formation in vascular allografts.
3.2. Apoptotic exosome-like vesicles trigger the recruitment of gamma delta T helper 17 (γδTh17) cells to the allograft
Having shown increased allograft IL‐17 expression in allograft recipients injected with ApoExo, and taking into consideration previous work suggesting a central role for T helper 17 (Th17) cells in TLS neogenesis,1 we used flow cytometry analysis to evaluate the presence of Th17 cells within the allograft infiltrate. Although immunohistochemistry demonstrated a significant enhancement of infiltrating CD3+ T cells in allografts from mice injected with ApoExo compared to those injected with the vehicle (Figure 1E), flow cytometry analysis showed that the number of allograft infiltrating CD3+TCRβ+ T cells remained low upon injection of ApoExo compared to mice injected with vehicle (Figure 3A). Furthermore, the percentage of TCRβ+ cells from the overall CD3+ infiltrate dramatically decreased in the same conditions (Figure 3B). In addition, IL‐17 and RORγ, which are characteristic of Th17 cells, were not expressed by infiltrating CD3+TCRβ+ T cells (Figure S4). These results suggest a subset of CD3+ cells, other than Th17 cells, is responsible for the production of IL‐17 within the allograft of mice injected with ApoExo.
Figure 3.
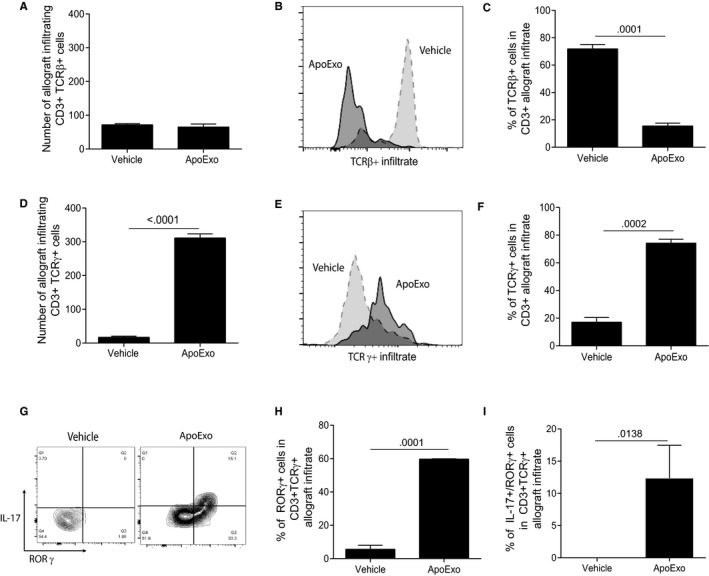
Apoptotic exosome‐like vesicles (ApoExo) triggers the recruitment of IL‐17+ RORγ+ γδT cells to vascular allografts. CD3+ T cell infiltrates in aortic allografts from allografted mice that underwent 3 weeks of injections with the vehicle (n = 7) or ApoExo (n = 8) were analyzed by flow cytometry for TCRβ, IL‐17, and RORγ expression. (A) Number of allograft infiltrating CD3+TCRβ+ cells for each condition. (B) Histogram showing detection of TCRβ+ cells. (C) Percentage of TCRβ+ cells in CD3+ allograft infiltrate for each condition. (D) Number of allograft infiltrating CD3+TCRγ+ cells for each condition. (E) Histogram showing detection of TCRγ+ cells. (F) Percentage of TCRγ+ cells in CD3+ allograft infiltrate for each condition. (G) Density plot for the detection of CD3+TCRγ+ cells that also express RORγ and/or IL‐17. (H) Percentage of RORγ+ cells in CD3+TCRγ+ allograft infiltrate for each condition. (I) Percentage of Il‐17+RORγ+ cells in CD3+TCRγ+ allograft infiltrate for each condition
We then considered the role of alternative T cell subsets. Gamma delta T (γδT) cells are unconventional T cells operating at the interface of innate and adaptive immunity, and are known to produce high levels of IL‐17.19 They have also been implicated in the generation of TLS.1 The number of allograft‐infiltrating CD3+ TCR gamma (TCRγ) + T cells was significantly higher in mice injected with ApoExo compared to those injected with the vehicle (Figure 3D). ApoExo injection also increased the percentage of γδT cells within the CD3+ allograft infiltrate (Figure 3E,F). Furthermore, a significant increase in the expression of IL‐17 and RORγ were found in infiltrating CD3+TCRγ+ T cells from mice injected with ApoExo (Figure 3F,G,H) hinting at a potential role for gamma delta T helper 17 (γδTh17) cells in the production of IL‐17 and in TLS neogenesis.
