

Short abstract
Linked Comment: https://doi.org/10.1111/bjd.18377.
https://doi.org/10.1111/bjd.18829 available online
1. Purpose and scope
The overall objective of this guideline is to provide the user with information on the laboratory diagnosis of inherited epidermolysis bullosa (EB) to improve outcomes (Table 1). An accurate diagnosis and subclassification of EB enables (i) early prognostication of disease severity, (ii) decision making for patient management, (iii) informed genetic counselling for the patient and family and DNA‐based prenatal or preimplantation genetic diagnosis, (iv) long‐term surveillance and management of possible complications, (v) inclusion in clinical trials and (vi) precision medicine.
Table 1.
Summary of key recommendations for laboratory diagnosis of epidermolysis bullosa (EB)
| No. | Recommendation | Grade of recommendation | Level of evidence | References |
|---|---|---|---|---|
| 1 | We strongly recommend that EB laboratory diagnosis should be performed; with the first clinical suspicion of EB an adapted diagnostic technique should be initiated | C | 2+ | 20, 22, 23, 24, 28, 34, 62, 77, 79, 112 |
| 2 | Early diagnosis by IFM and genetic testing is sufficient to provide prognosis and help decision making in most cases | C | IFM: 2+ | 20, 62, 77, 79 |
| B | Genetic testing: 2++ | 22, 23, 24, 28, 34, 112 | ||
| 3 | DNA‐based prenatal diagnosis is technically feasible for all EB subtypes and should be considered upon family request and according to national regulationsa | B | 2++ | 46, 47 |
| 4 | We strongly recommend that EB laboratory diagnosis should be performed in laboratories with documented specific expertise and experience in the field, preferably accredited | D | 4 | |
| 5 | Genetic testing is always recommended for the diagnosis of EB. The index case and, whenever possible, the parents should be tested in order to provide reliable genetic counselling and risk calculation for family members and offspring | B | 2++ | 22, 23, 24, 28, 34, 112 |
| 6 | Methods for genetic testing in EB include NGS with targeted EB gene panel, WES and SS. Additional methods to be applied in selected cases include SNP arrays for segregation analysis, MLPA, qPCR and RNA‐Seq, as well as homozygosity mapping in case of consanguinity. Hotspot and recurrent pathogenic variants can be tested in specific situations (population, clearly defined phenotype) to reduce costs and time | B | 2++ | 20, 21, 22, 23, 24, 28, 34, 112 |
| 7 | IFM is recommended to obtain a rapid diagnosis and prognosis, and to prioritize genetic testing and facilitate interpretation of genetic results | C | 2+ | 20, 62, 77, 79 |
| 8 | TEM is useful in a limited number of cases, and should be performed when IFM and genetic testing do not deliver conclusive results | C | 2+ | 77 |
| 9 | If appropriate EB laboratory diagnosis yields inconclusive results, the original diagnosis and the diagnostic strategy should be re‐evaluated and individualized strategies could be considered. In such cases further laboratory analyses imply additional expertise and high costs and are time consuming. This cannot be assured by all laboratories. EB is a rare disorder, therefore external, national and/or international collaboration is recommended to help solve such cases | C | 2+ | 57, 60, 61, 113, 114 |
| 10 | Results of the EB laboratory diagnosis should be communicated to the patient and family, preferably by geneticists and dermatologists with experience in the field, and according to national rules and regulations. Genetic counselling is always recommended | D | 4 |
IFM, immunofluorescence mapping; MLPA, multiplex ligation‐dependent probe amplification; NGS, next‐generation sequencing; qPCR, quantitative polymerase chain reaction; SNP, single‐nucleotide polymorphism; SS, Sanger sequencing; TEM, transmission electron microscopy; WES, whole‐exome sequencing. aDNA‐based prenatal diagnosis is only possible when familial mutation is known.
Users of the guideline will be dermatologists, neonatologists, paediatricians, geneticists and genetic counsellors, laboratory doctors and technicians, nurses, and people living with EB and their families. The target group consists of patients with skin blistering or fragility, suspected of having any type of EB.
2. Stakeholder involvement and peer review
In 2016, Dystrophic Epidermolysis Bullosa Research Association (DEBRA) International consulted with the international EB community and identified clinical practice guidelines (CPGs) for laboratory diagnosis of EB as a priority area ( http://www.debra-international.org/clinical-guidelines.html). This guideline was developed on behalf of DEBRA International with the financial support of DEBRA Austria, according to the DEBRA Guideline Development Standard. The CPG development group consisted of 16 international members representing 12 countries. The draft document was circulated to nine reviewers who are either internationally recognized experts in the field or people living with EB. On behalf of DEBRA International, a specialist in guideline development and coordinator of CPGs was appointed to guide the development panel through the entire process.
3. Methodology
The CPG development group consisted of dermatologists, paediatric dermatologists, geneticists, biologists and a nurse, and additionally patient representatives. All panel members completed written conflict of interest and code of conduct declarations. The evidence‐based development of clinical recommendations was led by two panel members (C.H. and L.L.). During the guideline development, the group met twice in face‐to‐face meetings (at least six members physically present) to discuss the clinical questions and methodology, to review the evidence and the recommendations, and to agree on structure and wording. Whenever input from the entire group was required, it was solicited via e‐mail. A research assistant (S.B.) coordinated communications and contributed to the preparation of the documents and manuscript.
To identify publications, a search of NCBI ‘All Databases’ and PubMed was performed using the terms ‘inherited EB and laboratory diagnosis’, ‘EB and mutation’ and ‘EB and prenatal diagnosis’, with the search period ending in December 2017. In addition, ‘epidermolysis bullosa’ was used to search articles in GeneReviews. In total 1485 articles were identified. In light of technological advances, articles published before 2010 were excluded from the appraisal, unless newer publications on a topic were lacking (e.g. prenatal diagnosis of EB). Case reports were only considered when they reported relevant methodology. Seven papers published between November 2017 and August 2018 were appraised, and many other recent publications were added because of their relevant contents.
Sixty‐four papers were appraised, each by two panel members, according to the Critical Appraisal Skills Programme1 and Scottish Intercollegiate Guidelines Network quality rating (Appendix S1; see Supporting Information).2 No meta‐analyses, systematic reviews or case–control studies were available. The highest level of evidence was achieved by high‐quality cohort studies.
4. Limitations of the guideline
The document has been prepared on behalf of DEBRA International and is based on the best data available at the time of the document preparation. EB is a rare disease and most of the subtypes are ultraorphan conditions (≤ 1 in 20 000). People living with EB may have their ‘private’ genetic variants and unusual genotype–phenotype correlations, which require individualized strategies for analysis. Moreover, experimental proof of pathogenicity of unclassified sequence variants (variants of uncertain significance, VUS) is performed in a basic research environment. Such situations are not covered by this guideline. Noninvasive prenatal diagnosis (PND) utilizing cell‐free fetal DNA and preimplantation genetic diagnosis are also not covered within this guideline. Detailed descriptions of the sequencing methods and their quality controls, and an introduction to good clinical practice of genetic counselling were beyond the scope of this guideline.
5. Plans for guideline revision
The proposed revision for this set of recommendations is scheduled for 2021.
6. Background
Inherited EB is a group of rare genetic disorders characterized by skin fragility and mechanically induced blistering. EB comprises four main types: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler syndrome (KS), with more than 30 clinical subtypes (Tables 2 and 3). EB is clinically heterogeneous, including a broad spectrum of severity. At one end of the spectrum, severe congenital cutaneous and mucosal fragility may be accompanied by extracutaneous involvement and complications, often resulting in a limited lifespan. In contrast, mild skin fragility may be localized to the extremities, begin later in life, or manifest only as nail dystrophy.3 In children and adults, clinical features may be typical and allow the clinical diagnosis of the EB type and subtype.4 In neonates and in individuals with mild clinical manifestations the determination of the EB type and subtype relies on laboratory diagnosis. In some situations, particularly in families with a first case of EB and apparent de novo occurrence, discrimination between autosomal dominant and recessive inheritance is not possible without genetic testing.5
Table 2.
Classification and molecular characteristics of epidermolysis bullosa (EB) including genes, proteins and types of pathogenic sequence variants: EB simplex (EBS)
| Gene | Level of skin cleavage and ultrastructural anomalies as assessed by TEM | Relative protein expression as assessed by IFM | Types of pathogenic sequence variantsa |
|---|---|---|---|
| Protein | |||
| Inheritance | |||
| KRT5 | Cleavage: basal keratinocyte cytoplasm; tonofilament clumping always present in EBS generalized severe and in some cases of EBS with mottled pigmentation | Unchanged | Missense, nonsense, splice site, frameshift, in‐frame (large) deletions or insertions |
| Keratin 5 | |||
| AD | |||
| KRT14 | Cleavage: basal keratinocyte cytoplasm; tonofilament clumping in EBS generalized severe; lack of tonofilaments in basal keratinocytes in AR EBS | Unchanged or absent | Missense, nonsense, splice site, frameshift, in‐frame deletion or duplications |
| Keratin 14 | |||
| AD, AR | |||
| PLEC | Cleavage: basal keratinocyte cytoplasm just above hemidesmosomes; diminutive hemidesmosomes | Plectin unchanged, absent or reduced with domain‐specific antibodies | Missense, nonsense, frameshift, splice site |
| Plectin | |||
| AD, AR | |||
| KLHL24 | Cleavage: basal keratinocyte cytoplasm; reduced tonofilaments in basal keratinocytes | Keratin 14 reduced or unchanged | Pathogenic variants in the translation initiation codon |
| Kelch‐like protein 24 | |||
| AD | |||
| DST | Cleavage: basal keratinocyte cytoplasm; diminutive hemidesmosomes lacking tonofilament attachment | BPAG1 (isoform e) absent | Nonsense, missense, frameshift, splice site |
| BPAG1 | |||
| AR | |||
| EXPH5 | Cleavage: basal keratinocyte cytoplasm; tonofilament aggregation in basal keratinocytes | Exophilin 5 absent | Nonsense, frameshift |
| Exophilin 5 | |||
| AR | |||
| CD151 | Cleavage: lower epidermis | CD151 absent | Frameshift, splice site |
| Tetraspanin 24 | |||
| AR | |||
| TGM5 | Cleavage: between stratum granulosum and corneum | Absent or reduced activity and expression of transglutaminase 5 | Missense, nonsense, frameshift, splice site |
| Transglutaminase 5 | |||
| AR | |||
| PKP1 | Cleavage: suprabasal epidermal layers; hypoplastic desmosomes | Plakophilin 1 absent | Nonsense, frameshift, splice site |
| Plakophilin 1 | |||
| AR | |||
| DSP | Cleavage: suprabasal epidermal layers; hypoplastic desmosomes | Desmoplakin reduced or absent | Nonsense, frameshift |
| Desmoplakin | |||
| AR | |||
| JUP | Cleavage: suprabasal epidermal layers; hypoplastic desmosomes | Plakoglobin absent | Nonsense |
| Plakoglobin | |||
| AR |
AD, autosomal dominant; AR, autosomal recessive; IFM immunofluorescence mapping. aTypes of sequence variants described in the literature according to the Human Gene Mutation Database 2018·3.
Table 3.
