

Abstract
The first examples of titanium‐catalyzed hydroaminoalkylation reactions of ethylene with secondary amines are presented. The reactions can be achieved with various titanium catalysts and they do not require the use of high pressure equipment. In addition, the first solid‐state structure of a titanapyrrolidine that is formed by insertion of an alkene into the Ti−C bond of a titanaaziridine is reported.
Keywords: amination, amines, ethylene, hydroaminoalkylation, titanium
No high pressure equipment is required for achieving the hydroaminoalkylation of ethylene with secondary amines in the presence of titanium catalysts (see scheme). This finding is in good agreement with the observation that the insertion of ethylene into the Ti−C bond of titanaaziridines, which are regarded to be the catalytically active species of hydroaminoalkylation reactions, takes place easily to give titanapyrrolidines.
![]()
Amines are among the most important classes of organic compounds and as a consequence, the development of efficient and sustainable methods for their synthesis is of exceptional interest in academic and industrial chemical research. While traditional synthetic pathways for the synthesis of amines usually rely on nucleophilic substitution, reductive amination, or cross‐coupling processes, the transition‐metal‐catalyzed hydroaminoalkylation of alkenes1 (Scheme 1) allows the direct addition of α‐C(sp3)−H bonds of amines across C−C double bonds of alkenes. Because corresponding reactions take place without the formation of any side products and inexpensive and widely available starting materials can be used it is not surprising that the development of hydroaminoalkylation processes has raised significant attention in recent years.2, 3, 4, 5 Although corresponding reactions can be achieved in the presence of selected late transition‐metal2 and group 3 metal catalysts,3 group 44 and 55 metal catalysts offer the broadest scope. Interestingly, in the latter cases, only liquid alkenes, such as 1‐hexene, 1‐octene, or styrene have extensively been used as substrates while the simplest alkene, ethylene, has largely been ignored. Only the first report on early transition metal‐catalyzed hydroaminoalkylation reactions, which was published in 1980 by Clerici and Maspero6 already describes two examples of [Nb(NMe2)5]‐ or [Nb(NEt2)5]‐catalyzed hydroaminoalkylation reactions of ethylene with dimethyl‐ or diethylamine. Unfortunately, these reactions were performed at elevated pressure (20 atm) and the hydroaminoalkylation products were only obtained in low yields between 13 and 28 %. In this context, it should be emphasized that in industry, ca. 150 million tons of ethylene are produced per year7 and as a consequence, ethylene is not only the most widely used alkene in industry, it must also be regarded as one of the most important organic compounds. For that reason, efficient catalytic procedures for the hydroaminoalkylation of ethylene could offer immense economic benefits in the chemical industry. The corresponding products, n‐propylamines, could be of interest as intermediates for the synthesis of pharmaceuticals or pesticides.
Scheme 1.

Transition‐metal‐catalyzed hydroaminoalkylation of alkenes.
Interestingly, the mechanism of the titanium‐catalyzed hydroaminoalkylation reaction (Scheme 2),4a, 8 which includes the insertion of the alkene substrate into the Ti−C bond of a catalytically active titanaaziridine as the C−C bond‐forming key step, suggests that ethylene should react smoothly because in comparison to larger 1‐alkenes or styrenes, steric hindrance of the insertion step is minimized in the case of ethylene. Based on this assumption, we recently decided to investigate the performance of a number of titanium catalysts developed in our group in hydroaminoalkylation reactions of ethylene.
Scheme 2.

Simplified mechanism of the titanium‐catalyzed hydroaminoalkylation of ethylene with secondary amines.
