

ABSTRACT
Loss of inner ear hair cells leads to incurable balance and hearing disorders because these sensory cells do not effectively regenerate in humans. A potential starting point for therapy would be the stimulation of quiescent progenitor cells within the damaged inner ear. Inner ear progenitor/stem cells, which have been described in rodent inner ears, would be principal candidates for such an approach. Despite the identification of progenitor cell populations in the human fetal cochlea and in the adult human spiral ganglion, no proliferative cell populations with the capacity to generate hair cells have been reported in vestibular and cochlear tissues of adult humans. The present study aimed at filling this gap by isolating colony‐forming progenitor cells from surgery‐ and autopsy‐derived adult human temporal bones in order to generate inner ear cell types in vitro. Sphere‐forming and mitogen‐responding progenitor cells were isolated from vestibular and cochlear tissues. Clonal spheres grown from adult human utricle and cochlear duct were propagated for a limited number of generations. When differentiated in absence of mitogens, the utricle‐derived spheres robustly gave rise to hair cell‐like cells, as well as to cells expressing supporting cell‐, neuron‐, and glial markers, indicating that the adult human utricle harbors multipotent progenitor cells. Spheres derived from the adult human cochlear duct did not give rise to hair cell‐like or neuronal cell types, which is an indication that human cochlear cells have limited proliferative potential but lack the ability to differentiate into major inner ear cell types. Anat Rec, 303:461–470, 2020. © 2019 The Authors. The Anatomical Record published by Wiley Periodicals, Inc. on behalf of American Association of Anatomists.
Keywords: progenitor cell, inner ear, utricle, organ of Corti, regeneration, hair cell
Loss of cochlear and vestibular hair cells leads to hearing loss and balance problems in humans. The main reason for the permanence of these chronic disorders is the fact that sensory hair cells do not spontaneously regenerate and that the limited regeneration observed in the vestibular system is not adequate to restore function (Forge et al., 1993; Warchol et al., 1993; Brigande and Heller, 2009).
A potential cure for these chronic disorders would either be the transplantation of substitutional cells into the damaged inner ear or the activation of a potential regenerative capacity of quiescent progenitor cells residing in the sensory epithelia of the inner ear to replace lost sensory hair cells. Inner ear self‐regeneration is prevalent in birds and other non‐mammalian vertebrates that are able to replace lost hair cells allowing them to restore the hearing function within a few weeks after deafening by acoustic trauma (Corwin and Cotanche, 1988; Ryals and Rubel, 1988; Rubel et al., 1991). A significant proportion of avian hair cell regeneration occurs via asymmetric division of presumably bona fide stem cells within the auditory epithelium, which are thought to be supporting cells or a subpopulation thereof (Stone and Cotanche, 2007).
Any transplantation or local replacement therapies in mammalians, however, would require the identification of suitable cells to start with. Inner ear stem cells, as described in the utricular sensory epithelia of adult mice (Li et al., 2003), would be good candidates for both strategies. Similar cells have also been found in the neonatal cochlea, but their stemness or ability to proliferate and differentiate into hair cell‐like cells when taken out of the epithelial context rapidly decreases after the second postnatal week (White et al., 2006; Oshima et al., 2007; Diensthuber et al., 2009). This loss of stemness, presumably of cochlear supporting cells, is accompanied by the upregulation of the cell cycle inhibitor p27kip‐1 (Lowenheim et al., 1999; White et al., 2006). Future efforts to cure hearing loss utilizing existing inner ear supporting cells would have to find a solution for the lack of stemness or regenerative potential in the adult cochlea.
Recent encouraging evidence indicates that the human cochlea may be different from the mouse cochlea. Mitogen‐responding, sphere‐forming progenitor cells were robustly isolated from spiral ganglion specimens that were harvested during transcochlear surgeries for lateral skull base tumors in adult humans (Rask‐Andersen et al., 2005). Nothing is known, however, about progenitor cells in vestibular sensory epithelia and the organ of Corti of humans. The present study was aimed at filling this gap.
To identify cells with proliferative potential in the human inner ear, tissues were harvested during translabyrinthine surgical procedures, and processed according to protocols that were successfully applied in rodents (Oshima et al., 2009). Due to the paucity of surgical procedures for the lateral skull base, autopsy‐derived postmortem human temporal bones were additionally used as a tissue source for the present study. Proliferative and multipotent progenitor/stem cells survive in the inner ear of mice even after protracted postmortem intervals (Senn et al., 2007). Autopsies are more frequently performed than lateral skull base surgeries and apart from this the use of postmortem temporal bones has the advantage that tissues can be obtained from healthy inner ears. However, the use of postmortem human inner ear tissues as a source for primary living cells has not been described earlier, and demonstration of the feasibility of this approach was the second major goal of the present study.
MATERIALS AND METHODS
Harvesting Human Inner Ear Tissues
The human inner ear tissues were either removed during lateral skull base surgeries at Stanford University Medical Center (n = 12) or removed from autopsy‐derived postmortem temporal bones (n = 27) at Stanford (n = 3) and Bern (n = 24). Table 1 shows a synopsis of the pertinent patient‐related data for surgical and postmortem donors. All donors of surgical specimens had no serviceable hearing on the operated side. The donors of autopsy temporal bones had no known history of ear diseases and died from various causes, which were not disclosed to us. However, donors who died from infectious diseases were excluded by the pathologists.
Table 1.