Classification and molecular characteristics of epidermolysis bullosa (EB) including genes, proteins and types of pathogenic sequence variants: junctional EB, dystrophic EB and Kindler syndrome
| Gene | Level of skin cleavage and ultrastructural anomalies as assessed by TEM | Relative protein expression as assessed by IFM | Types of pathogenic sequence variantsa |
|---|---|---|---|
| Protein | |||
| Inheritance | |||
| Junctional EB | |||
| LAMA3, LAMB3, LAMC2 | Cleavage: lamina lucida; rudimentary to hypoplastic hemidesmosomes in most cases | Laminin‐332 reduced or absent | Nonsense, frameshift, splice site, missense |
| Laminin‐332 | |||
| AR | |||
| COL17A1 | Cleavage: lamina lucida, very rarely within basal keratinocytes; hypoplastic hemidesmosomes in most cases | Type XVII collagen reduced or absent | Nonsense, frameshift, splice site, missense, large deletions |
| Type XVII collagen | |||
| AR | |||
| LAMA3A | Cleavage: usually not detectable; hypoplastic hemidesmosomes | No change in the relative protein expression | Frameshift, nonsense |
| Laminin chain α3A | |||
| AR | |||
| ITGA6, ITGB4 | Cleavage: lamina lucida, very rarely within basal keratinocytes; hypoplastic hemidesmosomes | Integrin α6β4 reduced or absent, rarely unchanged | Nonsense, frameshift, splice site, missense, large deletions |
| Integrin α6β4 | |||
| AR | |||
| ITGA3 | No data available | Integrin α3 subunit absent | Nonsense, frameshift, splice site, missense |
| Integrin α3 subunit | |||
| AR | |||
| Dystrophic EB | |||
| COL7A1 | Cleavage: sublamina densa, lack of anchoring fibrils in RDEB generalized severe, hypoplastic anchoring fibrils in the other subtypes | Type VII collagen reduced or absent, sometimes unchanged | Nonsense, frameshift, splice site, missenseb , c |
| Type VII collagen | |||
| AR | |||
| COL7A1 | Cleavage: sublamina densa, hypoplastic anchoring fibrils | Type VII collagen unchanged or reduced | Missense, splice site, (large) in‐frame deletionsd |
| Type VII collagen | |||
| AD | |||
| PLOD3 | Cleavage: sublamina densa, fragmentation of the lamina densa, variable number and altered morphology of anchoring fibrils | Type VII collagen reduced | Missense, frameshift |
| Lysyl hydroxylase 3 | |||
| AR | |||
| Kindler syndrome | |||
| FERMT1 | Cleavage: multiple levels (basal keratinocytes, lamina lucida, sublamina densa); lamina densa reduplications | Kindlin‐1 absent or reduced | Nonsense, splice site, frameshift, large deletions, regulatory, in frame, missense, deep intronic |
| Kindlin‐1 | |||
| AR | |||
AD, autosomal dominant; AR, autosomal recessive; IFM immunofluorescence mapping RDEB, recessive dystrophic EB; TEM, transmission electron microscopy. aTypes of sequence variants described in the literature according to the Human Gene Mutation Database 2018·3. bCases with compound heterozygosity for recessive and dominant sequence variants have been reported.41 cSomatic forward mosaicism has been reported.114 dGermline mosaicism has been reported.115
6.1. Classification of epidermolysis bullosa with genes and causative variants
Classification of EB into four main types is based on the ultrastructural level of skin cleavage.6 In EBS, splitting occurs within the epidermis (intraepidermal), in JEB within the lamina lucida (junctional) and in DEB below the basement membrane within the superficial dermis (dermal); in KS there is a mixed level of skin blistering (Tables 2 and 3). An EB classification scheme (onion skin) has been developed that sequentially takes into account the level of skin cleavage corresponding to the major EB type, the clinical severity, the inheritance pattern and the molecular defect, including the relative protein expression and the disease‐causing sequence variant(s).6 A detailed description of this EB classification system and the clinical subtypes has been reported by Fine et al.6
6.2. Clinical features of epidermolysis bullosa
6.2.1. Cutaneous and mucosal involvement in epidermolysis bullosa
Skin blistering on sites of mechanical trauma is the main clinical feature of EB. Depending on the level of skin cleavage, blisters may be superficial as with EBS and result in erosions, or they may be more profound such as with JEB, DEB and KS and lead to ulcerations. Blisters may be generalized, disseminated to different body sites, or localized to the extremities. Skin defects heal spontaneously by restitutio ad integrum, or with residual hypo‐ or hyperpigmentation, skin atrophy or scarring. Recurrent and chronic skin defects may result from permanent exposure of the fragile skin to mechanical trauma.
Oral, oesophageal, tracheal, genitourinary and ocular mucosal membranes may be affected by erosions, ulcerations and scarring. Fragility of the cutaneous adnexa may involve nails, which may become dystrophic or lost, and hair, leading to alopecia. These features are characteristic of specific EB subtypes.6
Progressive scarring results in contractures and/or mutilations of the extremities, microstomia, disfigurement and oesophageal stenosis, which are common in KS and in DEB, or dyspnoea with risk of suffocation in specific forms of JEB.
Teeth may be affected because of amelogenesis imperfecta (in JEB) or secondarily to the fragility and scarring of the oral mucosa leading to impaired oral hygiene (in DEB).
6.2.2. Extracutaneous involvement in epidermolysis bullosa
Due to the high caloric consumption and acquired complications in the context of permanent skin damage and regeneration, EB subtypes with generalized severe blistering are characterized by secondary involvement of other organs or systems.7, 8 This is mainly the case with generalized recessive DEB (RDEB), which may be accompanied by failure to thrive, anaemia, osteoporosis, joint contractures, cardiomyopathy or renal amyloidosis, for example.8
In syndromic EB types, expression of the affected genes in extracutaneous tissues leads to primary involvement of other organs or systems.9 Examples are muscular dystrophy in EBS with plectin deficiency; pyloric atresia in EBS with plectin deficiency and in JEB with integrin α6β4 deficiency; cardiomyopathy in EBS caused by KLHL24 or PLEC sequence variants and in skin fragility syndromes with DSP and JUP sequence variants;10 lung fibrosis and nephrotic syndrome in JEB with deficiency of the integrin α3 subunit;11 connective tissue abnormality in patients with PLOD3 gene mutation;12 or nephrotic syndrome in patients with CD151 deficiency.13 For detailed descriptions of the clinical features of EB, original and review articles are available.6, 7, 8, 14, 15
6.2.3. Molecular basis of epidermolysis bullosa
In EB, mucocutaneous fragility results from decreased resilience of the structures that confer mechanical stability to the epidermis (keratin cytoskeleton, desmosomes) and to the cutaneous basement membrane zone (BMZ) (hemidesmosomes, focal adhesions, anchoring filaments and anchoring fibrils) (Fig. 1). These multimolecular suprastructures link the keratinocytes to each other, the basal keratinocytes to the underlying basement membrane, and the basement membrane to the underlying connective tissue. Disease‐causing variants in at least 21 different genes account for the genetic and allelic heterogeneity of EB (Tables 2 and 3). These genes encode proteins that mainly play structural roles; their major characteristics, expression patterns and functions are summarized in Table S1 (see Supporting Information).
Figure 1.
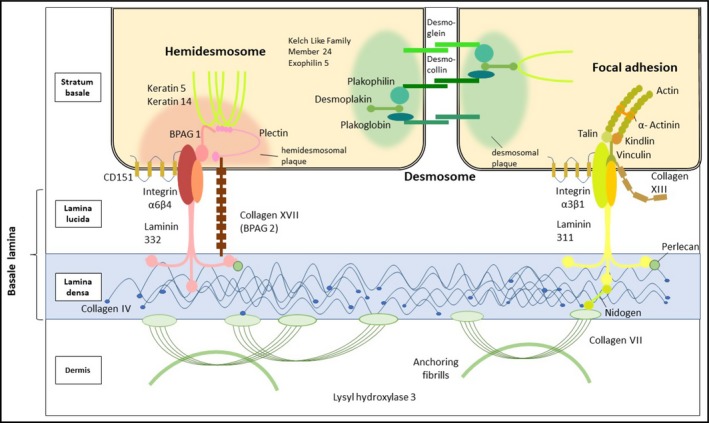
Schematic representation of intraepidermal and dermoepidermal adhesion structures with proteins relevant to epidermolysis bullosa.
7. Laboratory diagnosis of epidermolysis bullosa
7.1. Types of laboratory referral
This guideline provides the steps in making an accurate diagnosis in case of clinical suspicion of EB. It is therefore recommended that laboratories consider the testing criteria formulated and agreed by these guidelines. However, there are vast variations and differences among EB clinical and diagnostic centres around the world with respect to the diagnostic equipment and methods available, and also between the national health system regulations governing rare disease care and genetic testing, and reimbursement for these services. Therefore, one single guideline at this stage may not be able to cover all aspects related to laboratory diagnosis of EB. Such situation(s) may require EB clinicians and diagnostic scientists to make a reasonable adjustment, provided that such adjustment does not deviate from this guideline significantly.
7.1.1. Neonate with skin fragility
✓ A newborn baby showing congenital absence of skin, blistering or skin fragility should be referred to an EB diagnostic centre for diagnosis as soon as possible. In addition to a blood sample for the extraction of genomic DNA, a skin biopsy should be taken from the patient. The confirmation of diagnosis can be achieved (i) using the skin biopsy for immunohistochemistry (IHC) with fluorescence‐labelled secondary antibodies (immunofluorescence mapping, IFM); (ii) by skin ultrastructure examination by transmission electron microscopy (TEM) or (iii) by direct genetic testing, which is dependent on the facilities and resources available in the diagnostic centre. In some cases, all three approaches are necessary. Although genetic testing can make a definite diagnosis and its turnaround time is progressively shortening, IFM can provide the diagnosis within hours, thus ensuring appropriate neonatal management. While this will undoubtedly change in the coming years, IFM still remains the first method of choice.16
7.1.2. Paediatric and adult patients with skin fragility
✓ As the presentation of clinical manifestations may become clearer with a patient's age, any paediatric and/or adult patient with skin fragility who has already developed typical manifestations of the EB subtype can be referred directly to a diagnostic centre for genetic testing. Dependent on the situation, the method chosen can be next‐generation sequencing (NGS) or Sanger sequencing (SS). If both methods fail to provide a diagnosis, IFM and TEM may help to explain the molecular and ultrastructural basis of the skin fragility. The details of this part will be discussed later in this guideline.
7.1.3. Carrier testing
✓ The biological parents of a patient with EB, as well as biological siblings, can be referred to a diagnostic centre to test for carrier status when the genetic sequence variant has been confirmed in the index case (according to good clinical practice guidelines for genetic counselling). Under the dominant condition of the disease, this can act as a ‘sequence variant confirmation’. The segregation of pathogenic variant(s) in the parents and other family members is important in understanding the inheritance pattern (autosomal recessive, autosomal dominant, de novo) and the risk assessment for future pregnancies. Carrier screening for a person who is not connected to the patient through blood, or who is not from the same geographical area, can be recommended.17 According to the individual situation and national regulations, genetic testing of the partner should be performed after genetic counselling. Recurrence of the disease in the family is possible, even if the calculated risk is very low.18
7.1.4. Prenatal diagnosis
✓ When the carrier status of the familial sequence variant has been determined in both parties of an expecting couple, DNA‐based prenatal testing can be offered to the couple upon their request. Some countries may have their specific local regulations and ethical requirements, which need to be considered before a PND can take place. According to the national regulations, the test can be referred by a genetics counsellor (preferably with knowledge of EB) or a dermatologist specialized in EB. Referral for prenatal testing by linkage analysis would need to be discussed in detail with professionals in a genetic diagnostic centre, as such a situation usually requires substantial tests and knowledge of the index case.
7.2. Epidermolysis bullosa laboratory diagnostic flowchart
In cases with skin fragility and blistering, standard histopathological evaluation and direct immunofluorescence of skin samples, microbiological swabs and indirect immunofluorescence with patient's serum (and any other laboratory test required) are routinely indicated to rule out differential diagnoses of EB, such as infections (e.g. staphylococcal scalded skin syndrome, candidiasis, herpes simplex), autoimmune blistering disorders (e.g. bullous pemphigoid), mastocytosis or other genodermatoses (e.g. epidermolytic ichthyosis).
If the clinical features and family history are suggestive of EB, laboratory diagnosis is always indicated, after informed consent is given by the patient, parents or caregivers (Fig. 2).
Figure 2.