Initial experiments to achieve the hydroaminoalkylation of ethylene using a very simple experimental setup were performed with a selection of our recently developed titanium catalysts shown in Figure 1 or mentioned in Table 1. For that purpose, in each case, a Schlenk tube (V=100±5 mL) containing a solution of N‐methylaniline (1 a, 2 mmol) and the titanium catalyst (10 mol %) in toluene (1 mL) was first filled with ethylene (1.4 atm, ≈5.7 mmol) and sealed. Then the obtained mixture was stirred at temperatures between 105 and 160 °C for 3–48 h and finally, the crude product mixture was purified by column chromatography. Consistent with the 1980′s experiments,6 it was first found that tetrakis(dimethylamino)titanium does not show any catalytic activity up to temperatures of 140 °C (Table 1, entry 1). On the other hand, [Ind2TiMe2] (Ind=η5‐indenyl) does catalyze the reaction at a temperature of 105 °C and delivers the expected hydroaminoalkylation product N‐propylaniline (2 a) in 69 % yield after a reaction time of 24 h (Table 1, entry 3). Surprisingly, from this experiment, N‐(pentan‐3‐yl)aniline (3 a), which is formed by alkylation of the initially formed product 2 a with ethylene, could also be isolated in 23 % yield. In this context, it should be mentioned that N‐alkylanilines possessing an alkyl substituent larger than a methyl group usually react slowly in titanium‐catalyzed hydroaminoalkylation reactions4 (vide infra) and especially in the presence of the catalyst [Ind2TiMe2], they were reported to lack any reactivity.4b Interestingly, formation of the dialkylated product 3 a could not be suppressed by a shorter reaction time of 6 h and even in this case, 3 a was obtained in 17 % yield (Table 1, entry 2).
Figure 1.
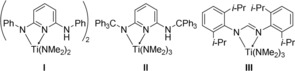
Selected titanium catalysts for hydroaminoalkylation reactions.
Table 1.
Catalyst screening for the hydroaminoalkylation of ethylene with N‐methylaniline (1 a).[a]
|
| |||||
|---|---|---|---|---|---|
|
Entry |
Catalyst |
T [°C] |
t [h] |
Yield 2 a [%] |
Yield 3 a [%] |
|
1 |
[Ti(NMe2)4] |
140 |
24 |
– |
– |
|
2 |
[Ind2TiMe2] |
105 |
6 |
66 |
17 |
|
3 |
[Ind2TiMe2] |
105 |
24 |
69 |
23 |
|
4 |
I |
140 |
24 |
48 |
4 |
|
5 |
II |
160 |
24 |
67 |
– |
|
6 |
III |
140 |
24 |
72 |
17 |
|
7 |
III |
160 |
3 |
72 |
9 |
|
8 |
III |
160 |
6 |
75 |
17 |
|
9[b] |
III |
160 |
12 |
78 |
16 |
|
10[c] |
III |
160 |
24 |
76 |
17 |
|
11 |
III |
160 |
48 |
78 |
19 |
|
12 |
I |
105 |
24 |
19 |
– |
|
13 |
II |
105 |
24 |
21 |
10 |
|
14 |
III |
105 |
24 |
10 |
5 |
|
15[d] |
[Ta(NMe2)5] |
160 |
24 |
68 |
7 |
|
16 |
[Nb(NMe2)5] |
160 |
24 |
20 |
3 |

[a] Reaction conditions: 1) N‐methylaniline (214 mg, 2.00 mmol), ethylene (1.4 atm), catalyst (0.20 mmol, 10 mol %), toluene (1 mL), T, t, isolated yields. [b] The reaction could also be achieved with reduced catalyst loadings. 5 mol % III gave 2 a in 77 % yield and 3 a in 9 % yield; 2.5 mol % III gave 2 a in 68 % yield and 3 a in 6 % yield; 1.25 mol % III gave 2 a in 33 % yield and 3 a in trace amounts. [c] The reaction could also be achieved with reduced catalyst loadings. 5 mol % III gave 2 a in 79 % yield and 3 a in 9 % yield; 2.5 mol % III gave 2 a in 68 % yield and 3 a in 4 % yield; 1.25 mol % III gave 2 a in 30 % yield and 3 a in trace amounts. [d] The reaction could also be achieved with a reduced catalyst loading. 5 mol % [Ta(NMe2)5] gave 2 a in 41 % and 3 a in trace amounts.