3.3. Apoptotic exosome-like vesicles favor γδT dependent tertiary lymphoid structure formation
To test the importance of γδT cells in TLS neogenesis, we performed aortic transplantation in γδT cell–knockout (γδKO) mice, followed by injection of ApoExo or the vehicle. γδKO mice injected with ApoExo showed similar levels of neointima formation as controls (Figure 4A,B). However, the number of adventitial TLS was significantly lower in γδKO mice injected with ApoExo (Figure 4C). This dramatic reduction in TLS formation was associated with significantly reduced numbers of CD3+ cells and AID+ cells in the perivascular area (Figure 4A,E,F). The number of CD20+ cells infiltrating the allograft either in the perivascular space or in the vessel wall was not modified in γδKO (Figure 4A,D). However, perivascular infiltration by CD20+ cells was less organized and did not result in the formation of nodular structures. CD3 and AID expression in the intima and media were similar in γδKO mice and wild‐type mice injected with ApoExo (Figure 4A,E,F). Importantly, γδKO mice showed reduced staining for IL‐17 throughout the vessel wall and in the perivascular area (Figure 4A,G). In addition, allografted γδKO mice injected with ApoExo showed reduced anti‐perlecan/LG3 and ANA levels when compared to allografted wild‐type mice injected with ApoExo (Figure 4H,I). Collectively, these results identify γδT cells as pivotal regulators of TLS neogenesis, coordinating the recruitment of T and B cells and their organization within TLS.
Figure 4.
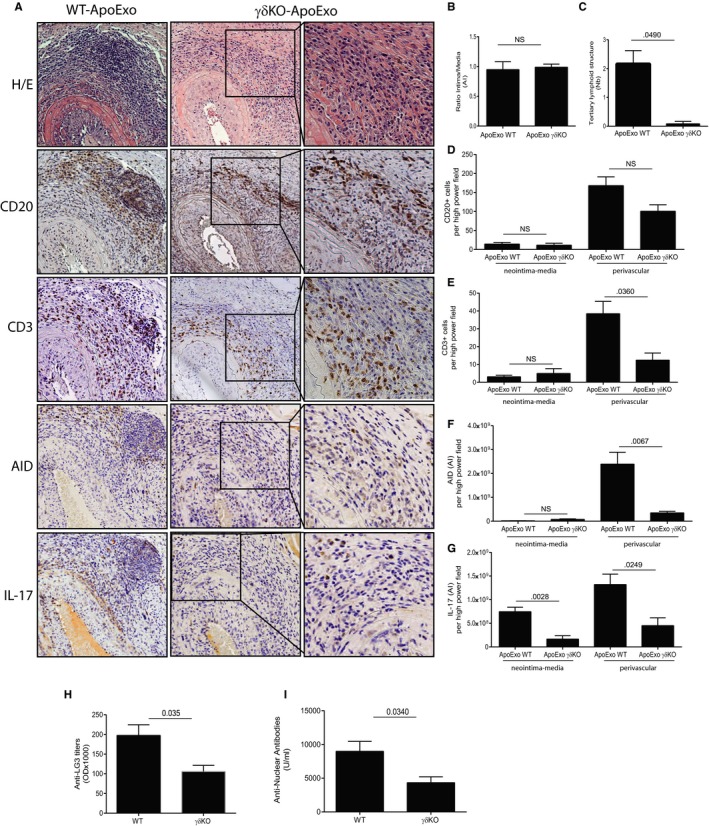
Absence of γδT cells reduces allograft CD3+ T cell infiltration and IL‐17 expression, abrogates tertiary lymphoid structure (TLS) formation and reduces the formation of anti‐perlecan/LG3 and ANA. γδKO (n = 4) or wild‐type (n = 4) mice were injected with apoptotic exosome‐like vesicles (ApoExo) for 3 weeks posttransplantation: (A) Aortic allograft sections stained with H&E, CD20, CD3, AID, and IL‐17 (magnification: 5×; magnification of right inset panels: 20×). (B) Ratio intima/media in the allografts. (C) Mean number of TLS per allograft. Neointima‐media and perivascular quantification of CD20+ B cells (D), CD3+ T cells (E), AID (f), and IL‐17 (g) staining in each high‐power field of the allografts. Anti‐LG3 (h) and ANA (i) IgG levels in sera from γδKO or wild‐type allografted mice after 3 weeks of intravenous injections with ApoExo. Data were pooled from two independent experiments and expressed as means ± SEM. Comparison with the vehicle was done with a Student's t test