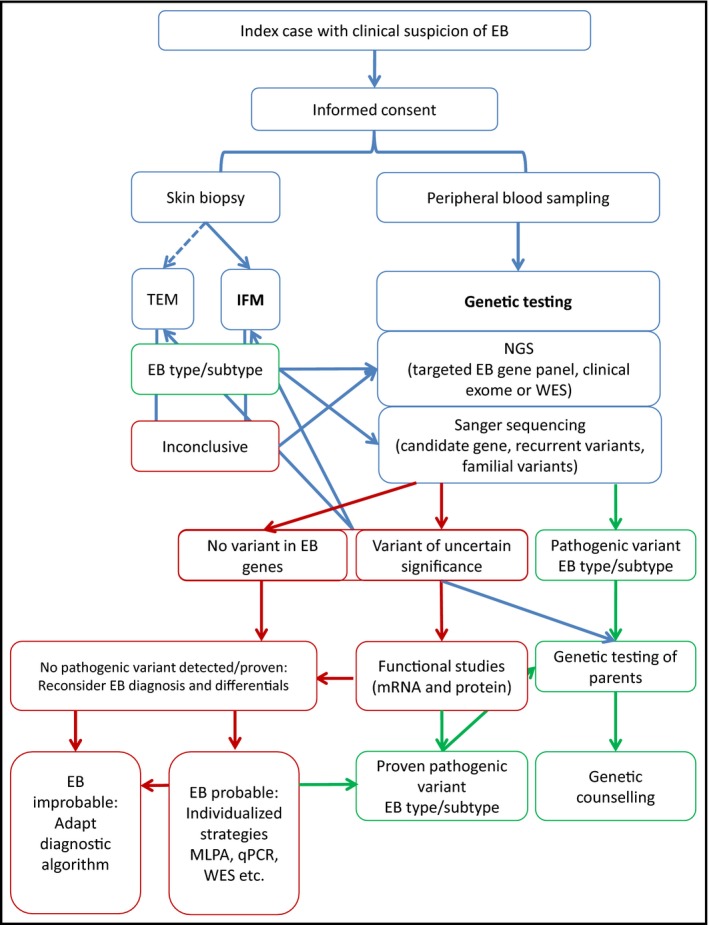
Flowchart of laboratory diagnosis of epidermolysis bullosa (EB). Schematic representation of the steps required to achieve a molecular diagnosis of EB. Steps shown in green lead to a clear diagnosis of the EB type or subtype, while steps shown in red may require individualized strategies in a research setting. IFM, immunofluorescence mapping; MLPA, multiplex ligation‐dependent probe amplification; NGS, next‐generation sequencing; qPCR, quantitative polymerase chain reaction; TEM, transmission electron microscopy; WES, whole‐exome sequencing.
✓ Ideally, both genetic testing and IFM should be performed to allow complete molecular characterization of EB, at both the DNA and protein levels. These methods provide complementary information that enables prediction of the consequences of novel sequence variants and genotype–phenotype correlations.
✓ However, the benefit for people with EB and their families, the availability of different methods, the national regulations and economic factors must be considered when EB laboratory diagnosis is planned. Prioritization of strategies can shorten the time to diagnosis and save resources, but it requires expertise of the clinicians and of the diagnostic scientists (Tables 4 and 5). In a clinical diagnostic setting, the following main prioritization strategies of EB laboratory diagnosis can be considered.
Table 4.
Comparisons of the main methods for laboratory diagnosis of epidermolysis bullosa (EB): genetic testing
| Method | Advantages | Disadvantages |
|---|---|---|
| Targeted NGS EB gene panel |
Relatively rapid and effective approach for EB diagnosis, in particular if clinical features, IFM and TEM findings do not indicate the candidate gene, or if such information is not available, or in situations with genetic heterogeneity Identifies disease‐causing pathogenic variant(s) In correlation with phenotypic information, identifies mode of inheritance Allows genetic counselling Allows DNA‐based prenatal diagnosis Detects mosaicism quantitatively Allows predictive diagnosis for partners with carrier statusa Level of evidence 2++ |
Not available in every country or healthcare setting Requires bioinformatics support Incidental findings (such as carrier status for autosomal recessive EB subtypes, other than expected) |
| Whole‐exome sequencing |
Effective approach if clinical features, IFM and TEM findings do not indicate the candidate gene, or if such information is not available Identifies disease‐causing pathogenic variant(s) May identify variants in new EB‐associated genes In correlation with phenotypic information, identifies mode of inheritance Allows genetic counselling Allows DNA‐based prenatal diagnosis Can detect mosaicism Allows predictive diagnosis for partners with carrier statusa Level of evidence 2+ |
Not available in every country or healthcare setting Requires bioinformatics support Analysis and interpretation of findings require expertise and are time consuming Incidental findingsb More expensive than targeted NGS |
| Candidate gene analysis by SS |
Straightforward approach if candidate genes are obvious or have been identified by IFM or TEM, or if the familial mutation is known Identifies disease‐causing pathogenic sequence variant(s) In correlation with phenotypic information, identifies mode of inheritance Allows genetic counselling Allows DNA‐based prenatal diagnosis Allows predictive diagnosis for partners with carrier status Level of evidence 2++ |
Will miss variations in other EB genes May be time consuming and more expensive if the ‘candidate’ gene is not correct and more genes have to be analysed |
NGS, next‐generation sequencing; IFM, immunofluorescence mapping; TEM, transmission electron microscopy; SS, Sanger sequencing. aIn most countries, NGS EB gene panel and whole‐exome sequencing are not indicated for carrier testing, and insurance companies do not cover the cost; before predictive genetic testing, individuals must undergo genetic counselling and must consent regarding communication of incidental findings. bSuch as pathogenic variants or variants of unknown significance in genes associated with cancer predisposition or genetic disorders with late onset, or carrier status for autosomal recessive disorders.
Table 5.
Comparison of the main methods for laboratory diagnosis of epidermolysis bullosa (EB): immunofluorescence mapping and transmission electron microscopy
| Method | Advantages | Disadvantages |
|---|---|---|
| Immunofluorescence mapping |
Easy technique Rapid result May indicate the candidate protein May indicate the consequence of the genetic variant(s) on the protein level Prognostic value May be helpful in interpretation of VUS May help in the identification of areas of revertant mosaicism Level of evidence 2+ |
Skin biopsy, a modestly invasive procedure, is required Possible artefacts (e.g. artificial junctional cleavage) May remain uninformative (no skin cleavage and no alteration of immunoreactivity) in mild EB subtypes (e.g. localized EBS or DEB) The delivered information depends on the quality and number of applied antibodies No information on the genetic defect Experience is required for interpretation of the results |
| Transmission electron microscopy |
Identifies ultrastructural anomalies that are specific for some types of EB Identifies ultrastructural anomalies that could help in validation of the pathogenic role of VUS Level of evidence 2+ |
Skin biopsy, a modestly invasive procedure, is required May remain uninformative (e.g. no skin cleavage, or presence of nonspecific alterations such as re‐epithelialization, or subtle changes in epithelial adhesion structures) Possible artefacts due to biopsy technique or processing (e.g. absence of epidermis or artefactual cleavage) No information on the genetic defect Expertise is required for both specimen processing and finding interpretation Time consuming |
VUS, variants of unknown significance; EBS, EB simplex; DEB, dystrophic EB.
In neonates, IFM should be the first diagnostic step as it delivers rapid results. In parallel, genetic testing should always be performed.
In cases with characteristic clinical features, including localized dominant EBS or DEB, for which IFM will frequently not deliver a useful result, genetic testing by NGS or SS can deliver a final diagnosis.
In EB (sub)types with genetic heterogeneity or in cases with uncharacteristic findings, without a clear candidate gene, genetic testing by NGS is recommended.
✓ If pathogenic variants are detected in the index case, the parents should be tested to determine the pattern of inheritance. Other family members can be tested to confirm segregation and allow genetic counselling.
✓ If no pathogenic variants are detected in the index case, the diagnostic algorithm must be adapted as described below.
7.3. Genetic testing for epidermolysis bullosa
The pathogenic sequence variants will provide clarity for the definitive diagnosis, prognosis and inheritance for the patient with EB and their family, and their identification is therefore essential. Moreover, it is the basis for the risk calculation of having affected offspring in the same generation by the same biological parents of the proband, and of his or her offspring being affected. Furthermore, it provides the basis for genetic prenatal or preimplantation diagnosis in subsequent pregnancies. With upcoming protein‐, RNA‐ and genomic DNA‐targeted therapies, finding the causative pathogenic sequence variant becomes even more important for personalized precision medicine. Therefore, every patient with an established or suspected diagnosis of EB is recommended to undergo genetic testing (level of evidence 2++, grade of recommendation B).
Genetic diagnosis of EB is recommended to be performed in laboratories with documented expertise in the field, preferably accredited (e.g. by ISO 15189 and ISO 17025 standards, Organization for Economic Co‐operation and Development guidelines or Clinical Laboratory Improvement Amendments certified) or certified (e.g. by ISO 9001), or participating in exchange of samples with other laboratories (in External Quality Assessment programmes, e.g. EMQN) (level of evidence 4, grade of recommendation D).
7.3.1. Methods
Genomic DNA isolated from peripheral blood leucocytes (ethylenediaminetetraacetic acid treated), saliva or buccal smear from patients and their parents is analysed. A brief summary on currently used genetic testing methods is provided here to allow understanding of the guideline; a detailed description of these techniques is beyond the scope of this article.
7.3.1a. Next‐generation sequencing‐targeted gene panel and whole‐exome sequencing in epidermolysis bullosa (level of evidence 2++, grade of recommendation B)
The term NGS describes the techniques used to analyse several genes and a large number of DNA samples in parallel using high‐throughput technology. NGS can be applied to sequence only defined DNA targets (e.g. targeted gene panels) or to sequence entire exomes (whole‐exome sequencing, WES), genomes (whole‐genome sequencing) or transcriptomes (RNA‐Seq), followed by post‐test filtering. Recently, NGS has been proved to be one of the most important tools for accurately and comprehensively identifying pathogenic variants in EB.3, 19, 20, 21, 22, 23, 24
Table S2 (see Supporting Information) summarizes the pros and cons of different genetic testing approaches. NGS platforms and subsequent data reporting are recommended to be in accordance with the guidelines published by the European Society of Human Genetics,25 as well as by the Human Genome Variation Society (HGVS; http://www.hgvs.org, http://varnomen.hgvs.org).26, 27
In EB (sub)types with genetic heterogeneity, in cases without a clear candidate gene, where candidate genes have been ruled out, or in cases when SS was the first chosen method and did not identify the pathogenic variant, targeted NGS with the 21 known EB genes or WES with targeted filtering for EB genes is recommended (level of evidence 2++, grade of recommendation B).19, 20, 21, 22, 23, 24, 28 Subsequently, confirmation of novel pathogenic variants found this way should be performed by SS (level of evidence 4, grade of recommendation D). Recent data showed that in clinically unaffected parents, mosaicism may be detected by NGS more often than expected (depending on the coverage of the NGS platform), which has important impacts on genetic counselling.29 The advantage of targeted EB gene panels is that they obviously have a much higher coverage per gene and base. However, current WES platforms should also provide sufficient coverage per gene and base to provide accurate results, but it is recommended to confirm this in individual laboratories. The regions where coverage is not reaching recommended values (at least 95% of bases more than 20 ×) should be analysed separately by SS. Finally, the power of WES in finding new genes, as well as multigene mutations, in patients with EB has been demonstrated.12, 19, 30, 31, 32, 33
7.3.1b. Sanger sequencing (level of evidence 2++, grade of recommendation B)
Direct bidirectional SS has been the first diagnostic method for identifying the pathogenic variants in EB. All over the world, similar Sanger‐based protocols have been successful for disclosing causative pathogenic sequence variants in EB genes.11, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43 Polymerase chain reaction (PCR) products (300–600 bp in size) are generated using gene‐specific primer pairs (sequences have been published for EB genes) covering the coding regions and the exon–intron boundaries. Subsequently these are examined by SS.
Direct SS is a rapid and cost‐ and time‐effective method for (i) genetic testing of small known candidates genes; (ii) carrier identification when the family's pathogenic variant is known; (iii) prevalent founder or ethnic pathogenic variant screening, with a remarkable impact in highly consanguineous populations associated with different EB genes;34, 36, 37, 40, 44 (iv) confirming pathogenic variants identified using other genetic techniques, as recommended by The American College of Medical Genetics and Genomics (ACMG);45 and (v) PND.46, 47
7.3.2. Results and interpretation
✓ There is strong evidence that both NGS‐ and SS‐based approaches are able to identify the pathogenic variants in the majority of cases of EB (level of evidence 2++, grade of recommendation B). Independently of the pathogenic variant detection technique the interpretation of genetic findings should correlate with the clinical and skin biopsy findings. Variants should be named according to the HGVS recommendations, with the proper reference sequence mentioned (RefSeq).48 The variant name should be checked online using Mutalyzer ( https://mutalyzer.nl).
Once a sequence variant is detected, it needs to be thoroughly evaluated in order to conclude its pathogenicity. It is recommended to classify all variants according to guidelines published by the ACMG.45 In 2015, the ACMG elaborated standards and guidelines for the interpretation of sequence variants, which provide a step‐by‐step procedure for consistent variant classification (Table S3; see Supporting Information). The scoring system enables separation of variants into five classes: 1, clearly benign; 2, likely benign; 3, uncertain significance (VUS); 4, likely pathogenic and 5, clearly pathogenic. This is based on the frequency in the population, testing of the probands’ parents, in silico predictive bioinformatic tools (Table S4; see Supporting Information), cosegregation with disease in more than one pedigree, and functional experimental evidence for the consequences at the mRNA and protein levels.
Genetic testing could identify one or more variants previously reported as ‘pathogenic’ in databases such as the Human Gene Mutation Database ( http://www.hgmd.cf.ac.uk), ClinVar ( http://www.ncbi.nlm.nih.gov/clinvar) and/or disease‐specific or locus‐specific databases (e.g. http://www.deb-central.org,38, 49 http://www.interfil.org,50 https://www.lovd.nl).51 In these cases interpretation is relatively straightforward. A clear positive result will be considered when the pathogenic variant(s) cosegregate(s) with the disease following an autosomal recessive or autosomal dominant inheritance pattern, as confirmed in the parents and/or other available family members.
✓ To prevent confusion about inheritance pattern definitions, in particular when the patient is the first affected in the family, it is recommended to test both parents for carrier status. Transmission of the information on the pathogenic variant and genetic counselling to the patient and parents should be done by an expert, and preferably by the combination of a clinical geneticist and dermatologist, according to national regulations (level of evidence 4, grade of recommendation D).
If a VUS is detected, interpretation of the results requires segregation analysis, predictive bioinformatics and additional analyses at the mRNA and protein levels. As more variants in EB genes are being identified, their disease‐causing roles must be interpreted in a clinical context or, if possible, by gene expression and functional studies. In such situations IFM provides valuable information on the consequences of genetic variants at the protein level, and biomaterial for further studies. In a research setting, new variants potentially affecting splicing should be confirmed for their consequences at the mRNA level. RNA‐Seq has been proved to be a reliable tool for identification of splicing errors.52 Finally, homozygosity mapping provides a tool for screening and evaluating homozygous recessive VUS in consanguineous families.53, 54, 55
7.3.3. Limitations and uncertainty
One of the major disadvantages of SS is that the preselection of a candidate gene is mandatory (Table 4). Even though this should be the scenario, a percentage of cases are not resolved by SS in most EB subtypes, for example up to 25% in EBS.56 SS is unable to detect large insertion and deletion variants, deep intronic or regulatory pathogenic variants located in uncovered regions and/or variants at low levels of mosaicism; these are frequently the reasons that no variant is found.34, 35, 41, 57, 58, 59, 60, 61 The use of complementary phylogenetic analyses and other genetic techniques has been the classical approach to circumvent these limitations of SS. Digenic inheritance33, 38, 39 and a growing number of EB‐causative genes need to be added to the genetic complexity of EB, and these will be missed by single‐gene SS. Finally, postzygotic (somatic) mosaicism for a de novo pathogenic variant may remain undetected with SS, and requires NGS with higher read coverage.
When no pathogenic variant is found with SS or targeted NGS, the diagnosis should be re‐evaluated. If SS and NGS do not detect the disease‐causing variant in a strong candidate gene (suggested by either clinical, IFM or TEM findings), additional techniques should be exploited. These are multiplex ligation‐dependent probe amplification, reverse‐transcriptase PCR, quantitative real‐time PCR, RNA‐Seq, single‐nucleotide polymorphism arrays or Western blotting, to gain evidence of larger rearrangements, splicing alterations, chromosomal rearrangements or gene expression alterations59 (level of evidence 2++, grade of recommendation B).
Open‐exome analysis (WES without targeted filters), in which a clinical exome could be the first step in screening all other disease‐associated genes (preferably in trio with the parents’ DNA), may be done in cases where candidates have been ruled out with the techniques described above (level of evidence 2+, grade of recommendation C).
Especially with the unbiased, hypothesis‐free WES, whole‐genome sequencing and RNA‐Seq, the major challenge will be the interpretation of all available data or ‘how to locate the needle in the haystack’ (noise). This requires robust and reliable data analysis pipelines that are commercially available or can be built in‐house. The latter approach necessitates a dedicated bioinformatics division, which at present is not available to most diagnostic service labs. However, with the ongoing progression of DNA diagnostics and generation of terabytes of data per patient, it is predicted that bioinformatics will become an increasingly important specialty for the diagnosis of the very rare molecularly unsolved patients with EB. Taken together, such analyses will mostly be done in a research setting; international collaboration in these cases is recommended (level of evidence 4, grade of recommendation D).
7.4. Immunofluorescence mapping (level of evidence 2+, grade of recommendation C)
IFM, also called antigen mapping, on frozen sections is a rapid technique for EB subtype diagnosis that is also feasible in resource‐limited settings.62 Variations of this technique include using different panels of antibodies or different IHC detection methods.62, 63, 64, 65 IHC on frozen sections is possible,62 but it requires nearly the same equipment as IFM, excluding the need for a fluorescence microscope. In formalin‐fixed, paraffin‐embedded samples antigen loss is a major problem for most molecules of interest for EB diagnosis, and it is therefore not recommended. Nevertheless, a high sensitivity and specificity can be reached at very low costs using two antibodies (antikeratin 14 and antitype IV collagen) on paraffin‐embedded sections.65 Haematoxylin and eosin staining may be useful in resource‐limited situations.66
7.4.1. Biopsy samples
✓ For the diagnosis of EB by IFM, a 4–6‐mm punch or shave cutaneous biopsy sample is necessary. In general, it is recommended to take the biopsy from an area of the body that is not exposed to the sun (i.e. the inner part of the upper arm), as skin exposed to the sun may create nonspecific background fluorescence, thus interfering with the interpretation. Application of a topical anaesthetic cream before taking the biopsy may induce artificial skin cleavage.67, 68 The biopsy should include perilesional (clinically normal appearing) skin as well as a small part of a fresh blister (< 12 h) (Fig. S1a; see Supporting Information). If no fresh blister is present, a new blister can be induced by rubbing the patient's skin adjacent to a lesional area until it becomes red or blistered.69, 70, 71, 72 An alternative method to induce a new blister after taking the biopsy is by suctioning the epidermal side of the biopsy with a 20‐mL syringe until a macroscopic blister appears.73 However, the quality and reliability of this technique have not been validated in additional publications. Usually the skin of patients with EB is extremely fragile and the trauma of the biopsy may by itself lead to dermoepidermal separation.
7.4.2. Handling of biopsy samples
✓ The biopsy sample for IFM can be either snap frozen in liquid nitrogen or placed in Michel's medium74, 75 (Table S5; see Supporting Information) and stored at room temperature until use or shipment. The samples stored in this medium can be sent worldwide to any specialized laboratory.76 However, it is advisable to ship them as soon as possible to the reference laboratory, as signs of epidermal cell cytolysis have been observed after just 48 h. Samples that are frozen in Michel's medium are deemed unusable for analytical purposes. Alternatively, samples can be shipped frozen in dry ice. Shipment in sterile saline, in Dulbecco's Modified Eagle Medium or in RPMI‐1640 medium is also possible, with arrival at the EB diagnosis centre within 1–3 days, as artificial junctional cleavage and other artefacts may occur.
7.4.3. Method
✓ A series of 4–6‐μm thin cryocut sections from patient's skin and normal (healthy) human skin (NHS) samples are used for IFM (Fig. S1b; see Supporting Information). A standard IFM protocol is provided in Table S6 (see Supporting Information). Depending on the availability of primary antibodies in different countries, we recommend performing the IFM with at least one antibody for each main type of EB, as well as with an antibody for type IV collagen to determine the level of blistering64, 69, 71 (Table S7; see Supporting Information). The routine internal positive control for each antibody is the simultaneous labelling on the same slide of NHS sections: one section each from the NHS sample and the patient's sample for each secondary antibody without primary antibody is recommended as negative controls for the staining method.
7.4.4. Results and interpretation
IFM allows visualization of the cleavage level in the blister of the patient's skin relative to the protein markers used. The presence of a detectable and consistent cleavage plane within the skin allows the diagnosis of the major EB type (Tables 2 and 3; and Fig. S1c; see Supporting Information). Briefly, type IV collagen can be used as a marker to delineate the plane of cleavage, as it is never affected in EB. Staining of type IV collagen to the floor of the blister is indicative of a junctional or an intraepidermal blister, whereas staining to the roof defines a dermal blister. In EBS, the cleavage occurs in the epidermis, either within the basal cell layer or above. Irregular keratin 14 labelling surrounding unstained areas is indicative of (micro)blistering within the basal cell layer. In KS, the plane of cleavage is variable. It can be intraepidermal, junctional or dermal, or can occur at multiple levels in the same specimen. Broad reticulated staining of type IV collagen, laminin‐332 and type VII collagen can be seen in KS.
The IFM staining result of NHS is compared with the IFM pattern and staining intensity of the patient's skin. This permits assessment of the presence, absence or reduced or altered expression of different proteins analysed in the skin of the patient. Absent, or reduced or altered expression of specific antigens (desmoplakin, plakoglobin, plakophilin 1, CD151, keratin 14, plectin, BPAG1, exophilin 5, laminin‐332, type XVII collagen, integrin α6β4, integrin α3 subunit, type VII collagen) is distinctive of specific EB types or subtypes. These findings also have prognostic value, as absence of specific proteins (e.g. type XVII collagen, laminin‐332, type VII collagen) is associated with severe phenotypes, while residual expression is associated with a milder clinical course.14
Lack of blistering and/or normal expression of tested antigens can be inconclusive and preclude a diagnosis of the EB type or subtype. In such cases TEM findings, if available, can be helpful for evaluation, and genetic testing should be carried out. In specific cases, the clinical diagnosis of EB should be reconsidered.
The sensitivity and specificity of IFM have been compared with those of TEM77, 78 or evaluated in relation to clinical diagnosis in a few case series of patients with all types of suspected EB.62, 73, 79 In only two of these studies the internal reference standard was genetic diagnosis.62, 77 Of note, the only prospective study, which used genetic testing as an independent standard criterion to measure the diagnostic accuracy of each test, reported that IFM is more sensitive and specific than TEM, although the difference did not reach statistical significance due to an insufficient number of samples evaluated.77
✓ If genetic testing identifies VUS, or no pathogenic variants in EB‐associated genes are found, alterations in the immunostaining pattern and intensity may provide valuable information on the affected protein.80, 81 Moreover, in such situations, obtaining keratinocytes and/or fibroblasts from a patient's skin sample enables expression and functional studies.
7.4.5. Limitations and uncertainty
There are a few limitations of IFM applied to EB diagnosis: (i) the presence of artificial splits or protein degradation or a sample denuded of epidermis, due to inappropriate sampling, transport and storage, can be confusing; (ii) the absence of blisters in sample sections and a normal immunoreactivity to the various markers tested are frequent in cases of mild skin fragility such as with localized EBS or DEB; (iii) changes in the expression pattern and intensity may be observed with multiple markers, making interpretation difficult; (iv) using an extended IFM panel can make the test expensive, particularly in resource‐limited settings; and (v) using an IFM panel with a limited number of antibodies can lead to an erroneous interpretation of the results and inconclusive or even incorrect diagnosis.
7.5. Electron microscopy (level of evidence 2+, grade of recommendation C)
Electron microscopy led to the initial classification of EB into three major types – simplex, junctional and dystrophic – based on the precise level of tissue separation.82, 83, 84
7.5.1. Method
When a biopsy for TEM is planned, the criteria for the choice of the skin biopsy site and the method used to acquire it are the same as described for IFM.85 The skin sample should be immediately immersed in an appropriate fixative for TEM, which usually contains both glutaraldehyde and formaldehyde (e.g. Karnovsky's fixative) and is suitable for sample shipment. Subsequent processing for TEM examination comprises cutting the sample into small pieces (0·5–1 mm thick), followed by further fixation, postfixation in OsO4, dehydration, epoxy resin embedding, and semithin section preparation according to standard TEM methods. Light microscopy examination of semithin sections will permit the selection of both fields containing blistering areas and intact skin for ultrathin section preparation and examination.
7.5.2. Results and interpretation
TEM examination allows the definition of the blister level within the skin to be defined, as detailed in Tables 2 and 3. Interpretation of TEM analysis requires a deep knowledge of epithelial cell–cell and cell–matrix adhesion structures and their appearance in normal and EB skin.
Under skin ultrastructure, the cleavage occurs (i) within the epidermis in EBS, (ii) at the level of the lamina lucida of the cutaneous BMZ in JEB, (iii) below the lamina densa of the BMZ in DEB and (iv) at multiple levels in KS.6
Overall, the most common EBS subtypes are due to sequence variants in the KRT5 and KRT14 genes, where the cleavage is within the cytoplasm of the epidermal basal cells, usually beneath the nucleus.85 Additional specific findings in these EBS subtypes include (i) aggregation and clumping of keratin tonofilaments within the basal keratinocytes, regularly detected in both lesional and perilesional skin in EBS generalized severe86 and in some cases of EBS with mottled pigmentation,87 and (ii) lack of keratin tonofilaments in basal keratinocytes in recessive EBS due to KRT14 sequence variants.88, 89 The ultrastructural characteristics of rare EBS subtypes are described in Tables 2 and 3.
Separation is through the lamina lucida of the BMZ in JEB subtypes due to pathogenic variants in the genes encoding laminin‐332, α6β4 integrin or type XVII collagen.85, 90, 91, 92, 93, 94, 95 Of note, pathogenic variants in type XVII collagen and the β4 integrin subunit can exceptionally be associated with intraepidermal cleavage,96, 97 and the split is usually undetectable in JEB–laryngo‐onycho‐cutaneous syndrome.98 JEB hemidesmosomes are usually hypoplastic and reduced in number, although they can appear normal in both structure and number, particularly in mild cases of JEB due to laminin‐332 or type XVII collagen gene pathogenic variants.90, 92, 93, 94, 95, 99, 100
RDEB generalized severe shows rudimentary or absent anchoring fibrils, in addition to subepidermal blistering.85, 101 Variably hypoplastic anchoring fibrils are usually observed also in the other RDEBs, and in dominant DEB subtypes. Finally, bullous dermolysis of the newborn is characterized by the presence of pathognomonic membrane‐bound inclusions containing amorphous material and rod‐like structures, named stellate bodies, in basal keratinocytes.102, 103, 104
The cleavage plane in KS may vary, being located either within the epidermis or the lamina lucida or beneath the lamina densa, with multiple separation levels frequently visible in the same specimen. Other characteristic findings include extensive reduplications of the lamina densa.105
As discussed earlier regarding the limitations of IFM and TEM, when neither blistering nor any typical finding can be detected by both IFM and TEM examination, genetic testing should be performed.
✓ TEM is presently still an important method for the early diagnosis of a limited number of EB subtypes, in particular EBS generalized severe, autosomal recessive EBS caused by EXPH5 or DST1, and, possibly, bullous dermolysis of the newborn. In these subtypes, IFM may not be able to provide a clear result. Thus, the early detection of specific ultrastructural features has direct prognostic and management implications.
✓ In cases in which VUS or no clear pathogenic variants are identified by genetic testing, TEM may provide valuable information on the underlying ultrastructural anomalies.59
7.5.3. Limitations and uncertainty
TEM is a more expensive, labour‐intensive and time‐consuming method than IFM. It requires both highly skilled technical work for specimen processing and preparation, and specific expertise for their observation and interpretation. Thus, TEM for EB diagnosis is performed at a limited number of centres. In addition, in several EB subtypes there are no specific ultrastructural findings and TEM does not allow direct identification and quantification of the defective protein. When blistering or adhesion structure abnormalities are not present or detectable in a TEM specimen, such as with localized EBS or very mild DEB subtypes, TEM findings can be inconclusive. Finally, the determination of subtle abnormalities of epithelial adhesion structures can require morphometric analysis, which is not feasible in a routine diagnostic setting.
7.6. Reporting scenarios
✓ When issuing the report, it should include as much patient information as possible, as it is often the case that the patient with EB is under the care of different medical professionals in diverse locations or facilities. The reason for referral should be restated, which at least specifies the type of test that was requested, for example diagnostic, carrier or prenatal test. Reference to the laboratory tests carried out must include brief mention of the method(s) used and details of what was tested. According to the settings in different EB diagnostic centres, the report can be issued by a laboratory scientist or consultant dermatologist, or sometimes by both. The report should be sent only to the referral physician, and the responsibility of the staff involved in the reporting should be clearly indicated.
7.6.1. Report for genetic testing
7.6.1a. Genetic testing in an affected individual (index case)
✓ The genetic testing report must provide a full and clear interpretation of the results, as the report may be read by a variety of professionals involved in the care of the patient, many of whom may not be familiar with genotyping results. It is also recommended to use HGVS nomenclature. For point sequence variants, the sequence change should be stated at the DNA level (assuming it has been characterized in DNA), and as predicted in the protein. Also for clarity it is useful to state in words what the change is, and its predicted effect. In addition, the paternal or maternal origin of the sequence variant, or its de novo occurrence, should be specified. In all mutation reports, it is essential to quote the accession number of the gene reference sequence that was used in classifying the mutation. When reporting a deletion or duplication, the report must clearly convey whether the end points of a deletion or duplication have been determined (this is not always possible in a routine diagnostic setting). The report should always mention that genetic consultation is recommended or legally obligated in most countries, and that screening for relatives is possible. Whenever appropriate, carrier risk and risk for having affected offspring should be calculated.106 If no clear diagnosis can be made from the evidence available this must be clearly stated in the report (ACMG guidelines).
✓ The report must state that the presence of the pathogenic variant confirms, or is consistent with, the diagnosis. The report must also clearly indicate whether the variants in the gene have previously been reported to cause the disease. For the report of VUS, the variant(s) may need to be discussed in relation to the variant database such as GnomAD, ExAc or ClinVar. For a negative result, the report should clearly indicate that ‘no clear pathogenic sequence variant was detected’ and conclude that ‘the clinical diagnosis of EB has not been explained at the molecular genetic level in this patient’. The report should also state the limitations of the technique, as well as the limitations in current understanding of the clinical manifestations of the disease. Depending on the local situation, the report may offer carrier testing and/or prenatal testing to the family, and/or suggest referral for genetic counselling.
7.6.1b. Carrier testing
✓ If the pathogenic variant is confirmed, the report should clearly state that the variant found in this individual is identical to the variant determined from the index case in the family, and therefore this individual is a carrier for this variant. Suggestion for genetic counselling and PND can also be made at this stage. For predictive diagnosis for a partner of a carrier, complete coverage can be achieved by diagnosis with an NGS EB gene panel; nevertheless, predictive genetic testing depends on national regulations and usually requires genetic counselling in advance.
7.6.1c. Prenatal diagnostic report
✓ The implication of the pathogenic variant present or absent in the PND must be clearly stated, and whether the fetus is clinically affected or unaffected must also be clearly stated. According to the local regulation, it should be stated whether the fetus is a carrier or not. The test results for maternal contamination also need to be clearly stated to confirm the validity of the result further. According to local regulation, the sex of the fetus can be indicated in the report if the person who requests the report feels such information might be of interest.
In addition to the aforementioned details of a report, when NGS analysis has been performed, the report must also include a list of genes tested within a particular panel. All variants reported need to be annotated according to HGVS nomenclature. The transcript being used to provide the c. and p. nomenclature and exon numbering should be provided in the report.
7.6.2. Report for IFM
✓ The report for IFM should include a list of the primary antibodies used, the expected staining pattern and the strength of signal observed in normal control skin for each antibody, compared with the staining pattern and signal strength in patient skin. A clear conclusion should be made from these observations when the result is conclusive. However, IFM may sometimes lead to an unclear or inconclusive result such as ‘no significant difference has been observed between normal control skin and patient skin, and no blister formation’. In these cases the report should suggest the possibility of further tests, or a differential diagnosis.
7.6.3. Report for TEM
✓ The report for TEM is usually done only with patient skin. This should include a description of the semithin section finding. The report of ultrastructural findings should concern the entire epidermis, from the horny layer to basal keratinocytes, and all the structural components of the cutaneous BMZ, from the hemidesmosomes with tonofilament attachment to anchoring filaments and anchoring fibrils. The conclusion should include whether the patient is affected with EB; if so which type and, if defined, which subtype; and what would be the next test in order to reach a final confirmation. Alternatively, the report should specify that the results are not conclusive and list further tests to be performed.
8. Recommendations for laboratory diagnosis of epidermolysis bullosa
Table 1 summarizes the recommendations for laboratory diagnosis of EB, with the levels of evidence and grades of recommendations based on the appraised literature.
9. How does the guideline work in practice
As this guideline is intended for international use, it is not possible to formulate a strategy for its implementation in all clinical centres. However, the activities of DEBRA International will aid in the dissemination of the guidelines and facilitate adoption by the proposed user groups. These guidelines will be translated into other languages, and a patient version will be made to aid accessibility. DEBRA International would value feedback on the guideline so they can continue to improve its quality and impact.
Two examples of how to use the guideline in practice are given below. They illustrate the limits, advantages and complementarity of the methods, as well as the crucial role of laboratory diagnosis in EB for outcomes such as prognostication, decision making, genetic counselling and PND. An example of a scientific report for case 1 is provided in Appendix S1 (see Supporting Information), and the flowchart applied to case 2 is illustrated in Figure S2 (see Supporting Information).
9.1. Case 1
Type of referral. At disease onset, at birth.
Clinical information. Female newborn with congenital skin defects on upper and lower limbs, mechanically induced skin blisters and milia. Family history was negative, parents were not related.
IFM. A skin biopsy was performed on the second day of life and analysed by IFM with an extended panel of 18 antibodies to proteins of the BMZ.64 Result: no skin cleavage, all markers stained comparable with the normal skin. TEM was not available.
Genetic testing. Genetic testing was performed with a targeted EB gene panel.20 Result: KRT5 (NCBI RefSeq NM_000424·3) c.548T>A, p.Ile183Asn, in a heterozygous state. This is a pathogenic variant previously reported in individuals with autosomal dominant EBS39 (class 5 according to ACMG). Genetic testing by SS excluded this pathogenic variant in the parents’ DNA.
Diagnosis. EBS caused by a de novo monoallelic KRT5 pathogenic variant. Based on clinical manifestations EBS was classified as severe generalized.
Comment. IFM was performed in the first days of life. It was not conclusive but excluded severe types of JEB and DEB, and recessive EBS. TEM was not available. In the absence of a candidate gene, genetic testing was performed by a targeted EB gene panel at the age of 3 months and enabled the diagnosis of EBS due to a de novo KRT5 pathogenic variant. As the girl was still in a life‐threatening condition, the diagnosis was important for prognosis, decision making and genetic counselling for the parents.
9.2. Case 2
Type of referral. An adult female patient at the age of 38 years, genetic counselling and PND envisaged for an eventual pregnancy.
Clinical information. Skin fragility manifestations at the age of 1 year, with pretibial and feet blistering, milia, dystrophic toenails and later loss of several toenails (Fig. S2; see Supporting Information). Family history: one similarly affected sibling, parents not affected, not related and from separate geographical areas.
IFM. Skin biopsy performed at the time that the diagnosis was requested. Result: skin cleavage at the dermal level. Type VII collagen staining reduced compared with the normal skin (clone LH7·2).
Genetic testing. Genetic testing was performed by direct bidirectional SS of COL7A1. Result: two heterozygous COL7A1 (NCBI RefSeq NM_000094·3) variants were identified in both the patient and her sibling, and recessive inheritance was confirmed in the progenitors.
COL7A1: c.6527dupC; p.Gly2177Trpfs*113 in exon 80 (paternal origin).
COL7A1: c.6341G>A; p.Gly2114Asp in exon 76 (maternal origin).
Partner of case 2: noncarrier of familial variants. Noncarrier of frequently reported pathogenic variants in COL7A1 exons 5, 23–25, 57–60, 76, 80–82, 105 and 106.
The pathogenic variant c.6527dupC in exon 80107 is the most frequent detected in Spanish108, 109, 110 and Chilean111 patients with RDEB (class 5 according to ACMG).
The variant c.6341G>A in exon 76 was not previously reported, either as a pathogenic variant ( http://www.hgmd.cf.ac.uk; http://www.deb-central.org; http://www.ncbi.nlm.nih.gov/clinvar) or as a single‐nucleotide polymorphism ( http://exac.broadinstitute.org; http://www.ncbi.nlm.nih.gov/snp; http://www.ensembl.org). A variant rs1285959723 affecting the same codon is reported (c.6340G>C; p.Gly2114Arg) with a highest population minor allele frequency < 0·01 (1000 Genomes, ESP, Exact, gnomAD), for which clinical data are not available. The variant in exon 76 cosegregates with the disease in the two affected siblings and was found in three other Spanish nonrelated cases of RDEB (unpublished data of the laboratory). In silico pathogenicity was predicted by standard computational programs: PolyPhen‐2 ( http://genetics.bwh.harvard.edu/pph2: probably damaging, 0·999), Mutation taster ( http://www.mutationtaster.org; disease causing, 0·999) and SIFT ( http://sift.bii.a-star.edu.sg; affect protein function score 0·00). Moreover, pathogenicity is supported by the location of p.Gly2114 in the collagenous domain of type VII collagen, in a conserved Gly‐X‐Y repeat (class 5 according to ACMG).
Diagnosis. RDEB with reduced type VII collagen and compound heterozygous COL7A1 pathogenic variants. Based on the clinical manifestations the subtype is pretibial RDEB.
Comments. COL7A1 genetic testing was performed by direct bidirectional SS to identify pathogenic variants and enable counselling for an eventual pregnancy. Suspected dominant DEB due to a de novo pathogenic variant was discarded. Causative pathogenic variants support IFM results and clinical manifestation. The partner was also tested, and no pathogenic variant was disclosed. Genetic counselling was provided.
10. Future research
Based on the literature research and appraisal, future research is needed to address the following issues regarding EB laboratory diagnosis.
Sensitivity, time to diagnosis and costs per patient for different EB laboratory diagnostic methods.
Preimplantation genetic diagnosis in EB.
Noninvasive PND utilizing cell‐free fetal DNA in EB.
Gene‐specific databases for interpretation of sequence variants, clinical trials and precision medicine.
Inter‐ and extrafamilial variability of the phenotype: coexpression factors.
Supporting information
Appendix S1 Example of the report for laboratory diagnosis of epidermolysis bullosa in case 1.
Fig S1. Biopsy and immunofluorescence mapping for laboratory diagnosis of epidermolysis bullosa.
Fig S2. Example of how the guideline works in clinical practice.
Table S1 Proteins involved in epidermolysis bullosa.
Table S2 Comparison of genetic testing techniques that can be used for laboratory diagnosis of epidermolysis bullosa.
Table S3 Classification of sequence variants.
Table S4 The most useful websites and online bioinformatics tools.
Table S5 The formula for Michel's medium.
Table S6 Standard protocol for immunofluorescence mapping for diagnosis of epidermolysis bullosa.
Table S7 Antibodies recommended for immunofluorescence mapping in epidermolysis bullosa.
Video S1 Author video.
Acknowledgments
This guideline were initiated by DEBRA International; financial support was provided by DEBRA Austria. The generous assistance of Rebecca Bodan, Lisa Brains, Sharon Cassidy and Kelsey Townsend‐Miller is gratefully acknowledged in providing patient or lay input into this guideline. The authors acknowledge the guidance of Kattya Mayre‐Chilton (DEDRA International). Johann Bauer (Paracelsus University and EB House, Salzburg, Austria), Christine Bodemer (Hôpital Universitaire Necker, Paris, France), Judith Fischer (Institute of Human Genetics, University of Freiburg, Germany), Jemima Mellerio (St John's Institute of Dermatology, Guy's and St Thomas’ NHS Foundation Trust, London, U.K.), Francis Palisson (Universidad del Desarrollo and DEBRA, Chile), Eli Sprecher (Department of Dermatology, Tel Aviv Sourasky Medical Center, Israel) and Jouni Uitto, Leila Youssefian and Hassan Vahidnezhad (all from the Department of Dermatology and Cutaneous Biology, Thomas Jefferson University, Philadelphia, PA, U.S.A.) are acknowledged as reviewers.
Appendix 1.
1.1.
Levels of evidence
| 1++ | High‐quality meta‐analyses, systematic reviews of randomized controlled trials (RCTs), or RCTs with a very low risk of bias |
| 1+ | Well‐conducted meta‐analyses, systematic reviews, or RCTs with a low risk of bias |
| 1− | Meta‐analyses, systematic reviews, or RCTs with a high risk of bias |
| 2++ | High‐quality systematic reviews of case–control or cohort studies |
| High‐quality case–control or cohort studies with a very low risk of confounding or bias and a high probability that the relationship is causal | |
| 2+ | Well‐conducted case–control or cohort studies with a low risk of confounding or bias and a moderate probability that the relationship is causal |
| 2− | Case–control or cohort studies with a high risk of confounding or bias and a significant risk |
| 3 | Nonanalytical studies, e.g. case reports, case series |
| 4 | Expert opinion |
Grades of recommendation made by the guideline panel
| Grade | Description |
|---|---|
| A | At least one meta‐analysis, systematic review or randomized controlled trial rated as 1++, and directly applicable to the target population; or |
| A body of evidence consisting principally of studies rated as 1+, directly applicable to the target population, and demonstrating overall consistency of results | |
| B | A body of evidence including studies rated as 2++, directly applicable to the target population, and demonstrating overall consistency of results; or |
| Extrapolated evidence from studies rated as 1++ or 1+ | |
| C | A body of evidence including studies rated as 2+, directly applicable to the target population and demonstrating overall consistency of results; or |
| Extrapolated evidence from studies rated as 2++ | |
| D | Evidence level 3 or 4; or |
| Extrapolated evidence from studies rated as 2+ |
Adapted from the SIGN 50 Guideline Developer's Handbook, National Health Service Scottish Intercollegiate Guidelines Network, revised edition January 2014.
Good practice points
| ✓ Recommended best practice based on the clinical experience of the guideline development group |
Funding sources Dystrophic Epidermolysis Bullosa Research Association Austria.
Conflicts of interest None to declare.
C.H., M.C.B., M.E.H. and G.Z. represent ERN‐Skin ( http://ern-skin.eu/about-the-ern-skin)
https://doi.org/10.1111/bjd.18829 available online
References
- 1. Zeng X, Zhang Y, Kwong JSW et al The methodological quality assessment tools for preclinical and clinical studies, systematic review and meta‐analysis, and clinical practice guideline: a systematic review. J Evid Based Med 2015; 8:2–10. [DOI] [PubMed] [Google Scholar]
- 2. Health Improvement Scotland . SIGN100: a handbook for patient and carer representatives. Available at: https://www.sign.ac.uk/sign-100-a-handbook-for-patient-and-carer-representatives.html (last accessed 2 July 2019).
- 3. Has C, Fischer J. Inherited epidermolysis bullosa: new diagnostics and new clinical phenotypes. Exp Dermatol 2019; 10.1111/exd.13668. [DOI] [PubMed] [Google Scholar]
- 4. Yenamandra VK, Moss C, Sreenivas V et al Development of a clinical diagnostic matrix for characterizing inherited epidermolysis bullosa. Br J Dermatol 2017; 176:1624–32. [DOI] [PubMed] [Google Scholar]
- 5. Almaani N, Liu L, Dopping‐Hepenstal PJC et al Identical glycine substitution mutations in type VII collagen may underlie both dominant and recessive forms of dystrophic epidermolysis bullosa. Acta Derm Venereol 2011; 91:262–6. [DOI] [PubMed] [Google Scholar]
- 6. Fine JD, Bruckner‐Tuderman L, Eady RA et al Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70:1103–26. [DOI] [PubMed] [Google Scholar]
- 7. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol 2009; 61:367–84. [DOI] [PubMed] [Google Scholar]
- 8. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol 2009; 61:387–402. [DOI] [PubMed] [Google Scholar]
- 9. Vahidnezhad H, Youssefian L, Saeidian AH, Uitto J. Phenotypic spectrum of epidermolysis bullosa, the paradigm of syndromic versus non‐syndromic skin fragility disorders. J Invest Dermatol 2019; 139:522–7. [DOI] [PubMed] [Google Scholar]
- 10. Schwieger‐Briel A, Fuentes I, Castiglia D et al Epidermolysis bullosa simplex with KLHL24 mutations is associated with dilated cardiomyopathy. J Invest Dermatol 2019; 139:244–9. [DOI] [PubMed] [Google Scholar]
- 11. Has C, Spartà G, Kiritsi D et al Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med 2012; 366:1508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vahidnezhad H, Youssefian L, Saeidian AH et al Mutations in PLOD3, encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa‐like blistering phenotype with abnormal anchoring fibrils and type VII collagen deficiency. Matrix Biol 2019; 81:91–106. [DOI] [PubMed] [Google Scholar]
- 13. Vahidnezhad H, Youssefian L, Saeidian AH et al Recessive mutation in tetraspanin CD151 causes Kindler syndrome‐like epidermolysis bullosa with multi‐systemic manifestations including nephropathy. Matrix Biol 2018; 66:22–33. [DOI] [PubMed] [Google Scholar]
- 14. Has C, Bruckner‐Tuderman L. The genetics of skin fragility. Annu Rev Genomics Hum Genet 2014; 15:245–68. [DOI] [PubMed] [Google Scholar]
- 15. McGrath JA. Recently identified forms of epidermolysis bullosa. Ann Dermatol 2015; 27:658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feinstein JA, Jambal P, Peoples K et al Assessment of the timing of milestone clinical events in patients with epidermolysis bullosa from North America. JAMA Dermatol 2019; 155:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henneman L, Borry P, Chokoshvili D et al Responsible implementation of expanded carrier screening. Eur J Hum Genet 2016; 24:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klausegger A, Pulkkinen L, Pohla‐Gubo G et al Is screening of the candidate gene necessary in unrelated partners of members of families with Herlitz junctional epidermolysis bullosa? J Invest Dermatol 2001; 116:474–5. [DOI] [PubMed] [Google Scholar]
- 19. Vahidnezhad H, Youssefian L, Saeidian AH et al Multigene next‐generation sequencing panel identifies pathogenic variants in patients with unknown subtype of epidermolysis bullosa: subclassification with prognostic implications. J Invest Dermatol 2017; 137:2649–52. [DOI] [PubMed] [Google Scholar]
- 20. Has C, Küsel J, Reimer A et al The position of targeted next‐generation sequencing in epidermolysis bullosa diagnosis. Acta Derm Venereol 2018; 98:437–40. [DOI] [PubMed] [Google Scholar]
- 21. Lucky AW, Dagaonkar N, Lammers K et al A comprehensive next‐generation sequencing assay for the diagnosis of epidermolysis bullosa. Pediatr Dermatol 2018; 35:188–97. [DOI] [PubMed] [Google Scholar]
- 22. Vahidnezhad H, Youssefian L, Zeinali S et al Dystrophic epidermolysis bullosa: COL7A1 mutation landscape in a multi‐ethnic cohort of 152 extended families with high degree of customary consanguineous marriages. J Invest Dermatol 2017; 137:660–9. [DOI] [PubMed] [Google Scholar]
- 23. Takeichi T, Liu L, Fong K et al Whole‐exome sequencing improves mutation detection in a diagnostic epidermolysis bullosa laboratory. Br J Dermatol 2015; 172:94–100. [DOI] [PubMed] [Google Scholar]
- 24. Yenamandra VK, Vellarikkal SK, Kumar M et al Application of whole exome sequencing in elucidating the phenotype and genotype spectrum of junctional epidermolysis bullosa: a preliminary experience of a tertiary care centre in India. J Dermatol Sci 2017; 86:30–6. [DOI] [PubMed] [Google Scholar]
- 25. Matthijs G, Souche E, Alders M et al Guidelines for diagnostic next‐generation sequencing. Eur J Hum Genet 2016; 24:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rehm HL, Berg JS, Plon SE. ClinGen and ClinVar – enabling genomics in precision medicine. Hum Mutat 2018; 39:1473–5. [Google Scholar]
- 27. Landrum MJ, Kattman BL. ClinVar at five years: delivering on the promise. Hum Mutat 2018; 39:1623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tenedini E, Artuso L, Bernardis I et al Amplicon‐based next‐generation sequencing: an effective approach for the molecular diagnosis of epidermolysis bullosa. Br J Dermatol 2015; 173:731–8. [DOI] [PubMed] [Google Scholar]
- 29. Jónsson H, Sulem P, Arnadottir GA et al Multiple transmissions of de novo mutations in families. Nat Genet 2018; 50:1674–80. [DOI] [PubMed] [Google Scholar]
- 30. Lin Z, Li S, Feng C et al Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat Genet 2016; 48:1508–16. [DOI] [PubMed] [Google Scholar]
- 31. McGrath JA, Stone KL, Begum R et al Germline mutation in EXPH5 implicates the Rab27B effector protein Slac2‐b in inherited skin fragility. Am J Hum Genet 2012; 91:1115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Y, Maier K, Leppert J et al Monoallelic mutations in the translation initiation codon of KLHL24 cause skin fragility. Am J Hum Genet 2016; 99:1395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vahidnezhad H, Youssefian L, Saeidian AH et al Next generation sequencing identifies double homozygous mutations in two distinct genes (EXPH5 and COL17A1) in a patient with concomitant simplex and junctional epidermolysis bullosa. Hum Mutat 2018; 39:1349–54. [DOI] [PubMed] [Google Scholar]
- 34. Mayer B, Silló P, Mazán M et al A unique LAMB3 splice‐site mutation with founder effect from the Balkans causes lethal epidermolysis bullosa in several European countries. Br J Dermatol 2016; 175:721–7. [DOI] [PubMed] [Google Scholar]
- 35. Youssefian L, Vahidnezhad H, Barzegar M et al The Kindler syndrome: a spectrum of FERMT1 mutations in Iranian families. J Invest Dermatol 2015; 135:1447–50. [DOI] [PubMed] [Google Scholar]
- 36. Takeichi T, Nanda A, Liu L et al Founder mutation in dystonin‐e underlying autosomal recessive epidermolysis bullosa simplex in Kuwait. Br J Dermatol 2015; 172:527–31. [DOI] [PubMed] [Google Scholar]
- 37. Szczecinska W, Nesteruk D, Wertheim‐Tysarowska K et al Under‐recognition of acral peeling skin syndrome: 59 new cases with 15 novel mutations. Br J Dermatol 2014; 171:1206–10. [DOI] [PubMed] [Google Scholar]
- 38. Wertheim‐Tysarowska K, Ołdak M, Giza A et al Novel sporadic and recurrent mutations in KRT5 and KRT14 genes in Polish epidermolysis bullosa simplex patients: further insights into epidemiology and genotype–phenotype correlation. J Appl Genet 2016; 57:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim EN, Harris AG, Bingham LJ et al A review of 52 pedigrees with epidermolysis bullosa simplex identifying ten novel mutations in KRT5 and KRT14 in Australia. Acta Derm Venereol 2017; 97:1114–19. [DOI] [PubMed] [Google Scholar]
- 40. Ben BA, Laroussi N, Mesrati H et al Genetic basis of dominant dystrophic epidermolysis bullosa in Tunisian families and co‐occurrence of dominant and recessive mutations. J Eur Acad Dermatol Venereol 2016; 30:155–7. [DOI] [PubMed] [Google Scholar]
- 41. Turczynski S, Titeux M, Pironon N et al Marked intrafamilial phenotypic heterogeneity in dystrophic epidermolysis bullosa caused by inheritance of a mild dominant glycine substitution and a novel deep intronic recessive COL7A1 mutation. Br J Dermatol 2016; 174:1122–5. [DOI] [PubMed] [Google Scholar]
- 42. Bolling MC, Jongbloed JDH, Boven LG et al Plectin mutations underlie epidermolysis bullosa simplex in 8% of patients. J Invest Dermatol 2014; 134:273–6. [DOI] [PubMed] [Google Scholar]
- 43. Kyrova J, Kopeckova L, Buckova H et al Epidermolysis bullosa simplex with muscular dystrophy. Review of the literature and a case report. J Dermatol Case Rep 2016; 10:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuentes I, Campos M, Repetto G et al Molecular epidemiology of junctional epidermolysis bullosa: discovery of novel and frequent LAMB3 mutations in Chilean patients with diagnostic significance. Br J Dermatol 2017; 176:1090–2. [DOI] [PubMed] [Google Scholar]
- 45. Richards S, Aziz N, Bale S et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pfendner EG, Nakanol A, Pulkkinen L et al Prenatal diagnosis for epidermolysis bullosa: a study of 144 consecutive pregnancies at risk. Prenat Diagn 2003; 23:447–56. [DOI] [PubMed] [Google Scholar]
- 47. Fassihi H, Eady RAJ, Mellerio JE et al Prenatal diagnosis for severe inherited skin disorders: 25 years’ experience. Br J Dermatol 2006; 154:106–13. [DOI] [PubMed] [Google Scholar]
- 48. den Dunnen JT, Dalgleish R, Maglott DR et al HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 2016; 37:564–9. [DOI] [PubMed] [Google Scholar]
- 49. Van den Akker PC, Jonkman MF, Rengaw T et al The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat 2011; 32:1100–7. [DOI] [PubMed] [Google Scholar]
- 50. Szeverenyi I, Cassidy AJ, Cheuk WC et al The human intermediate filament database: comprehensive information on a gene family involved in many human diseases. Hum Mutat 2008; 29:351–60. [DOI] [PubMed] [Google Scholar]
- 51. Has C, Castiglia D, del Rio M et al Kindler syndrome: extension of FERMT1 mutational spectrum and natural history. Hum Mutat 2011; 32:1204–12. [DOI] [PubMed] [Google Scholar]
- 52. Komori T, Dainichi T, Otsuka A et al Mild dystrophic epidermolysis bullosa associated with homozygous gene mutation c.6216+5G>T in type VII collagen ultrastructurally suggestive of the decreased number of anchoring fibrils. J Dermatol 2018; 45:e305–6. [DOI] [PubMed] [Google Scholar]
- 53. Vahidnezhad H, Youssefian L, Jazayeri A, Uitto J. Research techniques made simple: genome‐wide homozygosity/autozygosity mapping is a powerful tool for identifying candidate genes in autosomal recessive genetic diseases. J Invest Dermatol 2018; 138:1893–900. [DOI] [PubMed] [Google Scholar]
- 54. Vahidnezhad H, Youssefian L, Saeidian AH et al Genome‐wide single nucleotide polymorphism‐based autozygosity mapping facilitates identification of mutations in consanguineous families with epidermolysis bullosa. Exp Dermatol 2019; 10.1111/exd.13501. [DOI] [PubMed] [Google Scholar]
- 55. Mizrachi‐Koren M, Shemer S, Morgan M et al Homozygosity mapping as a screening tool for the molecular diagnosis of hereditary skin diseases in consanguineous populations. J Am Acad Dermatol 2006; 55:393–401. [DOI] [PubMed] [Google Scholar]
- 56. Bolling MC, Lemmink HH, Jansen GHL, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol 2011; 164:637–44. [DOI] [PubMed] [Google Scholar]
- 57. Chmel N, Danescu S, Gruler A et al a deep‐intronic FERMT1 mutation causes Kindler syndrome: an explanation for genetically unsolved cases. J Invest Dermatol 2015; 135:2876–9. [DOI] [PubMed] [Google Scholar]
- 58. Has C, Schumann H, Leppert J et al Monoallelic large intragenic KRT5 deletions account for genetically unsolved cases of epidermolysis bullosa simplex. J Invest Dermatol 2017; 137:2231–4. [DOI] [PubMed] [Google Scholar]
- 59. Chmel N, Bornert O, Hausser I et al Large deletions targeting the triple‐helical domain of collagen VII lead to mild acral dominant dystrophic epidermolysis bullosa. J Invest Dermatol 2018; 138:987–91. [DOI] [PubMed] [Google Scholar]
- 60. Fuchs‐Telem D, Nousbeck J, Singer A et al New intragenic and promoter region deletion mutations in FERMT1 underscore genetic homogeneity in Kindler syndrome. Clin Exp Dermatol 2014; 39:361–7. [DOI] [PubMed] [Google Scholar]
- 61. Gostyńska KB, Nijenhuis M, Lemmink H et al Mutation in exon 1a of PLEC, leading to disruption of plectin isoform 1a, causes autosomal‐recessive skin‐only epidermolysis bullosa simplex. Hum Mol Genet 2015; 24:3155–62. [DOI] [PubMed] [Google Scholar]
- 62. Yenamandra VK, Bhari N, Ray SB et al Diagnosis of inherited epidermolysis bullosa in resource‐limited settings: immunohistochemistry revisited. Dermatology 2017; 233:326–32. [DOI] [PubMed] [Google Scholar]
- 63. Leclerc‐Mercier S, Fraitag S. Histopathological diagnosis of inherited epidermolysis bullosa. Ann Dermatol Venereol 2011; 138:782–7. [DOI] [PubMed] [Google Scholar]
- 64. Has C, He Y. Research techniques made simple: immunofluorescence antigen mapping in epidermolysis bullosa. J Invest Dermatol 2016; 136:e65–71. [DOI] [PubMed] [Google Scholar]
- 65. Petronius D, Bergman R, Ben Izhak O et al A comparative study of immunohistochemistry and electron microscopy used in the diagnosis of epidermolysis bullosa. Am J Dermatopathol 2003; 25:198–203. [DOI] [PubMed] [Google Scholar]
- 66. Bergman R, Harel A, Sprecher E. Dyskeratosis as a histologic feature in epidermolysis bullosa simplex‐Dowling Meara. J Am Acad Dermatol 2007; 57:463–6. [DOI] [PubMed] [Google Scholar]
- 67. Schmutz JL, Trechot P. Note the irritant effect of Emla® cream potentially leading to diagnostic errors. Ann Dermatol Venereol 2012; 139:82–3. [DOI] [PubMed] [Google Scholar]
- 68. Kieliszak CR, Griffin JR, Pollinger TH, Junkins‐Hopkins JM. Pseudo‐bullous dermatosis induced by topical anesthetic agent – clues to this localized toxic reaction. Am J Dermatopathol 2017; 39:e19–22. [DOI] [PubMed] [Google Scholar]
- 69. Pohla‐Gubo G, Nischler E, Hintner H. Antigen mapping In: Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Care and Therapy (Fine JD, Hintner H, eds). New York: Springer, 2009; 35–42. [Google Scholar]
- 70. Intong LRA, Murrell DF. How to take skin biopsies for epidermolysis bullosa. Dermatol Clin 2010; 28:197–200. [DOI] [PubMed] [Google Scholar]
- 71. Pohla‐Gubo G, Cepeda‐Valdes R, Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa. Dermatol Clin 2010; 28:201–10. [DOI] [PubMed] [Google Scholar]
- 72. Pohla‐Gubo G, Kraus L, Hintner H. Role of immunofluorescence microscopy in dermatology. G Ital Dermatol Venereol 2011; 146:127–42. [PubMed] [Google Scholar]
- 73. Barzegar M, Asadi‐Kani Z, Mozafari N et al Using immunofluorescence (antigen) mapping in the diagnosis and classification of epidermolysis bullosa: a first report from Iran. Int J Dermatol 2015; 54:e416–23. [DOI] [PubMed] [Google Scholar]
- 74. Michel B, Milner Y, David K. Preservation of tissue‐fixed immunoglobulins in skin biopsies of patients with lupus erythematosus and bullous diseases. J Invest Dermatol 1973; 59:449–52. [DOI] [PubMed] [Google Scholar]
- 75. Vaughan Jones SA, Palmer I, Bhogal BS et al The use of Michel's transport medium for immunofluorescence and immunoelectron microscopy in autoimmune bullous diseases. J Cutan Pathol 1995; 22:365–70. [DOI] [PubMed] [Google Scholar]
- 76. Cepeda‐Valdés R, Pohla‐Gubo G, Borbolla‐Escoboza JR et al Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa. Actas Dermosifiliogr 2010; 101:673–82. [PubMed] [Google Scholar]
- 77. Yiasemides E, Walton J, Marr P et al A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. Am J Dermatopathol 2006; 28:387–94. [DOI] [PubMed] [Google Scholar]
- 78. Berk DR, Jazayeri L, Marinkovich MP et al Diagnosing epidermolysis bullosa type and subtype in infancy using immunofluorescence microscopy: the Stanford experience. Pediatr Dermatol 2013; 30:226–33. [DOI] [PubMed] [Google Scholar]
- 79. Hiremagalore R, Kubba A, Bansel S, Jerajani H. Immunofluorescence mapping in inherited epidermolysis bullosa: a study of 86 cases from India. Br J Dermatol 2015; 172:384–91. [DOI] [PubMed] [Google Scholar]
- 80. He Y, Balasubramanian M, Humphreys N et al Intronic ITGA3 mutation impacts splicing regulation and causes interstitial lung disease, nephrotic syndrome, and epidermolysis bullosa. J Invest Dermatol 2016; 136:1056–9. [DOI] [PubMed] [Google Scholar]
- 81. Yalcin EG, He Y, Orhan D et al Crucial role of posttranslational modifications of integrin α3 in interstitial lung disease and nephrotic syndrome. Hum Mol Genet 2015; 24:3679–88. [DOI] [PubMed] [Google Scholar]
- 82. Pearson RW. Studies on the pathogenesis of epidermolysis bullosa. J Invest Dermatol 1962; 39:551–75. [DOI] [PubMed] [Google Scholar]
- 83. Hashimoto I, Anton‐Lamprecht I, Gedde‐Dahl T, Schnyder UW. Ultrastructural studies in epidermolysis bullosa heriditaria. I. Dominant dystrophic type of Pasini. Arch Dermatol Res 1975; 252:167–78. [PubMed] [Google Scholar]
- 84. Rodeck C, Eady RA, Gosden C. Prenatal diagnosis of epidermolysis bullosa letalis. Lancet 1980; 1:949–52. [DOI] [PubMed] [Google Scholar]
- 85. Eady RA, Dopping‐Hepenstal PJ. Transmission electron microscopy for the diagnosis of epidermolysis bullosa. Dermatol Clin 2010; 28:211–22. [DOI] [PubMed] [Google Scholar]
- 86. McGrath JA, Ishida‐Yamamoto A, Tidman MJ et al Epidermolysis bullosa simplex (Dowling‐Meara). A clinicopathological review. Br J Dermatol 1992; 126:421–30. [DOI] [PubMed] [Google Scholar]
- 87. Irvine AD, Rugg EL, Lane EB et al Molecular confirmation of the unique phenotype of epidermolysis bullosa simplex with mottled pigmentation. Br J Dermatol 2001; 144:40–5. [DOI] [PubMed] [Google Scholar]
- 88. Rugg E, McLean W, Lane E et al A functional ‘knockout’ of human keratin 14. Genes Dev 1994; 8:2563–73. [DOI] [PubMed] [Google Scholar]
- 89. Chan YM, Anton‐Lamprecht I, Yu QC et al A human keratin 14 ‘knockout’: the absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev 1994; 8:2574–87. [DOI] [PubMed] [Google Scholar]
- 90. Tidman MJ, Eady RA. Hemidesmosome heterogeneity in junctional epidermolysis bullosa revealed by morphometric analysis. J Invest Dermatol 1986; 86:51–6. [DOI] [PubMed] [Google Scholar]
- 91. McGrath JA, Gatalica B, Christiano AM et al Mutations in the 180‐kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet 1995; 11:83–6. [DOI] [PubMed] [Google Scholar]
- 92. Nakano A, Chao SC, Pulkkinen L et al Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz versus non‐Herlitz phenotypes. Hum Genet 2002; 110:41–51. [DOI] [PubMed] [Google Scholar]
- 93. Yuen WY, Lemmink HH, Van Dijk‐Bos KK et al Herlitz junctional epidermolysis bullosa: diagnostic features, mutational profile, incidence and population carrier frequency in the Netherlands. Br J Dermatol 2011; 165:1314–22. [DOI] [PubMed] [Google Scholar]
- 94. Nakano A, Pulkkinen L, Murrell D et al Epidermolysis bullosa with congenital pyloric atresia: novel mutations in the β4 integrin gene (ITGB4) and genotype/phenotype correlations. Pediatr Res 2001; 49:618–26. [DOI] [PubMed] [Google Scholar]
- 95. Ruzzi L, Gagnoux‐Palacios L, Pinola M et al A homozygous mutation in the integrin α6 gene in junctional epidermolysis bullosa with pyloric atresia. J Clin Invest 1997; 99:2826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jonkman MF, Pas HH, Nijenhuis M et al Deletion of a cytoplasmic domain of integrin β4 causes epidermolysis bullosa simplex. J Invest Dermatol 2002; 119:1275–81. [DOI] [PubMed] [Google Scholar]
- 97. Huber M, Frenk E, Hohl D et al Deletion of the cytoplasmatic domain of BP180/collagen XVII causes a phenotype with predominant features of epidermolysis bullosa simplex. J Invest Dermatol 2002; 118:185–92. [DOI] [PubMed] [Google Scholar]
- 98. McLean WI, Irvine AD, Hamill KJ et al An unusual N‐terminal deletion of the laminin α3a isoform leads to the chronic granulation tissue disorder laryngo‐onycho‐cutaneous syndrome. Hum Mol Genet 2003; 12:2395–409. [DOI] [PubMed] [Google Scholar]
- 99. Posteraro P, De Luca N, Meneguzzi G et al Laminin‐5 mutational analysis in an Italian cohort of patients with junctional epidermolysis bullosa. J Invest Dermatol 2004; 123:639–48. [DOI] [PubMed] [Google Scholar]
- 100. Yuen WY, Pas HH, Sinke RJ, Jonkman MF. Junctional epidermolysis bullosa of late onset explained by mutations in COL17A1 . Br J Dermatol 2011; 164:1280–4. [DOI] [PubMed] [Google Scholar]
- 101. Tidman MJ, Eady RA. Evaluation of anchoring fibrils and other components of the dermal‐epidermal junction in dystrophic epidermolysis bullosa by a quantitative ultrastructural technique. J Invest Dermatol 1985; 84:374–7. [DOI] [PubMed] [Google Scholar]
- 102. Hashimoto K, Matsumoto M, Iacobelli D. Transient bullous dermolysis of the newborn. Arch Dermatol 1985; 121:1429–38. [PubMed] [Google Scholar]
- 103. Diociaiuti A, Castiglia D, Giancristoforo S et al Frequent occurrence of aplasia cutis congenita in bullous dermolysis of the newborn. Acta Derm Venereol 2016; 96:784–7. [DOI] [PubMed] [Google Scholar]
- 104. Heinecke G, Marinkovich MP, Rieger KE. Intraepidermal type VII collagen by immunofluorescence mapping: a specific finding for bullous dermolysis of the newborn. Pediatr Dermatol 2017; 34:308–14. [DOI] [PubMed] [Google Scholar]
- 105. Shimizu H, Sato M, Ban M et al Immunohistochemical, ultrastructural, and molecular features of Kindler syndrome distinguish it from dystrophic epidermolysis bullosa. Arch Dermatol 1997; 133:1111–17. [PubMed] [Google Scholar]
- 106. Claustres M, Kožich V, Dequeker E et al Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular genetic). Eur J Hum Genet 2014; 22:160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hovnanian A, Rochat A, Bodemer C et al Characterization of 18 new mutations in COL7A1 in recessive dystrophic epidermolysis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am J Hum Genet 1997; 61:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Escámez MJ, García M, Cuadrado‐Corrales N et al The first COL7A1 mutation survey in a large Spanish dystrophic epidermolysis bullosa cohort: c.6527insC disclosed as an unusually recurrent mutation. Br J Dermatol 2010; 163:155–61. [DOI] [PubMed] [Google Scholar]
- 109. Cuadrado‐Corrales N, Sánchez‐Jimeno C, García M et al A prevalent mutation with founder effect in Spanish recessive dystrophic epidermolysis bullosa families. BMC Med Genet 2010; 11:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sanchez‐Jimeno C, Cuadrado‐Corrales N, Aller E et al Recessive dystrophic epidermolysis bullosa: the origin of the c.6527insC mutation in the Spanish population. Br J Dermatol 2013; 168:226–9. [DOI] [PubMed] [Google Scholar]
- 111. Rodríguez FA, Gana MJ, Yubero MJ et al Novel and recurrent COL7A1 mutations in Chilean patients with dystrophic epidermolysis bullosa. J Dermatol Sci 2012; 65:149–52. [DOI] [PubMed] [Google Scholar]
- 112. Hammersen J, Has C, Naumann‐Bartsch N et al Genotype, clinical course, and therapeutic decision making in 76 infants with severe generalized junctional epidermolysis bullosa. J Invest Dermatol 2016; 136:2150–7. [DOI] [PubMed] [Google Scholar]
- 113. Schwieger‐Briel A, Weibel L, Chmel N et al A COL7A1 variant leading to in‐frame skipping of exon 15 attenuates disease severity in recessive dystrophic epidermolysis bullosa. Br J Dermatol 2015; 173:1308–11. [DOI] [PubMed] [Google Scholar]
- 114. Shipman AR, Liu L, Lai‐Cheong JE et al Somatic forward (nonrevertant) mosaicism in recessive dystrophic epidermolysis bullosa. JAMA Dermatol 2014; 150:1025–7. [DOI] [PubMed] [Google Scholar]
- 115. Cserhalmi‐Friedman PB, Garzon MC, Guzman E et al Maternal germline mosaicism in dominant dystrophic epidermolysis bullosa. J Invest Dermatol 2001; 117:1327–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Example of the report for laboratory diagnosis of epidermolysis bullosa in case 1.
Fig S1. Biopsy and immunofluorescence mapping for laboratory diagnosis of epidermolysis bullosa.
Fig S2. Example of how the guideline works in clinical practice.
Table S1 Proteins involved in epidermolysis bullosa.
Table S2 Comparison of genetic testing techniques that can be used for laboratory diagnosis of epidermolysis bullosa.
Table S3 Classification of sequence variants.
Table S4 The most useful websites and online bioinformatics tools.
Table S5 The formula for Michel's medium.
Table S6 Standard protocol for immunofluorescence mapping for diagnosis of epidermolysis bullosa.
Table S7 Antibodies recommended for immunofluorescence mapping in epidermolysis bullosa.
Video S1 Author video.