Subsequent experiments performed with the second‐generation titanium catalysts I,4d II,4f and III 4e (Figure 1) at temperatures between 140 and 160 °C then revealed that in comparison to [Ind2TiMe2], these catalysts favor the formation of the monoalkylated product 2 a with better selectivities (Table 1, entries 4–11). In this context, the finding that the dialkylated product 3 a was not formed at all in the presence of the sterically crowded trityl‐substituted mono(aminopyridinato) catalyst II deserves particular attention (Table 1, entry 5). In addition, it was also found that among the catalysts I–III, mono(formamidinate) catalyst III is the most active one (Table 1, compare entries 4 with 6 and entries 5 with 10). As a result, reactions performed with catalyst III gave access to 2 a in improved isolated yields between 72 and 78 % (Table 1, entries 6–11) and even after a relatively short reaction time of 3 h at 160 °C, it was possible to isolate 2 a in 72 % yield (Table 1, entry 7). Interestingly, the size of the Schlenk tube did not dramatically affect the isolated yields and the selectivity of the reaction. For example, a control experiment performed in a smaller Schlenk tube (V=75±5 mL, 10 mol % III, 160 °C, 24 h) gave 2 a and 3 a still in 71 and 17 % yield, respectively. On the other hand, under ambient pressure of ethylene, the formation of the dialkylated product 3 a was significantly suppressed and as a result, 3 a was only isolated in 3 % yield from a corresponding control experiment (V=100±5 mL, 10 mol % III, 160 °C, 24 h). Unfortunately, in this case, the yield of 2 a also decreased to 64 %. Additional control experiments, performed with the catalysts I–III at 105 °C (Table 1, entries 12–14) revealed that at this temperature, I–III do not show sufficient catalytic activity. On the other hand, at 160 °C, a reduction of the catalyst loading is possible and as a consequence, with 5 or 2.5 mol % III, 2 a could still be obtained in yields between 68 and 79 % (Table 1, entries 9 and 10, footnotes [b] and [c]). For a final direct comparison of our catalysts with the catalyst systems published by Clerici and Maspero,6 we also conducted experiments with 10 or 5 mol % of [Nb(NMe2)5] or [Ta(NMe2)5] under our reaction conditions. As can be seen from Table 1, entries 15 and 16 and footnote [d], [Ta(NMe2)5] turned out to be catalytically more active than [Nb(NMe2)5] while both catalysts deliver the products 2 a and 3 a in lower yields than catalyst III.
To further investigate the scope of the hydroaminoalkylation of ethylene, we then turned our attention towards reactions of a number of functionalized N‐methylanilines (Table 2). Because it was expected that some of the amines would react more slowly than N‐methylaniline (1 a), these reactions were usually performed under the conditions of entry 10 of Table 1 (10 mol % III, 160 °C, 24 h). As can be seen from Table 2, under these conditions, ethylene reacted smoothly with the N‐methylanilines 1 c–p to give the desired monoalkylated products 2 c–p in good to very good yields of 69–91 % while the dialkylated products 3 c–p were only obtained in yields of 6–16 % (Table 2, entries 3–16). Overall, it turned out that the reaction tolerates alkyl, ether, thioether, and fluorosubstitution as well as the presence of chloro and bromo substituents which offer various possibilities for further functionalization. As observed before in titanium‐catalyzed hydroaminoalkylation reactions, the reactivity of the ortho‐methyl‐substituted substrate 1 b was found to be significantly reduced compared with the meta‐ and para‐substituted analogues 1 c and 1 d and as a consequence, in this case, the reaction had to be stirred for 48 h at 160 °C to give the product 2 b in 55 % yield (Table 2, entry 2).
Table 2.
