

Summary
The landscape of medical sequencing has rapidly changed with the evolution of next generation sequencing (NGS). These technologies have contributed to the molecular characterization of the myelodysplastic syndromes (MDS) and chronic myelomonocytic leukaemia (CMML), through the identification of recurrent gene mutations, which are present in >80% of patients. These mutations contribute to a better classification and risk stratification of the patients. Currently, clinical laboratories include NGS genomic analyses in their routine clinical practice, in an effort to personalize the diagnosis, prognosis and treatment of MDS and CMML. NGS technologies have reduced the cost of large‐scale sequencing, but there are additional challenges involving the clinical validation of these technologies, as continuous advances are constantly being made. In this context, it is of major importance to standardize the generation, analysis, clinical interpretation and reporting of NGS data. To that end, the Spanish MDS Group (GESMD) has expanded the present set of guidelines, aiming to establish common quality standards for the adequate implementation of NGS and clinical interpretation of the results, hoping that this effort will ultimately contribute to the benefit of patients with myeloid malignancies.
Keywords: myelodysplastic syndromes, chronic myelomonocytic leukaemia, next generation sequencing, guidelines, molecular genetics
The development of next generation sequencing (NGS) techniques during the last decade has contributed to the molecular characterization of haematological malignancies, including myeloid neoplasms, such as the myelodysplastic syndromes (MDS) and chronic myelomonocytic leukaemia (CMML) (Arber et al, 2016). According to the World Health Organization (WHO) classification (Arber et al, 2016), MDS comprise a group of clonal haematopoietic stem cell disorders characterized by ineffective haematopoiesis in one or more myeloid cell lines, abnormal dysplastic cell morphology and potential for clonal evolution. On the other hand, CMML is included in the WHO group of myelodysplastic/myeloproliferative neoplasms (MDS/MPN) and is characterized by absolute peripheral monocytosis in conjunction with both effective (myeloproliferative variant, MP‐CMML ≥ 13 × 109/l leucocytes) and ineffective (myelodysplastic variant, MD‐CMML < 13 × 109/l leucocytes) haematopoiesis. MDS and CMML are very heterogeneous at the clinical, morphological and genetic level and both and have the risk of progressing to acute myeloid leukaemia (AML). The natural history of these diseases is highly variable and a risk‐adapted treatment strategy is mandatory.
Myeloid neoplasms arise through the serial acquisition of chromosomal alterations and somatic mutations that affect genes involved in several mechanisms. These mutations drive disease evolution from asymptomatic clonal haematopoiesis to overt disease and, eventually, progression to AML (Kennedy & Ebert, 2017). Recurrent mutations in more than 40 different genes have been identified in MDS and CMML and at least one mutation is detected at diagnosis in >80% of patients (Papaemmanuil et al, 2013; Haferlach et al, 2014; Elena et al, 2016). These mutations are related to the pathophysiological features of these diseases and play a role in their clinical heterogeneity. These molecular markers can complement current diagnostic criteria in MDS and CMML, as well as contribute to the risk assessment of these patients (Bejar et al, 2011; Haferlach et al, 2014; Elena et al, 2016). Therefore, diagnostic laboratories are including NGS genomic analyses in their routine clinical practice, in an effort to personalize the diagnosis, prognosis and treatment of MDS and CMML. Thorough evaluation of technical aspects, data analysis and variant interpretation is required for the correct implementation of NGS in routine diagnosis. Thus, the aim of this collaborative project within the Spanish MDS Group (GESMD) was to develop a set of guidelines, agreed by expert consensus panels, to standardize the use of targeted deep sequencing in the routine genetic testing of MDS and CMML patients, for the detection of single nucleotide variants (SNVs) and small insertions/deletions (indels). Our main objective is to harmonize analyses and variant interpretation, ensuring high standards in clinical reporting of genetic data. These consensually proposed NGS guidelines involve the following major sections, which will be further discussed in detail: (i) mutational landscape in MDS and CMML, (ii) sequencing workflow and quality control, (iii) data analysis and variant filtering, (iv) variant categorization/interpretation and (v) clinical reports.
Mutational landscape of MDS and CMML
Over 40 different genes are found recurrently mutated in MDS and CMML (Papaemmanuil et al, 2013; Haferlach et al, 2014; Elena et al, 2016). However, in contrast to other diseases, such as myeloproliferative neoplasms (MPN), none of them are specific to these disorders. Still, a few of these genes have been proved to be useful for diagnosis or to predict response to specific treatments (Papaemmanuil et al, 2011; Itzykson et al, 2011; Bejar et al, 2014; Traina et al, 2014). Moreover, some mutations are also associated with shorter survival and a higher risk of progression to AML and therefore can be used for prognostic stratification (Table 1) (Bejar et al, 2011, 2012; Papaemmanuil et al, 2013; Haferlach et al, 2014; Elena et al, 2016; Makishima et al, 2017). Thus, an MDS/CMML targeted sequencing panel should at least include all those genes that have been proven relevant for the disease (Malcovati et al, 2013; Arber et al, 2016; Greenberg et al, 2017) (Table 2). As NGS is a high throughput technique, it can be complemented with genes that are altered in other myeloid neoplasms to generate a pan‐myeloid sequencing panel, in view of a wider utility of the tool (e.g. for MDS, MPN and AML) (Table 3). In this regard, the Association for Molecular Pathology recently reviewed the clinical relevance of small DNA variants in chronic myeloid neoplasms, summarizing key findings that support clinical utility, and defining the need for gene inclusion in high‐throughput sequencing testing panels (McClure et al, 2018).
Table 1.
Clinical relevance of mutated genes in MDS and CMML (Malcovati et al, 2013; Arber et al, 2016; Greenberg et al, 2017).
| Gene | MDS | CMML | ||
|---|---|---|---|---|
| Incidence | Clinical impact | Incidence | Clinical impact | |
| ASXL1 | 5–25% | Unfavourable | 40–50% | Unfavourable |
| CSNK1A1 |
<1% 5–15% MDS with del(5q) |
Uncertain Associated with del(5q) |
<1% | Unknown |
| DNMT3A | 12–18% | Unfavourable in patients without SF3B1 mutations | 2–10% | Uncertain |
| EZH2 | 5–10% | Unfavourable | 5–12% | Unfavourable |
| IDH1 | <5% | Uncertain | <1% | Uncertain |
| IDH2 | <5% | Uncertain | 5–10% | Unfavourable |
| JAK2 | <5% | MDS with del(5q), 5–7% | 2–10% | Associated with MP‐CMML |
| KRAS | 5–10% | Uncertain | 10–20% |
Unfavourable Associated with MP‐CMML |
| NRAS | 5–10% | Unfavourable | 10–20% |
Unfavourable Associated with MP‐CMML |
| RUNX1 | 10–15% |
Unfavourable Can be of germline origin |
10–30% | Unfavourable |
| SETBP1 | <5% | Unfavourable | 5–10% | Unfavourable |
| SF3B1 |
20–30% 80% MDS‐RS |
Associated with RS Favourable |
5–10% | Unknown |
| SRSF2 | 10–15% | Unfavourable | 30–50% | Uncertain |
| STAG2 | 5–10% | Unfavourable | 5–10% | Unfavourable |
| TET2 | 20–25% | Uncertain | 45–60% | Uncertain |
| TP53 | 8–12% |
Unfavourable Associated with CK (50%), del(5q) (15–20%) Lower response rate to lenalidomide Can be of germline |
<5% | Unfavourable |
| U2AF1 | 8–12% | Unfavourable | 5–10% | Unfavourable |
| ZRSR2 | 5–10% | Unfavourable | 5–10% | Uncertain |
CK, complex karyotype; CMML, chronic myelomonocytic leukaemia; HMA, hypomethylating agents; MDS, myelodysplastic syndrome; MDS‐RS, MDS with ring sideroblasts; MP‐CMML; myeloproliferative CMML; NK, normal karyotype; RS, ring sideroblasts.
Table 2.
List of GESMD‐recommended genes for studying the clinical management of MDS and CMML.
| Gene | Region | Type of mutation | Frequency | |
|---|---|---|---|---|
| MDS | CMML | |||
| ASXL1 | Exons 10–13 | Nonsense, frameshift | 5–25% | 40–50% |
| Codons: all | ||||
| CSNK1A1 | Exons 2–4 | Missense | 5–10% | <1% |
| Codons: all | ||||
| DNMT3A | Complete coding region | All | 12–18% | 2–10% |
| Hotspot codon: R882 | ||||
| EZH2 | Complete coding region | Nonsense, frameshift | 5–10% | 5–12% |
| Codons: all | ||||
| IDH1 | Exon 4 | Missense | <5% | <1% |
| Hotspot codon: R132 | ||||
| IDH2 | Exon 4 | Missense | <5% | 5–10% |
| Hotspot codons: R140 and R172 | ||||
| JAK2 | Complete coding region | Missense | <5% | 2–10% |
| Hotspot codon: V617F | ||||
| KRAS | Exons 2 and 3 | Missense | 5–10% | 10–20% |
| Hotspot codons: G12, G13, Q61 and G146 | ||||
| NRAS | Exons 2 and 3 | Missense | 5–10% | 10–20% |
| Hotspot codons: G12, G13 and Q61 | ||||
| RUNX1 | Complete coding region | Nonsense, frameshift | 10–15% | 10–30% |
| Codons: all | ||||
| SETBP1 | Exon 4 | Missense | <5% | 5–10% |
| Codons: 858–870 | ||||
| SF3B1 | Exons 10–16 | Missense |
20–30% 80% RS |
5–10% |
| Codons: 622–781 | ||||
| SRSF2 | Complete coding region | Missense | 10–15% | 30–50% |
| Hotspot codon: P95 | ||||
| STAG2 | Complete coding region | Nonsense, frameshift, splicing | 5–10% | 5–10% |
| Codons: all | ||||
| TET2 | Complete coding region | All | 20–25% | 45–60% |
| Codons: 1134–1444 or 1842–1921 | ||||
| TP53 | Complete coding region | All | 8–12% | <5% |
| Codons: all | ||||
| U2AF1 | Exons 2–6 | Missense | 8–12% | 5–10% |
| Hotspot codons: S34 and Q157 | ||||
| ZRSR2 | Complete coding region | Nonsense, frameshift | 5–10% | 5–10% |
| Codons: all | ||||
GESMD, Spanish Group of MDS; CMML, Chronic Myelomonocytic Leukaemia; MDS, Myelodysplastic Syndrome; RS, ring sideroblasts.
Table 3.