Clinical data overview of temporal bone donors
| Human donors | N | Female | Male | Mean age (range) | Ear pathology (n) |
|---|---|---|---|---|---|
| Surgical | 12 | 7 | 5 | 49 (16–64) years |
Neuroma 10 Meningioma 1 Mb. Menière 1 |
| Postmortem | 27 | 9 | 18 | 72 (27–89) years | None |
Surgical specimens were obtained in the operation theater and immediately transferred to the laboratory in chilled DMEM/high‐glucose (4,500 mg/L dextrose) and F12 media (mixed 1:1, Invitrogen) supplemented with N2 and B27 supplements (Invitrogen). Postmortem temporal bones were obtained from the Pathology Department after a mean postmortem interval of 17.8 (±10.2) hr. The bones were transferred to the temporal bone laboratory in sterile saline solution and processed for isolation of inner ear tissues through the enlarged external ear canal (Fig. 1). The semicircular canals, the utricle, and the cochlear duct were removed and transferred in chilled DMEM/high‐glucose and F12 media (mixed 1:1, Invitrogen) supplemented with N2 and B27 supplements (Invitrogen) to the inner ear research laboratory. The spiral ganglion was not removed in this study. All experiments were fully approved by the ethical boards at Stanford (Certification of Human Subjects Approval, Protocol ID 96910) and Bern (KEK‐BE: 181/07).
Figure 1.
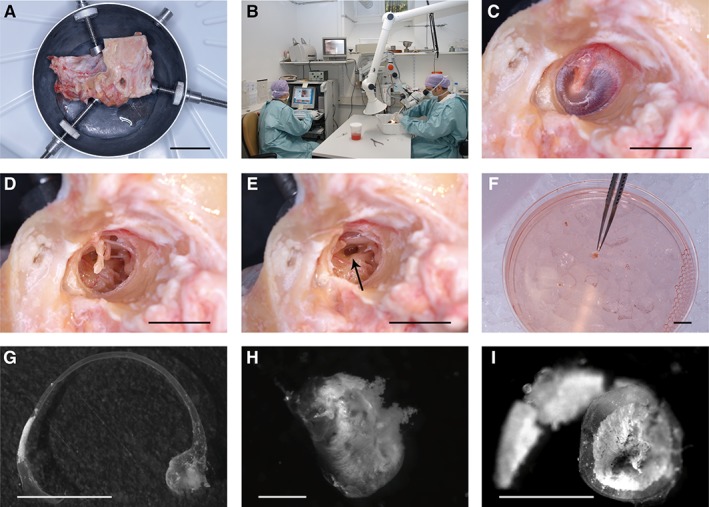
Tissue isolation from autopsy temporal bones. Unfixed human temporal bones (A) were obtained from the Pathology Department and dissected in the temporal bone laboratory (B). As a first step, the external ear canal is enlarged to allow visualization of the ear drum (C). After removal of the ear drum (D) and the ossicles (E), the inner ear was opened at the oval window (arrow in E) to remove the utricle and the membranous labyrinth, which are directly placed into cell culture medium (F). Further drilling was needed to remove the membranous part of the scala media (not shown). The membranous semicircular canal with the ampullary end (G), the utricle (H), and the membranous part of the cochlear scala media (I) are shown in higher magnification. Scale bars = 4 cm (A), 1 cm (C–F), 0.5 cm (G, I), and 0.2 cm (H).
Isolation of Inner Ear Progenitor Cells and Sphere Formation Assay
The ampullary maculae, utricular maculae, and cochlear ducts were carefully dissected and transferred into a small drop of sterile, chilled PBS and cleaned from surrounding tissues. All tissues were then subjected to a 5–10 min treatment with 0.125% trypsin in PBS at 37°C in a total volume of 100 μL for each organ. The enzymatic reaction was blocked by adding 100 μL of 0.5% soybean trypsin inhibitor (Worthington) and 2 mg/mL DNAse I in DMEM/high‐glucose (4,500 mg/L dextrose) and F12 media (mixed 1:1, Invitrogen). The cells were carefully triturated 20–50× with plastic pipette tips (epTIPS Filter 20–300 μL, Eppendorf). Cell separation was assured by microscopic inspection. Ninety‐five percent of the cell suspension was used for sphere formation assays in suspension cell culture, and 5 % of the undiluted cell suspension was used for determination of cell numbers immediately after dissection and trituration. The numbers of living and dead cells were assessed using a hemacytometer (Neubauer improved) after adding 10 μL of a 0.4% solution of trypan blue (Invitrogen) to 20 μL of the single cell solution. Exclusion of the dye from living cells allowed us to determine the numbers of dead and living cells.
Sphere formation was initiated by adding 2 mL DMEM/high‐glucose (4,500 mg/L dextrose) and F12 media (mixed 1:1) supplemented with N2 and B27 (Invitrogen), EGF (20 ng/mL), bFGF (10 ng/mL), IGF‐1 (50 ng/mL), and heparan sulfate (50 ng/mL) (all growth factors were obtained from R&D Systems and Sigma). The diluted suspension was then passed through a 70 μm cell strainer (BD Labware) directly into plastic Petri dishes (Greiner) resulting in a single cell solution virtually devoid of large cell aggregates. Cell aggregates of a few cells were observed in the suspension, but further trituration resulted in a reduction of survival. In three experiments, the cells were diluted to a low‐density of ≤0.2 cells/μL. In all other experiments, the density ranged from 1 to 50 cells/μL. The numbers of primary spheres were assessed after 7 days in culture in a humidified incubator at 37°C and 5% CO2.
