

Abstract
Objective
SCN8A encephalopathy is a developmental and epileptic encephalopathy (DEE) caused by de novo gain‐of‐function mutations of sodium channel Nav1.6 that result in neuronal hyperactivity. Affected individuals exhibit early onset drug‐resistant seizures, developmental delay, and cognitive impairment. This study was carried out to determine whether reducing the abundance of the Scn8a transcript with an antisense oligonucleotide (ASO) would delay seizure onset and prolong survival in a mouse model of SCN8A encephalopathy.
Methods
ASO treatment was tested in a conditional mouse model with Cre‐dependent expression of the pathogenic patient SCN8A mutation p.Arg1872Trp (R1872W). This model exhibits early onset of seizures, rapid progression, and 100% penetrance. An Scn1a +/− haploinsufficient mouse model of Dravet syndrome was also treated. ASO was administered by intracerebroventricular injection at postnatal day 2, followed in some cases by stereotactic injection at postnatal day 30.
Results
We observed a dose‐dependent increase in length of survival from 15 to 65 days in the Scn8a‐R1872W/+ mice treated with ASO. Electroencephalographic recordings were normal prior to seizure onset. Weight gain and activity in an open field were unaffected, but treated mice were less active in a wheel running assay. A single treatment with Scn8a ASO extended survival of Dravet syndrome mice from 3 weeks to >5 months.
Interpretation
Reduction of Scn8a transcript by 25 to 50% delayed seizure onset and lethality in mouse models of SCN8A encephalopathy and Dravet syndrome. Reduction of SCN8A transcript is a promising approach to treatment of intractable childhood epilepsies. Ann Neurol 2020;87:339–346
Developmental and epileptic encephalopathies (DEEs) are devastating early onset disorders characterized by seizures and developmental delay.1, 2 Monogenic causes have been identified for approximately one‐third of cases, with voltage‐gated neuronal sodium channels accounting for 2% of the total.3, 4, 5 SCN8A encephalopathy (MIM#614588) is a DEE caused by de novo missense mutations in the SCN8A gene that encodes the neuronal sodium channel Nav1.6.6, 7 Affected individuals are heterozygous for missense mutations that alter the biophysical properties of the channel, resulting in premature channel opening, delayed channel inactivation, or elevated persistent current.6, 8 Because Nav1.6 has a major role in excitatory neurons, elevated activity leads directly to increased neuronal excitability.6
The average age of seizure onset in individuals with SCN8A encephalopathy is 4 months. The clinical course is severe, and approximately 50% of affected individuals remain nonambulatory and nonverbal. Long‐term seizure control is rarely achieved with antiepileptic drugs.7 We and others have studied recurrent patient mutations at arginine codon 1872. Substitution of arginine 1872 by the uncharged amino acids leucine, glutamine, or tryptophan impairs inactivation of the Nav1.6 channel.8, 9, 10 We generated a conditional mouse model of p.Arg1872Trp that recapitulates the early seizure onset and susceptibility to premature death that are characteristic of DEE.11
Because the pathogenic mechanism of SCN8A encephalopathy is neuronal hyperexcitability due to gain‐of‐function mutations, reduction of transcript abundance is a logical therapeutic strategy. Antisense oligonucleotides (ASOs) hybridize by Watson–Crick base pairing to mRNAs, leading to degradation by ribonuclease H, inhibition of translation, or altered splicing. Dominant disorders can be treated with allele‐specific ASOs that specifically target the mutant transcript, or with non–allele‐specific ASOs that reduce both mutant and wild‐type transcript.12, 13 The application of ASO therapy to neurological disorders is receiving increasing attention,14, 15, 16 and the US Food and Drug Administration has recently approved treatment for spinal muscular atrophy that uses intrathecal administration of an ASO at 6‐month intervals.17 The goal of the current work was to evaluate ASO therapy for SCN8A encephalopathy. The results provide proof of principle that reduction of Scn8a transcript can reduce the severity of this devastating disorder as well as Dravet syndrome (DS), a DEE caused by haploinsufficiency of SCN1A.