3.4. The proteasome activity within ApoExo is required for induction of tertiary lymphoid structure formation
Because we previously showed that acceleration of rejection by ApoExo requires proteasome activity,15 we investigated whether ApoExo proteasome activity was required for induction of TLS neogenesis. We generated ApoExo in vitro from mECs treated with the proteasome inhibitor bortezomib. Bortezomib treatment of ECs does not reduce the production of ApoExo but prompts the production of proteasome‐inactive ApoExo (Figure S5).15 Allografted mice were injected with an equal number of either proteasome‐inactive ApoExo or control proteasome‐active ApoExo. Although neointima formation was similar in mice injected with proteasome‐inactive or proteasome‐active ApoExo (Figure 5A,B), TLS formation was significantly reduced in allograft recipients injected with proteasome‐inactive ApoExo (Figure 5A,C). This was associated with reduced perivascular allograft infiltration by CD3+ T cells and CD20+ B cells (Figure 5A,D,E). Similarly, perivascular IL‐17 and AID staining were significantly reduced in allograft recipients injected with proteasome‐inactive ApoExo (Figure 5A,F,G). Allograft recipients injected with proteasome‐inactive ApoExo also showed reduced anti‐perlecan/LG3, when compared to allograft recipients injected with control proteasome‐active ApoExo (Figure 5H). In addition, flow cytometry analysis of the T cell infiltrate showed reduced CD3+TCRγ+ T cell infiltration in allograft recipients injected with proteasome‐inactive ApoExo whereas CD3+TCRαβ infiltrate remained low in both conditions (Figure 6A). Again, IL‐17 and RORγ expression were identified only in infiltrating TCRγ+ T cells in allografts recipients injected with proteasome active ApoExo. Allografts of mice injected with proteasome inactive ApoExo were largely devoid of IL‐17+ and RORγ+T cells (Figure 6B,C).
