

Abstract
Objective
We explore factors that may have contributed to differences in treatment‐emergent adverse events in the phase 2 and phase 3 lasmiditan clinical trials.
Background
Phase 2 and phase 3 trials showed that the centrally penetrant 5‐HT1F agonist, lasmiditan, was effective; higher frequency and severity of adverse events (AEs) were seen in phase 2.
Methods
This work represents a hybrid of a review of primary documents and study reports with additional post hoc analyses. Protocols, informed consents, data collection forms, and methodologies were reviewed. This information was supplemented by results from the clinical study reports and post hoc analyses of individual patient data from each trial.
Results
For lasmiditan 100 and 200 mg, in phase 2, the incidence of ≥1 AE was 72‐86% (26% severe), while in phase 3 was 36‐43% (2% severe). The most common AEs in all studies were CNS‐related. The phase 2 consent form was more descriptive of AEs than phase 3. In phase 2, patients recorded AEs and severity in a paper diary that warned about drowsiness and dizziness. In phase 3, patients recorded in electronic diaries whether they experienced unusual feelings after dosing with lasmiditan that they had not felt with a migraine before, and were contacted to determine if an AE had occurred. In phase 2, the AE Schwindel was variably translated from German as “vertigo” or “dizziness,” while phase 3 vertigo cases were queried to ensure there was a sensation of rotation or movement. History of recurrent dizziness and/or vertigo was exclusionary in phase 3.
Conclusions
This work illustrates how informed consent wording, AE collection methods, translation, exclusion criteria, and other factors may be important determinants for reporting of the frequency and severity of AEs in clinical trials.
Keywords: episodic migraine, migraine headache, lasmiditan, adverse events, treatment‐emergent adverse event
Abbreviations
- AE
adverse event
- BMI
body mass index
- CI
confidence interval
- CNS
central nervous system
- CRF
case report form
- ICF
informed consent form
- ph2
phase 2
- ph3
phase 3
- TEAE
treatment‐emergent adverse event
- UK
United Kingdom
- US
United States
Introduction
Lasmiditan (previously known as Col‐144) is a high affinity, centrally penetrant, selective 5‐HT1F receptor agonist without significant pharmacological activity at 5‐HT1B or 5‐HT1D receptors. Evidence suggests that lasmiditan exerts its therapeutic effects in the treatment of migraine by decreasing neuropeptide release and modulating activity in pain pathways important in migraine, including the trigeminal nerve, without causing vasoconstriction.1, 2 Lasmiditan has shown efficacy in aborting migraine attacks in phase 2 (ph2)1 and phase 3 (ph3)3, 4 randomized clinical trials. Farkkila et al reported a relatively high incidence of central nervous system (CNS)‐related treatment‐emergent adverse events (TEAEs) in the ph2 study,1 which other authors have stated could affect acceptability of the drug.5, 6 However, Kuca et al and Goadsby et al have reported a lower incidence and severity of TEAEs in the ph3 studies.3, 4 In the present manuscript, we review the TEAEs from these lasmiditan studies. Then, we examine the different approaches related to informed consent, adverse event (AE) collection, language/linguistic differences, differences in exclusion criteria and baseline characteristics and migraine characteristics, and differences between geography that may have contributed to the difference in TEAEs in the ph2 versus ph3 trials. The goal of this project was to explore how various methodological issues might influence the experience and reporting of AEs. To accomplish this, data from lasmiditan studies were used, although the general principals may apply to other clinical development programs in migraine or other therapeutic areas.
Methods
During lasmiditan development, 1 placebo‐controlled oral ph2 and 2 placebo‐controlled ph3 trials were conducted; therefore, this review includes all ph2 and ph3 placebo‐controlled trials of oral lasmiditan as an acute treatment for migraine. Because TEAEs were more frequent and more severe in the ph2 study than in the ph3 trials, we reviewed the protocols, informed consent forms (ICF), data collection forms, and data collection methodologies between these trials in an effort to understand what factors may have contributed to the differences. The ph2 trial (NCT00883051) methodology and results have been previously published.1 The methodologies and results of the ph3 trials SAMURAI (NCT02439320) and SPARTAN (NCT02605174) have also been published.3, 4 IRB approvals were obtained for each of the original studies. This work represents a hybrid of a review of primary documents and study reports with additional post hoc analyses; therefore, additional IRB approval was exempted.
In these studies, oral lasmiditan was evaluated for the acute treatment of migraine. The study protocols were reviewed and approved by the appropriate institutional review board for each of the study sites, and the studies were conducted according to Good Clinical Practice and the Declaration of Helsinki guidelines. Patients provided written informed consent before undergoing study procedures. Investigators at each study site evaluated and confirmed eligibility, obtained consent, and enrolled the patients. Doses common to the studies were lasmiditan 100 and 200 mg, so those doses are the focus of this project.
All authors had access to the data and approved the final version of the manuscript prior to submission. All authors participated in drafting and/or critical revision of the manuscript.
Ph2 Study Design
Briefly, the ph2 study was conducted at 43 headache centers in 5 European countries (Belgium, Finland, France, Germany, and Spain) from July 2009 to February 2010. Eligible patients were male or female, between the ages of 18 and 65 years, and had at least a 1‐year history of episodic migraine with or without aura and 1‐8 migraine attacks per month.
Patients were randomized in a ratio of 1:1:1:1:1 to receive lasmiditan 50, 100, 200, or 400 mg, or placebo. The patients were instructed to treat their next moderate to severe migraine attack occurring within 8 weeks with study drug within 4 hours of onset. Rescue medications, excluding triptans or ergotamine, were permitted after 2 hours of study drug intake. Further study details, including the CONSORT diagram and the exclusion criteria, have been published.1
Phase 3 (SAMURAI and SPARTAN) Study Designs
Both the SAMURAI and SPARTAN clinical trials were prospective, randomized, double‐blind, and placebo‐controlled studies. The SAMURAI study was conducted from April 2015 to August 2016 at 99 sites in the United States (US), and the SPARTAN study was conducted from May 2016 to June 2017 at 125 sites, with 97 sites in the United States, 16 in Germany, and 12 in the United Kingdom (UK).