Materials and Methods
Mutant Mice
The conditional allele of Scn8a (Scn8a cond) on background strain C57BL/6J is activated by Cre recombinase to express the patient mutation R1872W.11 The EIIA‐Cre transgene on background strain C57BL/6J (JAX 003724) is expressed globally in preimplantation embryos and in mature oocytes. Homozygous male Scn8a cond/cond mice were crossed with female EIIA‐Cre mice to generate 50% affected animals (Scn8a cond/+, EIIA‐Cre) and 50% unaffected Scn8a cond/+ controls lacking Cre. Entire litters were randomly assigned to treatment with Scn8a or control ASO. Female EIIA‐Cre mice were used for breeding to avoid the mosaic Cre expression observed in offspring of male EIIA‐Cre mice.11 The Scn1a model of DS carries a deletion of exon 1 that is maintained in heterozygous state in strain 129S6/SvEvTac.18 Experiments were carried out on affected F1 mice generated by crossing with strain C57BL/6J.18 Mice were housed and cared for in accordance with NIH guidelines in a 12/12‐hour light/dark cycle with standard mouse chow and water available ad libitum. Experiments were approved by the Committee on the Use and Care of Animals at the University of Michigan. Open field activity and wheel running were assayed in the Michigan Mouse Metabolic Phenotyping Center (NIH U2CDK110768).
Antisense Oligonucleotides
ASOs were synthesized by Ionis Pharmaceuticals as described.19 The ASOs are 20 base pair (bp) gapmers with 5 2′‐O‐methoxyethyl‐modified nucleotides at each end and 10 DNA nucleotides in the center. The Scn8a ASO 5′ GACGA TTAGT GACAT AGGCT 3′ is complementary to the 3′ untranslated region (UTR) that does not differ between wild‐type and mutant transcript (Fig 1A). The control ASO 5′ CCTAT AGGAC TATCC AGGAA 3′ does not match any transcript encoded by the mouse genome and was shown to be well tolerated in the mouse.19
Figure 1.
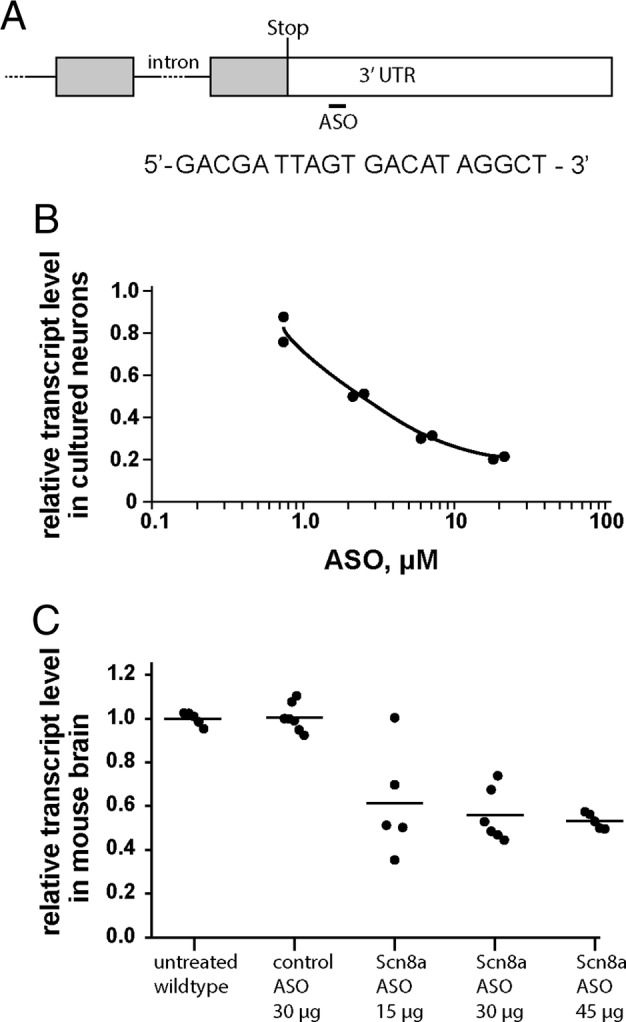
Scn8a antisense oligonucleotide (ASO) treatment reduces the abundance of Scn8a transcript in wild‐type neurons and brain. (A) Cartoon of the 3′ end of the Scn8a gene. The last 2 exons are represented by boxes separated by a line representing the last intron. Coding sequences within the exons are shaded. The position of the stop codon that terminates translation is marked as “stop.” The sequence of the 20 base pair (bp) Scn8a ASO is shown. The ASO sequence is identical to the genomic sequence of the 3′ untranslated region (UTR) at a position approximately 500 bp downstream from the translation stop codon within the last exon of the gene. (B) Primary cortical neurons from E14 embryos of strain C57BL/6J were cultured for 3 days with the indicated concentration of Scn8a ASO. (C) The indicated dose of ASO was administered to C57BL/6J mice on postnatal day 2, and the abundance of Scn8a transcript in brain RNA was measured at postnatal day 21. Five or 6 animals were treated with each dose; each symbol represents data from 1 animal.