Figure 5.
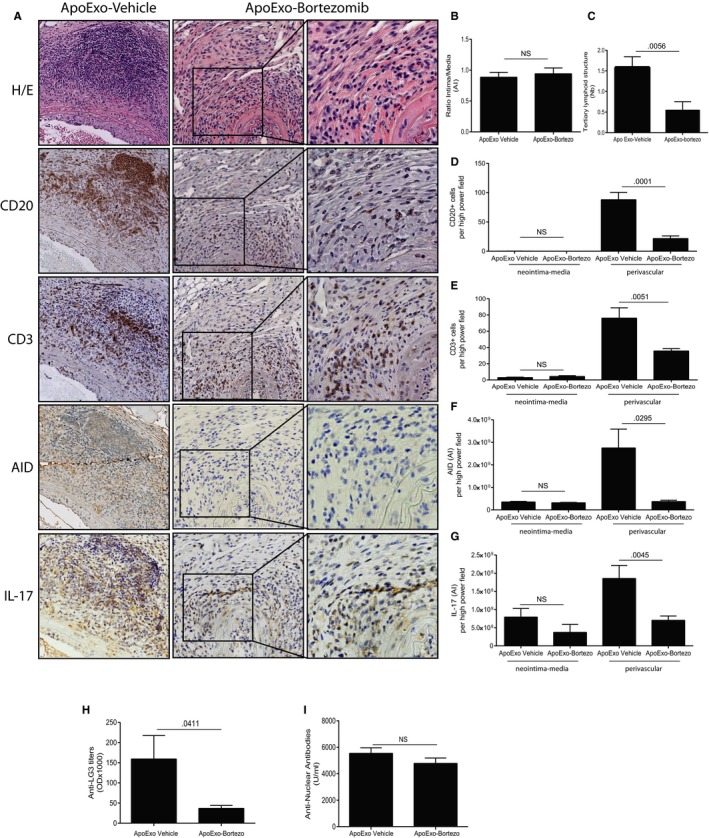
The proteasome activity within apoptotic exosome‐like vesicles (ApoExo) is required for induction of tertiary lymphoid structure (TLS) formation and for anti‐perlecan/LG3 production. Allografted mice were injected with ApoExo that were generated from serum‐starved murine epithelial cells (mECs) treated with bortezomib (n = 11) or the vehicle (n = 10) for 3 weeks posttransplantation: (A) Aortic allograft sections stained with H&E, CD20, CD3, AID, and IL‐17 (magnification: 5×, magnification of right inset panels: 20×). (B) Ratio intima/media ratio in the allografts. (C) Mean number of TLS per allograft. Neointima‐media and perivascular quantification of CD20+ B cells (D), CD3+ T cells (E), AID (F), and IL‐17 (G) staining in each high‐power field of the murine allografts. Anti‐LG3 (H) and ANA (I) IgG levels in sera from wild‐type allografted mice after 3 weeks of intravenous injections with ApoExo generated from serum‐starved mECs treated with bortezomib or the vehicle. Data were pooled from three independent experiments and expressed as means ± SEM. Comparison with the vehicle was done using a Student's t test
Figure 6.
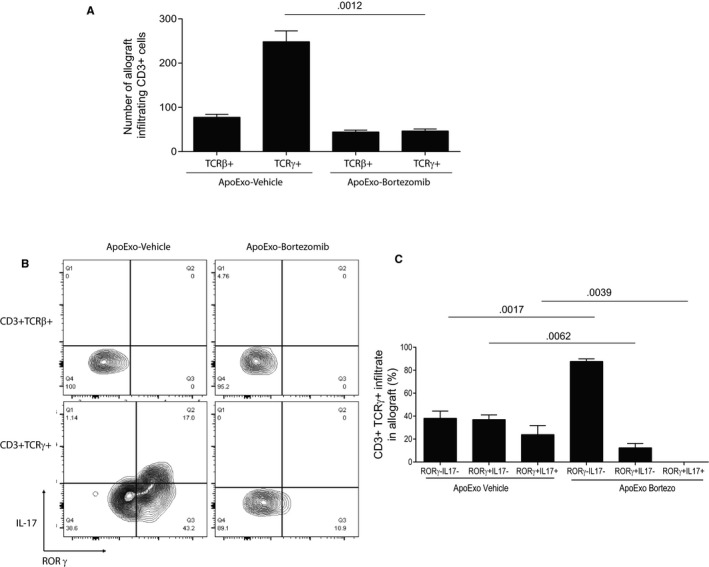
The proteasome activity within apoptotic exosome‐like vesicles (ApoExo) is required for the recruitment of RORγ+IL‐17+γδT cells to the allograft. Allografted mice were injected with ApoExo that were generated from serum‐starved murine epothelial cels treated with bortezomib (n = 10) or the vehicle (n = 9) for 3 weeks posttransplantation: CD3+TCRγ+ T cell aortic allograft infiltrates were analyzed by flow cytometry for IL‐17 and RORγ expression. (A) Number of allograft infiltrating of CD3+ expressing TCRβ or γ for each condition. (B) Density plot of CD3TCRβ+ and CD3+TCRγ+ cells expression of RORγ and IL‐17. (C) Percentage of CD3+TCRγ+ infiltrate expressing RORγ and or IL‐17 for each condition. Data were pooled from three independent experiments and expressed as means ± SEM
Collectively, these results identify the proteasome activity within ApoExo as a novel trigger for recruitment of γδT cells to the vascular allograft, therefore setting in motion IL‐17 overexpression and TLS neogenesis.