Progenitor Cell Propagation
The spheres were mechanically dissociated after a 5–10 min treatment with 0.125% trypsin in PBS at 37°C in a total volume of 100 μL for each organ. The enzymatic reaction was blocked by adding 100 μL of 0.5% soybean trypsin inhibitor (Worthington) and 2 mg/mL DNAse I in DMEM/high‐glucose (4,500 mg/L dextrose) and F12 media (mixed 1:1, Invitrogen). The cells were mechanically dissected using tungsten microneedles and carefully triturated 20–50× with plastic pipette tips (epTIPS Filter 20–300 μL, Eppendorf). The cell suspensions were replated into DMEM/high‐glucose (4,500 mg/L dextrose) and F12 media (mixed 1:1) supplemented with N2 and B27 (Invitrogen), EGF (20 ng/mL), bFGF (10 ng/mL), IGF‐1 (50 ng/mL), and heparan sulfate (50 ng/mL) in Petri dishes and the propagation procedure was repeated in 7‐day‐intervals. For long‐term propagation, we counted the number of spheres for each generation, related the number to the original sphere number and replated half of the spheres for producing the next generation.
Cell Differentiation
Second generation spheres were plated onto fibronectin‐coated Lab‐Tek®II slides (Chamber Slide™ system, Nalge Nunc International) or onto polylysine/fibronectin‐coated 12‐mm diameter glass cover slides (Assistent, Germany) in 24‐well plates (Multiwell 3047,14‐mm diameter per well; Falcon, Becton Dickinson, Franklin Lakes, NJ) in DMEM/high‐glucose (4,500 mg/L dextrose) and F12 medium (mixed 1:1) supplemented with N2 and B27 (Invitrogen). Eighty percentage of the medium was replaced twice a week. Differentiated cells were analyzed after 12–14 days by immunocytochemistry.
BrdU Labeling
The thymidine analog was added at 3 μg/mL final concentration to suspension cultures pulsed either for 24 hr, 48 hr, or continuously throughout the formation period of first or second generation spheres in n = 10 independent cultures. Incorporated BrdU was detected after a 20 min pretreatment with 2N HCL with a monoclonal mouse antibody (1:500, Sigma) 4 hr after adhering to the culture dish, at the end of the sphere formation period (n = 6), and after a 12‐day cell differentiation period (n = 4). In general, sphere formation as well as occurrence of hair cell‐like cells was greatly reduced in cultures treated with BrdU.
Immunocytochemistry
The attached cells were fixed for 5 min with 4% paraformaldehyde in PBS, washed with PBS, and nonspecific binding sites were blocked for 1 hr in 0.1% Triton‐100, 1% BSA (wt/vol), and 5% (wt/vol) heat‐inactivated goat serum in PBS (PBT1). The slides were incubated overnight at 4°C in PBT1 with diluted antibodies: 1:1,000 for either polyclonal guinea pig (Oshima, et al. 2006) or polyclonal rabbit antibody to murine myosin VIIa (Proteus), 1:2,000 for polyclonal rabbit antibody to parvalbumin III (Heller et al., 2002), 1:200 for monoclonal mouse antihuman cytokeratin (Dako), 1:500 for polyclonal antibody to glial fibrillary acidic protein (GFAP; Dako), 1:500 for monoclonal mouse antibody to neuron‐specific ß‐III tubulin (TuJ; Covance), and 1:500 for monoclonal mouse antibody to bromodeoxyuridine (BrdU, Sigma). Cy5‐, TRITC‐, and FITC‐conjugated secondary antibodies (Jackson Immuno Research) were diluted 1:400 in 0.1% Triton‐100 and 1% BSA (wt/vol) in PBS and used to detect primary antibodies. 4′,6‐Diamidino‐2‐phenylindole dihydrochloride (Invitrogen) was used to visualize nuclei. TRITC‐conjugated phalloidin (Sigma) was used to visualize F‐actin. The slides were analyzed using an epifluorescence microscope equipped with digital camera and image acquisition software (Zeiss Axio Imager with AxioCam HR and AxioVision 4.0 running on a Fujitsu‐Siemens PC in Stanford or Nikon Eclipse 800 with NIS‐Elements BR 3.0 running on a Dell Optiplex PC in Bern).
Statistical Analysis
The software package GraphPad InStat (http://www.graphpad.com) was used for statistical analysis.
RESULTS
Sphere‐Forming Progenitor Cells from the Adult Human Inner Ear
Spheres were successfully generated from suspensions derived from utricles, ampullae, and one cochlea (Fig. 2). The success rate for primary sphere generation was highest for the utricle and lowest for the cochlea, where only 1 out of 10 attempts with postmortem specimens was successful (Fig. 2D). Similarly, the number of primary spheres was highest for utricle‐derived cultures with an average incidence of 13 (±19; n = 22) spheres per 104 primary living cells, although the variability was high (Fig. 2E). When grown in low‐density (<0.2 cells/μL), the average sphere number was 20 (±2.6; n = 3) spheres per 104 primary living cells. Spheres were solid in appearance and average sizes ranged from 30 to 86 μm (Fig. 2F.) In general, surgical versus postmortem tissues were associated with higher success rates for sphere generation, higher numbers of primary spheres, and smaller size of primary spheres, although no statistical evaluation was performed. Hollow spheres that were previously described in rodent inner ear suspension cultures (Oshima et al., 2007; Senn et al., 2007; Diensthuber et al., 2009) were rarely observed in 7‐day cultures of human origin (<5%, data not shown).
Figure 2.
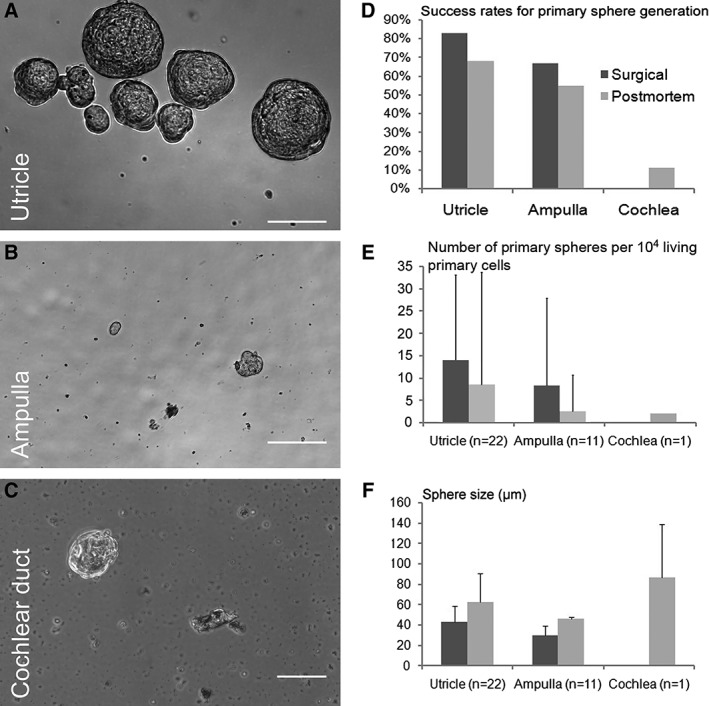
Primary spheres derived from the human utricle (A), ampulla (B), and the cochlear duct with the organ of Corti (C). The success rates for primary sphere generation (number of specimens that generate spheres per total number of specimens obtained) are shown for surgical and postmortem specimens (D). Because only one postmortem specimen and zero surgical specimens generated primary cochlear spheres, no error bars are indicated. The mean number of primary spheres (E) and the mean sphere sizes (F) are shown with one positive standard deviation for surgical and postmortem specimens. Scale bars = 50 μm.
Adult Human Inner Ear Progenitor Cells Show Limited Propagation Potential
Spheres of surgical and postmortem origin were routinely propagated into the third generation (two passages) in utricle‐derived (Fig. 3A), ampulla‐derived (Fig. 3B), and cochlea‐derived cultures (Fig. 3C). In the best conditions, we were able to propagate utricle‐derived spheres into the sixth generation (five passages) with maximal propagation ratios (number of daughter spheres per mother spheres) of ≈4 in the first two passages. However, these results were obtained in isolated experiments and overall propagation and expansion of human inner ear progenitor cells proved to be increasingly challenging with rising passage numbers.
Figure 3.
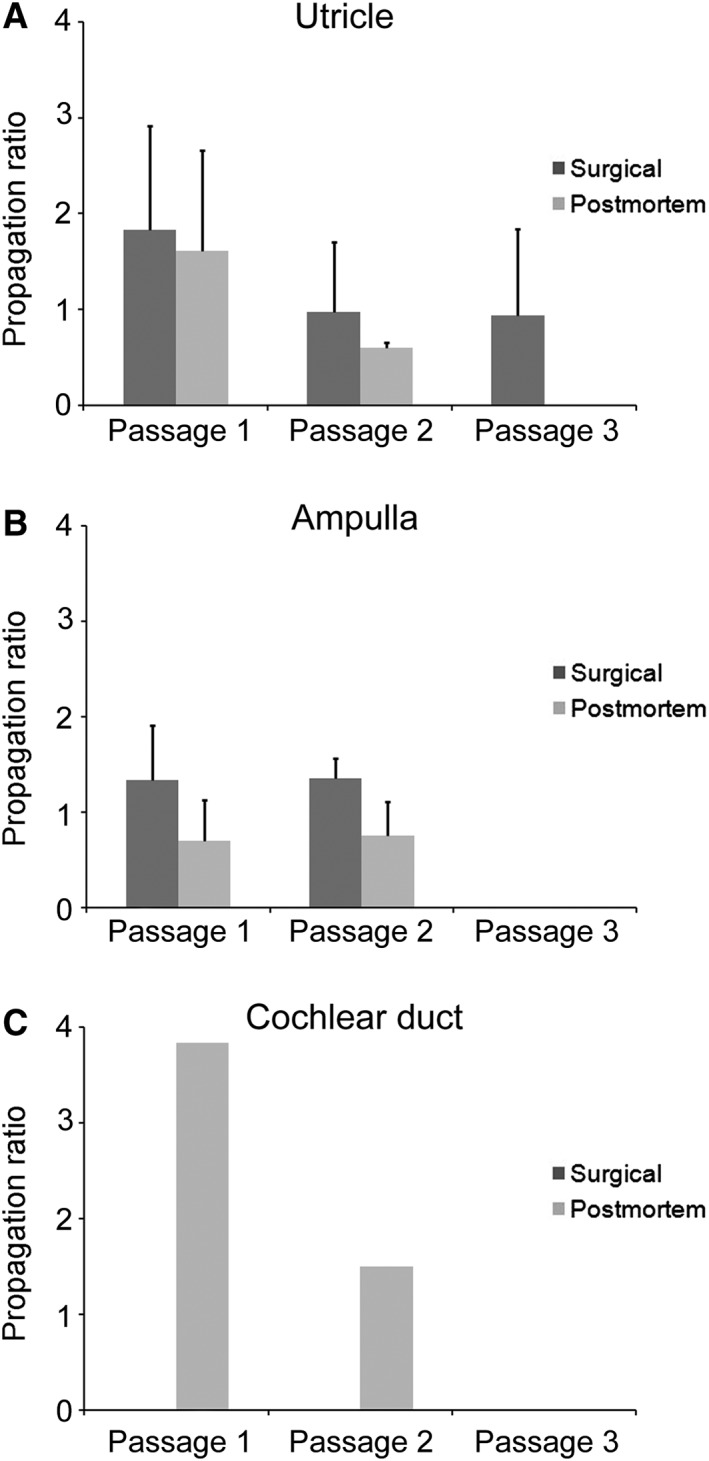
Mean propagation ratios (number of daughter spheres per one mother sphere) for utricle (A), ampulla (B), and cochlear duct (C) are shown with one positive standard deviation for surgical and postmortem specimens.
Adult Human Inner Ear Progenitor Cells Are Multipotent
For cell differentiation, we used second and third generation spheres from utricle and cochlear duct that were attached to fibronectin‐coated cell culture dishes and allowed to differentiate for 2 weeks in mitogen‐deprived medium. After culturing the cells in these conditions, which were previously utilized to induce hair cell and neuronal differentiation in rodent protocols (Li et al., 2003; Oshima et al., 2007; Senn et al., 2007), we analyzed the cells for the expression of cell type‐specific markers. Spheres from the utricle robustly gave rise to hair cell‐like cells, defined as monoexpression of myosin VIIa (Fig. 4A,B), or coexpression of myosin VIIa with either filamentous actin (Fig. 4D,H,F) or parvalbumin III (data not shown). Hair cell‐like cells were found in surgical and postmortem‐derived cultures and occurred in small patches (<100 cells per patch, Fig. 4D) or large patches (>100 cells per patch, Fig. 4A) of differentiated cells, of which a significant proportion was labeled with pancytokeratin, a protein expressed in supporting cells (Fig. 4A,C). In large patches, the incidence of hair cell‐like cells was low (1.8%), whereas in small patches the incidence was almost 10 times higher (18%, Fig. 4E). Spheres derived from surgical specimens gave more frequently rise to large, confluent patches of differentiated cells covering wide areas of the culture dish. Postmortem‐derived spheres gave predominantly rise to smaller patches (Fig. 4A) with a higher percentage of hair cell‐like cells per patch. However, the variability was high and about a third of utricle‐derived specimens that generated spheres failed to generate hair cell‐like cells at the end of the differentiation period. No hair cell‐like cells were differentiated from second or third generation spheres derived from the single sphere‐forming cochlear duct specimen. These spheres generated homogenous appearing fibroblast‐like cell types that stained positive for β‐III tubulin without displaying neural morphology (Fig. 5B).
Figure 4.
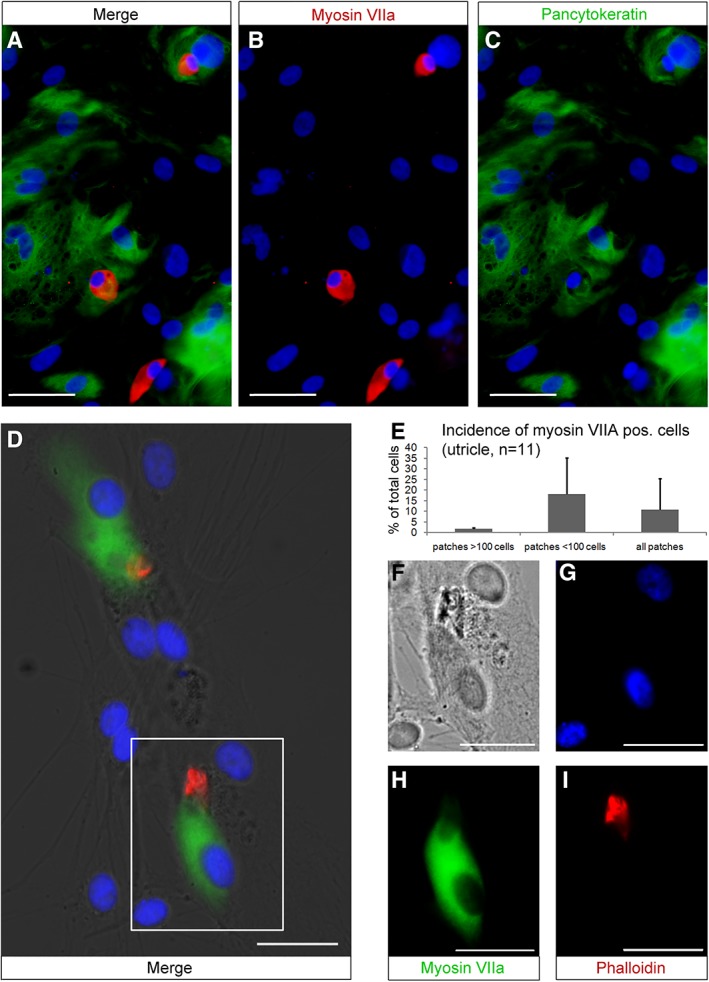
Utricle‐derived spheres generate hair cell‐like cells expressing myosin VIIA (red in A, B) or coexpress myosin VIIA (green in D, H) and the hair bundle marker phalloidin (red, D, I). Hair cell‐like cells are distributed among cells labeled with the supporting cell marker pancytokeratin (green, A and C). Nuclei are visualized with 4′,6‐diamidino‐2‐phenylindole hydrochloride (DAPI) (blue). The mean percentage of myosin VIIa positive cells per patch is indicated with one positive standard deviation (E). The box in D corresponds to the merged individual channels DIC (F), DAPI (G), myosin VIIA (H), and phalloidin (I). The inner ear cell types were generated from postmortem utricles of a 27‐year old (A–C) and a 72‐year old male donor (D, F–I). Scale bars = 50 μm (A–C) and 20 μm (D, F–I).
Figure 5.
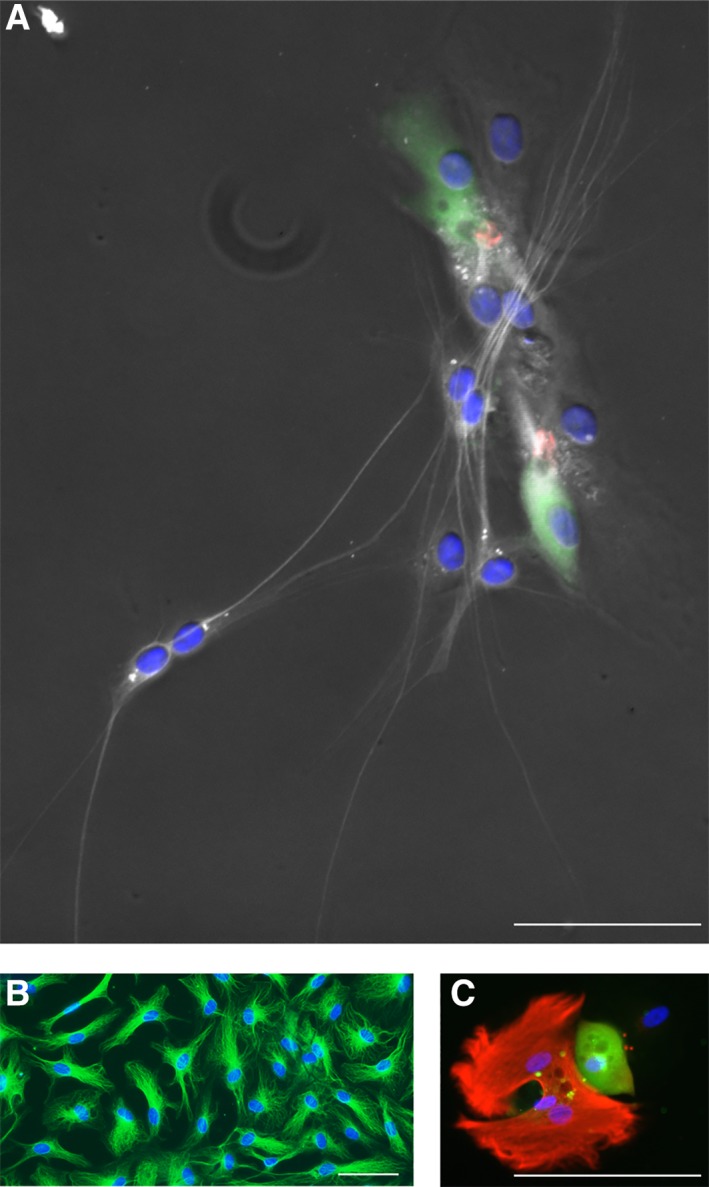
Neuronal and glial cell types. Cell types with neuronal morphology are labeled with a marker for ß‐III tubulin (TUJ, white, A). These cells were generated from postmortem utricle‐derived spheres of a 72‐year‐old male donor (same patch of cells as in Fig. 4C). Homogeneous appearing cells with no neural morphology (TUJ, green, B) were obtained from cochlear duct‐derived spheres of an 80‐year‐old male postmortem donor. A glial cell type, characterized by GFAP staining (red, C) is shown next to a hair cell‐like cell (myosin VIIA, green) in a patch of utricle‐sphere derived cells. Cell nuclei are visualized using 4′,6‐diamidino‐2‐phenylindole hydrochloride (blue, A–C). Scale bars = 50 μm.
β‐III tubulin positive cell types were found in some utricle‐derived cultures, of which isolated cells had a neural morphology (Fig. 5A). The majority of β‐III tubulin positive cell types, however, had no neural morphology (Fig. 5B), and the identity of these cells remained unclear. Other cell types stained positive for GFAP (Fig. 5C). The incidence of neuronal and glial cell types, however, was too low for a meaningful quantification. Overall, we note that utricle‐derived spheres were able to give rise to cells that displayed markers for hair cells, neurons, and glial cells.
We attempted (n = 10) to label sphere cells and differentiated cell types including hair cell‐like cells with BrdU, which would have been an indication that the cells were derived from progenitors that went through an S‐phase. Nevertheless, we found that exposure to BrdU was detrimental to sphere formation and to formation of hair cell‐like cells, which made it difficult to maintain healthy cultures due to important toxicity. Overall, we were not able to detect hair cell‐like cells that were co‐labeled with BrdU, although three different administration methods were tried (pulsed 24 hr, pulsed 48 hr, or chronic administration).
DISCUSSION
Our results show that sphere‐forming multipotent progenitor cells reside in the vestibular and cochlear sensory epithelia of the adult human inner ear, and that these cells can be isolated from surgery‐ and autopsy‐derived inner ear tissues. The isolation of sphere‐forming cells differed across inner ear compartments with higher success rates (>50%) for the vestibular sensory epithelia and a low success rate (10%) for the cochlear duct. This finding corroborates earlier findings from rodents, where vestibular stem cells were isolated throughout the life span of the animal in contrast to cochlear progenitor/stem cells, which could only be detected during the first few weeks of life (White et al., 2006; Oshima et al., 2007; Diensthuber et al., 2009). Our results show that the presumably quiescent adult human inner ear tissues, when taken out of the context of the epithelial organization, and supplied with appropriate growth factors and culture conditions, were able to proliferate. Using a sphere formation assay, which promotes the proliferation of progenitor/stem cells and clonal floating colony formation, we found that a small number of cells with progenitor‐features exist in the adult human utricle and cochlear duct.
To demonstrate the self‐renewal capacity and potency of human inner ear progenitor cells, we propagated the spheres and tested second and third generation spheres for their ability to generate different cell types. Utricle‐derived spheres routinely gave rise to hair cell‐like cells and occasionally to neuronal or glial cell types (Figs. 4 and 5). Cochlear duct‐derived spheres, however, failed to generate hair cell‐like cells and gave rise to morphologically homogenous‐appearing cell populations consisting of β‐III tubulin positive cells without neural morphology (Fig. 5B). The failure of generating hair cell‐like cells from adult cochlear tissue in these experiments is a strong reminder of the challenges that lie ahead with respect to curing human hearing loss by stimulation of presumptive residual regenerative potential of the human organ of Corti. It is also possible that with improved propagation or differentiation methods a better yield of spheres or hair cell‐like cells could be obtained in the future.
Our study also demonstrated the feasibility of using human postmortem autopsy temporal bones for harvesting living inner ear cells with progenitor cell characteristics. This approach is in agreement with earlier reports of stem cell retrieval from the human brain (Palmer et al., 2001) and the human eye (Mayer et al., 2005). The use of postmortem human temporal bones is important for practical reasons because autopsies are more frequently performed compared to lateral skull base surgeries and because tissues from healthy inner ears can be obtained. In comparison to surgical specimens, however, the success rates of postmortem specimens for sphere generation are lower. We believe that this is mainly due to an increase in fungal and bacterial infections, which particularly occurred after protracted postmortem intervals. Postmortem infections mainly ascended through the internal auditory canal into the spiral ganglion and cochlear turns and in a second phase into the vestibule. This route of infection is plausible because the pathologists excised temporal bones in a non‐sterile way, and we believe that the internal auditory canal has a high chance of becoming contaminated during this procedure because it lies at the outer border of the specimen. Vestibular tissues, however, were shielded in the otic capsule behind the intact eardrum and were further away from the contaminated surface (Fig. 1). Another possible argument for higher success rates in surgical specimens is the fact that the donors of surgical specimens were substantially younger compared to the donors of postmortem temporal bones (mean age of 49 years vs. mean age of 72 years; Table 1) and we may hypothesize that stemness of inner ear progenitors may be reduced with increasing age, as observed in rodents (Oshima et al., 2007).
The lack of BrdU labeling of hair cell‐like cells of sphere‐derived origin leaves room for discussion. Our results cannot prove that hair cell marker‐positive cells were generated following S‐phase and mitotic divisions of progenitor cells. We observed a strong effect of BrdU on sphere formation, which made it impossible to generate and propagate spheres for the complete 7‐day sphere formation periods. We have experienced similar detrimental effects of BrdU and EdU on cultures derived from the mouse utricle and organ of Corti, and we find that the differentiation of hair cell marker‐expressing cells is substantially suppressed in presence of BrdU and EdU (data not shown). When we treated fibronectin‐attached cells with BrdU, we observed a steady decrease of cell numbers and increased vacuolization of surviving cells often accompanied by cytoplasmic localization of BrdU immunoreactivity, indicative of toxicity (data not shown). Nevertheless, generation of hair cell‐like cells from progenitor/stem cells has been shown in adult mice (Li et al., 2003; Oshima et al., 2007) and isolation of dividing progenitor cells with the ability to generate new neuronal cell types has been shown in adult human spiral ganglion (Rask‐Andersen et al., 2005). Three additional arguments for the formation of new hair cell‐ and neuronal cell types in our cultures are the following: first, the primary spheres that arose from isolated inner ear progenitor cells are clonal because previous studies have shown that a sphere assay, when conducted at <2 cells per μL, is highly clonal (Kalani et al., 2008). Our clonality tests were done with 10× more diluted cultures, which makes it virtually impossible that primary spheres formed from cell aggregates and not mitotically; nevertheless, we cannot exclude that some spheres grew from small cell clusters that were present after trituration. Consequently, all cells that made up a sphere after the 7‐day sphere formation period arose either from a single proliferating progenitor cell or a small cluster of cells with at least one proliferating progenitor cell. A second argument is that even if an adult hair cell would initially survive and attach to a growing sphere, it is highly unlikely that the hair cell would survive a second trypsinization and trituration followed by a prolonged incubation period in serum‐free media. Finally, the occurrence of ß‐III tubulin‐expressing cells with neuronal morphology as well as GFAP‐positive cells is highly indicative of neurogenesis; the fact that the starting material did not contain vestibular‐ and spiral ganglion tissue let us to conclude that the neuronal cell types that we detected were newly generated.
Although the hair cell‐like cells that are presented in this study are the first from mature human inner ears, they are not the first culture‐generated human hair cells reported. One group has repeatedly been able to generate hair cell‐like cells and neurons from fetal cochleae (Chen et al., 2007, 2009). The advantage of using fetal sources seems to be the fact that higher numbers of progenitor cells exist, which leads to a much more robust generation of differentiated inner ear cells. Our own recent studies corroborate the fact that cochlear hair cell‐like cells and neuron‐like cells can be robustly generated from human fetal sources (Roccio et al., 2018).
CONCLUSIONS
With the present study, we demonstrate that sphere‐forming progenitor cells can be harvested from adult human inner ears. We further show that the spheres grown from these progenitor cells can be propagated, albeit with limited potential, and that spheres grown from utricle‐derived progenitors gave rise to hair cell‐like cells and neuronal or glial cell types. Based on this finding, we conclude that the adult human utricle harbors multipotent progenitor cells. For the cochlea, sphere‐forming stem cells were only found in one specimen and despite the fact that these spheres could be propagated and expanded, we did not observe differentiation of hair cell‐like cells. These shortcomings may be the result of intrinsic regenerative inability of the human cochlear duct, or of suboptimal culture conditions needed to provide the cells with appropriate stimulation to reveal an in vitro potential to generate inner ear cell types. Finally, the feasibility of using human autopsy temporal bones as a progenitor cell source could be demonstrated.
ACKNOWLEDGEMENTS
Surgery‐derived inner ear tissues could not have been obtained without the highly appreciated help by Saumil Merchant and Ronald de Venecia at the Massachusetts Eye and Ear Infirmary who were instrumental in initial pilot studies that were conducted in collaboration with Huawei Li, Karen Watters, and C. Eduardo Corrales. We thank Sumit Agrawal, Anh Nguyen, and Robert Jackler (Stanford) for surgical specimens, as well as Alfred Wüthrich, Thomas Schaffner, and Aurel Perren of the Pathology Department, University of Bern for autopsy‐derived temporal bones, and Christoph Stieger, Rudolf Häusler, and Marco Caversaccio (Bern) for providing infrastructure at the temporal bone laboratory. The authors further thank Marta Roccio, Markus Huth, Angélique Ducray, Stefano di Santo, and Hans Rudolf Widmer of the Inner Ear Research Laboratory and the Laboratory for Neural Repair at the Departments of ORLHNS and Neurosurgery (Bern) for generous technical, logistical and intellectual support. Begun with pilot experiments at the Massachusetts Eye & Ear Infirmary, continued at the Department for OHNS of the Stanford Medical school, and concluded at the University Department of OHNS in Bern, Switzerland, this work was supported in part by Grants PBBEB‐105075 and 1226/PASMA‐111607/1 from the Swiss National Science Foundation and by the “Stiftung für Hörgeschädigte” Luzern, Switzerland, to P.S., by grants from the National Institutes of Health DC007132 and DC006167 to S.H., by funds of the MEDEL company (Austria, Innsbruck) to A.M. and S.V. and by funds from the DAAD and from the Stanford Dean's Office to V.S.
Literature Cited
- Brigande JV, Heller S. 2009. Quo vadis, hair cell regeneration? Nat Neurosci 12(6):679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Cacciabue‐Rivolta DI, Moore HD, Rivolta MN. 2007. The human fetal cochlea can be a source for auditory progenitors/stem cells isolation. Hear Res 233(1–2):23–29. [DOI] [PubMed] [Google Scholar]
- Chen W, Johnson SL, Marcotti W, Andrews PW, Moore HD, Rivolta MN. 2009. Human fetal auditory stem cells can be expanded in vitro and differentiate into functional auditory neurons and hair cell‐like cells. Stem Cells 27(5):1196–1204. [DOI] [PubMed] [Google Scholar]
- Corwin JT, Cotanche DA. 1988. Regeneration of sensory hair cells after acoustic trauma. Science 240(4860):1772–1774. [DOI] [PubMed] [Google Scholar]
- Diensthuber M, Oshima K, Heller S. 2009. Stem/progenitor cells derived from the cochlear sensory epithelium give rise to spheres with distinct morphologies and features. J Assoc Res Otolaryngol 10(2):173–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A, Li L, Corwin JT, Nevill G. 1993. Ultrastructural evidence for hair cell regeneration in the mammalian inner ear. Science 259(5101):1616–1619. [DOI] [PubMed] [Google Scholar]
- Heller S, Bell AM, Denis CS, Choe Y, Hudspeth AJ. 2002. Parvalbumin 3 is an abundant Ca2+ buffer in hair cells. J Assoc Res Otolaryngol 3(4):488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R. 2008. Wnt‐mediated self‐renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A 105(44):16970–16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liu H, Heller S. 2003. Pluripotent stem cells from the adult mouse inner ear. Nat Med 9(10):1293–1299. [DOI] [PubMed] [Google Scholar]
- Lowenheim H, Furness DN, Kil J, Zinn C, Gultig K, Fero ML, Frost D, Gummer AW, Roberts JM, Rubel EW, et al. 1999. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc Natl Acad Sci U S A 96(7):4084–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer EJ, Carter DA, Ren Y, Hughes EH, Rice CM, Halfpenny CA, Scolding NJ, Dick AD. 2005. Neural progenitor cells from postmortem adult human retina. Br J Ophthalmol 89(1):102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K, Grimm CM, Corrales CE, Senn P, Martinez Monedero R, Geleoc GS, Edge A, Holt JR, Heller S. 2007. Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J Assoc Res Otolaryngol 8(1):18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K, Senn P, Heller S. 2009. Isolation of sphere‐forming stem cells from the mouse inner ear. Methods Mol Biol 493:141–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH. 2001. Progenitor cells from human brain after death. Nature 411(6833):42–43. [DOI] [PubMed] [Google Scholar]
- Rask‐Andersen H, Bostrom M, Gerdin B, Kinnefors A, Nyberg G, Engstrand T, Miller JM, Lindholm D. 2005. Regeneration of human auditory nerve. In vitro/in video demonstration of neural progenitor cells in adult human and Guinea pig spiral ganglion. Hear Res 203(1–2):180–191. [DOI] [PubMed] [Google Scholar]
- Roccio M, Perny M, Ealy M, Widmer HR, Heller S, Senn P. 2018. Molecular characterization and prospective isolation of human fetal cochlear hair cell progenitors. Nat Commun 9(1):4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubel EW, Oesterle EC, Weisleder P. 1991. Hair cell regeneration in the avian inner ear. Ciba Found Symp 160:77–96. discussion 96‐102. [DOI] [PubMed] [Google Scholar]
- Ryals BM, Rubel EW. 1988. Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240(4860):1774–1776. [DOI] [PubMed] [Google Scholar]
- Senn P, Oshima K, Teo D, Grimm C, Heller S. 2007. Robust postmortem survival of murine vestibular and cochlear stem cells. J Assoc Res Otolaryngol 8(2):194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JS, Cotanche DA. 2007. Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol 51(6–7):633–647. [DOI] [PubMed] [Google Scholar]
- Warchol ME, Lambert PR, Goldstein BJ, Forge A, Corwin JT. 1993. Regenerative proliferation in inner ear sensory epithelia from adult Guinea pigs and humans. Science 259(5101):1619–1622. [DOI] [PubMed] [Google Scholar]
- White PM, Doetzlhofer A, Lee YS, Groves AK, Segil N. 2006. Mammalian cochlear supporting cells can divide and trans‐differentiate into hair cells. Nature 441(7096):984–987. [DOI] [PubMed] [Google Scholar]