Intracerebroventricular Administration of ASOs
At postnatal day 2 (P2) mice received 2μl of ASO in phosphate‐buffered saline (PBS) injected manually into the cerebral ventricle.20 Adult mice received 2.5μl of ASO in PBS by stereotaxic injection into the left ventricle using an automated injector pump (Stoelting, Wood Dale, IL) connected to a 5μl Hamilton syringe.21 Coordinates for stereotaxic injection into the cerebral ventricle were mediolateral = 1.0mm, anteroposterior = 0.3mm, and ventrolateral = 3.0mm.
Quantitative Reverse Transcription Polymerase Chain Reaction
Scn8a transcripts in mouse brain and cultured neurons were quantified using TaqMan gene expression assays (Applied Biosystems, Foster City, CA).11 Total Scn8a transcript was detected with the FAM‐labelled assay Mm00488110_m1 using as endogenous control the Tata binding protein mRNA (VIC labelled Mm01277042_m1). Relative transcript quantity was calculated by the ΔΔCT method.22 Data represent the average result from 2 independent cDNA preparations for each animal.
Western Blotting
Purified membrane protein was prepared from whole brain and analyzed by Western blotting essentially as described.23 The primary antibodies were rabbit anti‐Nav1.6 (Millipore, Billerica, MA; # 5580, lot 3188705) diluted 1:500 and mouse antitubulin (Millipore, # MAB 1637, lot 2080833) diluted 1:1,000.
Results
The Scn8a ASO targets a sequence in the proximal 3′ UTR of mouse Scn8a (see Fig 1). Addition of ASO to the culture medium of wild‐type primary mouse neurons generates a dose‐dependent reduction in transcript abundance. Wild‐type mice of strain C57BL/6J were treated on P2 by intracerebroventricular (ICV) injection of ASO, and the abundance of Scn8a transcript in brain RNA was measured at P21. Control ASO did not reduce the Scn8a transcript, but a dose‐dependent reduction of Scn8a mRNA was obtained in mice treated with 15 to 45μg of Scn8a ASO. Spinal cord Scn8a transcript was also reduced to 0.6 ± 0.1 relative to wild type (n = 5). Treatment with 45μg of ASO did not interfere with weight gain in the postnatal period, with no evidence of muscle wasting; at P21, body weight was 9.7 ± 1.4g (mean ± standard deviation) in untreated wild‐type mice (n = 21) and 10.6 ± 1.2 (n = 18) in mice treated with 45μg of ASO at P2 (p = 0.2, unpaired t test) (Fig 1C).
The therapeutic effect of the ASO was evaluated in mice expressing a conditional allele of Scn8a‐p.Arg1872Trp.11 Homozygous conditional mice were crossed with mice expressing the global EIIA‐Cre transgene to activate the conditional allele. The resulting (untreated) Scn8a cond/+, EIIA‐Cre offspring exhibited sudden onset of convulsive seizures at P14 to P16, followed by death within 24 hours.11 Prior to seizure onset, these mice develop normally and do not exhibit epileptiform electroencephalographic (EEG) discharges.11
ASO was administered by ICV injection at P2 to litters containing 50% affected Scn8a cond/+, EIIA‐Cre mice and 50% unaffected Scn8a cond/+ controls lacking Cre. Treatment of Scn8a cond/+, EIIA‐Cre mice with control ASO did not prevent seizure onset and death at P14 to P16 (Fig 2). Administration of 15 to 45μg of Scn8a ASO resulted in a dose‐dependent increase in survival up to 7 weeks of age. To test the efficacy of repeated doses of ASO, mice were treated with 30μg of ASO on P2 followed by a second dose of 100μg on P30, administered by stereotactic injection. The second treatment further increased median survival from 6 weeks for 30μg ASO to approximately 9 weeks, indicating that repeated ASO administration might be effective as a long‐term treatment, as is the case for spinal muscular atrophy.17
Figure 2.