Hydroaminoalkylation of ethylene with various N‐methylanilines (1 a–p).[a]
|
| ||||
|---|---|---|---|---|
|
Entry |
R (1) |
Yield 2 [%] |
Yield 3 [%] |
Yield 2+3 [%] |
|
1 |
H (1 a) |
76 (2 a) |
17 (3 a) |
93 |
|
2[b] |
o‐Me (1 b) |
55 (2 b) |
5[c] (3 b) |
60 |
|
3 |
m‐Me (1 c) |
81 (2 c) |
14 (3 c) |
95 |
|
4 |
p‐Me (1 d) |
75 (2 d) |
14 (3 d) |
89 |
|
5 |
p‐i‐Pr (1 e) |
77 (2 e) |
11 (3 e) |
88 |
|
6 |
m‐F (1 f) |
82 (2 f) |
16 (3 f) |
98 |
|
7 |
p‐F (1 g) |
71 (2 g) |
15 (3 g) |
86 |
|
8 |
m‐Cl (1 h) |
69 (2 h) |
12 (3 h) |
81 |
|
9 |
p‐Cl (1 i) |
78 (2 i) |
12 (3 i) |
90 |
|
10 |
m‐Br (1 j) |
72 (2 j) |
9 (3 j) |
81 |
|
11 |
p‐Br (1 k) |
79 (2 k) |
12 (3 k) |
91 |
|
12 |
p‐OMe (1 l) |
91 (2 l) |
6[c] (3 l) |
97 |
|
13 |
p‐OPh (1 m) |
76 (2 m) |
11 (3 m) |
87 |
|
14 |
p‐SMe (1 n) |
81 (2 n) |
9 (3 n) |
90 |
|
15 |
p‐OCF3 (1 o) |
77 (2 o) |
13 (3 o) |
90 |
|
16 |
p‐SCF3 (1 p) |
80 (2 p) |
13 (3 p) |
93 |

[a] Reaction conditions: 1) amine (2.00 mmol), ethylene (1.4 atm), III (109 mg, 0.20 mmol, 10 mol %), toluene (1 mL), 160 °C, 24 h, isolated yields. [b] 160 °C, 48 h. [c] The product could not be isolated in pure form.
To further study the scope and especially the limitations of the hydroaminoalkylation of ethylene, we next performed a number of corresponding experiments with secondary amines, which are all known to usually react sluggishly with sterically more demanding 1‐alkenes or styrenes (Table 3).4a, 4b, 4d–4f Unfortunately, during this study, it was first found that reactions of ethylene with the N‐alkylanilines 2 a and 4 possessing alkyl substituents larger than a methyl group do not give better results than reported for corresponding reactions of other alkenes (Table 3, entries 1 and 2). However, interesting is the fact that 3 a which had already been obtained in 17 % yield as the dialkylation side product of the hydroaminoalkylation of ethylene with N‐methylaniline (1 a, Table 1, entry 10) was only isolated in 9 % yield when 2 a was used as the starting material (Table 3, entry 1). From a mechanistic point of view, this finding strongly suggests that once 2 a is formed under catalytic conditions from ethylene and N‐methylaniline (1 a), it stays bonded or at least in close proximity to the titanium center which facilitates the C−H activation that initiates the second alkylation step (Scheme 2). The fact that N‐benzylaniline (5) and N‐methylbenzylamine (6) gave the desired hydroaminoalklyation products 14 and 15 9 in significantly better yields of 45 and 69 % (Table 3, entries 3 and 4) can simply be explained by the increased reactivity of the benzylic position which selectively undergoes C−H activation during the course of the reaction. However, it should be noted that even in these cases, it was beneficial to expand the reaction time to 48 h. Under identical conditions, the hydroaminoalkylation of N‐methylcyclohexylamine (7) with ethylene took place regioselectively at the sterically less hindered methyl group to give the corresponding product 16 9 in 71 % yield (Table 3, entry 5). While this result suggests that dialkylamines can in principal, be regarded as suitable substrates for the titanium‐catalyzed hydroaminoalkylation of ethylene, nitrogen‐containing heterocycles were found to be problematic substrates. Among the heterocycles 8–12 (Table 3, entries 6–10), only 1,2,3,4‐tetrahydroquinoline (11) gave the desired hydroaminoalkylation product 20 in acceptable yield (61 %, Table 3, entry 9). In contrast, pyrrolidine (8) and indoline (10) were found to be completely unreactive and piperidine (9) could only be converted into product 18 9 in poor yield (7 %, Table 3, entry 7). The fact that the hydroaminoalkylation of ethylene with 1,2,3,4‐tetrahydroisoquinoline (12) did deliver the expected product 21 9 in 19 % yield (Table 3, entry 10) deserves particular attention because, to the best of our knowledge, this reaction represents the first successful example of a titanium‐catalyzed alkene hydroaminoalkylation that involves 1,2,3,4‐tetrahydroisoquinoline (12) as a substrate. A simple explanation for the observation that 12 reacts more sluggishly than 1,2,3,4‐tetrahydroquinoline (11) is the fact that 12 is not an aniline derivative.5a In comparison to N‐methylbenzylamine (6), 12 gives a significantly lower yield because the benzylic position in which the reaction takes place is part of a piperidine ring and piperidine (9) was found to be a poor substrate (Table 3, entry 7).