For both studies, eligible patients were males and females at least 18 years of age with an onset of migraine with or without aura prior to the age of 50 years. In addition, patients were required to have at least moderately disabling migraine, indicated by a total Migraine Disability Assessment Score (MIDAS) ≥11, and a history of 3‐8 migraine attacks per month. Further study details, including the CONSORT diagram and the exclusion criteria, have been published recently.3, 4 Patients were randomized equally to receive a first dose of lasmiditan 50 mg (SPARTAN only), 100 or 200 mg. Patients were also randomized to a second dose of lasmiditan or placebo for rescue or recurrence of migraine. The patients were instructed to treat their next migraine attack within 4 hours of onset, provided that the headache severity was either moderate or severe and not improving. Patients were requested to avoid using any other medication until at least 2 hours after intake of study drug. If the migraine did not respond (defined as pain freedom) at 2 hours, or if there was a response and then a recurrence after 2 hours but before 24 hours, then an optional second dose of study drug could be taken if no other acute migraine medication had been used. Alternatively, patients could take other migraine medications excluding triptans, ergotamines, opioids, and barbiturates.
Statistics
Information from the clinical study reports was supplemented with post hoc analyses of individual patient data from each trial. Patient demographics and clinical characteristics are from the safety populations of the studies. The reported TEAEs are from the clinical study reports. Forest plots showing percentages and 95% confidence intervals (CIs) for the incidence of any TEAE and of the most common individual TEAEs in each trial are presented for placebo, lasmiditan 100 mg, and lasmiditan 200 mg to facilitate identification of findings with non‐overlapping CIs.
For baseline and migraine characteristics that differed between the ph2 and the ph3 studies, post hoc logistic regression analyses were conducted to assess the potential impact of these factors on the incidence of any TEAE. Separate models using 2 predictor variables were constructed to explore the impact of ph2 versus ph3 and of each individual factor that appeared to differ between phase 2 and phase 3; these factors were average migraines per month, baseline migraine severity (severe = Y), body mass index (BMI), nausea as a baseline symptom (Y/N), and time to dose from migraine pain onset (hours). Wald chi‐square statistics and odds ratios with 95% CIs are reported. Additionally, a post hoc analysis of TEAEs by geography was performed to enable a comparison of the results for German patients who participated in the ph2 study versus the SPARTAN study. Analyses were conducted using SAS Version 9.1 or higher. Results were considered statistically significant if the 2‐sided P value was <.05.
Results
Patient Demographics and Clinical Characteristics
The patient demographics, clinical characteristics, and migraine characteristics for the safety populations of the trials are shown in Table 1. Patients were predominantly Caucasian females and had a mean age ranging from 40 to 43 years. Patients in the ph3 studies had a higher BMI (30 kg/m2) than those in the ph2 study (24 kg/m2). The average years of history of migraine and the percentage of patients with history of aura were similar among the 3 studies, but the average number of migraine attacks per month was lower in the ph2 study (3.2) than in the ph3 studies (5.1 and 5.3). Patients in the ph2 study had a longer time to dosing from onset of migraine and there was a higher percentage of patients with severe migraine than in the ph3 studies. Photophobia and phonophobia were similar among the 3 studies, but the ph2 study had more patients with nausea (60%) than the SAMURAI and SPARTAN studies (39% and 39%, respectively).
Table 1.
Demographics, Clinical, and Migraine Characteristics
| Ph2 (N = 391) | SAMURAI (N = 1856)† | SPARTAN (N = 2583)† | |
|---|---|---|---|
| Female, n (%) | 342 (87.5) | 1552 (83.6) | 2174 (84.2) |
| Age, years, mean (SD) | 40.3 (10.8) | 42.0 (12.0) | 42.7 (12.8) |
| Caucasian, n (%) | 387 (99.0) | 1400 (75.4) | 2071 (80.2) |
| BMI (kg/m2), mean (SD) | 24.1 (4.1) | 30.4 (7.9) | 30.1 (8.9) |
| Time to dosing (hours), mean (SD) | 2.9 (3.6) | 1.9 (4.3) | 1.6 (8.1) |
| Migraine history, years, mean (SD) | 19.9 (12.1) | 19.3 (12.9) | 18.3 (13.0) |
| Migraine attacks/month, mean (SD) | 3.2 (1.7) | 5.1 (1.9) | 5.3 (2.1) |
| Severe headache, n (%) | 164 (41.9) | 466 (25.1) | 681 (26.4) |
| Moderate headache, n (%) | 224 (57.3) | 1174 (63.3) | 1594 (61.7) |
| Mild headache†, n (%) | 0 | 31 (1.7) | 34 (1.3) |
| Baseline symptoms, n (%) | |||
| Phonophobia | 260 (66.5) | 1033 (55.7) | 1454 (56.3) |
| Photophobia | 297 (76.0) | 1291 (69.6) | 1768 (68.4) |
| Nausea | 234 (59.8) | 723 (39.0) | 1009 (39.1) |
| Accompanying aura at first dose, n (%) | 33 (8.4) | 42 (2.3) | 62 (2.4) |
Patients were encouraged not to take their dose until the migraine was either moderate or severe as per the study protocol; however, following US Food and Drug Administration advice, mild headache was included as an option to choose in the electronic diary – these patients are shown here although they deviated from the protocol.
BMI = body mass index; Ph2 = phase 2; SD = standard deviation.
TEAEs in ph2 and ph3 Clinical Studies
In each study, the proportion of patients reporting TEAEs was higher in the lasmiditan compared to the placebo group, and the proportion of patients with TEAEs tended to increase with increasing dose of lasmiditan. The most frequently reported TEAEs were dizziness, paresthesia and somnolence. The proportion of patients reporting TEAEs was similar for the same doses of lasmiditan in the SAMURAI and the SPARTAN studies. In particular, the proportion of patients reporting at least 1 TEAE in the SAMURAI and SPARTAN studies receiving 100 mg of lasmiditan was 229/630 (36%) and 229/635 (36%), respectively, and for those receiving 200 mg was 260/609 (43%) and 253/649 (39%). In contrast, the proportion of patients in the ph2 study reporting TEAEs receiving 100 and 200 mg of lasmiditan was 59/82 (72%) and 61/71 (86%), respectively. Similar results were seen when the most frequent TEAEs, defined as those occurring ≥2% in any treatment group, were compared separately. Thus, dizziness, paresthesia, somnolence, fatigue, nausea, and lethargy occurred in similar proportions of patients at the same doses in the ph3 studies, but in higher proportions in the ph2 study. The numbers and proportions of patients reporting each of these TEAEs are summarized in Table 2, and the percentages of patients, with 95% CIs, are depicted in Figure 1.
Table 2.
Most Commonly Reported Treatment‐Emergent Adverse Events
| Preferred Term | Ph2 | SAMURAI | SPARTAN | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lasmiditan | Placebo | Lasmiditan | Placebo | Lasmiditan | Placebo | |||||||
| 50 mg (n = 82) | 100 mg (n = 82) | 200 mg (n = 71) | 400 mg (n = 70) | (n = 86) | 100 mg (n = 630) | 200 mg (n = 609) | (n = 617) | 50 mg (N = 654) | 100 mg (N = 635) | 200 mg (N = 649) | (N = 645) | |
| Patients with ≥1 TEAE, n (%) | 53 (65) | 59 (72) | 61 (86) | 59 (84) | 19 (22) | 229 (36) | 260 (43) | 101 (16) | 166 (25) | 229 (36) | 253 (39) | 75 (12) |
| Dizziness, n (%) | 19 (23) | 21 (26) | 27 (38) | 26 (37) | 0 | 79 (13) | 99 (16) | 21 (3.4) | 56 (8.6) | 115 (18) | 117 (18) | 16 (2.5) |
| Paresthesia, n (%) | 2 (2.4) | 9 (11) | 12 (17) | 14 (20) | 2 (2.3) | 36 (5.7) | 48 (7.9) | 13 (2.1) | 16 (2.4) | 37 (5.8) | 43 (6.6) | 6 (0.9) |
| Somnolence, n (%) | 8 (9.8) | 10 (12) | 8 (11) | 8 (11) | 2 (2.3) | 36 (5.7) | 33 (5.4) | 14 (2.3) | 35 (5.4) | 29 (4.6) | 42 (6.5) | 13 (2.0) |
| Fatigue, n (%) | 10 (12) | 17 (21) | 15 (21) | 17 (24) | 2 (2.3) | 26 (4.1) | 19 (3.1) | 2 (0.3) | 18 (2.8) | 26 (4.1) | 31 (4.8) | 6 (0.9) |
| Nausea, n (%) | 4 (4.9) | 8 (10) | 2 (2.8) | 5 (7.1) | 0 | 19 (3.0) | 32 (5.3) | 12 (1.9) | 18 (2.8) | 21 (3.3) | 17 (2.6) | 8 (1.2) |
| Lethargy†, n (%) | 4 (4.9) | 4 (4.9) | 9 (13) | 5 (7.1) | 1 (1.2) | 12 (1.9) | 15 (2.5) | 2 (0.3) | 8 (1.2) | 8 (1.3) | 14 (2.2) | 1 (0.2) |
| Vertigo, n (%) | 8 (9.8) | 12 (15) | 12 (17) | 17 (24) | 1 (1.2) | 6 (1.0) | 2 (0.3) | 0 | 2 (0.3) | 5 (0.8) | 4 (0.6) | 1 (0.2) |
Lethargy includes sensation of heaviness.
Figure 1.
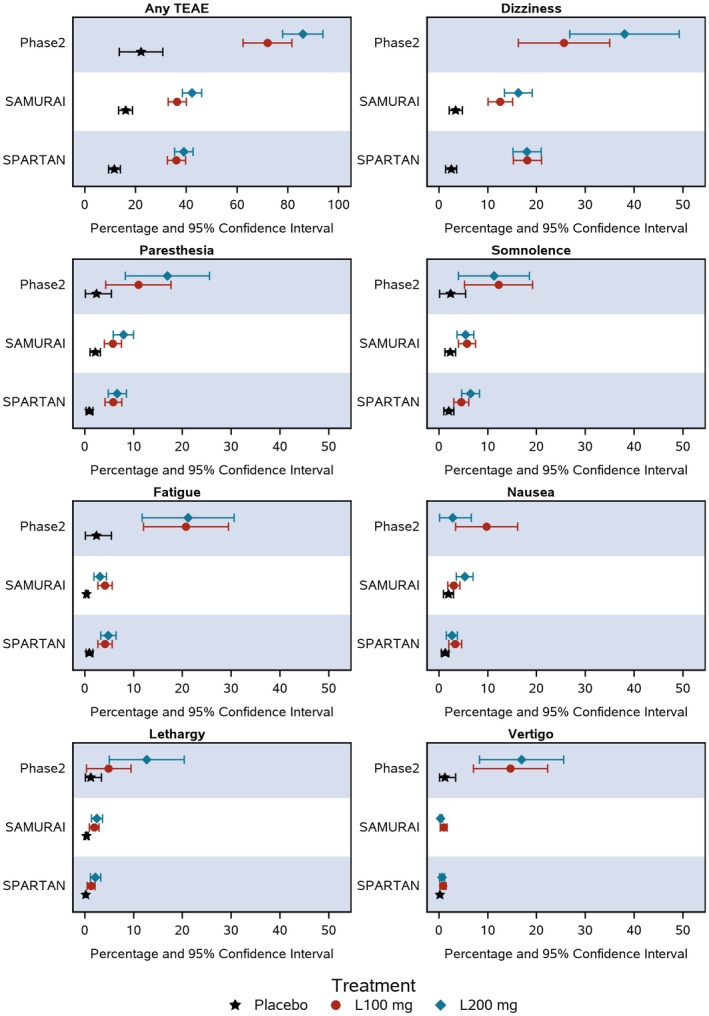
The proportion of patients (as %) and the 95% confidence intervals are shown in a forest plot for the most commonly reported treatment‐emergent adverse events (TEAEs).
The ph2 study reported a total of 102/391 (26%) patients with 1 or more severe TEAEs, whereas the SAMURAI and SPARTAN studies each reported 44/1856 (2.4%) and 43/2583 (1.7%) patients with 1 or more severe TEAEs, respectively. Moreover, there were higher percentages of patients with severe dizziness, paresthesia, somnolence, fatigue, nausea, lethargy, and vertigo in the ph2 study than in either the SAMURAI or SPARTAN studies (Table 3).
Table 3.
Severe Treatment‐Emergent Adverse Events (TEAEs)
| Preferred Term | Ph2 | SAMURAI | SPARTAN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lasmiditan | Placebo | Lasmiditan | Placebo | Lasmiditan | Placebo | ||||||||
| 50 mg (n = 82) | 100 mg (n = 82) | 200 mg (n = 71) | 400 mg (n = 70) | (n = 86) | 100 mg (n = 630) | 200 mg (n = 609) | (n = 617) | 50 mg (N = 654) | 100 mg (N = 635) | 200 mg (N = 649) | (N = 645) | ||
| Patients with ≥1 Severe TEAE, n (%) | 16 (20) | 22 (27) | 28 (39) | 31 (44) | 5 (5.8) | 15 (2.4) | 21 (3.4) | 7 (1.1) | 10 (1.5) | 10 (1.6) | 19 (2.9) | 4 (0.6) | |
| Dizziness, n (%) | 1 (1.2) | 8 (9.8) | 11 (16) | 12 (17) | 0 | 6 (1.0) | 6 (1.0) | 1 (0.2) | 2 (0.3) | 4 (0.6) | 11 (1.7) | 0 | |
| Paresthesia, n (%) | 1 (1.2) | 2 (2.4) | 4 (5.6) | 5 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Somnolence, n (%) | 3 (3.7) | 2 (2.4) | 2 (2.8) | 2 (2.9) | 1 (1.2) | 3 (0.5) | 2 (0.3) | 0 | 1 (0.2) | 1 (0.2) | 1 (0.2) | 1 (0.2) | |
| Fatigue, n (%) | 5 (6.1) | 7 (8.5) | 11 (16) | 8 (11) | 1 (1.2) | 3 (0.5) | 0 | 0 | 2 (0.3) | 1 (0.2) | 3 (0.5) | 1 (0.2) | |
| Nausea, n (%) | 2 (2.4) | 0 | 2 (2.8) | 0 | 0 | 1 (0.2) | 4 (0.7) | 1 (0.2) | 1 (0.2) | 1 (0.2) | 1 (0.2) | 0 | |
| Lethargy†, n (%) | 3 (3.7) | 1 (1.2) | 2 (2.8) | 3 (4.3) | 1 (1.2) | 0 | 0 | 0 | 0 | 2 (0.3) | 0 | 0 | |
| Vertigo, n (%) | 1 (1.2) | 3 (3.7) | 3 (4.2) | 8 (11) | 0 | 0 | 0 | 0 | 0 | 2 (0.3) | 2 (0.3) | 0 | |
Lethargy includes sensation of heaviness.
In the forest plots showing percentages and 95% CIs for all TEAEs and for the most frequent TEAEs (Fig. 1), there was no overlap between CIs for the lasmiditan dose groups of the ph2 vs the ph3 studies for any TEAE, fatigue, and vertigo, suggesting significant differences for these outcomes. However, there was some overlap for other TEAEs owing to wide CIs around the ph2 findings.
Logistic regression analysis was conducted to assess potential impact of baseline characteristics and migraine characteristics on TEAE frequency; results are shown in Table 4. In particular, the impact of ph2 versus ph3 and potential predictors BMI, baseline migraine severity (severe = Y), time to dose from migraine pain onset (hours), average migraines per month, and nausea as a baseline symptom (Y/N), for all TEAEs were analyzed. In each analysis, ph2 versus ph3 was highly significant with odds ratios ranging from 3.87 to 4.45. The various predictors were also statistically significant except for time to dose from migraine pain onset; however, the odds ratios were relatively modest, ranging from 0.74 to 1.15.
Table 4.
Logistic Regression Analyses for Baseline and Migraine Characteristics on Incidence of Any TEAE
| Event | Factor | Phase 2 vs Phase 3 | Factor | ||
|---|---|---|---|---|---|
| P Value | Odds Ratio (95% CI) | P Value | Odds Ratio (95% CI) | ||
| Any TEAE | Body mass index | <.001 | 3.87 (3.10, 4.84) | <.001 | 0.99 (0.98, 0.99) |
| Severe migraine | <.001 | 4.45 (3.57, 5.55) | <.001 | 0.74 (0.64, 0.86) | |
| Time to dose from migraine pain onset (hours) | <.001 | 4.26 (3.43, 5.31) | .869 | 1.00 (0.99, 1.02) | |
| Average migraines per month | <.001 | 3.98 (3.18, 4.99) | .021 | 0.96 (0.93, 0.99) | |
| Nausea as a baseline symptom | <.001 | 4.17 (3.36, 5.19) | .027 | 1.15 (1.02, 1.31) | |
CI = confidence interval; TEAE = treatment‐emergent adverse event.
TEAEs by country of patient participation are shown in Table 5 to facilitate a comparison of German patients from the ph2 study with those from the SPARTAN trial. Consistent with the overall results, there was a numerically higher percentage of German patients reporting dizziness, paresthesia, somnolence, fatigue, nausea, lethargy, and vertigo in the ph2 study than in the ph3 study. Overall, there was no obvious difference in the pattern of reported rates of TEAEs among patients in the UK, Germany, and the US within the SPARTAN ph3 study.
Table 5.
Adverse Effects by Country
| Ph2 | SPARTAN | |||||||
|---|---|---|---|---|---|---|---|---|
| Preferred Term | Germany (n = 173) | Finland (n = 124) | France (n = 36) | Spain (n = 31) | Belgium (n = 27) | Germany (n = 255) | UK (n = 174) | US (n = 2154) |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Dizziness | 48 (27.7) | 33 (26.6) | 1 (2.8) | 5 (16.1) | 6 (22.2) | 42 (16.5) | 29 (16.7) | 233 (11) |
| Paresthesia | 19 (11.0) | 11 (8.9) | 2 (5.6) | 2 (6.5) | 5 (18.5) | 12 (4.7) | 13 (7.5) | 77 (3.6) |
| Somnolence | 14 (8.1) | 5 (4.0) | 8 (22.2) | 4 (12.9) | 5 (18.5) | 4 (1.6) | 3 (1.7) | 112 (5.2) |
| Fatigue | 35 (20.2) | 22 (17.7) | 0 | 1 (3.2) | 2 (7.4) | 15 (5.9) | 8 (4.6) | 58 (2.7) |
| Nausea | 9 (5.2) | 8 (6.5) | 2 (5.6) | 0 | 0 | 16 (6.3) | 3 (1.7) | 45 (2.1) |
| Lethargy† | 16 (9.2) | 4 (3.2) | 0 | 0 | 1 (3.7) | 5 (2.0) | 5 (2.9) | 21 (1.0) |
| Vertigo | 34 (19.7) | 11 (8.9) | 13 (36.1) | 0 | 0 | 8 (3.1) | 0 | 4 (0.2) |
Lethargy includes sensation of heaviness.
UK = United Kingdom; US = United States.
Assessment of Methodological Differences Between the ph2 and ph3 Clinical Studies
Ph2 Study
The ICF provided in the ph2 study informed patients of previous intravenous infusion and oral administration studies and that no serious AEs had occurred. However, the ICF for the ph2 study included the following text:
The following adverse events were observed in participants after administration of COL‐144: feeling of dizziness (46x), drowsiness and fatigue (43x), paresthesias (abnormal sensation on the skin, 41x), hot flashes (14x), feeling of heaviness (9x), nausea (6x), feeling of tension (5x), vertigo (4x), blurry vision (4x), scotoma (partial visual field loss, 3x), headaches (3x), asthenia (weakness, 2x), abdominal pain (2x), vomiting (2x), shortness of breath (2x), diarrhea (1x), sluggishness (1x), impaired alertness (1x), itching (1x), hypotension (low blood pressure, 1x), slow heart rate (1x) and collapse (1x).
There was also a description of another study with additional AEs:
In the most recent study on healthy volunteers who were given COL‐144 as an oral liquid formulation, 14 volunteers received the highest dose of 400 mg COL‐144. They experienced drowsiness and fatigue (7x), feelings of dizziness (3x), paresthesias (3x), nausea, headaches, nasopharyngitis, tonsillitis and blurred vision (1x each).
Patients used a paper diary to record information about the migraine attack prior to and at 0.5, 1, 1.5, 2, 3, 4, and 24 hours after taking the investigational product. At the top of the paper diary, the following text was in large white font within a black background:
Due to the possible known adverse effects such as drowsiness and dizziness in the 12 hours after administration of the study drug, ….
The AE page of the paper diary asked the patient to take these actions:
“Please tell us about any unusual symptoms you experienced” with directions: “Did you have any unusual symptoms at any time after taking the study medication? If so, please record symptoms below until you return to the clinic.”
The patient diary included space to record details about 4 symptoms. Patients were to record the start date and time and the stop date and time of their symptoms and rate them as mild, moderate, or severe, but these descriptors were not defined.
The protocol stated that when the patient returned to the site, the investigator would determine whether any AEs had occurred, and that patients would be questioned generally and specific symptoms would not be suggested. However, the protocol also specified that the AE information from the patient diary would be transcribed into the case report form (CRF) by the investigator. Investigators were to record AEs and their severity (mild – the AE is easily tolerated and does not interfere with normal daily activities, moderate – the AE causes some interference with daily activities, and severe – the AE causes all normal daily activities to be completely halted). The CRF for AEs included a space for a description of each AE, start date and time, stop date and time, severity, relationship to study medication, action taken, outcome, and whether the AE was serious. Notably, at the German‐speaking ph2 study sites, the word “Schwindel” was variably translated to English as “vertigo” or “dizziness,” and translations to vertigo were apparently not assessed for presence of a movement or rotational component.
Ph3 Studies SAMURAI and SPARTAN
In the ph3 clinical studies, responses to the first dose and, if taken, to the second dose of study drug were recorded by the patient in an electronic diary at baseline and 0.5, 1, 1.5, 2, 3, 4, 24, and 48 hours after dosing. At each time point, the electronic diary included a question: “Do you feel anything unusual since you took the Study Medication that you have not felt with a migraine before?” A “yes” response to this question triggered an email alert to the site to contact the subject and assess the AE in terms of what the subject was experiencing, how long the symptoms lasted and how much the symptom(s) impacted them. Severity of AEs were assessed as mild (awareness of sign or symptom, but easily tolerated), moderate (discomfort enough to cause interference with usual activity), severe (incapacitating with inability to work or perform usual activity), or life threatening (no life threatening AEs were reported). All AEs reported from the time of dosing up to 48 hours afterward were considered to be TEAEs. Patients were provided with an instruction sheet for their electronic diary with some white space where they were advised to write down, in their own words, what was happening during an AE in order to facilitate discussion, and were to be called within 48 hours by the site, so that any potential recall bias might be minimized. Differences in collection of TEAEs between the ph2 and ph3 studies are summarized in Table 6.
Table 6.
Differences† in Phase 2 vs Phase 3 Collection of Adverse Events
| Ph2 | SAMURAI, SPARTAN | |
|---|---|---|
| History consideration | Diagnoses, not symptoms | Diagnoses, symptoms |
| Ascertainment of AEs | Patient could write down on an AE page | Daily question “How are you feeling today” and questions during migraine |
| Prompting | Patient AE form instructions warned patients about drowsiness, dizziness, and restricted driving or operating heavy machinery | No specific warnings about particular AEs, sites asked open‐ended question |
| Diary | Paper | Electronic with alarms |
| Site participation or discussion with patient about each event | Site instructed to both transcribe information from patient journal as well as determine whether AEs occurred | Site determined if there was a new issue or previous issues had worsened or changed |
| Vertigo | Not managed with German word Schwindel sometimes translated to “vertigo” and sometimes to “dizziness” | Cases of vertigo were queried regarding whether there was a movement or rotational component; if not the site was asked whether dizziness might be more appropriate |
Patients with a history of recurrent dizziness and/or vertigo including benign paroxysmal positional vertigo (BPPV), Meniere’s disease, vestibular migraine, and other vestibular disorders were excluded from the SAMURAI and SPARTAN studies.
AE = adverse event; Ph2 = phase 2.
In the ph3 studies, the ICF informed patients as follows: “You may experience some or none of these side effects: dizziness, fatigue, sleepiness, tingling sensation in hands and feet, generalized weakness, unsteadiness, orthostatic hypotension (drop in blood pressure when you stand up) with or without dizziness. This is very transient and resolves quickly. Because lasmiditan (the study drug) may cause dizziness or sleepiness, do not drive a car, use machinery or do anything where you need to be alert until you know how you will react to lasmiditan.”
For all studies, AEs were entered into the CRF. The English translations of the reported TEAEs were then coded using classifications and preferred terms from the Medical Dictionary for Regulatory Activities, version 18.0. In the ph3 studies, if a case of vertigo was reported, the site was queried about whether there was a movement or rotational component; if not, the site was asked if they thought the AE should be changed to dizziness.
Discussion
Previous studies established that lasmiditan was superior to placebo in relieving migraine headache pain and associated symptoms.1, 3, 4 However, published reviews discussing the ph2 study of oral lasmiditan expressed some concern about the relatively high incidence of CNS‐related TEAEs (especially dizziness, vertigo, and fatigue).5, 6 However, the results of 2 ph3 studies have now been reported with the incidence and severity of these TEAEs being relatively lower.3, 4 We explored various factors which may have contributed to this difference between ph2 and ph3 TEAE reporting and found that the best explanation for the difference in the rates of TEAEs may have been related to differences in ICF documents and the methods used to record and code data. Other factors that may have contributed to the differences include an exclusion related to recurrent dizziness or vertigo and baseline characteristics of patients and their behavior. Geography did not appear to explain the differences.
Regarding the ICF, patients participating in the ph2 study received information about potential AEs such as “feeling of dizziness (46x).” The meaning of “46x” was not defined, so patients may have interpreted this variably (eg, instances of the event or fold increase in risk). In addition, the patients in the ph2 were warned about drowsiness and dizziness in the paper diary, while those in the ph3 studies who used the electronic diaries were asked open‐ended questions about unusual symptoms. Although it might be supposed that the repeated warnings regarding side effects in ph2 might lead to higher expectations of efficacy, review of the efficacy data from the ph2 study does not show increased efficacy relative to the ph3 studies (data not shown).
Several reviews provide evidence that the wording of ICFs can influence patients’ reported AEs.7, 8, 9, 10 Notably, nonspecific symptoms, such as drowsiness, nausea, dizziness, and fatigue, which were among the most common TEAEs observed in the studies, are most susceptible to a possible nocebo response.10 An example of how ICFs can influence outcomes is provided by a placebo‐controlled trial of aspirin, where the ICF used at 2 centers included a statement that “side effects are not anticipated beyond occasional gastrointestinal irritation and, rarely, skin rash.”11 This statement was not included in the ICF at the third study center. Patients at the centers informing versus those at centers not informing about gastrointestinal irritation reported significantly higher gastrointestinal symptoms and 6 times higher withdrawals from the study because of gastrointestinal distress.11 That ICFs may themselves influence patients’ experiences during clinical trials is a concern, and strategies to mitigate this influence have been discussed in the literature.7, 10 Suggestions include informing patients of the percentage who do not report certain side effects (“positive” communication) rather than the percentage of those who do (“negative” communication).7 In addition, the concept of “contextualized informed consent” has been proposed, where the possible side effects, the characteristics of the patient, and the disease involved are considered when formulating the informed consent.10 One suggestion is for the patient to contact the study site if they experience “any new or unusual symptoms,” so that the site can then evaluate the patient and determine if the patient’s event is related to the study drug.9 This strategy differs from what was employed in ph3 studies described here, where patients were asked at 8 post‐baseline time points whether they were feeling anything unusual, and if they recorded yes in the electronic diary, then the site contacted the patient. While this approach might detect clinically relevant TEAEs, it might also result in underreporting, as some patients might be reluctant to call the investigative site.
A possible factor that may have influenced the AE reporting from these clinical trials is the nocebo (“I will harm”) effect.7, 8, 12 Although it has not been as well studied as the placebo (“I will please”) response in clinical trials, evidence suggests that its impact may be as important.7, 8 Nocebo effect has been demonstrated in several studies,8, 9, 10 including one where an innocuous stimulus produced a perception of pain,13 and another where a potent analgesic (ie, remifentanil) was rendered ineffective,14 due to the subjects’ negative expectations. Supportive of a possible nocebo effect, the placebo group in the ph2 study showed an incidence of ≥1 TEAE in 22% of patients (5.8% severe) while SAMURAI showed 16% (1.1% severe) and SPARTAN showed 12% (0.6% severe). However, the rates of TEAEs were considerably lower in the placebo than lasmiditan groups in all studies, indicating that a drug effect is present but that the nocebo effect may have influenced the frequency and severity of reported TEAEs.
Collection methodology can influence the frequency of AE reporting. For example, a prospective study of 415 patients with migraine who were on a triptan for ≥3 months assessed how the means of collecting AEs affected outcome.15 In that study, patients filled out one part of a questionnaire that asked if they had any AEs while taking triptans (ie, unprompted), and if yes, to list and grade the severity of the AEs. They then received a second form that listed 49 possible AEs, most of which were known side effects of triptans (ie, prompted). Most patients (75% in United States, 66% in Italy) reported no AEs on the unprompted form, yet a majority (63% United States, 54% Italy) also reported at least 1 AE on the prompted form. Most patients who reported unprompted AEs only indicated 1, whereas with the prompted form, most patients indicated 2 or more AEs. This finding is consistent with the results of the present investigation, where the ph2 study, including some prompting about potential AEs, had higher numbers of reported AEs when compared to the ph3 studies, where an unprompted collection method was used.
The paper patient diary for the ph2 study asked patients, “Did you have any unusual symptoms at any time after taking the study medication?” and the electronic diary used in the ph3 studies asked patients, “Do you feel anything unusual since you took the study medication that you have not felt with a migraine before?” The difference between these prompts may be relevant since the ph2 study may have included more reporting of symptoms related to migraine than the ph3 study. However, the ph3 studies included this question at 8 post‐baseline time points, and this frequency of questioning may have facilitated more complete ascertainment of AEs.
Consistent with overall results, German patients reported TEAEs at higher frequency in the ph2 study compared to in the SPARTAN study. Because the German patients in the ph2 study may have been relatively similar to those in SPARTAN, this finding strengthens the argument that ICF and diary language may significantly affect reporting of AE symptoms by patients within a relatively similar cultural and linguistic context.
There were methodological differences between the ph2 and the ph3 studies regarding the collection of the TEAE vertigo. Some patients at German speaking ph2 sites were recorded to have the TEAE Schwindel; this German word was translated to English variably as “vertigo” or “dizziness,” apparently without regard to whether there was a movement or rotational component suggesting actual vertigo. In contrast, in the ph3 studies, AEs of vertigo were queried and if there was no movement or rotational component, the site was asked to consider whether reporting dizziness might be more appropriate. However, reports of dizziness were not similarly queried, possibly reducing the number of cases of vertigo reported during the ph3 study. Thus, both translation issues as well as querying sites about particular TEAEs may have markedly influenced the observed rate of particular TEAEs such as vertigo.
A previous 5‐HT1F agonist, LY334370, underwent ph2 testing and showed TEAEs, including dizziness in 35%, asthenia in 27%, somnolence in 23%, and paresthesia in 19% in the 200‐mg dose group.16 This ph2 study also included a paper diary and might have served as a model for the conduct of the lasmiditan ph2 study, raising the possibility that similar methods may have contributed to the relatively high rate of TEAEs in the LY33470 and lasmiditan ph2 studies.
In contrast to the lasmiditan development program, development of other drugs with more similar methodology in the ph2 and ph3 studies has shown more similar results. For example, the ph2 study for galcanezumab for the preventive treatment of episodic migraine reported at least 1 TEAE in 72% of patients receiving galcanezumab and 67% patients receiving placebo.17 In the EVOLVE‐1 ph3 study, between 60% and 68% of patients reported at least 1 TEAE,18 and in the EVOLVE‐2 ph3 study, at least 1 TEAE was reported by between 62% and 72% of patients.19
The most marked differences in populations and behavior between these studies were examined as potential factors explaining the differences in reporting of any TEAE. In those analyses, while ph2 versus ph3 was highly predictive (odds ratios 3.87 to 4.45), the impact of other factors such as BMI, baseline migraine severity, average migraines per month, and nausea as a baseline symptom were statistically significant but relatively less predictive (odds ratios 0.74 to 1.15).
The exclusion of patients with history of recurrent dizziness and/or vertigo in the ph3 studies but not in the ph2 study may have impacted the observed frequencies of those events. For dizziness, this exclusion does not appear to be a major factor since the proportions of patients with dizziness and with any TEAE decreased approximately similarly in ph3 studies relative to the ph2 study. However, this exclusion may have contributed to vertigo decreasing relatively more than the decrease in any TEAE in the ph3 studies. While this is possible, other factors, as discussed above, may also have contributed to the relatively low incidence of vertigo in ph3 versus ph2, including differences in the question about AEs, site queries, and translation.
Conclusion
Several factors likely contributed to the relatively higher incidence and severity of TEAEs in the lasmiditan ph2 study compared with the ph3 studies. The ph2 study included more description of potential TEAEs in the ICF, and the paper diary included warnings about dizziness and somnolence that could have contributed to a possible nocebo effect. Additional studies could help to further explore the importance of the nocebo effect in randomized clinical trials of acute treatments for migraine. In the ph2 study, sites were instructed to both transcribe TEAEs recorded by patients and also to determine whether TEAEs had occurred. The transcription of TEAEs recorded by patients in the ph2 study may have included some symptoms of migraine rather than drug effects. Recording of TEAEs by trained investigative personnel asking open‐ended questions may lead to more consistent reporting of TEAEs across sites and patients. In addition, linguistic and cultural issues may also influence the reporting of TEAEs. These can be minimized by employing uniform, internally consistent translations and coding that account for vagaries of culture, language, and dialects. Finally, patients had a lower BMI, migraine headache was more severe, average migraines per month was lower, and nausea was higher in the ph2 compared to the ph3 studies, and these factors significantly influenced the occurrence of TEAEs. Moreover, the ph3 study excluded patients with history of disorders that result in recurrent dizziness and/or vertigo. In conclusion, although the particular common TEAEs were similar, differences in the methods of conducting clinical trials of an acute treatment for migraine along with some differences in patient population and behavior influenced the frequency and severity of the observed TEAEs. These considerations are relevant to the design and conduct of clinical trials of acute treatments for migraine, and the general principals may apply to clinical trials in other therapeutic areas.
Statement of Authorship
Category 1
(a) Conception and Design
David Kudrow, John H. Krege, Hans P. Hundemer, Paul H. Berg, Rashna Khanna, Michael H. OssipovPatricia Pozo‐Rosich
(b) Acquisition of Data
David Kudrow, John H. Krege, Hans P. Hundemer, Paul H. Berg, Rashna Khanna, Michael H. OssipovPatricia Pozo‐Rosich
(c) Analysis and Interpretation of Data
David Kudrow, John H. Krege, Hans P. Hundemer, Paul H. Berg, Rashna Khanna, Michael H. OssipovPatricia Pozo‐Rosich
Category 2
(a) Drafting the Manuscript
John Krege and Michael Ossipov
(b) Revising It for Intellectual Content
David Kudrow, John H. Krege, Hans P. Hundemer, Paul H. Berg, Rashna Khanna, Michael H. OssipovPatricia Pozo‐Rosich
Category 3
(a) Final Approval of the Completed Manuscript
David Kudrow, John H. Krege, Hans P. Hundemer, Paul H. Berg, Rashna Khanna, Michael H. OssipovPatricia Pozo‐Rosich
Acknowledgments
The authors thank the patients who participated in the studies. The authors also thank Teri Tucker, ELS of Syneos Health for her help with writing, editing, and formatting the manuscript and Gina Moore of Syneos health for her help with writing the manuscript. Eli Lilly and Company contracted Syneos Health for writing and editorial services.
Conflict of Interest: JHK, HPH, PHB, and RK are employees of Eli Lilly and Company and own stock in the company. PP‐R has received honoraria for medical education programs and consultancy from Allergan, Almirall, Chiesi Spain, Eli Lilly, Novartis, and Teva. DK Advisory Board: Alder, Eli Lilly, Amgen, Novartis, Biohaven, Promius. Research Investigator: Alder, Eli Lilly, Amgen, Novartis, Teva, Biohaven, Roche‐Genentech, VM Biopharma, UCB, Allergan. Speaker’s Bureau: Amgen/Novartis, Eli Lilly.
Funding: These trials were funded by Eli Lilly and Company.
References
- 1. Färkkilä M, Diener HC, Géraud G, et al. Efficacy and tolerability of lasmiditan, an oral 5‐HT(1F) receptor agonist, for the acute treatment of migraine: A phase 2 randomised, placebo‐controlled, parallel‐group, dose‐ranging study. Lancet Neurol. 2012;11:405‐413. [DOI] [PubMed] [Google Scholar]
- 2. Ferrari MD, Färkkilä M, Reuter U, et al. Acute treatment of migraine with the selective 5‐HT1F receptor agonist lasmiditan – A randomised proof‐of‐concept trial. Cephalalgia. 2010;30:1170‐1178. [DOI] [PubMed] [Google Scholar]
- 3. Kuca B, Silberstein SD, Wietecha L, Berg PH, Dozier G, Lipton RB. Lasmiditan is an effective acute treatment for migraine: A phase 3 randomized study. Neurology. 2018;91:e2222‐e2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goadsby PJ, Wietecha LA, Dennehy EB, et al. Phase 3 randomized, placebo‐controlled, double‐blind study of lasmiditan for acute treatment of migraine. Brain. 2019;142:1894‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitsikostas DD, Tfelt‐Hansen P. Targeting to 5‐HT1F receptor subtype for migraine treatment: Lessons from the past, implications for the future. Cent Nerv Syst Agents Med Chem. 2012;12:241‐249. [DOI] [PubMed] [Google Scholar]
- 6. Tfelt‐Hansen PC, Olesen J. The 5‐HT1F receptor agonist lasmiditan as a potential treatment of migraine attacks: A review of two placebo‐controlled phase II trials. J Headache Pain. 2012;13:271‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benedetti F, Shaibani A. Nocebo effects: More investigation is needed. Expert Opin Drug Saf. 2018;17:541‐543. [DOI] [PubMed] [Google Scholar]
- 8. Glick M. Placebo and its evil twin, nocebo. J Am Dent Assoc. 2016;147:227‐228. [DOI] [PubMed] [Google Scholar]
- 9. Barsky AJ, Saintfort R, Rogers MP, Borus JF. Nonspecific medication side effects and the nocebo phenomenon. JAMA. 2002;287:622‐627. [DOI] [PubMed] [Google Scholar]
- 10. Wells RE, Kaptchuk TJ. To tell the truth, the whole truth, may do patients harm: The problem of the nocebo effect for informed consent. Am J Bioeth. 2012;12:22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Myers MG, Cairns JA, Singer J. The consent form as a possible cause of side effects. Clin Pharmacol Ther. 1987;42:250‐253. [DOI] [PubMed] [Google Scholar]
- 12. Carlino E, Vase L. Can knowledge of placebo and nocebo mechanisms help improve randomized clinical trials? Int Rev Neurobiol. 2018;138:329‐357. [DOI] [PubMed] [Google Scholar]
- 13. Schweiger A, Parducci A. Nocebo: The psychologic induction of pain. Pavlov J Biol Sci. 1981;16:140‐143. [DOI] [PubMed] [Google Scholar]
- 14. Bingel U, Wanigasekera V, Wiech K, et al. The effect of treatment expectation on drug efficacy: Imaging the analgesic benefit of the opioid remifentanil. Sci Transl Med. 2011;3:70ra14. [DOI] [PubMed] [Google Scholar]
- 15. Sheftell FD, Feleppa M, Tepper SJ, Rapoport AM, Ciannella L, Bigal M. Assessment of adverse events associated with triptans – Methods of assessment influence the results. Headache. 2004;44:978‐982. [DOI] [PubMed] [Google Scholar]
- 16. Goldstein DJ, Roon KI, Offen WW, et al. Selective seratonin 1F (5‐HT(1F)) receptor agonist LY334370 for acute migraine: A randomised controlled trial. Lancet. 2001;358:1230‐1234. [DOI] [PubMed] [Google Scholar]
- 17. Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene‐related peptide, for the prevention of migraine: A phase 2, randomised, double‐blind, placebo‐controlled study. Lancet Neurol. 2014;13:885‐892. [DOI] [PubMed] [Google Scholar]
- 18. Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR. Evaluation of galcanezumab for the prevention of episodic migraine: The EVOLVE‐1 randomized clinical trial [published erratum in: JAMA Neurol. 2019 May 28. 10.1001/jamaneurol.2019.1177]. JAMA Neurol. 2018;75:1080‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE‐2 phase 3 randomized controlled clinical trial. Cephalalgia. 2018;38:1442‐1454. [DOI] [PubMed] [Google Scholar]