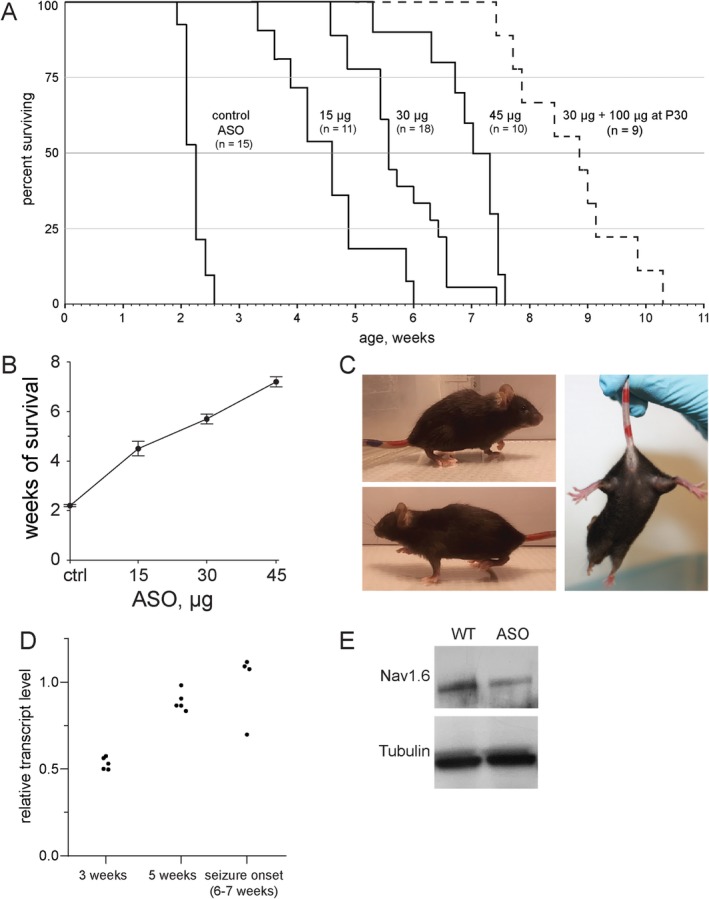
Scn8a antisense oligonucleotide (ASO) delays seizure onset and prolongs survival of mutant mice expressing the pathogenic mutation SCN8A‐R1872W. (A) Scn8a cond/+, EIIA‐CRE mice were treated on postnatal day 2 by intracerebroventricular injection with the indicated mount of control or Scn8a ASO. A second dose of 100μg of Scn8a ASO at postnatal day 35 further extended survival (p < 0.001) (dotted line). (B) Dose dependence of mean survival; values are mean ± standard error of the mean. (C) Scn8a cond/+, EIIA‐CRE mice treated with 45μg exhibit normal posture without hind limb clasping at P21. (D) Duration of the effect of Scn8a ASO on transcript level. Brain RNA was prepared from Scn8a cond/+, EIIA‐CRE mice treated with 45μg of ASO and untreated age‐matched wild‐type mice and analyzed by quantitative reverse transcription polymerase chain reaction. (E) Reduction of Nav1.6 protein in brain at 3 weeks of age in mice treated with 45μg of ASO as in (D).
In mice treated with 45μg ASO on P2, seizure onset began at 6 weeks of age (see Fig 2). To determine the duration of the effect of ASO treatment on transcript level and the relationship between transcript level and seizure onset, we measured Scn8a transcripts at 3 time points after treatment at P2 with 45μg ASO. Transcript levels in treated mice at each point were compared with age‐matched untreated wild‐type controls. The level of transcript at 3 weeks of age was 50% of control (Fig 2D and Fig 1C). At 5 weeks posttreatment, Scn8a transcript level was increased to 80% of control, but no seizures were observed. At the time of seizure onset, transcripts in the mice with seizures had reached 100% of the level of untreated controls (Fig 2D). Thus, the reduction of Scn8a transcripts by ASO treatment persists for approximately 6 weeks and is correlated with seizure protection. The corresponding reduction of Nav1.6 protein at 3 weeks posttreatment is demonstrated in Figure 2E.
To investigate the possibility that treated mice experience subthreshold electrographic seizures during the period before seizure onset, mice were injected with 45μg of ASO at P2 and monitored by EEG recording continuously for 8 days from P31 to P39. Five of the 6 treated mice exhibited normal EEGs throughout this period, which ended near the age of seizure onset (Fig 3). One treated mouse developed a convulsive seizure at the age of 37 days, followed within 30 seconds by death. The EEG recording of this seizure (see Fig 3) is similar to that previously reported for untreated mutant mice at P14 to P16.11 Thus treatment with SCN8A ASO delayed seizure onset but did not prevent the very rapid progression from initial seizure to death that is characteristic of mice expressing the R1872W mutation.
Figure 3.