Other myeloid‐related genes more frequent in AML, MPN and other MDS/MPN.
| Gene |
Region Frequent mutations |
Type of mutation | Frequency | |
|---|---|---|---|---|
| MDS (%) | CMML (%) | |||
| Frequent in myeloproliferative neoplasms | ||||
| CALR |
Exon 9 Codons: all |
Frameshift | <1 | <1 |
| CBL |
Exons 8 and 9 Codons: 366–420 |
Missense | <5 | 8–18 |
| CSF3R |
Complete coding region Hotspot codons: 618 |
Missense | <1 | 3–4 |
| GATA2 |
Exons 2 and 6 Codons: all |
Missense, frameshift | <5 | <1 |
| MPL |
Complete coding region Codons 505 and 515 |
Missense | <1 | <1 |
| NF1 |
Complete coding region Codons: all |
Nonsense, frameshift, splicing | <5 | <5 |
| PTPN11 |
Exons 3 and 7 Codons: all |
Missense | <1 | 4 |
| Frequent in acute myeloid leukaemia | ||||
| BCOR |
Complete coding region Codon: all |
All | <5 | <5 |
| BCORL1 |
Complete coding region Codons: all |
Nonsense, frameshift | <1 | <1 |
| CEBPA |
Complete coding region Codons: all |
Missense, frameshift | <5 | <5 |
| ETV6 |
Complete coding region Codons: all |
Nonsense, frameshift | <5 | <1 |
| FLT3 |
Exons 13–15 and 20 Hotspot codons: FLT3‐ITD and D835 |
Missense, frameshift | <5 | <5 |
| KIT |
Exons 2, 8–11, 13 and 17 Codons: all |
Missense, frameshift | <3 | <1 |
| NPM1 |
Exon 11 Hotspot codons: W288 |
Frameshift | <5 | <5 |
| WT1 |
Exons 7 and 9 Codons: all |
Missense, frameshift | <3 | <3 |
CMML, chronic myelomonocytic leukaemia; ITD, internal tandem duplication; MDS, myelodysplastic syndrome.
Driver genes in MDS and CMML, which are common among the spectrum of myeloid malignancies, affect specific cellular processes and can be categorized according to their function (Greenberg et al, 2017; Kennedy & Ebert, 2017; McClure et al, 2018) (Fig 1).
Figure 1.

Classification of frequently mutated genes in MDS and CMML according to their functional category (modified from Kennedy & Ebert, 2017; Kennedy & Ebert, 2017).
Mutations in RNA‐splicing machinery are the most frequent type of somatic mutations detected in MDS. Recurrent mutations have been reported in the spliceosome components of the SF3B1, SRSF2, U2AF1 and ZRSR2 genes, while mutations in PRPF40B, SF3A1, SF1 and U2AF2 (U2AF65) genes have been described in only 1–2% of MDS patients (Yoshida et al, 2011; Makishima et al, 2012; Papaemmanuil et al, 2013). SF3B1 mutations are associated with a higher overall survival rate and an inferior risk of AML progression, as they are the only ones that are clearly associated with a better prognosis, especially as a single mutation (Papaemmanuil et al, 2011; Malcovati et al, 2014). SRSF2 mutations are associated with monocytosis and marked thrombocythaemia and are especially frequent in CMML (Yoshida et al, 2011; Meggendorfer et al, 2012). Mutations in SRSF2 and U2AF1 have been associated with less favorable outcomes in some studies (Graubert et al, 2011; Thol et al, 2012; Meggendorfer et al, 2012; Haferlach et al, 2014; Makishima et al, 2017).
Epigenetic regulators are the second most common class of mutation in MDS, and can be divided into DNA methylation enzymes (TET2, DNMT3A and IDH1/2 genes) and chromatin modifiers (ASXL1 and EZH2 genes) (Delhommeau et al, 2009; Walter et al, 2011; Gelsi‐Boyer et al, 2012; Shih et al, 2012; Ganguly & Kadam, 2016). TET2 mutations are very common in CMML and the combination of TET2 and SRSF2 is highly associated with this disorder (Malcovati et al, 2014). TET2 mutations do not have a prognostic impact but they are associated with an increased response rate to hypomethylating agents (Itzykson et al, 2011; Bejar et al, 2014) (Table 3). DNMT3A mutations are associated with a more aggressive clinical course and rapid progression to AML in both MDS and CMML patients (Walter et al, 2011; Bejar et al, 2014; Patnaik et al, 2017). ASXL1 mutations are independently associated with a poor outcome in the spectrum of myeloid diseases, including a shorter overall survival and a higher risk of progression to AML (Boultwood et al, 2010; Gelsi‐Boyer et al, 2012; Itzykson et al, 2013a). EZH2 mutations are associated with worsened overall survival in both low and high risk MDS and CMML (Bejar et al, 2011; Grossmann et al, 2011; Bejar et al, 2012).
The multiprotein cohesin complex is involved in the cohesion of sister chromatids and the post‐replicative DNA repair system, and it is encoded by the genes STAG1/2, SMC1A, SMC3 and RAD21, which are mutated in up to 11% of low risk and 17% of high risk MDS patients (Kon et al, 2013; Thota et al, 2014). Mutations in STAG2 are more frequent in patients with multilineage dysplasia and excess of blasts and seem to be associated with shorter overall survival (Thota et al, 2014).
Loss‐of‐function mutations in a transcription factor gene, such as RUNX1, GATA2 or ETV6, are detected in approximately 20% of MDS (Chen et al, 2007; Bejar et al, 2011; Wall et al, 2012). RUNX1 mutations are associated with a high risk phenotype, thus being more frequent in high risk MDS and CMML and in secondary AML (Bejar et al, 2011; Cazzola et al, 2013; Elena et al, 2016).
MDS and CMML patients frequently harbour mutations in signal transduction genes, which constitutionally activate a signalling pathway involved in cell proliferation, apoptosis and cell differentiation (CBL, CSNK1A1, JAK2, N/KRAS, MPL, NF1, PTPN11, KIT, and FLT3 genes). These mutations are more frequent in myeloproliferative CMML cases and usually correspond to secondary events that may lead to AML progression (Pich et al, 2009; Kohlmann et al, 2010; Ricci et al, 2010; Schwaab et al, 2012; Itzykson et al, 2013b; Smith et al, 2015; Elena et al, 2016).
Other genes recurrently mutated in MDS and CMML that are involved in other cellular processes, include TP53, SETBP1, BCOR and BCORL1 (Bejar et al, 2011; Jädersten et al, 2011; Damm et al, 2013a,2013b; Bejar et al, 2014; Elena et al, 2016). TP53 mutations are more frequent in high risk patients and are associated with adverse prognosis features, including advanced disease, complex karyotypes, monosomal karyotypes, excess of blasts, severe thrombocytopenia and therapy‐related MDS (Kita‐Sasai et al, 2001; Bejar et al, 2011; Kulasekararaj et al, 2013; Ok et al, 2015). TP53 mutations are associated with a very poor prognosis in all MDS subtypes, including patients who have undergone an haematopoietic stem cell transplantation (Bejar et al, 2011; Kulasekararaj et al, 2013; Bejar et al, 2014). TP53 mutations are also associated with the presence of del(5q) and with a lower response rate to lenalidomide treatment in these patients (Jädersten et al, 2011; Meggendorfer et al, 2017).
Myeloid neoplasms with germline predisposition
Although most cases of MDS or AML are sporadic diseases, a number of cases arise as the result of genetic predisposition due to the presence of inherited germline mutations, which has led the WHO classification to include a new section on myeloid neoplasms with germline predisposition. This section comprises cases of MDS, MDS/MPN and AML that arise on the background of a predisposing germline mutation (Arber et al, 2016). Several genes have been associated with a genetic predisposition to myeloid malignancies, including ANKRD26, CBL, CEBPA, DDX41, ELANE, ETV6, GATA2, GSKIP, MPL, NF1, SAMD9, SAMD9L, PTPN11, RUNX1, SRP72 and TP53, among others (Kennedy & Shimamura, 2019). The majority of these genes are included in myeloid‐related targeted panels focused on somatic mutations. In addition, recent studies suggest that the frequency of germline variants in some of these genes, like DDX41, may be underappreciated, suggesting that they should also be considered for inclusion in a panel of recommended genes (Berger et al, 2017; Tawana et al, 2018). This may be notably relevant in the context of the search for a related donor. Considering all the above, when performing targeted sequencing in MDS patients, the possibility of detecting a germline mutation must be considered. Although a variant allele frequency (VAF) close to 50% or 100% may be suggestive of a germline mutation, its nature should be confirmed using a control germline sample before acting on it, and should always be evaluated in the clinical and familial context of each patient. Even though germline genetic predisposition is frequently associated with MDS with early onset (children and young adults), it is increasingly recognized also in MDS cases diagnosed in advanced ages. The prevalence of hereditary myeloid malignancies has not been clearly established but is suspected to concern at least 5% of adult patients with MDS/AML (Tawana et al, 2018). In addition, currently known MDS/AML predisposition genes account for only 25% of familial cases, so other risk loci remain to be discovered (Tawana et al, 2018). When a germline mutation is confirmed, referral of patients to professionals with expertise in cancer predisposing syndromes and in genetic counselling is recommended, due to the need for highly specialized and rapidly advancing evaluations and tailored treatments for these patients (Godley & Shimamura, 2017).
Clonal haematopoiesis of uncertain significance
Recurrent somatic mutations in MDS‐associated genes, such as DNMT3A, TET2, ASLX1, TP53 and JAK2 genes, have been identified in the peripheral blood of healthy individuals with advanced age and normal blood counts (Genovese et al, 2014; Jaiswal et al, 2014; Xie et al, 2014). These mutations, usually detected at very low VAFs (<10%), provide a selective advantage to the haematopoietic stem cells in which they occur, but cells still maintain the ability to differentiate into circulating leucocytes (Steensma et al, 2015). This phenomenon has been defined as clonal haematopoiesis of indeterminate potential (CHIP) (Table 4) (Steensma et al, 2015). The incidence of CHIP increases with age, with more than 10% of healthy people older than 70 years of age carrying a mutation in one of those genes (Genovese et al, 2014; Jaiswal et al, 2014). These individuals have an increased risk of cardiovascular disease and a higher risk of developing an haematological malignancy, although thankfully the rate of malignant transformation is very low (0·5–1% per year) (Jaiswal et al, 2014, 2017). This risk increases when the VAFs are higher (>10%) or more than one mutation is detected. In addition, recent studies have shown that patients with solid tumours who have CHIP before being exposed to treatment, are at increased risk of developing a therapy‐related myeloid neoplasm (TRMN) (Takahashi et al, 2013; Gillis et al, 2017). Factors that influence the natural history of CHIP are currently under investigation, but it is known that the acquisition of further mutations drives the progression of CHIP to overt malignancy.