Table 3.
Hydroaminoalkylation of ethylene with various secondary amines.[a]
|
| ||||
|---|---|---|---|---|
|
Entry |
Starting Material |
Product |
t [h] |
Yield [%] |
|
1 |
|
|
24 |
9 |
|
2 |
|
|
24 |
15 |
|
3 |
|
|
48 |
45 |
|
4[b] |
|
|
48 |
69 |
|
5[b] |
|
|
48 |
71 |
|
6[b] |
|
|
24 |
– |
|
7[b] |
|
|
48 |
7 |
|
8 |
|
|
24 |
– |
|
9 |
|
|
48 |
61 |
|
10[b] |
|
|
48 |
19 |



















[a] Reaction conditions: 1) amine (2.00 mmol), ethylene (1.4 atm), III (109 mg, 0.20 mmol, 10 mol %), toluene (1 mL), 160 °C, t, isolated yields. [b] The product was tosylated after the hydraminoalkylation to simplify the purification.
Because dimethylamine (22) is one of the most important amines in the chemical industry,10 we finally attempted to prove that this amine can also be reacted with ethylene in the presence of catalyst III (Scheme 3). For that purpose, a solution of dimethylamine in toluene4g was used and as reported before,4g monoalkylation as well as dialkylation at both α‐positions of 22 occurred at 160 °C. Subsequent tosylamide formation delivered the corresponding products 23 9 and 24 9, 11 in 16 and 29 % yield, respectively and in good agreement with the observations made with N‐methylaniline (1 a, Table 1), the trialkylated product 25 9 could also be isolated in 12 % yield.
Scheme 3.

Hydroaminoalkylation of ethylene with dimethylamine (22).
Although the insertion of an alkene into the Ti−C bond of a catalytically active titanaaziridine is generally accepted to be the C−C bond‐forming key step of the hydroaminoalkylation (Scheme 2),4c, 8 to the best of our knowledge, only one corresponding insertion product has already been isolated and characterized by IR, NMR, and MS techniques8 while no information about its solid‐state structure is available. To close this gap in knowledge, we performed a couple of insertion reactions between ethylene (1.4 atm) and the titanaaziridines 26 a–c 8 which smoothly gave access to the expected titanapyrrolidines 27 a–c in 78–85 % yield (Scheme 4). In this context, it should be mentioned that titanium complexes with sterically demanding adamantyl‐substituted cyclopentadiene ligands like the titanaaziridines 26 a–c are known to lack any catalytic activity in hydroaminoalkylation reactions of alkenes12 and for that reason, they are perfectly suited as model compounds for the isolation and characterization of stable intermediates of the catalytic cycle.8, 13 Slow evaporation of a solution of 27 b in n‐hexane/C6D6 then delivered crystals suitable for X‐ray single‐crystal analysis. As can be seen from the corresponding solid‐state structure (Figure 2), the titanapyrrolidine ring of 27 b adopts an open envelop conformation that includes a trigonal planar geometry around the nitrogen center. Finally, it should be mentioned that the hydroaminoalkylation product 2 a could easily be released from titanapyrrolidine 27 a by simple hydrolysis. On the other hand, attempts to regenerate 26 a from 27 a by the addition of one equivalent of N‐methylaniline (1 a) and heating to 90 °C failed.14
Scheme 4.