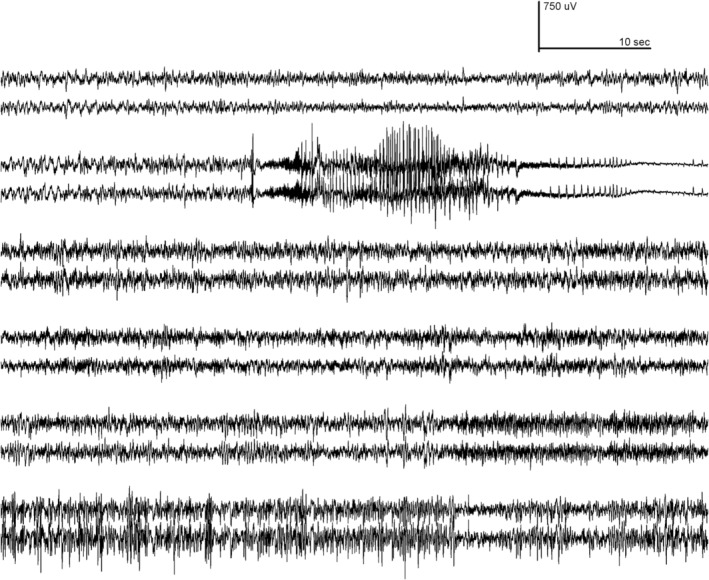
Antisense oligonucleotide (ASO) treatment protects against electrographic seizures. Mutant mice of genotype Scn8a cond/+ , EIIA Cre were treated with 45μg Scn8a ASO by intracerebroventricular injection at postnatal (P) day 2 (P2) and monitored from the age of P31 to P39 by 24‐hour video electroencephalographic (EEG) recording. Traces recorded from right and left cortical electrodes of 6 mice are shown. Five mice were completely protected from electrographic and behavioral seizures. Mouse 2 exhibited a single, fatal, generalized tonic–clonic seizure at P37 that closely resembled the EEG profile in untreated mutants at P15.11 (P37 corresponds to the age of the earliest seizure in the cohort of 10 mice treated with 45μg of ASO in Fig 2.)
Because SCN8A is an essential gene,24 it was important to evaluate potential adverse effects of ASO treatment. From previous work in the mouse, we know that reduction of brain transcripts below 5% of the wild‐type level is lethal,24, 25 and reduction to 10% of wild‐type level results in dystonia, muscle wasting, and reduced body weight.25, 26 In contrast, reduction to 50% of wild‐type level is well tolerated in the mouse, with only mild behavioral abnormalities.27 Similarly, many haploinsufficient patients with 50% of wild‐type expression have mild intellectual disability.28, 29 These earlier observations predicted that the ASO treatment that prevented seizures, with approximately 50% of wild‐type expression remaining in brain (see Fig 1), would have only small effects on movement.
To detect potential impairment of motor function, we compared ASO‐treated mutant mice with untreated wild‐type littermates. We examined motor activity during the period of extended survival after ASO treatment. In the open field test, mice treated with 45μg of ASO exhibited normal activity at 6 weeks of age. Total distance travelled, percentage time spent in the center of the open field, and number of rearing events did not differ from wild‐type controls (Fig 4A).
Figure 4.
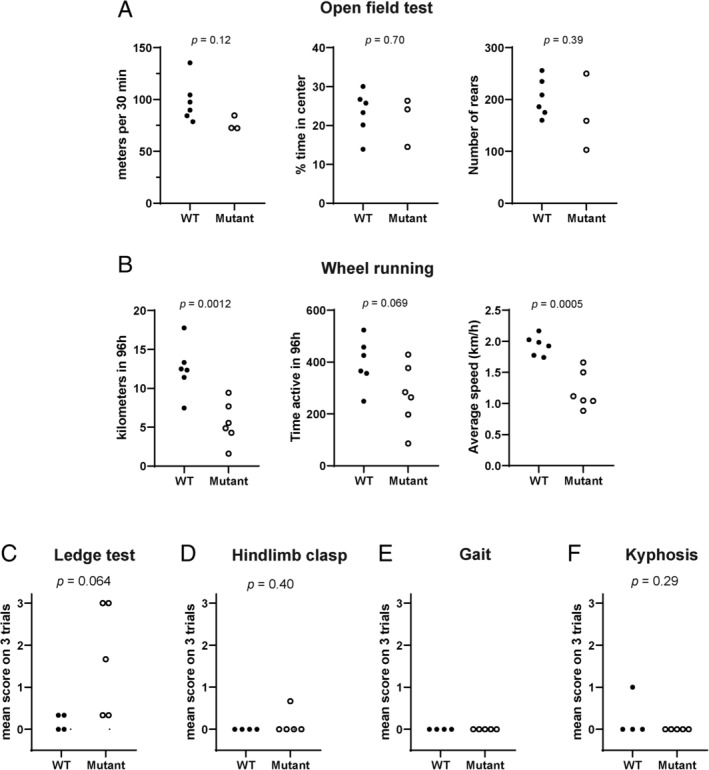
Motor activity of antisense oligonucleotide (ASO) treated mice. Scn8a cond/+ , EIIA Cre mice were treated with 45μg Scn8a ASO at postnatal (P) day 2 (P2). (A) Activity in an open field was monitored for 30 minutes at 45 days of age. Treated mutant mice did not differ significantly from wild‐type mice in average running speed (Student t test, p = 0.12) or percentage of time spent in the center of the open field (2‐way analysis of variance, p = 0.83). Rearing behavior of mutant and wild‐type mice was also comparable. (B) Wheel running activity was monitored 24 h/day during the 4‐day interval between P31 and P39, after habituation to the running cage for 10 days. The distance travelled by the treated mutant mice during the 96 hours of monitoring was significantly smaller than for wild‐type mice. There was no significant difference in the time spent running, but the average speed was lower for the mutant mice. (C–F) Cerebellar function was assessed at 5 weeks of age by analysis of ledge walking, hind limb clasping, gait, and kyphosis as described.30 The only deficit observed was impaired ledge walking in 3 of the 5 treated mutant mice. Each symbol represents 1 animal, and values are the mean of triplicate assays. The p values are for the comparisons between wild‐type and mutant mice (Student t test). WT = wild‐type C57BL/6J mice untreated with ASO; Mutant = Scn8a cond/+ , EIIA Cre mice treated with 45μg ASO.
In the wheel running test, the ASO‐treated mutant mice were slower than the wild‐type mice (see Fig 4B). There was no difference between the 2 groups in the time spent running, but the distance covered was reduced by approximately one‐third due to a slower average speed of 1.2km/h for treated mutant mice compared with 2.0km/h for wild‐type controls. Cerebellar function was assessed by analysis of ledge walking, hind limb clasping, gait, and kyphosis.30 The only deficit observed was impaired ledge walking in 3 of the 5 treated mutant mice (see Figs 4C–F). The normal posture and home cage activity of ASO‐treated mice is demonstrated in Figure 2 and Supplementary Videos 1 to 3. Thus, reduction of Scn8a transcript to a level sufficient for delay of seizure onset results in only minor effects on movement. Overall, the ASO‐treated mice remained alert and active until the onset of seizures.
Because Nav1.6 is a major determinant of the firing properties of excitatory neurons, reduction of Scn8a expression could have a general ameliorating effect on seizure disorders of various causes.31 To test this possibility, we examined the effect of the Scn8a ASO in a mouse model of DS, a DEE caused by loss‐of‐function mutations of SCN1A. Reduction of Scn8a by other methods was previously shown to be protective against acutely induced seizures in a Dravet mouse.32, 33 We used the Kearney mouse model of DS that is haploinsufficient due to deletion of exon 1 of Scn1a.18 The untreated DS mice exhibited onset of spontaneous seizures by 4 weeks of age and 50% penetrance of seizures and lethality (Fig 5), consistent with previous reports.18 In contrast, DS mice treated with a single dose of 45μg of Scn8a ASO on P2 survived beyond 5 months of age without behavioral seizures. The Scn8a ASO resulted in 50% reduction of Scn8a transcript in brain and spinal cord of DS mice with no effect on the Scn1a transcript (Fig 5B). Five consecutive days of EEG recordings of ASO‐treated DS mice recorded at 5 months of age did not detect any subthreshold electrographic abnormalities or seizures (Fig 5C). This extended period of protection after a single treatment suggests that ASO administration during a critical period of postnatal development might give long‐term seizure control in Dravet patients. A transient developmental window of interneuron dysfunction has been described in Dravet mice.34 Expansion of future testing of the Scn8a ASO to other seizure models will be of great interest.
Figure 5.
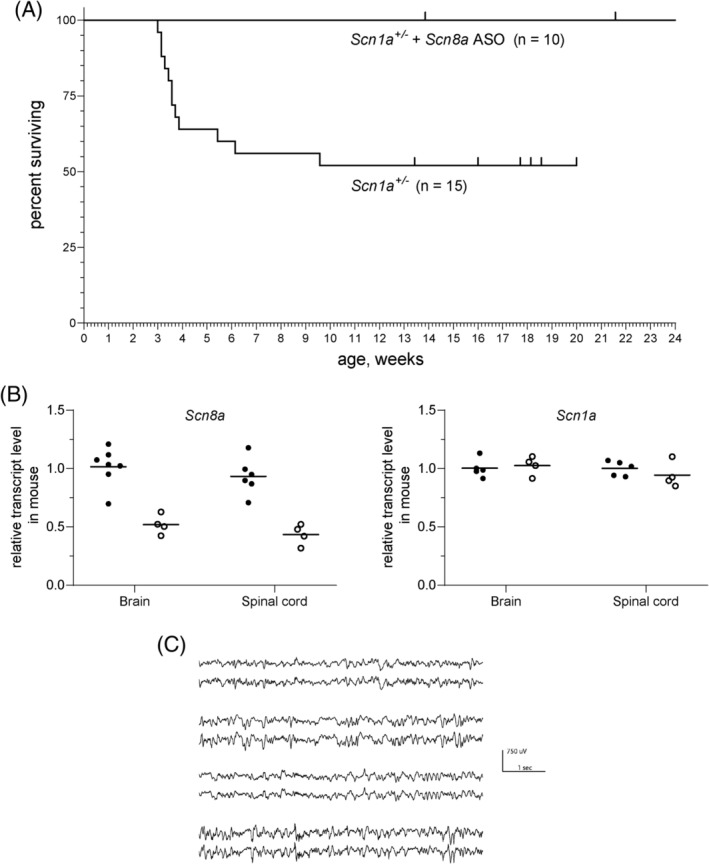
Scn8a antisense oligonucleotide (ASO) rescues survival of Dravet syndrome mice. Scn1a +/− mice were treated on postnatal (P) day 2 (P2) with 45μg Scn8a ASO. (A) For untreated Scn1a +/− mice, median survival was 26 days with 50% penetrance of the lethal phenotype, consistent with the original description of this Dravet model.18 In contrast, all of the ASO treated mice have survived beyond 5 months of age (p = 0.004, Cox–Mantel log‐rank test). Tick marks represent living mice. (B) The Scn8a ASO reduces Scn8a transcript level with no effect on Scn1a transcript level. Brain and spinal cord RNA was prepared from untreated wild‐type P21 (solid symbols) or mice treated with 45μg Scn8a ASO (open symbols). (C) The Scn8a ASO protects against electrographic seizures in Scn1a +/− mice. Four ASO treated mice at 5 months of age were monitored for 5 successive days with 24‐hour electroencephalographic (EEG) recording. No electrographic seizures or other EEG abnormalities were observed (2 traces for each animal, from left and right cortex).
Discussion
The experiments described here provide the first preclinical evidence that therapeutic reduction of Scn8a transcript could delay the onset of spontaneous convulsive seizures in patients with SCN8A encephalopathy. ASO treatment did not result in major adverse effects, suggesting that it may be well tolerated in patients with SCN8A encephalopathy or Dravet Syndrome. Further investigations will be required to determine whether initiation of ASO treatment after seizure onset is also effective. In these animal models, death follows within minutes to days of the first seizure and the resulting inability to evaluate postonset treatment is a limitation of the present study. Clinical application will also require prevention of the deleterious effects associated with more extensive reduction of SCN8A expression, for example, by using haplotype‐specific ASOs targeting synonymous SNPs such as rs303815. Our observations on the DS mouse support the exciting possibility that treatment with SCN8A ASO could also be protective in Dravet Syndrome and other seizure disorders with a variety of underlying etiologies.
Author Contributions
G.M.L., P.J.‐N., J.L.W., R.J.G., F.R., and M.H.M. contributed to the conception and design of the study; G.M.L., P.J.‐N., L.D.H., S.F.H., C.E.S., J.L.W., H.P., W.Y., K.B., J.Z., J.P., and R.J.G. contributed to the acquisition and analysis of data; G.M.L., S.F.H., W.Y., J.P., R.J.G., and M.H.M. contributed to drafting the text and preparing figures.
Potential Conflicts of Interest
P.J.‐N. and F.R. are employed by Ionis Pharmaceuticals, a for‐profit company that develops ASO therapies.
Supporting information
Video 1. Normal gait and movement, side view. 30 sec.
Video 2. Normal gait and activity, overhead view.
Video 3. Absence of hind‐limb clasping in the treated mutant mice.
Appendix S1: Supporting Information
Acknowledgment
This study was supported by NIH (NINDS) R01 NS34509.
We thank H. McLoughlin and L. Isom for valuable advice; J. Kearney for providing the Dravet mice; and S. Whitesall, M. Schmitt, and D. Michele of the Michigan Mouse Metabolic Phenotyping Center (NIH U2CDK110768) for assistance with wheel running and open field. We thank the two anonymous reviewers for their valuable suggestions.
References
- 1. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 2. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meisler MH, Kearney KJ. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 2005;115:2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015;84:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018;59:1062–1071. [DOI] [PubMed] [Google Scholar]
- 6. Meisler MH, Helman G, Hammer MF, et al. SCN8A encephalopathy: research progress and prospects. Epilepsia 2016;57:1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gardella E, Marini C, Trivisano M, et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018;91:e1112–e1124. [DOI] [PubMed] [Google Scholar]
- 8. Wagnon JL, Barker BS, Hounshell JA, et al. Pathogenic mechanisms of recurrent epileptogenic mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 2016;3:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wagnon J, Meisler MH. Recurrent and non‐recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 2015;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Schubert J, Sonnenberg L, et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 2019;142:376–390. [DOI] [PubMed] [Google Scholar]
- 11. Bunton‐Stasyshyn RKA, Wagnon JL, Wengert ER, et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019;142:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Southwell AL, Kordasiewicz HB, Langbehn D, et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington's disease. Sci Transl Med 2018;10:eaar3959. [DOI] [PubMed] [Google Scholar]
- 13. McCampbell A, Cole T, Wegener AJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest 2018;128:3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med 2019;70:307–321. [DOI] [PubMed] [Google Scholar]
- 15. Roovers J, De Jonghe P, Weckhuysen S. The therapeutic potential of RNA regulation in neurological disorders. Expert Opin Ther Targets 2018;22:1017–1028. [DOI] [PubMed] [Google Scholar]
- 16. Bennett CF, Krainer AR, Cleveland D. Antisense oligonucleotide therapies for neurodegenerative disorders. Ann Rev Neurosci 2019;42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med 2017;377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 18. Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 2014;13:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swayze EE, Siwkowski AM, Wancewicz EV, et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 2007;35:687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin‐2 extends lifespan and reduces pathology in TDP‐43 mice. Nature 2017;544:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duan Y, Wang SH, Song J, et al. Semaphorin 5A inhibits synaptogenesis in early postnatal‐ and adult‐born hippocampal dentate granule cells. Elife 2014;3:e04390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative CT method. Nat Protoc 2008;3:1101–1108. [DOI] [PubMed] [Google Scholar]
- 23. Wagnon JL, Korn MJ, Parent R, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A related epileptic encephalopathy. Human Molec Genet 2015;24:506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgess DL, Kohrman DC, Galt J, et al. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant 'motor endplate disease'. Nat Genet 1995;10:461–465. [DOI] [PubMed] [Google Scholar]
- 25. Kearney JA, Buchner DA, de Haan G, et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Nav1.6). Hum Mol Genet 2002;11:2765–2775. [DOI] [PubMed] [Google Scholar]
- 26. Sprunger LK, Escayg A, Tallaksen‐Greene S, et al. Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Hum Mol Genet 1999;8:471–479. [DOI] [PubMed] [Google Scholar]
- 27. McKinney BC, Chow CY, Meisler MH, Murphy GG. Exaggerated emotional behavior in mice heterozygous for the sodium channel Scn8a (Na(v)1.6). Genes Brain Behav 2008;7:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagnon JL, Barker BS, Ottolini M, et al. Loss‐of‐function variants of SCN8A in intellectual disability without seizures. Neurol Genet 2017;3:e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Møller R, Wengert ER, Tronhjem CE, et al. Biallelic variants of SCN8A with reduced channel activity in two families with developmental and epileptic encephalopathy. Epilepsia 2019;60:2277–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guyenet SJ, Furrer SA, Damian VM, et al. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 2010;39:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isom LL. Is targeting of compensatory ion channel gene expression a viable therapeutic strategy for Dravet syndrome? Epilepsy Curr 2019;19:193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong JC, Makinson CD, Lamar T, et al. Selective targeting of Scn8a prevents seizure development in a mouse model of mesial temporal lobe epilepsy. Sci Rep 2018;8:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin MS, Tang B, Papale LA, et al. The voltage‐gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 2007;16:2892–2899. [DOI] [PubMed] [Google Scholar]
- 34. Favero M, Sotuyo NP, Lopez E, et al. A transient developmental window of fast‐spiking interneuron dysfunction in a mouse model of Dravet syndrome. J Neurosci 2018;38:7912–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Normal gait and movement, side view. 30 sec.
Video 2. Normal gait and activity, overhead view.
Video 3. Absence of hind‐limb clasping in the treated mutant mice.
Appendix S1: Supporting Information