Table 4.
Characteristics of CHIP, ICUS, CCUS and MDS.
| CHIP | ICUS | CCUS | MDS | |
|---|---|---|---|---|
| Cytopenia | No | Yes (≥1) | Yes (≥1) | Yes (≥1) |
| Dysplasia | No | No, or minimum | No, or minimum | Yes |
| Mutations | Yes (n = 1) | No | Yes (n ~ 1, 6) | Yes >85% (n ~ 2, 6) |
| VAF | 2–12% | – | 30–40% | 30–50% |
| Progression Risk | 0.5–1% per year | >10% in 5 years | >85% in 5 years |
CCUS, clonal cytopenia of uncertain significance; CHIP, clonal haematopoiesis of uncertain significance; ICUS, idiopathic cytopenia of uncertain significance; MDS, myelodysplastic syndrome; VAF, variant allele frequency.
Apart from healthy individuals, mutations in these MDS‐associated genes can also be detected in 35% of patients with persistent blood cytopenias for which no explanation is apparent, usually called idiopathic cytopenias of undetermined significance (ICUS) (Table 4). These cases, that present with both clonal haematopoiesis and cytopenias, are designated clonal cytopenias of uncertain significance (CCUS). Patients with CCUS are at a higher risk of developing a myeloid malignancy compared to those individuals with CHIP not presenting any cytopenia, especially in the presence of two or more mutations or if a single mutation in a splicing gene is detected (Arber et al, 2016; Bejar, 2017; Jaiswal et al, 2017) (Table 4). In addition, average VAFs in patients with CCUS (30–40%) are higher compared to those found in CHIP.
Taking all these observations into account, when performing NGS routine tests in a patient with an indication of MDS, diagnosis should always be performed in the context of other laboratory and clinical data.
Sequencing workflow and quality criteria
Targeted NGS panels commonly include a selected number of specific genes, or coding regions within genes that are clinically‐actionable in the disease of interest, or that are known to harbour mutations that contribute to the pathogenesis of the disease. Focusing on a limited set of genes allows greater depth of coverage for increased analytical sensitivity and specificity. Thus, targeted sequencing is often used in clinical care because, while keeping the cost relatively low, it provides greater confidence and facilitates the interpretation of the findings in their clinical context. The choice of sequencing strategy will depend on several factors, including size of the panel, sequencing platform, turnaround time (TAT) for reporting the results and cost per sample.
Panel design strategy
When designing a custom panel, it is recommended to select targets and transcripts only from genes with potential clinical relevance. There are several available online tools that can be used to design the specific primers/probes to enrich the targets of interest. Although custom panels are usually flexible and can be modified over time, they require a long process of optimization and validation, which is skipped when using off‐the‐shelf panels. On the other hand, commercial panels offer a closed design, but do not require a previous design phase and are already optimized. Of note, these panels are commercially available as ready‐to‐use, but quite often they still require validation in each laboratory.
Library preparation
In MDS and CMML, as in most haematological neoplasms, it is recommended to perform molecular analyses on bone marrow‐derived cells. Some studies suggest that, due to the high sensitivity of NGS, there is little difference between bone marrow and peripheral blood samples (Duncavage et al, 2017; Martin et al, 2018). Therefore, if bone marrow is not available, peripheral blood can be useful, especially for molecular monitoring or in myeloproliferative disorders like CMML. Although DNA from fresh samples is the optimal input material, targeted NGS can be performed on any DNA sample as long as the quality and quantity are sufficient. DNA purity can be measured by spectrophotometry (i.e. Nanodrop device, Thermo Fisher Scientific, Waltham, MA, USA), while DNA integrity can be assessed by capillary electrophoresis (i.e. Tape Station or Bioanalyzer devices, Agilent, Santa Clara, CA, USA). Input DNA and DNA libraries should be quantified using a fluorometric assay (i.e. Qubit or Quant‐iT PicoGreen, Thermo Fisher Scientific) that can distinguish and measure the concentration of double‐stranded DNA. The manufacturer’s recommendations should be strictly followed during library preparation.
Two main strategies for library preparation are widely used in targeted NGS: amplicon‐based and hybrid‐capture‐based. Amplicon panels are based on the use of specific primers and the amplification of the regions of interest by polymerase chain reaction (PCR)‐based methods (single or multiplex PCR, or emulsion PCR). On the other hand, target enrichment is performed in capture panels using probe hybridization‐based methods. In both cases, DNA fragments generated during library preparation contain adapter sequences on both ends, which are complementary to platform‐specific sequencing primers. Samples can be tagged with a unique oligonucleotide barcode that enables pooling of different patient samples in the same sequencing run (Table S1). In addition, some strategies use unique molecular barcodes that tag each original DNA fragment and allow identification and removal of PCR duplicates during bioinformatic processing of sequencing data.
Sequencing platforms
Sequencers for NGS, based on the ability to perform an elevated number of chemical reactions in parallel, are increasingly being used in the clinical setting. Sequencing instruments can be classified according to the mean read length (short or long), the type of sequencing (single‐end and paired‐end sequencing) and the sequencing chemistry (mainly by synthesis, ligation or hybridization). Multiple factors should be considered when choosing a sequencing platform, such as run time, size of sequenced region, required depth of coverage, read length, TAT requirements and cost per sample. According to the GESMD, Illumina (San Diego, CA, USA) and Thermo‐Fisher Scientific, short‐read sequencing platforms are ideal for the implementation of targeted deep sequencing in diagnostic laboratories (Table S2).
There is a number of sequencing parameters that define the scope and quality of data output of a given NGS technology: sequencing capacity, type and length of the reads, sequencing output, depth of coverage, percentage of reads mapped correctly and uniformity of the reads (Table S3). Some of these parameters can be influenced by other variables, such as input DNA quality, adequate library preparation, correct design of the primers/probes, choice of sequencing platform, and the initial estimation of the number of required reads. Regarding the depth of coverage, we recommend that ≥95% of the bases targeted in the panel design are covered by at least 100 reads (≥100×), and that the mean coverage for each sample is ≥1000×. This will enable the identification of variants with a sensitivity of at least 5% in most regions, as long as variant quality criteria are fulfilled (we recommend the presence of at least 25 reads for the alternative allele). Of note, even though this is our recommendation, less conservative approaches are also likely to be highly valid, especially for certain hotspot locations, and should be validated in each laboratory. The minimum output required per library can be estimated by taking into account the intended depth of coverage together with the size of the panel (including on‐ and off‐target regions). It is recommended that the percentage of on‐target mapped reads is higher than 70%.
Test validation
During test development, iterative cycles of performance optimization should be executed until all the different components of the assay are optimized, including panel design, library preparation conditions, sequencing parameters, number of samples that should be pooled in order to achieve the desired coverage level, as well as analysis settings and pipeline. TAT for data reports need to be estimated and should be clinically appropriate. Once the entire panel conditions have been established, the assay needs to be validated, and performance characteristics (such as sensitivity, specificity and reproducibility) should be determined.
Two rounds of validation should be consecutively performed. Firstly, a technical validation round aimed at evaluating the quality of the design and identifying the regions with poor coverage. For technical validation, input DNA requirements, library generation and sequencing should be performed, strictly following manufacturer’s recommendations. During this process, the percentage of bases covered at different depths of coverage should be determined, as well as the regions with low or no coverage, which should be then redesigned or removed from the design. Biological replicates should also be assessed, including sequencing two libraries generated from the same input DNA and sequencing two libraries from two different DNA extractions of the same sample. Recurrent artefacts should also be identified. For example, variants that appear at greater frequency than expected in a large cohort or that have reads supporting them in a large number of samples, especially if they all cluster at similar VAFs, are likely to be technical artefacts. Generation of internal databases with known technical artefacts can be helpful in order to filter these artefacts out during the analysis‐filtering steps. Secondly, a clinical validation round should also be performed with the aim of determining if the designed test is able to detect known alterations associated with the disease of interest. In order to do so, previously characterized samples with known SNVs and/or indels, should be analysed with the new test. Whenever possible, previously undescribed variants and complex indels should be validated by other technologies, such as Sanger sequencing, amplification refractory mutation system (ARMS)‐PCR, allele‐specific oligonucleotide (ASO)‐PCR, digital PCR or other NGS platforms. Finally, sequencing of serial dilutions from the same sample will help to determine the sensitivity of the test.
In NGS, standards for TAT should be established by each laboratory based on the clinically proper indication for testing. Multiplexing samples from different diseases and using small‐scale sequencing systems can help reduce TAT. In real world scenario of diagnosis of MDS and CMML, these TAT should be consistent with other cytogenetic tests (karyotype and fluorescence in situ hybridisation), and should not exceed, for most cases, 15 working days (Rack et al, 2019). Urgent referrals should be prioritized and, for these cases, results should try to be reported within 10 days.
Data analysis and variant filtering
Data analysis
The goal of data analysis is to use bioinformatic tools within an analysis pipeline in order to transform the raw data coming from the sequencer into a list of variants that can be visualized, filtered and interpreted. The challenge of data analysis includes the huge number of available tools, the constantly evolving analysis pipelines and the lack of consensus regarding which tools to use. Typical NGS data analysis workflows include base calling, read alignment variant calling and variant annotation. Data analysis steps and tools that are recommended, and most commonly used, by the GESMD are summarized in (Data S1, Tables S4, S5). It should be noted that several open‐source and commercial tools have been developed to facilitate NGS data analysis in the clinical setting, in the form of user‐friendly interfaces. Some of these tools, used by the GESMD, include MiSeq Reporter (Illumina), Variant Studio (Illumina), IonReporter (Thermo‐Fisher Scientific), DNAnexus (DNAnexus, Santa Clara, CA, USA) and Sophia DDM (Sophia Genetics, Lausanne, Vaud, Switzerland).
Variant filtering
After data analysis, variant filtering should then be performed in order to obtain a list of candidate variants that will later be categorized, interpreted and reported. Of note, variant filtering criteria should always be updated according to the advances made in NGS technologies and in the disease of interest. The variant filtering workflow proposed by the GESMD is described in Table 5 and Fig 2.
Table 5.
Variant filtering workflow detailed information proposed by the GESMD.
| Step | Description |
|---|---|
| Variant pre‐filtering | |
| Filter according to variant region |
|
| Remove sequencing errors |
|
| Variant filtering | |
| Polymorphisms |
|
| Synonymous variants |
|
| Variants in UTR regions |
|
| Quality criteria | |
| Coverage |
|
BAM, binary alignment map; bp, base pairs; CMML, chronic myelomonocytic leukaemia; GESMD, Spanish MDS Group; IGV, Integrative Genomics Viewer; MAF, minor allele frequency; MDS, myelodysplastic syndrome; SAM, sequence alignment map; SNV, single nucleotide variant; UTR, untranslated region.
Figure 2.
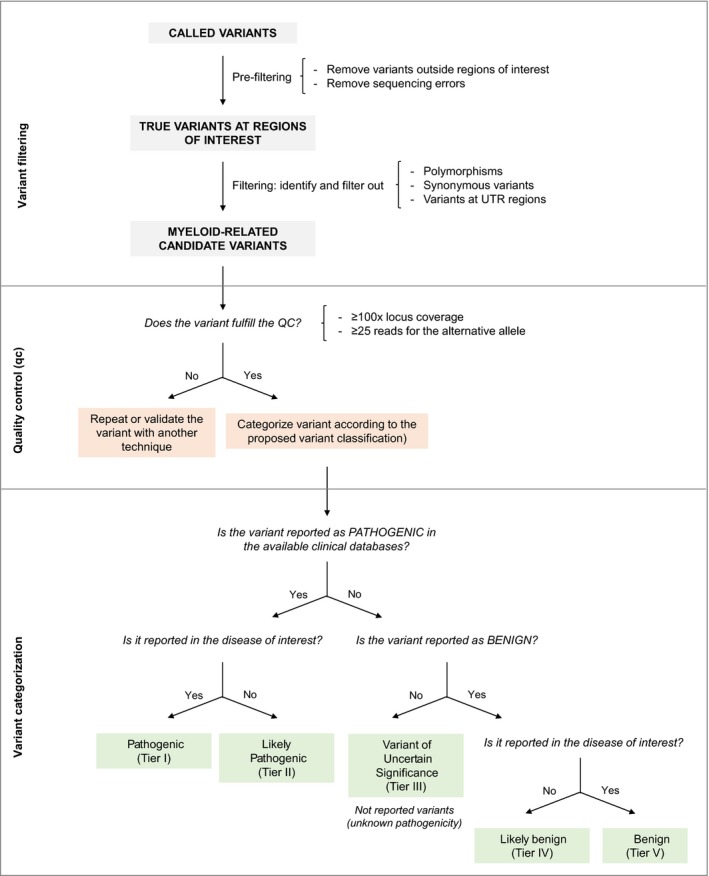
Proposed workflow for variant filtering and categorization. UTR, untranslated region.
Variant interpretation and categorization
Interpretation systems for classifying variants are useful to standardize the way in which variants are reported to clinicians. In response to the classification discrepancies that often exist between laboratories, the use of unified guidelines is highly recommended. On one hand, germline variants classification systems aim to classify variants according to their pathogenicity in a given disease (Richards et al, 2015; Matthijs et al, 2016). A joint consensus between the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, for example, proposes updated standards for variant assessment and promotes the systematic evidence‐based classification of variants found in Mendelian disorders (Richards et al, 2015). On the other hand, in cancer, somatic variants are assessed for diagnostic, prognostic, predictive and/or therapeutic impact in the context of tumour site and histology (Sukhai et al, 2016). Given their relevance, a system to prioritize variants detected by whole exome sequencing (WES) as well as two somatic tier‐based variant classification systems have been reported in the literature (Van Allen et al, 2014; Sukhai et al, 2016; Li et al, 2017). Sukhai et al (2016) propose a classification scheme in which variants are categorized from Class 1 (highest priority) to Class 5 depending on the influence that the variant shows on diagnosis, prognosis or treatment of the specific tumour being evaluated. Similarly, Li et al (2017) classify variants from Tier I (Variants with Strong Clinical Significance) to Tier IV (Benign or Likely Benign Variants) based on the clinical impact of a given variant, which is determined according to currently available evidence. It is of high importance to include a separate category for benign variants, to inform clinicians and patients and to reduce the burden on laboratories (Hoskinson et al, 2017). Based on our experience, the use of specific standard terminology (pathogenic, likely pathogenic, uncertain significance, likely benign and benign) facilitates the use of classification systems, especially for clinicians.
In light of this, and based on the same tier‐based classification system of the American Association for Molecular Pathology, American Society of Clinical Oncology and College of American Pathologists (Li et al, 2017), we propose a system to classify somatic variants detected in myeloid neoplasms (Fig 2).
Tools for variant interpretation
In order to properly classify and annotate the detected somatic variants, several currently available repositories and genomic databases can be used. Before using these databases, clinical laboratories should make an effort to understand their content and note their limitations (Li et al, 2017). Likewise, establishing and consulting an in‐house database of variants identified in the laboratory is recommended. The tools recommended by the GESMD are listed in Table 6.
Table 6.
List of web resources useful for variant interpretation.
Reference sequence databases
Reference sequence databases encompass the human reference genome and related information for the unequivocal localization of a variant in the genome. These databases annotate variant location (coding, non‐coding, splicing site or untranslated region) and the strand representation (positive or negative) within the corresponding gene. Frequently consulted resources include Ensembl [European Molecular Biology Laboratory‐ European Bioinformatics Institute (EMBL‐EBI) and the Wellcome Trust Sanger Institute; http://www.ensembl.org/index.htm], RefSeq [National Center for Biotechnology Information (NCBI) Reference Sequence Database; https://www.ncbi.nlm.nih.gov/refseq] and Locus Reference Genomic (LRG, NCBI and EMBL‐EBI; https://www.lrg-sequence.org/). This information is essential for the correct nomenclature of the variants according to the Human Genome Variation Society (HGVS) (Li et al, 2017).
Population databases
Population databases are useful for obtaining the frequency of variants at a given locus in a variety of geographically distinct populations (Cottrell et al, 2014; Richards et al, 2015). They are commonly used to filter out polymorphic variants based on an established cut‐off of minor allele frequency (MAF). Although a standardized MAF to discriminate between polymorphic or benign variants has not been established yet, current practice within GESMD sets 1% as the cut‐off point in studies of somatic variants. Additionally, an ethnic specificity of the variant should be considered based on the ethnic background of the patient. Finally, we would emphasise a cautionary note when consulting these databases, as they were built under the assumption that the population included in them is healthy, but some somatic variants may accidentally have been included as well. For example, common hotspot mutations, such as DNMT3A R882 or JAK2 V617F, can be found in some of these databases and therefore a whitelist of known hotspot mutations could be useful to rescue these variants.
Recommended population databases include The Database of Short Genetic Variation (dbSNP, the NCBI database of genetic variation; https://www.ncbi.nlm.nih.gov/snp/), Exome Variant Server (EVS, NHLBI GO Exome Sequencing Project; http://evs.gs.washington.edu/EVS/), 1000 Genomes Project (International Genome Sample Resource, IGSR; http://browser.1000genomes.org), Exome Aggregation Consortium (ExAC, Broad Institute; http://exac.broadinstitute.org/) and Genome Aggregation Database (gnomAD, Broad Institute; http://gnomad.broadinstitute.org/). Databases such as ExAC or gnomAD are more accurate than dbSNP when it comes to filtering polymorphisms and neutral variants.
Somatic and germline variants in clinical databases
Both somatic and germline variants are included in clinical databases. The incidence and prevalence of a variant is often detailed in different subtypes of cancers and functional prediction algorithms are provided to categorize the variants according to their pathogenicity. Frequently, clinical databases also include information for accurate annotation and prioritization of somatic variants; bibliographic references with or without systematic review; additional information about the tissue in which the variant has been described; outcome disease data or targeted therapies, among others. An example is the National Cancer Institute's Genome Data Commons (https://gdc.cancer.gov), which contains all the genomic data generated at the National Cancer Institute, and which includes the well‐known databases of The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/), the Therapeutically Applicable Research to Generate Effective Therapies (TARGET; https://ocg.cancer.gov/programs/target), and the Cancer Genome Characterization Initiative (CGCI; https://ocg.cancer.gov/programs/cgci). Another public database is the Catalog of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) from the Wellcome Sanger Institute, which contains millions of somatic variations described in numerous types of tumours and has also recently included intron variants. However, the information provided in these databases is not always up‐to‐date. In addition, a poor representation of pathological diagnostic standards is detected as well as a lack of an exhaustive validation of the findings and/or the sources from which the included variants were extracted.
Germline variants are also usually detected upon tumour sequencing. Such variants may be associated with cancer predisposition syndromes. To evaluate them, there are several germline mutations databases, such as Human Gene Mutation Database (Institute of Medical Genetics; http://www.hgmd.cf.ac.uk/ac/index.php) or ClinVar (NCBI; http://www.ncbi.nlm.nih.gov/clinvar).
Finally, for TP53 gene variant interpretation, the International Agency for Research on Cancer (IARC) TP53 Database (WHO; http://p53.iarc.fr/TP53GeneVariations.aspx) and TP53 Website (Centre de Recherche des Cordeliers and Karolinska Institute; http://p53.fr/) are useful resources for evaluating their impact in haematological malignancies, because they include the frequencies of the variants observed in both the somatic and germinal contexts.
In‐house laboratory databases
We recommend that each clinical laboratory should establish an in‐house database to provide consistent variant annotation and categorization, to determine the frequency of mutations and to identify platform‐specific artefacts or potential false‐positive variants. A continuous review of new publications, clinical trials and databases used in variant interpretation, which are regularly updated, should be carried out to ensure that variant annotation is up‐to‐date. To increase the quality of interpretation, we recommend that in‐house databases are complemented with further relevant annotation, including results from other techniques such as morphology, cytogenetic and immunophenotyping, because NGS results should always be combined and interpreted in the context of other laboratory and clinical diagnostic data. Moreover, it would be extremely useful to share those internal databases with other centres/laboratories that perform similar determinations. Indeed, the GESMD is making that effort by building the RESMDmol database (https://www.gesmd.es/carrerasresearch/index.php), in view of facilitating the homogenization of results informed in the clinical reports.
Functional consequence of the variant
In silico predictive algorithms estimate the possible deleterious consequences of a variant on the encoded protein of a given gene. The criteria used to determine this effect varies across different tools. Algorithms designed to predict the functional consequence of a missense variant are commonly based on the evolutionary conservation of an amino acid or nucleotide, the location and context within the protein sequence and the biochemical consequence of the amino acid substitution (Brunak et al, 1991; Vreeswijk et al, 2009; Thusberg et al, 2011). In silico tools have also been developed to evaluate the consequence of splicing variants related to the creation or loss of splice sites at the exonic or intronic regions (Thusberg et al, 2011; Houdayer et al, 2012). However, in the context of cancer, the interpretation of these predictions is usually not easy, especially for gain‐of‐function mutations. Therefore, it is recommended that least four different prediction algorithms should be used to analyse each variant, although the obtained output should never be used as sole evidence for clinical decision making, as, to date, there is no consensus about the relevance of each predictor, nor about how to combine outputs from different prediction algorithms (Li et al, 2017).
In the case of variants with available information about their functional effect, two scenarios can be considered. On one hand, when the pathogenicity of the affected gene is associated with a loss of function, frameshift variants must be interpreted as probably pathogenic, as long as the variant does not affect the last exon of the gene, unless the pathogenicity of those variants affecting the last exon has been validated (i.e. variants in ASXL1 and CALR). Loss of function variants located at the last exon of a gene will need evaluation according to other parameters, such as the type of protein domain that is altered and the effect of other proximity variants. Accordingly, these will be classified as variants of uncertain significance or probably pathogenic. On the other hand, when the pathogenicity of the affected gene is associated with a gain‐of‐function, and a frameshift variant is detected, this will be interpreted as a variant of uncertain significance unless there is strong evidence of pathogenicity based on functional studies in the literature.
Criteria for variant classification
We recommend to interpret and report variants that fulfil all three following criteria: (i) MAF < 1%, (ii) VAF ≥ 5% and (iii) minor allele coverage ≥25 reads. Although we recommend that cut‐off of VAF ≥ 5% should be used, the analyst should consider informing variants with lower allele frequencies in particular cases, such as hotspot regions in genes with clear clinical relevance, such as TP53 or JAK2. However, in order to report variants with a VAF below 5%, the depth of coverage must be increased to maintain, at least, the minor allele coverage over 25 reads.
The GESMD proposes that variants should be categorised according to the following criteria: (i) relevance of the altered gene based on its actionability, defined as being relevant for the diagnosis/classification, prognosis and/or treatment (therapeutic target or related to sensitivity, toxicity or resistance to therapy) of the disease; (ii) presence of the variant in clinical databases and published literature that indicates the recurrence or pathogenicity of the identified variant; (iii) tissue and/or tumour histology in which the variant has been described; and (iv) in silico predictive algorithms and functional studies.
Therefore, the categorization of the variant must be based on its clinical impact on the specific disease and tissue under study. Due to the importance of the definition of the actionability of a gene, we strongly recommend following the evidence defined by Sukhai et al (2016). Of note, determining the actionability of a gene will be particularly relevant when using pan‐haematological or oncological targeted NGS panels that include genes which have not been previously described in the disease of interest, as well as for WES and whole genome sequencing.
Variant classification system
According to our categorization criteria, variants will be classified into one of the following classes (Fig 2, Table 7):
Table 7.
Classification variant system.
| Category | Criterion | ||||
|---|---|---|---|---|---|
| Gene actionability | Variant clinical significance | Variant recurrence in databases | Tissue and/or tumour histology | Predictive algorithms and functional studies | |
| Pathogenic (Tier I) | Actionable | Diagnostic, prognostic and/or treatment significance in in the disease of interest (Biomarker) | Described and confirmed as pathogenic | Disease or tissue of interest | NA |
| Likely pathogenic (Tier II) | Actionable | Clinical significance in other haematological neoplasms or solid tumors | Described as pathogenic | Other haematological neoplasms or solid tumors | NA |
| Variant of uncertain significance (VUS) (Tier III) | Actionable/not actionable | Of uncertain significance | Unknown pathogenicity | NA | Likely pathogenic |
| Likely benign (Tier IV) | Actionable/not actionable | Clinically irrelevant | Described as benign | Other haematological neoplasms or solid tumors | Probably benign |
| Benign (Tier V) | Actionable/not actionable | Clinically irrelevant | Described and confirmed as benign | Disease or tissue of interest | Probably benign |
NA, not applicable.
Pathogenic (Tier I)
Described in the literature as relevant for the clinical management of MDS/CMML or other myeloid neoplasms. The variant affects an actionable gene and has been established as a pathogenic variant in myeloid neoplasms.
Likely pathogenic (Tier II)
Described in the literature but with no established relevance in the clinical management of MDS or other myeloid neoplasms. The variant affects an actionable gene but has been established as a pathogenic variant in solid tumours or in non‐myeloid haematological neoplasms. This category also includes variants previously not described for which predictive algorithms strongly classify them as pathogenic.
Variant of uncertain significance (VUS) (Tier II)
Previously described but with no sufficient evidence of its pathogenicity on actionable MDS genes, other haematological neoplasms or solid tumours. This category also includes variants not previously described for which predictive algorithms cannot strongly classify them into pathogenic or benign.
Likely benign (Tier IV) or benign (Tier V)
Described in the literature as clinically insignificant because it has no effect on the protein. Frequently, these variants present MAF > 1% and, sometimes, VAF of approximately 50% or 100%. These levels of VAF may be indicative of the germline nature of the variant and, therefore, are neither acquired nor tumour‐specific. However, the VAF criterion should be considered with caution, as there are somatic variants present in major clones with high VAF. This category also includes variants previously not described for which predictive algorithms strongly classify them as benign. We recommend that, to avoid confusion, benign and likely benign variants should not be included in the clinical report.
Clinical report
Next generation sequencing clinical reports should follow the general professional organizations’ recommendations and guidelines (Richardson, 2002; Rack et al, 2019), such as avoiding long reports for the benefit of clarity, inclusion of pagination, and reviewing and signing by, preferably, two geneticists with the relevant expertise on haematological malignancies. We recommend that the report includes (at least) the information detailed below.
Patient identification
As in current standard tests, proper patient identification should be incorporated in the clinical report, including two unique patient identifiers, demographic data and referral information (reason for referral, suspected or confirmed diagnosis and clinical time point). Sample details should also be provided (type, source and received date) as well as hospital and referring physician. A statement of signed informed consent should also be made.
Methodology
Details on the methodology used should be specified, including library preparation chemistry and sequencing instrument. Regions included in the panel (genes according to the Human Genome Organisation nomenclature, exons and hotspots) and low covered regions should be detailed. Analysis settings, filters and cut‐off values (such as minimum coverage or VAF) should be specified. The version of the human reference sequence to which sequence reads are aligned should also be included, as well as the clinical databases used for annotation. Key quality control metrics and limitations need also be listed.
Results and interpretation
A list of identified variants should be included in the results section, annotated in accordance to the HGVS mutation nomenclature. For each variant, we recommend including the following information: gene, exon, nucleotide change, protein change, type of variant (missense, frameshift, nonsense, splicing), transcript RefSeq ID, variant ID from clinical databases, VAF and position depth of coverage. When using a variant classification system, the categorization of each variant should be included and the used classification system should be described. Regarding variant interpretation, we recommend including a critical summary of the clinical relevance of the variant in the disease of interest, including treatment or referenced clinical trials information, if applicable. For the categorization of the disease, the WHO 2017 nomenclature should be used (Arber et al, 2016). Further studies to validate the significance of the results obtained should be conducted when appropriate.
Conclusion
We have long awaited the introduction of NGS into the routine diagnostic armamentarium of myeloid malignancies. Currently, an increasing number of laboratories are implementing NGS procedures into the diagnostic algorithms of patients with haematological malignancies, opening new horizons for individualized clinical management of these patients. It is therefore of major importance to standardize the generation, analysis and clinical interpretation of NGS data. To that end, the GESMD has expanded the present set of guidelines, aiming at stablishing common quality standards for the adequate clinical interpretation of NGS results, hoping that this effort will ultimately contribute to the benefit of patients with myeloid malignancies.
Author contribution
All authors contributed in the development of these guidelines and in the revision of the manuscript.
Supporting information
Table S1. Main characteristics and differences between amplicon‐ and capture‐based panels.
Table S2. Comparison of Illumina and Thermo Fisher NGS platforms.
Table S3. Main sequencing parameters.
Table S4. List of most commonly used alignment tools.
Table S5. List of most commonly used somatic variant callers.
Data S1. Supplementary methods.
Acknowledgements
This work was supported by a grant from the Spanish Group of MDS (GESMD, 2017). LP, FF, PA and FS research is supported by a grant from 2017 SGR288 (GRC) Generalitat de Catalunya, economical support from CERCA Programme/Generalitat de Catalunya, Fundació Internacional Josep Carreras and from Celgene International. LP and JMHS are supported by a research grant from FEHH (Fundación Española de Hematología y Hemoterapia, 2017). IV acknowledges support from Pethema. MC and LZ research is supported by a grant from Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo, Spain (PI 11/02519). MFM and her research is supported by the Spanish Association against Cancer (AECC, AIO2014), and the Ministerio de Economía y Competitividad of Spanish Central Government (PI16/00159).
References
- Van Allen, E.M. , Wagle, N. , Stojanov, P. , Perrin, D.L. , Cibulskis, K. , Marlow, S. , Jane‐Valbuena, J. , Friedrich, D.C. , Kryukov, G. , Carter, S.L. , McKenna, A. , Sivachenko, A. , Rosenberg, M. , Kiezun, A. , Voet, D. , Lawrence, M. , Lichtenstein, L.T. , Gentry, J.G. , Huang, F.W. , Fostel, J. , Farlow, D. , Barbie, D. , Gandhi, L. , Lander, E.S. , Gray, S.W. , Joffe, S. , Janne, P. , Garber, J. , MacConaill, L. , Lindeman, N. , Rollins, B. , Kantoff, P. , Fisher, S.A. , Gabriel, S. , Getz, G. & Garraway, L.A. (2014) Whole‐exome sequencing and clinical interpretation of formalin‐fixed, paraffin‐embedded tumor samples to guide precision cancer medicine. Nature Medicine, 20, 682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber, D.A. , Orazi, A. , Hasserjian, R. , Thiele, J. , Borowitz, M.J. , Le Beau, M.M. , Bloomfield, C.D. , Cazzola, M. & Vardiman, J.W. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 127, 2391–2405. [DOI] [PubMed] [Google Scholar]
- Bejar, R. (2017) CHIP, ICUS, CCUS and other four‐letter words. Leukemia, 31, 1869–1871. [DOI] [PubMed] [Google Scholar]
- Bejar, R. , Stevenson, K. , Abdel‐Wahab, O. , Galili, N. , Nilsson, B. , Garcia‐Manero, G. , Kantarjian, H. , Raza, A. , Levine, R.L. , Neuberg, D. & Ebert, B.L. (2011) Clinical effect of point mutations in myelodysplastic syndromes. The New England Journal of Medicine, 364, 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar, R. , Stevenson, K.E. , Caughey, B.A. , Abdel‐Wahab, O. , Steensma, D.P. , Galili, N. , Raza, A. , Kantarjian, H. , Levine, R.L. , Neuberg, D. , Garcia‐Manero, G. & Ebert, B.L. (2012) Validation of a prognostic model and the impact of mutations in patients with lower‐risk myelodysplastic syndromes. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 30, 3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar, R. , Stevenson, K.E. , Caughey, B. , Lindsley, R.C. , Mar, B.G. , Stojanov, P. , Getz, G. , Steensma, D.P. , Ritz, J. , Soiffer, R. , Antin, J.H. , Alyea, E. , Armand, P. , Ho, V. , Koreth, J. , Neuberg, D. , Cutler, C.S. & Ebert, B.L. (2014) Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem‐cell transplantation. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 32, 2691–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, G. , van den Berg, E. , Sikkema‐Raddatz, B. , Abbott, K.M. , Sinke, R.J. , Bungener, L.B. , Mulder, A.B. & Vellenga, E. (2017) Re‐emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia, 31, 520–522. [DOI] [PubMed] [Google Scholar]
- Boultwood, J. , Perry, J. , Pellagatti, A. , Fernandez‐Mercado, M. , Fernandez‐Santamaria, C. , Calasanz, M.J. , Larrayoz, M.J. , Garcia‐Delgado, M. , Giagounidis, A. , Malcovati, L. , Della Porta, M.G. , Jädersten, M. , Killick, S. , Hellström‐Lindberg, E. , Cazzola, M. & Wainscoat, J.S. (2010) Frequent mutation of the polycomb‐associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia, 24, 1062–1065. [DOI] [PubMed] [Google Scholar]
- Brunak, S. , Engelbrecht, J. & Knudsen, S. (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. Journal of Molecular Biology, 220, 49–65. [DOI] [PubMed] [Google Scholar]
- Cazzola, M. , Della Porta, M.G. & Malcovati, L. (2013) The genetic basis of myelodysplasia and its clinical relevance. Blood, 122, 4021–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.‐Y. , Lin, L.‐I. , Tang, J.‐L. , Ko, B.‐S. , Tsay, W. , Chou, W.‐C. , Yao, M. , Wu, S.‐J. , Tseng, M.‐H. & Tien, H.‐F. (2007) RUNX1 gene mutation in primary myelodysplastic syndrome–the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. British Journal of Haematology, 139, 405–414. [DOI] [PubMed] [Google Scholar]
- Cottrell, C.E. , Al‐Kateb, H. , Bredemeyer, A.J. , Duncavage, E.J. , Spencer, D.H. , Abel, H.J. , Lockwood, C.M. , Hagemann, I.S. , O’Guin, S.M. , Burcea, L.C. , Sawyer, C.S. , Oschwald, D.M. , Stratman, J.L. , Sher, D.A. , Johnson, M.R. , Brown, J.T. , Cliften, P.F. , George, B. , McIntosh, L.D. , Shrivastava, S. , Nguyen, T.T. , Payton, J.E. , Watson, M.A. , Crosby, S.D. , Head, R.D. , Mitra, R.D. , Nagarajan, R. , Kulkarni, S. , Seibert, K. , Virgin, H.W. , Milbrandt, J. & Pfeifer, J.D. (2014) Validation of a next‐generation sequencing assay for clinical molecular oncology. The Journal of Molecular Diagnostics: JMD, 16, 89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm, F. , Chesnais, V. , Nagata, Y. , Yoshida, K. , Scourzic, L. , Okuno, Y. , Itzykson, R. , Sanada, M. , Shiraishi, Y. , Gelsi‐Boyer, V. , Renneville, A. , Miyano, S. , Mori, H. , Shih, L.‐Y. , Park, S. , Dreyfus, F. , Guerci‐Bresler, A. , Solary, E. , Rose, C. , Cheze, S. , Prébet, T. , Vey, N. , Legentil, M. , Duffourd, Y. , de Botton, S. , Preudhomme, C. , Birnbaum, D. , Bernard, O.A. , Ogawa, S. , Fontenay, M. & Kosmider, O. (2013a) BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood, 122, 3169–3177. [DOI] [PubMed] [Google Scholar]
- Damm, F. , Itzykson, R. , Kosmider, O. , Droin, N. , Renneville, A. , Chesnais, V. , Gelsi‐Boyer, V. , de Botton, S. , Vey, N. , Preudhomme, C. , Clavert, A. , Delabesse, E. , Park, S. , Birnbaum, D. , Fontenay, M. , Bernard, O.A. & Solary, E. (2013b) SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia, 27, 1401–1403. [DOI] [PubMed] [Google Scholar]
- Delhommeau, F. , Dupont, S. , Della Valle, V. , James, C. , Trannoy, S. , Massé, A. , Kosmider, O. , Le Couedic, J.‐P. , Robert, F. , Alberdi, A. , Lécluse, Y. , Plo, I. , Dreyfus, F.J. , Marzac, C. , Casadevall, N. , Lacombe, C. , Romana, S.P. , Dessen, P. , Soulier, J. , Viguié, F. , Fontenay, M. , Vainchenker, W. & Bernard, O.A. (2009) Mutation in TET2 in myeloid cancers. The New England Journal of Medicine, 360, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Duncavage, E.J. , Uy, G.L. , Petti, A.A. , Miller, C.A. , Lee, Y.‐S. , Tandon, B. , Gao, F. , Fronick, C.C. , O’Laughlin, M. , Fulton, R.S. , Wilson, R.K. , Jacoby, M.A. , Cashen, A.F. , Wartman, L.D. , Walter, M.J. , Westervelt, P. , Link, D.C. , DiPersio, J.F. , Ley, T.J. & Welch, J.S. (2017) Mutational landscape and response are conserved in peripheral blood of AML and MDS patients during decitabine therapy. Blood, 129, 1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena, C. , Gallì, A. , Such, E. , Meggendorfer, M. , Germing, U. , Rizzo, E. , Cervera, J. , Molteni, E. , Fasan, A. , Schuler, E. , Ambaglio, I. , Lopez‐Pavia, M. , Zibellini, S. , Kuendgen, A. , Travaglino, E. , Sancho‐Tello, R. , Catricalà, S. , Vicente, A.I. , Haferlach, T. , Haferlach, C. , Sanz, G.F. , Malcovati, L. & Cazzola, M. (2016) Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood, 128, 1408–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly, B.B. & Kadam, N.N. (2016) Mutations of myelodysplastic syndromes (MDS): an update. Mutation Research. Reviews in Mutation Research, 769, 47–62. [DOI] [PubMed] [Google Scholar]
- Gelsi‐Boyer, V. , Brecqueville, M. , Devillier, R. , Murati, A. , Mozziconacci, M.‐J. & Birnbaum, D. (2012) Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. Journal of Hematology & Oncology, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese, G. , Kähler, A.K. , Handsaker, R.E. , Lindberg, J. , Rose, S.A. , Bakhoum, S.F. , Chambert, K. , Mick, E. , Neale, B.M. , Fromer, M. , Purcell, S.M. , Svantesson, O. , Landén, M. , Höglund, M. , Lehmann, S. , Gabriel, S.B. , Moran, J.L. , Lander, E.S. , Sullivan, P.F. , Sklar, P. , Grönberg, H. , Hultman, C.M. & McCarroll, S.A. (2014) Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. The New England Journal of Medicine, 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, N.K. , Ball, M. , Zhang, Q. , Ma, Z. , Zhao, Y. , Yoder, S.J. , Balasis, M.E. , Mesa, T.E. , Sallman, D.A. , Lancet, J.E. , Komrokji, R.S. , List, A.F. , McLeod, H.L. , Alsina, M. , Baz, R. , Shain, K.H. , Rollison, D.E. & Padron, E. (2017) Clonal haemopoiesis and therapy‐related myeloid malignancies in elderly patients: a proof‐of‐concept, case‐control study. The Lancet. Oncology, 18, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley, L.A. & Shimamura, A. (2017) Genetic predisposition to hematologic malignancies: management and surveillance. Blood, 130, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert, T.A. , Shen, D. , Ding, L. , Okeyo‐Owuor, T. , Lunn, C.L. , Shao, J. , Krysiak, K. , Harris, C.C. , Koboldt, D.C. , Larson, D.E. , McLellan, M.D. , Dooling, D.J. , Abbott, R.M. , Fulton, R.S. , Schmidt, H. , Kalicki‐Veizer, J. , O’Laughlin, M. , Grillot, M. , Baty, J. , Heath, S. , Frater, J.L. , Nasim, T. , Link, D.C. , Tomasson, M.H. , Westervelt, P. , DiPersio, J.F. , Mardis, E.R. , Ley, T.J. , Wilson, R.K. & Walter, M.J. (2011) Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nature Genetics, 44, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, P.L. , Stone, R.M. , Al‐Kali, A. , Barta, S.K. , Bejar, R. , Bennett, J.M. , Carraway, H. , De Castro, C.M. , Deeg, H.J. , DeZern, A.E. , Fathi, A.T. , Frankfurt, O. , Gaensler, K. , Garcia‐Manero, G. , Griffiths, E.A. , Head, D. , Horsfall, R. , Johnson, R.A. , Juckett, M. , Klimek, V.M. , Komrokji, R. , Kujawski, L.A. , Maness, L.J. , O'Donnell, M.R. , Pollyea, D.A. , Shami, P.J. , Stein, B.L. , Walker, A.R. , Westervelt, P. , Zeidan, A. , Shead, D.A. & Smith, C. (2017) Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network: JNCCN, 15, 60–87. [DOI] [PubMed] [Google Scholar]
- Grossmann, V. , Kohlmann, A. , Eder, C. , Haferlach, C. , Kern, W. , Cross, N.C.P. , Haferlach, T. & Schnittger, S. (2011) Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia, 25, 877–879. [DOI] [PubMed] [Google Scholar]
- Haferlach, T. , Nagata, Y. , Grossmann, V. , Okuno, Y. , Bacher, U. , Nagae, G. , Schnittger, S. , Sanada, M. , Kon, A. , Alpermann, T. , Yoshida, K. , Roller, A. , Nadarajah, N. , Shiraishi, Y. , Shiozawa, Y. , Chiba, K. , Tanaka, H. , Koeffler, H.P. , Klein, H.‐U. , Dugas, M. , Aburatani, H. , Kohlmann, A. , Miyano, S. , Haferlach, C. , Kern, W. & Ogawa, S. (2014) Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia, 28, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskinson, D.C. , Dubuc, A.M. & Mason‐Suares, H. (2017) The current state of clinical interpretation of sequence variants. Current Opinion in Genetics & Development, 42, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer, C. , Caux‐Moncoutier, V. , Krieger, S. , Barrois, M. , Bonnet, F. , Bourdon, V. , Bronner, M. , Buisson, M. , Coulet, F. , Gaildrat, P. , Lefol, C. , Léone, M. , Mazoyer, S. , Muller, D. , Remenieras, A. , Révillion, F. , Rouleau, E. , Sokolowska, J. , Vert, J.‐P. , Lidereau, R. , Soubrier, F. , Sobol, H. , Sevenet, N. , Bressac‐de Paillerets, B. , Hardouin, A. , Tosi, M. , Sinilnikova, O.M. & Stoppa‐Lyonnet, D. (2012) Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Human Mutation, 33, 1228–1238. [DOI] [PubMed] [Google Scholar]
- Itzykson, R. , Kosmider, O. , Renneville, A. , Gelsi‐Boyer, V. , Meggendorfer, M. , Morabito, M. , Berthon, C. , Adès, L. , Fenaux, P. , Beyne‐Rauzy, O. , Vey, N. , Braun, T. , Haferlach, T. , Dreyfus, F. , Cross, N.C.P. , Preudhomme, C. , Bernard, O.A. , Fontenay, M. , Vainchenker, W. , Schnittger, S. , Birnbaum, D. , Droin, N. & Solary, E. (2013a) Prognostic score including gene mutations in chronic myelomonocytic leukemia. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 31, 2428–2436. [DOI] [PubMed] [Google Scholar]
- Itzykson, R. , Itzkson, R. , Fenaux, P. & Solary, E. (2013b) Chronic myelomonocytic leukemia: myelodysplastic or myeloproliferative? Best Practice & Research . Clinical Haematology, 26, 387–400. [DOI] [PubMed] [Google Scholar]
- Itzykson, R. , Kosmider, O. , Cluzeau, T. , Mansat‐De Mas, V. , Dreyfus, F. , Beyne‐Rauzy, O. , Quesnel, B. , Vey, N. , Gelsi‐Boyer, V. , Raynaud, S. , Preudhomme, C. , Adès, L. , Fenaux, P. & Fontenay, M. ; Groupe Francophone des Myelodysplasies (GFM) . (2011) Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia, 25, 1147–1152. [DOI] [PubMed] [Google Scholar]
- Jädersten, M. , Saft, L. , Smith, A. , Kulasekararaj, A. , Pomplun, S. , Göhring, G. , Hedlund, A. , Hast, R. , Schlegelberger, B. , Porwit, A. , Hellström‐Lindberg, E. & Mufti, G.J. (2011) TP53 mutations in low‐risk myelodysplastic syndromes with del(5q) predict disease progression. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 29, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Jaiswal, S. , Fontanillas, P. , Flannick, J. , Manning, A. , Grauman, P.V. , Mar, B.G. , Lindsley, R.C. , Mermel, C.H. , Burtt, N. , Chavez, A. , Higgins, J.M. , Moltchanov, V. , Kuo, F.C. , Kluk, M.J. , Henderson, B. , Kinnunen, L. , Koistinen, H.A. , Ladenvall, C. , Getz, G. , Correa, A. , Banahan, B.F. , Gabriel, S. , Kathiresan, S. , Stringham, H.M. , McCarthy, M.I. , Boehnke, M. , Tuomilehto, J. , Haiman, C. , Groop, L. , Atzmon, G. , Wilson, J.G. , Neuberg, D. , Altshuler, D. & Ebert, B.L. (2014) Age‐related clonal hematopoiesis associated with adverse outcomes. The New England Journal of Medicine, 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, S. , Natarajan, P. , Silver, A.J. , Gibson, C.J. , Bick, A.G. , Shvartz, E. , McConkey, M. , Gupta, N. , Gabriel, S. , Ardissino, D. , Baber, U. , Mehran, R. , Fuster, V. , Danesh, J. , Frossard, P. , Saleheen, D. , Melander, O. , Sukhova, G.K. , Neuberg, D. , Libby, P. , Kathiresan, S. & Ebert, B.L. (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. The New England Journal of Medicine, 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, J.A. & Ebert, B.L. (2017) Clinical implications of genetic mutations in myelodysplastic syndrome. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 35, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, A.L. & Shimamura, A. (2019) Genetic predisposition to MDS: clinical features and clonal evolution. Blood, 133, 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita‐Sasai, Y. , Horiike, S. , Misawa, S. , Kaneko, H. , Kobayashi, M. , Nakao, M. , Nakagawa, H. , Fujii, H. & Taniwaki, M. (2001) International prognostic scoring system and TP53 mutations are independent prognostic indicators for patients with myelodysplastic syndrome. British Journal of Haematology, 115, 309–312. [DOI] [PubMed] [Google Scholar]
- Kohlmann, A. , Grossmann, V. , Klein, H.‐U. , Schindela, S. , Weiss, T. , Kazak, B. , Dicker, F. , Schnittger, S. , Dugas, M. , Kern, W. , Haferlach, C. & Haferlach, T. (2010) Next‐generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 28, 3858–3865. [DOI] [PubMed] [Google Scholar]
- Kon, A. , Shih, L.‐Y. , Minamino, M. , Sanada, M. , Shiraishi, Y. , Nagata, Y. , Yoshida, K. , Okuno, Y. , Bando, M. , Nakato, R. , Ishikawa, S. , Sato‐Otsubo, A. , Nagae, G. , Nishimoto, A. , Haferlach, C. , Nowak, D. , Sato, Y. , Alpermann, T. , Nagasaki, M. , Shimamura, T. , Tanaka, H. , Chiba, K. , Yamamoto, R. , Yamaguchi, T. , Otsu, M. , Obara, N. , Sakata‐Yanagimoto, M. , Nakamaki, T. , Ishiyama, K. , Nolte, F. , Hofmann, W.K. , Miyawaki, S. , Chiba, S. , Mori, H. , Nakauchi, H. , Koeffler, H.P. , Aburatani, H. , Haferlach, T. , Shirahige, K. , Miyano, S. & Ogawa, S. (2013) Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nature Genetics, 45, 1232–1237. [DOI] [PubMed] [Google Scholar]
- Kulasekararaj, A.G. , Smith, A.E. , Mian, S.A. , Mohamedali, A.M. , Krishnamurthy, P. , Lea, N.C. , Gäken, J. , Pennaneach, C. , Ireland, R. , Czepulkowski, B. , Pomplun, S. , Marsh, J.C. & Mufti, G.J. (2013) TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. British Journal of Haematology, 160, 660–672. [DOI] [PubMed] [Google Scholar]
- Li, M.M. , Datto, M. , Duncavage, E.J. , Kulkarni, S. , Lindeman, N.I. , Roy, S. , Tsimberidou, A.M. , Vnencak‐Jones, C.L. , Wolff, D.J. , Younes, A. & Nikiforova, M.N. (2017) Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. The Journal of molecular diagnostics: JMD, 19, 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima, H. , Visconte, V. , Sakaguchi, H. , Jankowska, A.M. , Abu Kar, S. , Jerez, A. , Przychodzen, B. , Bupathi, M. , Guinta, K. , Afable, M.G. , Sekeres, M.A. , Padgett, R.A. , Tiu, R.V. & Maciejewski, J.P. (2012) Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood, 119, 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima, H. , Yoshizato, T. , Yoshida, K. , Sekeres, M.A. , Radivoyevitch, T. , Suzuki, H. , Przychodzen, B. , Nagata, Y. , Meggendorfer, M. , Sanada, M. , Okuno, Y. , Hirsch, C. , Kuzmanovic, T. , Sato, Y. , Sato‐Otsubo, A. , LaFramboise, T. , Hosono, N. , Shiraishi, Y. , Chiba, K. , Haferlach, C. , Kern, W. , Tanaka, H. , Shiozawa, Y. , Gómez‐Seguí, I. , Husseinzadeh, H.D. , Thota, S. , Guinta, K.M. , Dienes, B. , Nakamaki, T. , Miyawaki, S. , Saunthararajah, Y. , Chiba, S. , Miyano, S. , Shih, L.Y. , Haferlach, T. , Ogawa, S. & Maciejewski, J.P. (2017) Dynamics of clonal evolution in myelodysplastic syndromes. Nature Genetics, 49, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati, L. , Hellström‐Lindberg, E. , Bowen, D. , Adès, L. , Cermak, J. , Del Cañizo, C. , Della Porta, M.G. , Fenaux, P. , Gattermann, N. , Germing, U. , Jansen, J.H. , Mittelman, M. , Mufti, G. , Platzbecker, U. , Sanz, G.F. , Selleslag, D. , Skov‐Holm, M. , Stauder, R. , Symeonidis, A. , van de Loosdrecht, A.A. , de Witte, T. & Cazzola, M. (2013) Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood, 122, 2943–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati, L. , Papaemmanuil, E. , Ambaglio, I. , Elena, C. , Gallì, A. , Della Porta, M.G. , Travaglino, E. , Pietra, D. , Pascutto, C. , Ubezio, M. , Bono, E. , Da Vià, M.C. , Brisci, A. , Bruno, F. , Cremonesi, L. , Ferrari, M. , Boveri, E. , Invernizzi, R. , Campbell, P.J. & Cazzola, M. (2014) Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood, 124, 1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, R. , Acha, P. , Ganster, C. , Palomo, L. , Dierks, S. , Fuster‐Tormo, F. , Mallo, M. , Ademà, V. , Gómez‐Marzo, P. , De Haro, N. , Solanes, N. , Zamora, L. , Xicoy, B. , Shirneshan, K. , Flach, J. , Braulke, F. , Schanz, J. , Kominowski, A. , Stromburg, M. , Brockmann, A. , Trümper, L. , Solé, F. & Haase, D. (2018) Targeted deep sequencing of CD34+ cells from peripheral blood can reproduce bone marrow molecular profile in myelodysplastic syndromes. American Journal of Hematology, 93, E152–E154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijs, G. , Souche, E. , Alders, M. , Corveleyn, A. , Eck, S. , Feenstra, I. , Race, V. , Sistermans, E. , Sturm, M. , Weiss, M. , Yntema, H. , Bakker, E. , Scheffer, H. & Bauer, P. (2016) Guidelines for diagnostic next‐generation sequencing. European journal of human genetics: EJHG, 24, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure, R.F. , Ewalt, M.D. , Crow, J. , Temple‐Smolkin, R.L. , Pullambhatla, M. , Sargent, R. & Kim, A.S. (2018) Clinical significance of DNA variants in chronic myeloid neoplasms: a report of the association for molecular pathology. The Journal of molecular diagnostics: JMD, 20, 717–737. [DOI] [PubMed] [Google Scholar]
- Meggendorfer, M. , Roller, A. , Haferlach, T. , Eder, C. , Dicker, F. , Grossmann, V. , Kohlmann, A. , Alpermann, T. , Yoshida, K. , Ogawa, S. , Koeffler, H.P. , Kern, W. , Haferlach, C. & Schnittger, S. (2012) SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood, 120, 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggendorfer, M. , Haferlach, C. , Kern, W. & Haferlach, T. (2017) Molecular analysis of myelodysplastic syndrome with isolated deletion of the long arm of chromosome 5 reveals a specific spectrum of molecular mutations with prognostic impact: a study on 123 patients and 27 genes. Haematologica, 102, 1502–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ok, C.Y. , Patel, K.P. , Garcia‐Manero, G. , Routbort, M.J. , Fu, B. , Tang, G. , Goswami, M. , Singh, R. , Kanagal‐Shamanna, R. , Pierce, S.A. , Young, K.H. , Kantarjian, H.M. , Medeiros, L.J. , Luthra, R. & Wang, S.A. (2015) Mutational profiling of therapy‐related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leukemia Research, 39, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil, E. , Cazzola, M. , Boultwood, J. , Malcovati, L. , Vyas, P. , Bowen, D. , Pellagatti, A. , Wainscoat, J.S. , Hellstrom‐Lindberg, E. , Gambacorti‐Passerini, C. , Godfrey, A.L. , Rapado, I. , Cvejic, A. , Rance, R. , McGee, C. , Ellis, P. , Mudie, L.J. , Stephens, P.J. , McLaren, S. , Massie, C.E. , Tarpey, P.S. , Varela, I. , Nik‐Zainal, S. , Davies, H.R. , Shlien, A. , Jones, D. , Raine, K. , Hinton, J. , Butler, A.P. , Teague, J.W. , Baxter, E.J. , Score, J. , Galli, A. , Della Porta, M.G. , Travaglino, E. , Groves, M. , Tauro, S. , Munshi, N.C. , Anderson, K.C. , El‐Naggar, A. , Fischer, A. , Mustonen, V. , Warren, A.J. , Cross, N.C. , Green, A.R. , Futreal, P.A. , Stratton, M.R. & Campbell, P.J. (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England Journal of Medicine, 365, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil, E. , Gerstung, M. , Malcovati, L. , Tauro, S. , Gundem, G. , Van Loo, P. , Yoon, C.J. , Ellis, P. , Wedge, D.C. , Pellagatti, A. , Shlien, A. , Groves, M.J. , Forbes, S.A. , Raine, K. , Hinton, J. , Mudie, L.J. , McLaren, S. , Hardy, C. , Latimer, C. , Della Porta, M.G. , O'Meara, S. , Ambaglio, I. , Galli, A. , Butler, A.P. , Walldin, G. , Teague, J.W. , Quek, L. , Sternberg, A. , Gambacorti‐Passerini, C. , Cross, N.C. , Green, A.R. , Boultwood, J. , Vyas, P. , Hellstrom‐Lindberg, E. , Bowen, D. , Cazzola, M. , Stratton, M.R. , Campbell, P.J. (2013) Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood, 122, 3616–3627; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik, M.M. , Barraco, D. , Lasho, T.L. , Finke, C.M. , Hanson, C.A. , Ketterling, R.P. , Gangat, N. & Tefferi, A. (2017) DNMT3A mutations are associated with inferior overall and leukemia‐free survival in chronic myelomonocytic leukemia. American Journal of Hematology, 92, 56–61. [DOI] [PubMed] [Google Scholar]
- Pich, A. , Riera, L. , Sismondi, F. , Godio, L. , Davico Bonino, L. , Marmont, F. & Francia di Celle, P. (2009) JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. Journal of Clinical Pathology, 62, 798–801. [DOI] [PubMed] [Google Scholar]
- Rack, K.A. , van den Berg, E. , Haferlach, C. , Beverloo, H.B. , Costa, D. , Espinet, B. , Foot, N. , Jeffries, S. , Martin, K. , O’Connor, S. , Schoumans, J. , Talley, P. , Telford, N. , Stioui, S. , Zemanova, Z. & Hastings, R.J. (2019) European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia, 33, 1851–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, C. , Fermo, E. , Corti, S. , Molteni, M. , Faricciotti, A. , Cortelezzi, A. , Lambertenghi Deliliers, G. , Beran, M. & Onida, F. (2010) RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 16, 2246–2256. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W.W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H.L. ; ACMG Laboratory Quality Assurance Committee . (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, H. (2002) Medical laboratories–requirements for quality and competence: an ISO perspective. Vox Sanguinis, 83(Suppl. 1), 333–335. [DOI] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. & Mesirov, J.P. (2011) Integrative genomics viewer. Nature Biotechnology, 29, 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaab, J. , Ernst, T. , Erben, P. , Rinke, J. , Schnittger, S. , Ströbel, P. , Metzgeroth, G. , Mossner, M. , Haferlach, T. , Cross, N.C.P. , Hochhaus, A. , Hofmann, W.‐K. & Reiter, A. (2012) Activating CBL mutations are associated with a distinct MDS/MPN phenotype. Annals of Hematology, 91, 1713–1720. [DOI] [PubMed] [Google Scholar]
- Shih, A.H. , Abdel‐Wahab, O. , Patel, J.P. & Levine, R.L. (2012) The role of mutations in epigenetic regulators in myeloid malignancies. Nature Reviews. Cancer, 12, 599–612. [DOI] [PubMed] [Google Scholar]
- Smith, A.E. , Kulasekararaj, A.G. , Jiang, J. , Mian, S. , Mohamedali, A. , Gaken, J. , Ireland, R. , Czepulkowski, B. , Best, S. & Mufti, G.J. (2015) CSNK1A1 mutations and isolated del(5q) abnormality in myelodysplastic syndrome: a retrospective mutational analysis. The Lancet. Haematology, 2, e212–221. [DOI] [PubMed] [Google Scholar]
- Steensma, D.P. , Bejar, R. , Jaiswal, S. , Lindsley, R.C. , Sekeres, M.A. , Hasserjian, R.P. & Ebert, B.L. (2015) Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 126, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhai, M.A. , Craddock, K.J. , Thomas, M. , Hansen, A.R. , Zhang, T. , Siu, L. , Bedard, P. , Stockley, T.L. & Kamel‐Reid, S. (2016) A classification system for clinical relevance of somatic variants identified in molecular profiling of cancer. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 18, 128–136. [DOI] [PubMed] [Google Scholar]
- Takahashi, K. , Pemmaraju, N. , Strati, P. , Nogueras‐Gonzalez, G. , Ning, J. , Bueso‐Ramos, C. , Luthra, R. , Pierce, S. , Cortes, J. , Kantarjian, H. & Garcia‐Manero, G. (2013) Clinical characteristics and outcomes of therapy‐related chronic myelomonocytic leukemia. Blood, 122, 2807–2811; quiz 2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawana, K. , Drazer, M.W. & Churpek, J.E. (2018) Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: are we there yet? Leukemia, 32, 1482–1492. [DOI] [PubMed] [Google Scholar]
- Thol, F. , Kade, S. , Schlarmann, C. , Löffeld, P. , Morgan, M. , Krauter, J. , Wlodarski, M.W. , Kölking, B. , Wichmann, M. , Görlich, K. , Göhring, G. , Bug, G. , Ottmann, O. , Niemeyer, C.M. , Hofmann, W.‐K. , Schlegelberger, B. , Ganser, A. & Heuser, M. (2012) Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood, 119, 3578–3584. [DOI] [PubMed] [Google Scholar]
- Thota, S. , Viny, A.D. , Makishima, H. , Spitzer, B. , Radivoyevitch, T. , Przychodzen, B. , Sekeres, M.A. , Levine, R.L. & Maciejewski, J.P. (2014) Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood, 124, 1790–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg, J. , Olatubosun, A. & Vihinen, M. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Human Mutation, 32, 358–368. [DOI] [PubMed] [Google Scholar]
- Traina, F. , Visconte, V. , Elson, P. , Tabarroki, A. , Jankowska, A.M. , Hasrouni, E. , Sugimoto, Y. , Szpurka, H. , Makishima, H. , O’Keefe, C.L. , Sekeres, M.A. , Advani, A.S. , Kalaycio, M. , Copelan, E.A. , Saunthararajah, Y. , Olalla Saad, S.T. , Maciejewski, J.P. & Tiu, R.V. (2014) Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia, 28, 78–87. [DOI] [PubMed] [Google Scholar]
- Vreeswijk, M.P.G. , Kraan, J.N. , van der Klift, H.M. , Vink, G.R. , Cornelisse, C.J. , Wijnen, J.T. , Bakker, E. , van Asperen, C.J. & Devilee, P. (2009) Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice‐site prediction programs. Human Mutation, 30, 107–114. [DOI] [PubMed] [Google Scholar]
- Wall, M. , Rayeroux, K.C. , MacKinnon, R.N. , Zordan, A. & Campbell, L.J. (2012) ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica, 97, 1933–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, M.J. , Ding, L. , Shen, D. , Shao, J. , Grillot, M. , McLellan, M. , Fulton, R. , Schmidt, H. , Kalicki‐Veizer, J. , O’Laughlin, M. , Kandoth, C. , Baty, J. , Westervelt, P. , DiPersio, J.F. , Mardis, E.R. , Wilson, R.K. , Ley, T.J. & Graubert, T.A. (2011) Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia, 25, 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, M. , Lu, C. , Wang, J. , McLellan, M.D. , Johnson, K.J. , Wendl, M.C. , McMichael, J.F. , Schmidt, H.K. , Yellapantula, V. , Miller, C.A. , Ozenberger, B.A. , Welch, J.S. , Link, D.C. , Walter, M.J. , Mardis, E.R. , Dipersio, J.F. , Chen, F. , Wilson, R.K. , Ley, T.J. & Ding, L. (2014) Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine, 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, K. , Sanada, M. , Shiraishi, Y. , Nowak, D. , Nagata, Y. , Yamamoto, R. , Sato, Y. , Sato‐Otsubo, A. , Kon, A. , Nagasaki, M. , Chalkidis, G. , Suzuki, Y. , Shiosaka, M. , Kawahata, R. , Yamaguchi, T. , Otsu, M. , Obara, N. , Sakata‐Yanagimoto, M. , Ishiyama, K. , Mori, H. , Nolte, F. , Hofmann, W.K. , Miyawaki, S. , Sugano, S. , Haferlach, C. , Koeffler, H.P. , Shih, L.Y. , Haferlach, T. , Chiba, S. , Nakauchi, H. , Miyano, S. & Ogawa, S. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 478, 64–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Main characteristics and differences between amplicon‐ and capture‐based panels.
Table S2. Comparison of Illumina and Thermo Fisher NGS platforms.
Table S3. Main sequencing parameters.
Table S4. List of most commonly used alignment tools.
Table S5. List of most commonly used somatic variant callers.
Data S1. Supplementary methods.