Insertion of ethylene into the Ti−C bond of titanaaziridines 26 a–c and subsequent hydrolysis of titanapyrrolidine 27 a to 2 a.
Figure 2.

Molecular structure of 27 b. Hydrogen atoms are omitted for clarity except H6 and H21. Thermal ellipsoids are drawn at the 50 % probability level. Selected bond lengths [Å] and angles [°]: Ti1−N1: 2.068(2), N1−C33: 1.472(3), C33−C32: 1.515(4), C32−C31: 1.523(3), C31−Ti1: 2.192(2), C1−C6: 1.516(3), C16−C21: 1.513(3); N1‐Ti1‐C31: 82.29(9), Ct1‐Ti1‐Ct2: 131.28 Definitions: Ct1, centroid C1–C5; Ct2, centroid C16–C20.
In summary, we have presented the first examples of titanium‐catalyzed hydroaminoalkylation reactions of ethylene with secondary amines. The new reaction does not require the use of high pressure equipment and it tolerates the presence of alkyl, ether, thioether, fluoro, chloro, and bromo substitution in the amine substrates. In addition, it was possible for the first time to determine the solid‐state structure of a titanapyrrolidine that is formed by insertion of an alkene into the Ti−C bond of a titanaaziridine.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Research Training Group “Chemical Bond Activation” (GRK 2226) funded by the Deutsche Forschungsgemeinschaft for financial support of our research and Jessica Reimer for experimental assistance.
M. Rosien, I. Töben, M. Schmidtmann, R. Beckhaus, S. Doye, Chem. Eur. J. 2020, 26, 2138.
Contributor Information
Prof. Dr. Rüdiger Beckhaus, Email: ruediger.beckhaus@uni-oldenburg.de.
Prof. Dr. Sven Doye, Email: doye@uni-oldenburg.de.
References
- 1.For reviews on hydroaminoalkylation reactions of alkenes, see:
- 1a. Chong E., Garcia P., Schafer L. L., Synthesis 2014, 46, 2884–2896; [Google Scholar]
- 1b. Hannedouche J., Schulz E., Organometallics 2018, 37, 4313–4326; [Google Scholar]
- 1c. Edwards P. M., Schafer L. L., Chem. Commun. 2018, 54, 12543–12560. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples of late transition-metal-catalyzed hydroaminoalkylation reactions of alkenes, see:
- 2a. Tran A. T., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 10530–10534; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10666–10670; [Google Scholar]
- 2b. Spettel M., Pollice R., Schnürch M., Org. Lett. 2017, 19, 4287–4290; [DOI] [PubMed] [Google Scholar]
- 2c. Thullen S. M., Rovis T., J. Am. Chem. Soc. 2017, 139, 15504–15508; [DOI] [PubMed] [Google Scholar]
- 2d. Lahm G., Opatz T., Org. Lett. 2014, 16, 4201–4203; [DOI] [PubMed] [Google Scholar]
- 2e. Schinkel M., Wang L., Bielefeld K., Ackermann L., Org. Lett. 2014, 16, 1876–1879; [DOI] [PubMed] [Google Scholar]
- 2f. Kulago A. A., Van Steijvoort B. F., Mitchell E. A., Meerpoel L., Maes B. U. W., Adv. Synth. Catal. 2014, 356, 1610–1618. [Google Scholar]
- 3.For selected examples of group 3 metal-catalyzed hydroaminoalkylation reactions of alkenes with tertiary amines, see:
- 3a. Nako A. E., Oyamada J., Nishiura M., Hou Z., Chem. Sci. 2016, 7, 6429–6434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Liu F., Luo G., Hou Z., Luo Y., Organometallics 2017, 36, 1557–1565; [Google Scholar]
- 3c. Gao H., Su J., Xu P., Xu X., Org. Chem. Front. 2018, 5, 59–63. [Google Scholar]
- 4.For selected examples of group 4 metal-catalyzed hydroaminoalkylation reactions of alkenes, see:
- 4a. Kubiak R., Prochnow I., Doye S., Angew. Chem. Int. Ed. 2009, 48, 1153–1156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1173–1176; [Google Scholar]