4. DISCUSSION
Although significant progress has been made over the last decades in understanding the molecular mechanisms responsible for the organization and maintenance of secondary lymphoid organs, the processes regulating TLS formation in allogeneic organs remain to be elucidated. Here we identify ApoExo, extracellular vesicles derived from injured vascular tissue, as initiators of allograft TLS neogenesis. The proteasome activity of endothelial ApoExo favors the recruitment of γδTh17 cells to the allograft, which in turn orchestrates the organization of infiltrating T and B cells into polarized structures positive for germinal center activity. TLS are organized accumulations of T and B cells at nonlymphoid sites within structures reminiscent of secondary lymphoid organs. They are thought to play a role in governing local immune responses against self‐antigens.20, 21 When developing within the vasculature, TLS have been associated with persistent inflammation and abnormal vascular remodelling.2, 22, 23
Our results shed new light on the molecular and cellular pathways supporting TLS formation. Previous reports have suggested a critical role for IL‐17 in driving lymphoid neogenesis.22, 24, 25, 26, 27 Here we show that γδTh17 cells play a critical role in IL‐17 overexpression, which in turn drives lymphoid neogenesis. IL‐17–producing T cells, including Th17 cells and γδT cells, were found to be necessary for TLS formation in models of pulmonary infections.24, 28 In the experimental autoimmune encephalomyelitis model, TLS formation in the central nervous system was also shown to require IL‐17 production.29 The importance of IL‐17 in the activation of autoimmune responses in the context of transplantation appears to stem from its capacity to initiate recruitment of immune cells to sites of injury and promote maturation of antigen‐presenting cells. For example, in lung transplant recipients, IL‐17 expression has been associated with autoimmune collagen V–specific responses and increased incidence and severity of bronchiolitis obliterans syndrome (BOS), a form of chronic rejection of lung transplants.30, 31 In addition, peripheral blood mononuclear cells from lung transplant recipients with BOS contain lower numbers of cells producing IL‐10 and higher numbers of cells producing IL‐17 and interferon gamma in response to K‐alpha‐1 tubulin and collagen V self‐antigens.32 In turn, immunoreactivity toward K‐alpha‐1 tubulin and collagen V self‐antigens has been closely associated with the development of BOS.33 Furthermore, in a murine model of lung chronic rejection, anti–IL‐17 therapy reduced autoantibody levels and lesions associated with chronic rejection.34 Similarly, heart transplant recipients with cardiac allograft vasculopathy demonstrate increased frequency of T cells producing IL‐17 and decreased frequency of T cells producing IL‐10 specific to myosin, vimentin, collagen V, and K‐alpha‐1 tubulin.35
As Th17 cells are the classic producers of IL‐17, they are suggested to play a pivotal role in autoimmune pathways triggered following transplantation. Intriguingly, our findings demonstrate the importance of γδT cells, rather than Th17 cells, in coordinating the IL‐17 response triggered by ApoExo. Although ApoExo robustly induced prominent allograft IL‐17 overexpression in transplanted mice, Th17 cell allograft infiltration was not elevated in allografted mice injected with ApoExo. Instead, allograft leukocyte infiltrates were dramatically enriched with γδT cells. γδT cell invalidation abrogated TLS formation triggered by ApoExo, demonstrating the pivotal role of γδT cells in sensing ApoExo‐dependent autoimmune signals. Indeed, γδKO allograft recipients formed fewer autoantibodies upon ApoExo injection and showed lower expression of IL‐17 within vascular allografts and reduced TLS formation. These observations are in line with previous studies showing that human IL‐17‐producing γδT cells are generated in the periphery and can be recruited to inflamed tissues where they accumulate.36, 37 This process takes place more rapidly compared to the activation of conventional T lymphocytes38 as γδT cells can be activated in the absence of a cognate TCR ligand.38 The mechanism by which ApoExo activate γδT cells is still ill defined, but our results identify the proteasome activity of ApoExo as a pivotal signal regulating trafficking of γδT cells to sites of vascular injury. The scope of future investigations will be to identify whether activation of γδT cells by ApoExo is antigen specific or derives from innate signaling triggered by Toll‐like receptor ligands or nonprotein mediators.
In the present work, we show that ApoExo injection in transplanted mice triggered perlecan/LG3 and ANA autoantibodies production but did not increase the production of other types of autoantibodies implicated in allograft dysfunction, such as anti‐AT1R, anti‐vimentin, and anti‐fibronectin. This is not surprising given that the protein content of endothelial ApoExo, as characterized by unbiased large‐scale proteomics, contains LG3 but does not include fibronectin, vimentin, and AT1R.15 These results are also consistent with the lack of correlation between anti‐AT1R, anti‐vimentin, and anti‐perlecan/LG3 antibody levels observed in human trials.39, 40 Also our results are in line with previous reports suggesting that autoimmune pathways contributing to rejection are rather innocuous in the normal context. Indeed, our results further demonstrate that ApoExo trigger anti‐LG3 and ANA production independently of alloimmunity but that their pathophysiological impact (TLS formation and vascular remodeling) is optimal develops only in the context of ongoing alloimmune‐dependent vascular injury.
Recent findings suggest that the protein content of ApoExo may differ depending on the type of dying parental cells. For example, lung epithelial cells release exosome‐like vesicles bearing the autoantigens collagen V and K‐alpha‐1 tubulin.41 These vesicles released from stressed and/or dying lung epithelial cells were also found to be associated with occurrence of bronchiolitis obliterans and negative long‐term outcomes in lung transplant patients.41, 42 Whether exosome‐like vesicles that stem from cell types other than endothelial cells can trigger TLS neogenesis remains to be determined and will be the scope of further investigation.
Our results are consistent with findings highlighting a dual role for apoptosis in dampening inflammation while concomitantly promoting different types of immune responses. The importance of apoptosis in the release of calreticulin, heat shock protein 90, type‐I interferon, and adenosine triphosphate has been shown to play an important role in cancer immunogenicity through the activation of infiltrating dendritic cells.43 In the present work, the injection of ApoExo recapitulates the secretion of circulating ApoEXo as a result of ischemia reperfusion injury as previously demonstrated using two ischemia reperfusion injury models.15 Hence, our results add to this emerging field of research and suggest that, following transplantation, ApoExo, released by the graft at the time of ischemia reperfusion or released by the recipient's own vasculature if injured, can contribute to alerting the immune system. Chronic vascular damage leading to prolonged release of ApoExo would foster TLS formation, vascular inflammation, and autoimmunity.
In conclusion, our results identify ApoExo, extracellular vesicles derived from injured vascular tissue, as initiators of TLS formation and provide novel insights into the cellular and molecular pathways regulating TLS formation after transplantation. They demonstrate the pivotal role of γδTh17 cells in coordinating TLS formation and autoantibody production. Finally, they identify proteasome inhibition with bortezomib as a potential option for controlling TLS formation in rejected allografts.
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
AUTHOR CONTRIBUTIONS
MD, JT, and MJH developed concepts, designed the experiments, and wrote the manuscript. MD, JT, AKR, DB, and SQ performed the experiments and analyzed the data. AKR, NP, LAG, and EB analyzed the data, gave technical support and conceptual advice, and edited the manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors acknowledge support from the Canadian National Transplantation Research Program (CNTRP) (MD, EB, MJH), the Canadian Institutes of Health Research (CIHR) (MOP‐15447, PJT‐148884 (MJH); Foundation grant (EB) and the Kidney Foundation of Canada (MJH). MJH is the holder of the Shire Chair in Nephrology, Transplantation and Renal Regeneration of the Université de Montréal. We thank the J.‐L. Lévesque Foundation for renewed support. EB is recipient of a new investigator award from the CIHR. The views expressed herein do not necessarily represent the view of the Canadian federal government. The Institute for Research in Immunology and Cancer (IRIC) receives infrastructure support from the Canadian Centre of Excellence in Commercialization and Research, the Canadian Foundation for Innovation, and the Fonds de Recherche du Québec ‐ Santé (FRQS). We wish to thank Yves Durocher for providing recombinant LG3, Geneviève Marcoux from Centre de Recherche du CHU de Québec for small particle flow cytometry expertise, Melina Narlis, Micheline Fortin and Julie Hinsinger from IRIC histology platform and Dominique Gauchat from CRCHUM cytometry platform.
Dieudé M, Turgeon J, Karakeussian Rimbaud A, et al. Extracellular vesicles derived from injured vascular tissue promote the formation of tertiary lymphoid structures in vascular allografts. Am J Transplant. 2020;20:726–738. 10.1111/ajt.15707
Mélanie Dieudé and Julie Turgeon contributed equally to the manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol. 2018;9:1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thaunat O, Patey N, Caligiuri G, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J Immunol. 2010;185(1):717‐728. [DOI] [PubMed] [Google Scholar]
- 3. Huibers MMH, Gareau AJ, Vink A, et al. The composition of ectopic lymphoid structures suggests involvement of a local immune response in cardiac allograft vasculopathy. J Heart Lung Transplant. 2015;34(5):734‐745. [DOI] [PubMed] [Google Scholar]
- 4. Dragun D, Müller DN, Bräsen JH, et al. Angiotensin II type 1‐receptor activating antibodies in renal‐allograft rejection. N Engl J Med. 2005;352(6):558‐569. [DOI] [PubMed] [Google Scholar]
- 5. Kalache S, Dinavahi R, Pinney S, Mehrotra A, Cunningham MW, Heeger PS. Anticardiac myosin immunity and chronic allograft vasculopathy in heart transplant recipients. J Immunol. 2011;187(2):1023‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joosten SA, Sijpkens YWJ, van Ham V, et al. Antibody response against the glomerular basement membrane protein agrin in patients with transplant glomerulopathy. Am J Transplant. 2005;5(2):383‐393. [DOI] [PubMed] [Google Scholar]
- 7. Rose ML. Role of anti‐vimentin antibodies in allograft rejection. Hum Immunol. 2013;74(11):1459‐1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Linke AT, Marchant B, Marsh P, Frampton G, Murphy J, Rose ML. Screening of a HUVEC cDNA library with transplant‐associated coronary artery disease sera identifies RPL7 as a candidate autoantigen associated with this disease. Clin Exp Immunol. 2001;126(1):173‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porcheray F, DeVito J, Yeap BY, et al. Chronic humoral rejection of human kidney allografts associates with broad autoantibody responses. Transplantation. 2010;89(10):1239‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warraich RS, Pomerance A, Stanley A, Banner NR, Dunn MJ, Yacoub MH. Cardiac myosin autoantibodies and acute rejection after heart transplantation in patients with dilated cardiomyopathy. Transplantation. 2000;69(8):1609‐1617. [DOI] [PubMed] [Google Scholar]
- 11. Cardinal H, Dieudé M, Brassard N, et al. Antiperlecan antibodies are novel accelerators of immune‐mediated vascular injury. Am J Transplant. 2013;13(4):861‐874. [DOI] [PubMed] [Google Scholar]
- 12. Angaswamy N, Klein C, Tiriveedhi V, et al. Immune responses to collagen‐IV and fibronectin in renal transplant recipients with transplant glomerulopathy. Am J Transplant. 2014;14(3):685‐693. [DOI] [PubMed] [Google Scholar]
- 13. Bharat A, Saini D, Steward N, et al. Antibodies to self‐antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg. 2010;90(4):1094‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subramanian V, Ramachandran S, Banan B, et al. Immune response to tissue‐restricted self‐antigens induces airway inflammation and fibrosis following murine lung transplantation. Am J Transplant. 2014;14(10):2359‐2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dieude M, Bell C, Turgeon J, et al. The 20S proteasome core, active within apoptotic exosome‐like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med. 2015;7(318):318ra200. [DOI] [PubMed] [Google Scholar]
- 16. Arismendi M, Giraud M, Ruzehaji N, et al. Identification of NF‐kappaB and PLCL2 as new susceptibility genes and highlights on a potential role of IRF8 through interferon signature modulation in systemic sclerosis. Arthritis Res Ther. 2015;17:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soulez M, Sirois I, Brassard N, et al. Hébert, Epidermal growth factor and perlecan fragments produced by apoptotic endothelial cells co-ordinately activate ERK1/2-dependent antiapoptotic pathways in mesenchymal stem cells. Stem Cells. 2010;28:810–820. [DOI] [PubMed] [Google Scholar]
- 18. Gjurich BN, Taghavie‐Moghadam PL, Galkina EV. Flow cytometric analysis of immune cells within murine aorta. Methods Mol Biol. 2015;1339:161‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paul S, Shilpi LG. Role of gamma‐delta (gammadelta) T cells in autoimmunity. J Leukoc Biol. 2015;97(2):259‐271. [DOI] [PubMed] [Google Scholar]
- 20. Moyron‐Quiroz JE, Rangel‐Moreno J, Hartson L, et al. Persistence and responsiveness of immunologic memory in the absence of secondary lymphoid organs. Immunity. 2006;25(4):643‐654. [DOI] [PubMed] [Google Scholar]
- 21. Hsiao HM, Li W, Gelman AE, Krupnick AS, Kreisel D. The role of lymphoid neogenesis in allografts. Am J Transplant. 2016;16(4):1079‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deteix C, Attuil‐Audenis V, Duthey A, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. 2010;184(9):5344‐5351. [DOI] [PubMed] [Google Scholar]
- 23. Akhavanpoor M, Gleissner CA, Akhavanpoor H, et al. Adventitial tertiary lymphoid organ classification in human atherosclerosis. Cardiovasc Pathol. 2018;32:8‐14. [DOI] [PubMed] [Google Scholar]
- 24. Rangel‐Moreno J, Carragher DM, de la Luz Garcia‐Hernandez M, et al. The development of inducible bronchus‐associated lymphoid tissue depends on IL‐17. Nat Immunol. 2011;12(7):639‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X, Lu B. IL‐17 initiates tertiary lymphoid organ formation. Cell Mol Immunol. 2012;9(1):9‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cupedo T. An unexpected role for IL‐17 in lymphoid organogenesis. Nat Immunol. 2011;12(7):590‐592. [DOI] [PubMed] [Google Scholar]
- 27. Patakas A, Benson RA, Withers DR, et al. Th17 effector cells support B cell responses outside of germinal centres. PLoS ONE. 2012;7(11):e49715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moyron‐Quiroz JE, Rangel‐Moreno J, Kusser K, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med. 2004;10(9):927‐934. [DOI] [PubMed] [Google Scholar]
- 29. Peters A, Pitcher L, Sullivan J, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35(6):986‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burlingham WJ, Love RB, Jankowska‐Gan E, et al. IL‐17‐dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117(11):3498‐3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saini D, Weber J, Ramachandran S, et al. Alloimmunity‐induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. 2011;30(6):624‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K‐alpha1 tubulin‐specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180(7):4487‐4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hachem RR, Tiriveedhi V, Patterson GA, Aloush A, Trulock EP, Mohanakumar T. Antibodies to K‐alpha 1 tubulin and collagen V are associated with chronic rejection after lung transplantation. Am J Transplant. 2012;12(8):2164‐2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182(1):309‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nath DS, Ilias Basha H, Tiriveedhi V, et al. Characterization of immune responses to cardiac self‐antigens myosin and vimentin in human cardiac allograft recipients with antibody‐mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant. 2010;29(11):1277‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Papotto PH, Reinhardt A, Prinz I, Silva‐Santos B. Innately versatile: gammadelta17 T cells in inflammatory and autoimmune diseases. J Autoimmun. 2018;87:26‐37. [DOI] [PubMed] [Google Scholar]
- 37. Caccamo N, La Mendola C, Orlando V, et al. Differentiation, phenotype, and function of interleukin‐17‐producing human Vgamma9Vdelta2 T cells. Blood. 2011;118(1):129‐138. [DOI] [PubMed] [Google Scholar]
- 38. Shiromizu CM, Jancic CC. γδ T lymphocytes: an effector cell in autoimmunity and infection. Front Immunol. 2018;9(2389). 10.3389/fimmu.2018.02389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hönger G, Cardinal H, Dieudé M, et al. Human pregnancy and generation of anti‐angiotensin receptor and anti‐perlecan antibodies. Transpl Int. 2014;27(5):467‐474. [DOI] [PubMed] [Google Scholar]
- 40. Yang B, Dieudé M, Hamelin K, et al. Anti‐LG3 Antibodies aggravate renal ischemia‐reperfusion injury and long‐term renal allograft dysfunction. Am J Transplant. 2016;16(12):3416‐3429. [DOI] [PubMed] [Google Scholar]
- 41. Gunasekaran M, Xu Z, Nayak DK, et al. Donor‐derived exosomes with lung self‐antigens in human lung allograft rejection. Am J Transplant. 2017;17(2):474‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeo JG, Leong J, Lu J. Complement the cell death. Cell Death Dis. 2016;7(11):e2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Land WG, Agostinis P, Gasser S, Garg AD, Linkermann A. DAMP‐induced allograft and tumor rejection: the circle is closing. Am J Transplant. 2016;16(12):3322‐3337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.