- 4b. Kubiak R., Prochnow I., Doye S., Angew. Chem. Int. Ed. 2010, 49, 2626–2629; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2683–2686; [Google Scholar]
- 4c. Prochnow I., Zark P., Müller T., Doye S., Angew. Chem. Int. Ed. 2011, 50, 6401–6405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6525–6529; [Google Scholar]
- 4d. Dörfler J., Preuß T., Schischko A., Schmidtmann M., Doye S., Angew. Chem. Int. Ed. 2014, 53, 7918–7922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8052–8056; [Google Scholar]
- 4e. Dörfler J., Preuß T., Brahms C., Scheuer D., Doye S., Dalton Trans. 2015, 44, 12149–12168; [DOI] [PubMed] [Google Scholar]
- 4f. Lühning L. H., Brahms C., Nimoth J. P., Schmidtmann M., Doye S., Z. Anorg. Allg. Chem. 2015, 641, 2071–2082; [Google Scholar]
- 4g. Bielefeld J., Doye S., Angew. Chem. Int. Ed. 2017, 56, 15155–15158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15352–15355. [Google Scholar]
- 5.For selected examples of group 5 metal-catalyzed hydroaminoalkylation reactions of alkenes, see:
- 5a. Herzon S. B., Hartwig J. F., J. Am. Chem. Soc. 2007, 129, 6690–6691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Reznichenko A. L., Emge T. J., Audörsch S., Klauber E. G., Hultzsch K. C., Schmidt B., Organometallics 2011, 30, 921–924; [Google Scholar]
- 5c. Reznichenko A. L., Hultzsch K. C., J. Am. Chem. Soc. 2012, 134, 3300–3311; [DOI] [PubMed] [Google Scholar]
- 5d. Lauzon J. M., Eisenberger P., Roşca S.-C., Schafer L. L., ACS Catal. 2017, 7, 5921–5931; [Google Scholar]
- 5e. Brandt J. W., Chong E., Schafer L. L., ACS Catal. 2017, 7, 6323–6330; [Google Scholar]
- 5f. Edwards P. M., Schafer L. L., Org. Lett. 2017, 19, 5720–5723; [DOI] [PubMed] [Google Scholar]
- 5g. DiPucchio R. C., Roşca S.-C., Schafer L. L., Angew. Chem. Int. Ed. 2018, 57, 3469–3472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3527–3530; [Google Scholar]
- 5h. DiPucchio R. C., Roşca S.-C., Athavan G., Schafer L. L., ChemCatChem 2019, 11, 3871–3876; [Google Scholar]
- 5i. Braun C., Nieger M., Bräse S., Schafer L. L., ChemCatChem 2019, 11, 5264–5268. [Google Scholar]
- 6. Clerici M. G., Maspero F., Synthesis 1980, 305–306. [Google Scholar]
- 7.Research and Markets, “The Ethylene Technology Report 2016”, can be found under: https://www.researchandmarkets.com.
- 8. Manßen M., Lauterbach N., Dörfler J., Schmidtmann M., Saak W., Doye S., Beckhaus R., Angew. Chem. Int. Ed. 2015, 54, 4383–4387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4458–4462. [Google Scholar]
- 9.The initially formed hydroaminoalkylation product was converted into the corresponding tosylamide to simplify the work up procedure and the final chromatographic purification.
- 10. Weissermel K., Arpe H.-J., Industrial Organic Chemistry, 4th ed., Wiley-VCH, Weinheim, 2003, 51–52. [Google Scholar]
- 11.The initially formed product, di-n-propylamine, is an intermediate for the industrial production of herbicides (e.g. oryzalin).
- 12.In a control experiment, complex 26 a (10 mol %) did not catalyze the hydroaminoalkylation of ethylene with N-methylaniline (1 a) at 140 °C.
- 13.All attempts to isolate or at least to characterize intermediates of the proposed catalytic cycle from catalytically active complexes such as Ind2TiMe2 or III, have failed so far.
- 14.The experiment was performed in [D8]toluene and followed by 1H NMR. At temperatures above 90 °C, slow decomposition of 27 a was observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary