

Abstract
Angioimmunoblastic T‐cell lymphoma (AITL) is a neoplastic proliferation of T follicular helper cells with clinical and histological presentations suggesting a role of antigenic drive in its development. Genetically, it is characterized by a stepwise acquisition of somatic mutations, with early mutations involving epigenetic regulators (TET2, DNMT3A) and occurring in haematopoietic stem cells, with subsequent changes involving signaling molecules (RHOA, VAV1, PLCG1, CD28) critical for T‐cell biology. To search for evidence of potential oncogenic cooperation between genetic changes and intrinsic T cell receptor (TCR) signaling, we investigated somatic mutations and T‐cell receptor β (TRB) rearrangement in 119 AITL, 11 peripheral T‐cell lymphomas with T follicular helper phenotype (PTCL‐TFH), and 25 PTCL‐NOS using Fluidigm polymerase chain reaction (PCR) and Illumina MiSeq sequencing. We confirmed frequent TET2, DNMT3A, and RHOA mutations in AITL (72%, 34%, 61%) and PTCL‐TFH (73%, 36%, 45%) and showed multiple TET2 mutations (2 or 3) in 57% of the involved AITL and PTCL‐TFH. Clonal TRB rearrangement was seen in 76 cases with multiple functional rearrangements (2–4) in 18 cases (24%). In selected cases, we confirmed bi‐clonal T‐cell populations and further demonstrated that these independent T‐cell populations harboured identical TET2 mutations by using BaseScope in situ hybridization, suggesting their derivation from a common TET2 mutant progenitor cell population. Furthermore, both T‐cell populations expressed CD4. Finally, in comparison with tonsillar TFH cells, both AITL and PTCL‐TFH showed a significant overrepresentation of several TRB variable family members, particularly TRBV19*01. Our findings suggest the presence of parallel neoplastic evolutions from a common TET2 mutant haematopoietic progenitor pool in AITL and PTCL‐TFH, albeit to be confirmed in a large series of cases. The biased TRBV usage in these lymphomas suggests that antigenic stimulation may play an important role in predilection of T cells to clonal expansion and malignant transformation. © 2019 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: AITL, clonality, TET2 mutation, progenitor cells, lymphoma genesis
Introduction
Patients with angioimmunoblastic T‐cell lymphoma (AITL) often present with clinical and laboratory autoimmune features 1. Histologically, the lymphoma is characterised by a polymorphous infiltrate with the neoplastic T cells typically forming clusters in the vicinity of prominent arborising high endothelial venules and expanded follicular dendritic cells 2. The lymphoma cells originate from T follicular helper (TFH) cells and possess their cardinal phenotype, albeit showing variable CD10, CXCL13, ICOS, PD‐1, and BCL6 expression. The lymphoma cells also preserve the major function of TFH cells, for example, helping B cells in their antibody production 3, 4. Because high‐affinity TCR is essential for the commitment of CD4+ T cells to differentiate into TFH cells as well as their maintenance and survival, active TCR signaling most likely plays an important role in the pathogenesis of AITL. This is also supported indirectly by the finding of a number of somatic genetic changes, which involve molecules downstream of TCR signaling.
Although exome and targeted sequencing have identified a wide spectrum of genetic changes in AITL, and also demonstrated a remarkable similarity in the mutation profile between AITL and peripheral T‐cell lymphoma (PTCL) with a TFH cell phenotype, suggesting their close relationship 5, 6, 7, 8, 9, 10, 11, 12. In addition, these studies revealed distinct classes of genetic changes that occur at different stages of AITL development.
Class I genetic changes include mutations in epigenetic (DNA methylation) regulators namely TET2, DNMT3A, and IDH2, with the TET2 gene frequently affected by more than one mutation 7, 13, 14, 15, 16, 17. Mutations in these genes are found in a range of haematological malignancies with IDH2 mutation additionally seen in several types of solid tumours 15, 18, 19, 20. In patients with AITL, the lymphoma associated TET2 and DNMT3A mutations most likely occur at an early stage of haematopoiesis, as they are also observed in several lineages of non‐neoplastic cells including non‐neoplastic B and CD8+ T cells 7, 13, 14, 16, 21, 22. Thus, TET2 and DNMT3A mutations are initiating events, promoting ‘clonal haematopoiesis’ and increasing the risk of lymphomagenesis 10, 23, 24.
Class II genetic changes include mutation in RHOA, VAV1, PLCG1, and CD28, as well as the CTLA4‐CD28, ITK‐SYK, and VAV1‐STAP2 fusion 6, 7, 10, 17, 25, 26, 27, 28. These genetic changes are secondary events, and they involve molecules critical for the biology of T cells, thus most likely promoting malignant transformation and clonal expansion, consequently generating the malignant phenotype of AITL.
Among the above genetic changes, mutations in TET2, DNMT3A, and RHOA are highly frequent and often concurrent in AITL, arguing for their potential cooperation in lymphoma development. This is supported by several mouse model studies, which demonstrate oncogenic cooperation between TET2 inactivation and DNMT3A mutation 29, and also between TET2 inactivation and RHOA mutation 30, 31. It is pertinent to speculate that these genetic changes may also cooperate with the intrinsic TCR signaling in clonal evolution and malignant transformation. To search for such evidence, we investigated TCR gene usage and somatic mutations in 155 cases of AITL and PTCL by targeted sequencing. Our findings suggest the presence of multiple independent neoplastic evolutions from a common TET2 mutant haematopoietic progenitor pool in AITL and PTCL‐TFH.
Materials and methods
Tissue materials and DNA extraction
The use of archival tissues for research was approved by the ethics committees of the involved institutions.
A total of 155 cases of AITL and PTCL were successfully investigated and they were from Department of Pathology, West China Hospital, Chengdu, PR China (n = 91); Department of Histopathology, Addenbrooke's Hospital, Cambridge, UK (n = 32); Department of Cellular Pathology, Southampton University Hospitals, Southampton, UK (n = 21); Department of Pathology, Chi‐Mei Foundation Hospital, Taiwan (n = 8); and Department of Histopathology, Royal‐Marsden Hospital, UK (n = 3). Formalin‐fixed paraffin‐embedded diagnostic tissue biopsies were available in each case, and additionally fresh frozen specimens and high molecular weight (HMW) DNA samples were available in nine cases of AITL that were previously investigated by whole exome sequencing 10, with 92% of these specimens being lymph node biopsies. The histology and immunophenotype of these cases were reviewed by specialized haematopathologists, where necessary additional immunohistochemistry for CD4 (mouse monoclonal 4B12, Leica Biosystems, Milton Keynes, UK), CD10 (mouse monoclonal 56C6, Leica Biosystems), PD1 (mouse monoclonal NAT105, Sigma, Gillingham, UK), BCL6 (mouse monoclonal LN22, Leica Biosystems), and CXCL13 (mouse polyclonal, R&D Systems, Abingdon, UK) was performed using an automatic Bond‐III platform (Leica Biosystems). The final lymphoma diagnoses were established according to the 2016 WHO classification of tumours of haematopoietic and lymphoid tissues 2, and they included 119 AITL, 11 PTCL‐TFH (including one follicular T‐cell lymphoma), and 25 PTCL‐NOS.
DNA was extracted from enriched tumour cell population (>30%) and where possible non‐neoplastic cells by crude microdissection using the QIAamp DNA‐MicroKit. The quality of DNA from tumour cells was assessed by PCR and those showing amplification of ≥300 bp genomic fragments were used for mutation screening by targeted sequencing 32, while those from non‐neoplastic cells were used to exclude germline variants.
Targeted sequencing using Fluidigm Access Array and Illumina MiSeq
This was performed as described previously with each of the DNA samples analysed in duplicate for mutations in TET2, DNMT3A, IDH2, RHOA, PLCG1, CCND3, CD28, and TNFRSF21 32. In brief, genomic DNA (50 ng) was pre‐amplified with a cocktail of all primer pairs to enrich targets, then subjected to Fluidigm Access Array multiplex PCR (up to four primer pairs combined), and followed by barcoding and Illumina MiSeq sequencing 32. Primers and PCR conditions are described in supplementary material, Table S1.
For single nucleotide variant (SNV) detection, bam files were processed using a pipeline based on GATK v3.6 best practices including indel realigned and recalibration steps. The calling variant was run using UnifiedGenotyper with 10 000 to prevent downsampling 33, 34. As UnifiedGenotyper was unable to call SNVs at <8% AAF (alternative allele frequency) reliably, MuTect2 was additionally employed for detection of hotspot mutations at low AAF values (down to 2%). Indel detection was separately carried out on the recalibrated bam files using Pindel v0.2.5 35. Variant call files were concatenated to produce one library vcf each for the SNV and Indel pipelines. These library files were then filtered using a combination of vcftools v0.1.15 and bedtools v2.25 for read depth, quality score, and known PCR/sequence artefacts. Further filtering was accomplished using an in‐house script to remove variants in intronic regions outside essential splicing sites, SNPs with a minor allele frequency ≥ 1% (1000 Genomes Project, European super population) and synonymous changes. In addition, missense variants predicted to be benign by seven or more of nine functional prediction tools (SIFT, Polyphen2 HDIV, Polyphen2 HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, SVM score, and LR score) were excluded. The resulting novel variants were further scrutinized by reviewing the bam file to eliminate any potential PCR/sequence artefacts. Only the variants that appeared in both replicates, with novel changes ≥8% AAF and known hotspot changes ≥2% AAF, were regarded as a true change. Where possible, variants were confirmed as somatic by using Sanger sequencing analysis of corresponding non‐neoplastic DNA or assumed to be somatic if previously confirmed as somatic in the COSMIC database.
Sequencing analysis of the rearranged TCR genes using Illumina MiSeq
The rearranged TRB (T‐cell receptor beta) genes were amplified using the BIOMED‐2 protocols and then sequenced with Illumina MiSeq platform. In brief, genomic DNA (10 and 20 ng) was amplified in duplicate for the rearranged TRB genes (TCRB tube A and B) according to the BIOMED‐2 protocols with the exception that the primers were tagged with a common sequence (CS1: 5'‐ACACTGACGACATGGTTCTACA‐3′, or CS2:5'‐TACGGTAGCAGAGACTTGGTCT‐3′) 36. The amplified products were routinely purified using AMPure XP beads (Beckman Coulter, UK), barcoded and pooled for Illumina MiSeq sequencing as described previously 32. The sequences obtained were analysed using the online software VIDJIL 37, and only productive rearrangements were included in further analyses to search for evidence of biased TCR gene usage in the T‐cell lymphoma entities investigated. The clonal size was calculated for each unique VDJ rearrangement as percentage of the total TRB sequence reads from the corresponding PCR tube, and the mean between the two replicates was used for data presentation, with ≥10% reads used as cutoff value for defining clonal TRB rearrangement.
BaseScope in situ hybridization (ISH)
On selected cases, specific DNA probes were designed to target the unique TRB VDJ junctional sequence or TET2 mutations and used for ISH to visualize the corresponding clonal T cells and the cells carrying the TET2 mutation, respectively. The BaseScope ISH was carried out according to the manufacturer's instructions (Advanced Cell Diagnostics, Newark, CA, USA). In brief, formalin‐fixed paraffin embedded tissue sections were routinely dewaxed, treated with targeted retrieval reagents at 100 °C for 15 min in a steamer, and digested with protease IV for 15 min at 40 °C in a hybridization oven. The slides were hybridized with a BaseScope probe for 2 h at 40 °C, followed by a serial amplification steps at 40 °C in a hybridization oven and at room temperature (the last two amplification steps), and finally incubated with the Fast Red substrate at room temperature to visualise the hybridization signals.
For double BaseScope ISH, the TRB‐VDJ and TET2 mutation probes were labeled in different ‘channels,’ allowing independent signal amplification, with the TRB‐VDJ signal detected using Fast Red and the TET2 mutation signal using the kit's Green reagents. The double ISH was similarly performed using the BaseScope Duplex detection kit according to the manufacturer's instructions.
For TRB‐VDJ and CD4/CD8 double staining, TRB‐VDJ BaseScope ISH was performed as described earlier, and the slide was then routinely stained with a CD4 or CD8 antibody using a Bond‐III platform with Bond Polymer Refine Detection reagents (Leica Biosystems).
Statistical analysis
Fisher's exact test was used to examine potential associations between categorical variables. A chi‐square with Yate's correction test was used to compare the TRBV usage between AITL/PTCL‐TFH and normal tonsillar follicular helper T cells 38, and between PTCL‐NOS and peripheral blood T cells (a total of 22 704 rearranged TRB sequences from 550 healthy donors) 39.
Results
Mutation profile in AITL, PTCL‐TFH, and PTCL‐NOS
In general, the mutation profile was broadly similar between AITL and PTCL‐TFH, with the most frequent mutation being TET2 (72%, 73% respectively), followed by RHOA (61%, 45% respectively) and DNMT3A (34%, 36% respectively) (Figure 1). IDH2 and CD28 mutations were seen in only AITL (25%, 4% respectively), and not in PTCL‐TFH, with mutation in the remaining genes being at low frequencies in both groups. In contrast, PTCL‐NOS had a much lower frequency of mutation in TET2 (24%) and DNMT3A (12%), and no mutation in RHOA, IDH2, and CD28.
Figure 1.
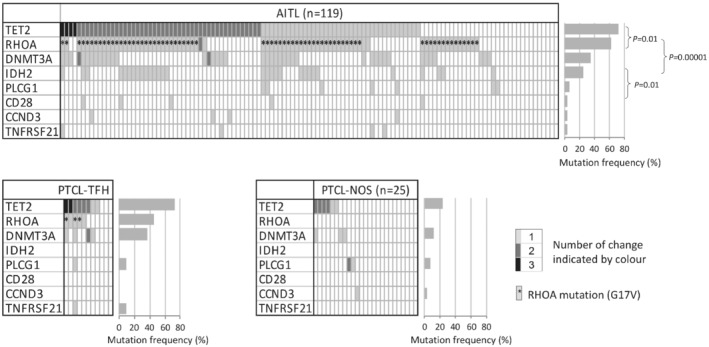
Mutation profile in AITL, PTCL‐TFH, and PTCL‐NOS.
Correlation analysis demonstrated a significant association between TET2 and RHOA (P = 0.01), RHOA and IDH2 (P = 0.00001), and IDH2 and CD28 (P = 0.01) mutation in AITL. Overall, there was a tendency of mutual exclusion among PLCG1, CD28, CCND3, and TNFRSF21 mutations.
Mutation characteristics
Because the mutation profile was broadly similar between AITL and PTCL‐FTH, the characteristics of mutations from both groups were presented together below (supplementary material, Figures S1 and S2, Table S2).
TET2 mutation
A total of 154 TET2 mutations were seen in 94 cases of AITL and PTCL‐FTH, and 118 (77%) of these mutations were frameshift indels or nonsense changes, and rather widely distributed, predicting variably truncated protein products (supplementary material, Figure S1). A small proportion of the mutations were missense changes and they were clustered in the cysteine rich and DSBH (double‐stranded β helix) domains, which were essential for the integrity of the overall structure and the catalytic activity of TET2 40. These mutational features are similar to those previously reported 6, 7.
Multiple TET2 mutations ranging from 2 and 3 were seen in 54 cases, accounting for 57% of the cases investigated. Among the cases with multiple TET2 mutations, 31 cases had two deleterious changes (frameshift indels, nonsense changes, or mutation at the essential splicing site), further highlighting potential inactivation of both TET2 alleles.
DNMT3A mutation
Sixty percent of the DNMT3A mutations identified were missense changes that occurred nearly exclusively in the methyltransferase domain, with R882 being a hotspot accounting for 25% of all mutations (supplementary material, Figure S1). The remaining mutations were frameshift indels, nonsense changes, and substitutions at the essential splicing site, which were widely distributed (supplementary material, Figure S1).
RHOA mutation
This was overwhelmingly dominated by changes in G17, with G17 V being the most frequent, accounting for 91% of all RHOA mutations (supplementary material, Figure S1). The remaining mutations were S26R and C20W, each seen in one case. The majority of cases with RHOA mutation had TET2 mutation (Figure 1), underpinning their oncogenic cooperation in AITL development 30, 31.
IDH2 mutation occurred exclusively in R172, with R172G being the most frequent change, and the majority of cases with IDH2 mutations also had both TET2 and RHOA mutations (Figure 1 and supplementary material, Figure S1). IDH R172 mutant confers a neomorphic enzymatic activity, capable of converting α‐ketoglutarate (αKG) to the D form of 2 hydroxyglutarate (2HG), which inhibits TET and Jumonji (JMJ) family histone demethylases, thereby promoting lymphomagenesis 21, 41.
PLCG1 mutation was seen in eight cases, with the missense changes including R48W, S345F, G869E, and D1165H being reported previously and shown to be gain of function changes (supplementary material, Figure S1) 17, 25.
CD28 mutation was observed in five cases, with four being missense changes at T195 in its cytoplasmic domain (supplementary material, Figure S1). These mutations have been shown to be an activating change, enhancing CD28 downstream signaling 17, 27.
CCND3 mutation was found in four cases, with three mutations including two nonsense, one frameshift indel, and P284S at the C‐terminal region downstream of the Cyclin‐C domain, which was a mutation hotspot (supplementary material, Figure S1) 42, 43. Mutations in this C‐terminal region affect a phosphorylation motif important for polyubiquitination and proteasome‐mediated degradation, consequently enhancing CCND3 stability and its function 44.
High mutation allele frequency in TET2 and DNMT3A
In support of the previous observations that TET2 and DNMT3A mutations occur at an early stage of haematopoiesis, and were seen in non‐neoplastic B and CD8+ T cells in patients with AITL 7, 13, 14, 15, 16, we found that the mutation allele frequencies of both TET2 and DNMT3A were significantly higher than those of RHOA and IDH2 (supplementary material, Figure S3). In contrast, there was no difference in the mutation allele frequencies between TET2 and DNMT3A, nor between RHOA and IDH2 (supplementary material, Figure S3). Of interest, the mean age of patients with TET2, RHOA, DNMT3A, or TNFRSF21 mutation was significantly higher than those without TET2 (66 versus 60 years, P = 0.003), RHOA (66 versus 61, P = 0.01), DNMT3A (68 versus 62 years, P = 0.001), or TNFRSF21 (77 versus 63, P = 0.01) mutation respectively.
Evidence of more than one clonal T‐cell population
A total of 112 cases were successfully investigated for TRB rearrangements by the BIOMED‐2 TRB PCR (tube A and B) and Illumina MiSeq sequencing, 76 (68%) of these cases yielded a clonal result using ≥10% as a cut‐off. Of the 76 informative cases, 69 cases (62%) showed functional clonal TRB rearrangements, and they included 56 AITL, 4 PTCL‐TFH and 9 PTCL‐NOS. Multiple functional TRB rearrangements (2–4) were seen in 18 cases (26%) including 16 AITL, one PTCL‐TFH, and one PTCL‐NOS (Figure 2 and supplementary material, Figure S4). The number of cases with multiple functional TRB rearrangements was much higher in cases with multiple TET2 mutations (11/13 = 85%) than those with a single TET2 mutation (21/38 = 55%), albeit the difference was not statistically significant. Among these cases, one case (PTCL‐TFH010), which harboured two TET2 and two DNMT3A mutations, had four functional TRB rearrangements. These observations suggest possible bi‐ or oligo‐clonal T‐cell populations rather than bi‐allelic functional rearrangements (Figure 2).
Figure 2.
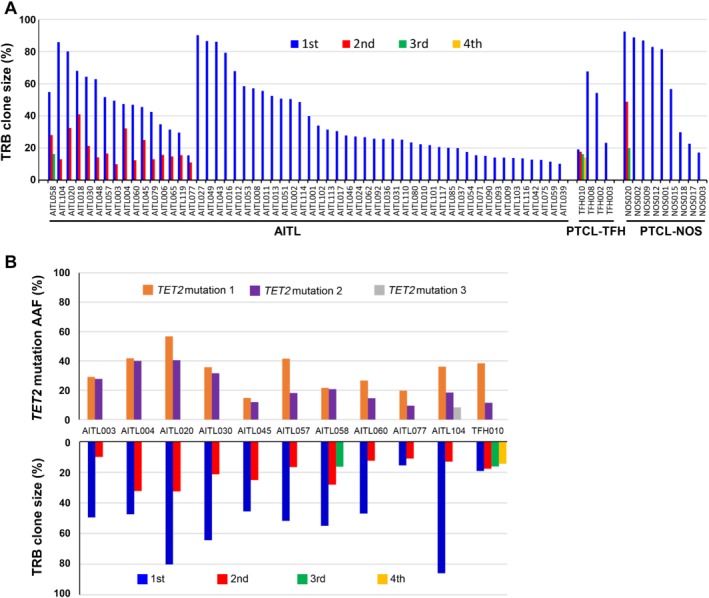
TRB functional rearrangements in AITL, PTCL‐TFH, and PTCL‐NOS. (A) Number of functional TRB rearrangements and their clonal sizes. (B) Comparison between the loads of TET2 mutations and the size of clonal TRB rearrangements on select cases.
Confirmation of two clonal T‐cell populations that carry the same TET2 mutations
To investigate the above speculation, we performed BaseScope ISH using specific probes targeting the unique TRB VDJ junctional sequence in two cases (one AITL and one PTCL‐TFH). As shown in case AITL030 (Figure 3), the V7‐J1 probe identified a diffuse cell population, while the V27‐J2 probe revealed only scattered cells, confirming that they represented two independent clonal T‐cell populations. To examine whether both clonal T‐cell populations carry the same TET2 mutations, we further designed the BaseScope probes for the two TET2 mutations in AITL030 and performed double BaseScope ISH by combining the TRB VDJ probe with each of the TET2 mutation probes. The results showed co‐localization of the TRB‐VDJ and each of the two TET2 mutation probe signal in both T‐cell populations (Figure 4A), indicating that both clonal T‐cell populations harboured the same TET2 mutations.
Figure 3.
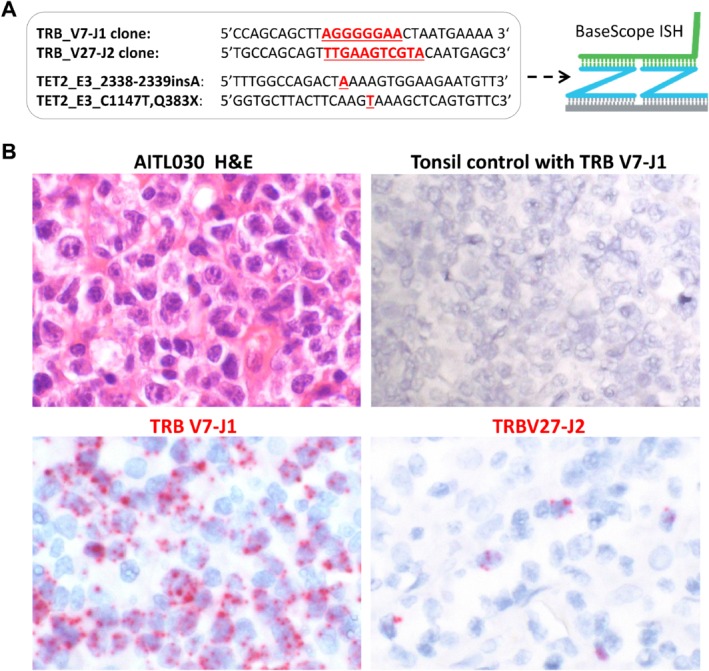
Confirmation of two clonal T‐cell populations by BaseScope ISH with TRB clone specific probes. (A) BaseScope probe design with the unique VDJ junctional sequence and TET2 mutation highlighted in red. (B) BaseScope in situ hybridization in case AITL030 with tonsil serving as a negative control. The TRB V7‐J1 probe identifies a diffuse cell population, whereas the TRB V27‐J2 probe reveals only scattered cells, confirming that they represent two independent clonal T‐cell populations.
Figure 4.
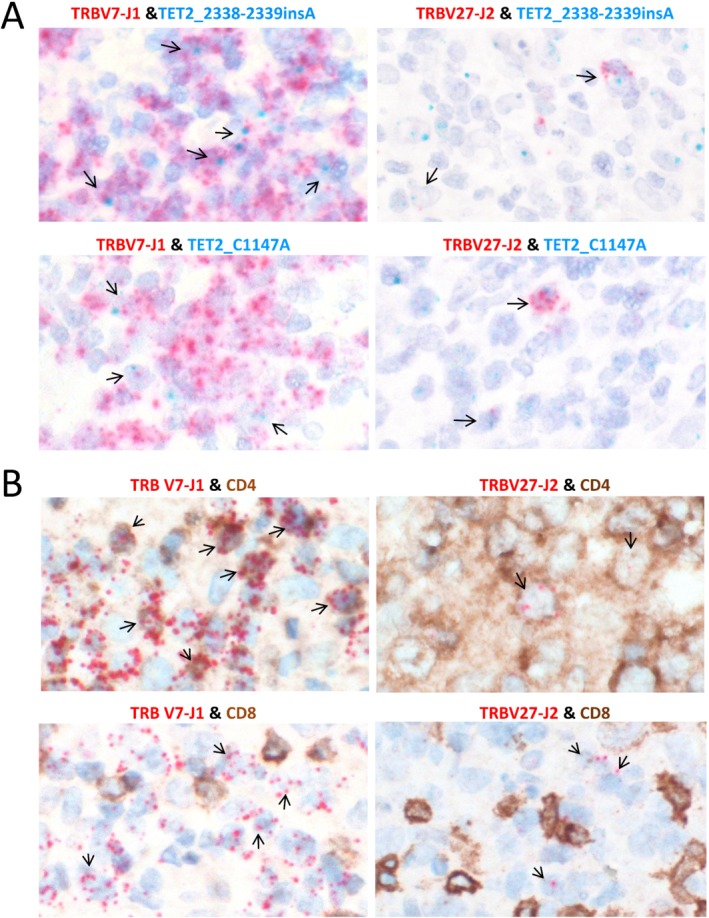
Demonstration of TET2 mutations and CD4 positivity in two clonal T‐cell populations of AITL030. (A) Double BaseScope in situ hybridization shows the presence of both TET2 mutations in the two clonal T‐cell populations of AITL030. Please note that the BaseScope probes were designed to hybridize to mRNA, thus yielding stronger signals for highly expressed (TRB) than lowly expressed (TET2) genes. (B) Double BaseScope ISH and immunohistochemistry demonstrate that both clonal T‐cell populations are CD4 positive, but CD8 negative.
To further investigate the immunophenotype, particularly that of the minor V27‐J2 clone, we combined BaseScope ISH with immunohistochemistry, and demonstrated that both of the clonal T‐cell populations are CD4 positive, but CD8 negative (Figure 4B).
Biased TRB gene usage in AITL and PTCL‐TFH
In comparison with human tonsillar TFH cells (1987 TRB rearrangements from three tonsils) 38, both AITL and PTCL‐TFH showed an over‐representation of several TRB variable family members, for example, TRBV19*01 in AITL, and TRBV27*01 in PTCL‐TFH (Figure 5A). Despite there were three TRBV19 polymorphic alleles, only TRBV19*01 was used in AITL. Among the 18 TRBV19*01 functional rearrangements seen in AITL and PTCL‐TFH, 8 involved TCR βJ2‐1. In two cases (AITL031 and AITL036), the VDJ junctional sequence was nearly identical with difference only at a single amino acid, strongly arguing for their similarity in antigen recognition (Figure 5B).
Figure 5.
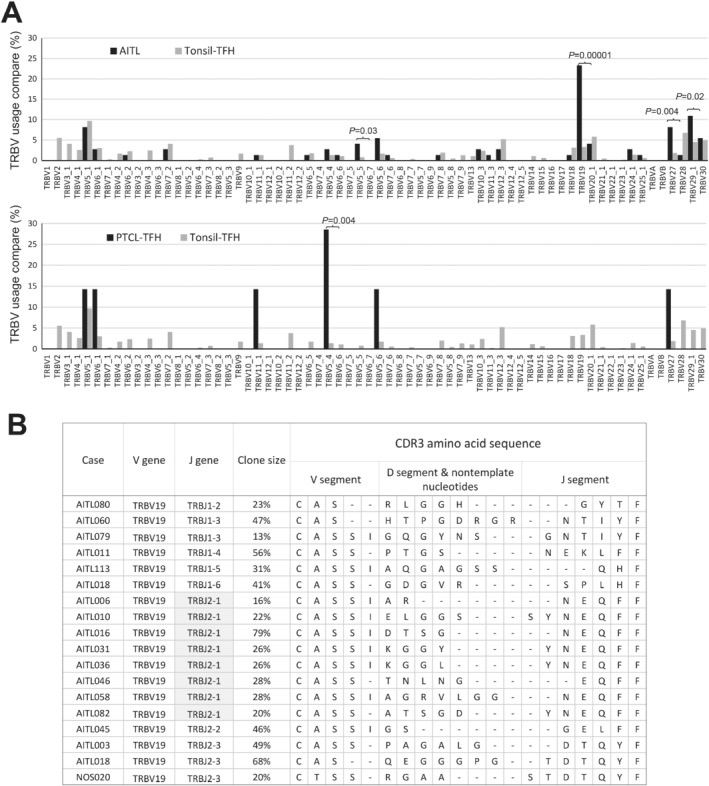
Biased usage of TRB variable genes in AITL and PTCL‐TFH. (A) Comparison of TRB variable gene usage between AITL/PTCL‐TFH and tonsillar TFH. (B) Comparison of CDR3 sequences among cases harbouring TRBV19 functional rearrangements. AITL031 and AITL036 have a nearly identical CDR3 sequence, suggesting possible recognition of a common antigen.
Discussion
This study reports several novel findings that expand our knowledge on the pathological features of AITL and PTCL‐TFH, and also offer fresh insights into their multistage development and pathogenesis. We confirmed a similar mutation profile between AITL and PTCL‐TFH as demonstrated by previous studies 11, 17. We showed evidence of bi‐ or oligoclonal functional TRB rearrangements in 26% of these T‐cell lymphomas and confirm two independent clonal T‐cell populations for selected cases. We demonstrated that the two independent T‐cell clones in the same case share the same TET2 mutations, suggesting derivation from a common precursor cell population. Finally, we found evidence of significantly biased usage of TRB variable genes in both AITL and PTCL‐TFH, with high identity of VDJ junctional sequence in some cases.
In the 2016 WHO lymphoma classification, a broad category of nodal lymphoma of follicular helper cell origin, including AITL, PTCL‐TFH, and follicular T‐cell lymphoma, was introduced due to their overlapping morphological, immunophenotypic, genetic, and clinical presentations, while also bearing certain distinct features. Several studies show that RHOA and IDH2 mutations are strongly associated with the TFH immunophenotype and more extensive characteristics typical of AITL such as proliferation of follicular dendritic cells, high endothelial vessels, and presence of clear cells, with IDH2 mutation defining a unique subgroup of AITL 12, 45, 46, 47. In support of these observations, we found RHOA mutation only in AITL and PTCL‐TFH but not in PTCL‐NOS, and IDH2 mutation exclusively in AITL in the present study.
PCR analysis of the rearranged TCR genes is commonly used in aid of routine diagnosis of T‐cell lymphoma. However, the amplified PCR products are not routinely sequenced, thus not providing any definite data on the number of functional TCR rearrangements. By sequencing the PCR products, we showed evidence of bi‐ or oligo‐clonal functional TRB rearrangements in 26% of AITL and PTCL‐TFH. Although the clonal size could not be accurately measured by the adopted BIOMED‐2 assay as it was carried out in two different reaction tubes, there was a considerable difference in many cases between the clonal size of the different TRB rearrangements, with one being more predominant than the other, suggesting they may represent different T‐cell populations, rather than bi‐allelic rearrangements. This was confirmed by ISH using BaseScope probes to the unique TRB VDJ junctional sequence in selected cases.
By using double BaseScope ISH, we were able to confirm that both clonal T‐cell populations carried the same TET2 mutations in a representative case. These findings imply that these different T‐cell clones originated from a common precursor cell population that harboured the TET2 mutations, and this common precursor cell population is most likely at a differentiation stage before TCR gene rearrangement. In patients with AITL, the lymphoma‐associated TET2 mutations are frequently seen in haematopoietic progenitor cells and several lineages of non‐neoplastic cells including B and CD8+ T‐cells, which are polyclonal as reported to date 7, 13, 14, 16, 21. In this context, our finding of the lymphoma‐associated TET2 mutation in an independent minor T‐cell population is not totally a surprise, but this raises an interesting question about whether the same TET2 mutation may initiate an independent lymphomagenic process (Figure 6). Nonetheless, it remains to be investigated whether the above finding represents a common feature of AITL, or some of the minor T‐cell clones might be the consequence of immune reaction to immunodeficiency and EBV reactivation in patients with AITL, rather than due to TET2 or DNMT3A mutation (Figure 6).
Figure 6.
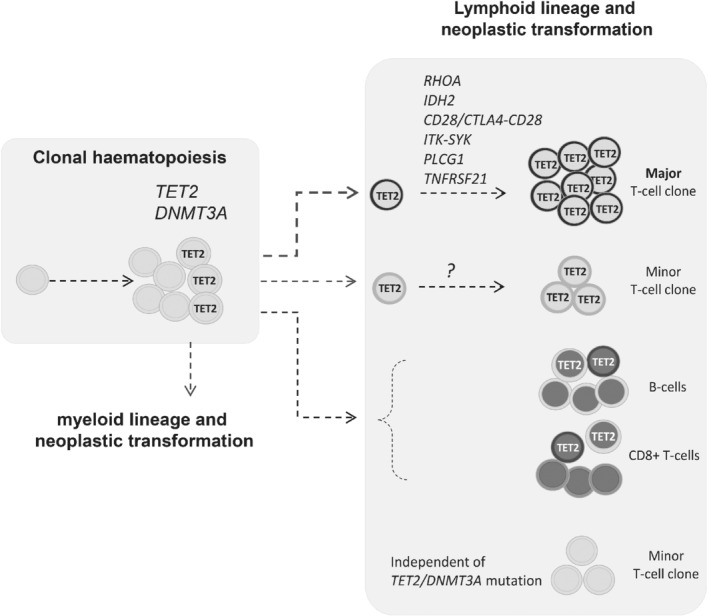
The proposed model of multistage development in AITL. Mutations in DNA methylation regulators—namely TET2, DNMT3A, and IDH2—are early events, with TET2 and DNMT3A mutation occurring in hematopoietic stem cells. These mutations enhance the self‐renewal of haematopoietic stem cells, increase the risk of lymphomagenesis, and may frequently generate bi‐ or oligoclonal T‐cell populations in AITL or PTCL‐TFH following acquisition of additional genetic changes in genes important for T‐cell biology. The TET2 mutant hematopoietic progenitor cells also give rise to non‐neoplastic polyclonal B and CD8 positive T‐cells as reported in several previous studies 7, 13, 14, 16, 21. Finally, there is also a possibility that minor T‐cell clones are not related to TET2 or DNMT3A mutation but are the consequence of immunodeficiency and EBV reactivation in patients with AITL.
To investigate the above speculation, we performed double BaseScope ISH and immunohistochemistry, and demonstrated that both T‐cell populations in the index case are positive for CD4, but not CD8. Based on BaseScope ISH, the minor V27‐J2 clone was sparsely distributed in all the three independent tissue biopsies available for study, without showing any clusters. The nature and malignant potential of this minor T‐cell clone is unclear. Nonetheless, parallel malignant processes, such as development of AITL and myeloid leukaemia from the same TET2 mutant haematopoietic progenitor population have been elegantly illustrated by recent case studies 48, 49.
The clinical relevance of the finding of an additional minor clonal T‐cell population in AITL and PTCL‐TFH remains to be investigated. It is essential to accurately determine the clonal size of such minor clonal T‐cell population and their evolution during disease progression using more established next‐generation sequencing––based TRB rearrangement analysis. In particular, it is important to investigate both metachronous and synchronous lesions to assess whether such minor clonal T‐cell population may become a major pathogenic player. This will help to identify suitable samples containing more expanded cell population for further in‐depth phenotypic and genetic investigations. Specimens differentially involved by distinct T‐cell clones would be highly valuable for further characterization of potential genetic changes in minor T‐cell clones, particularly RHOA and IDH2 mutations, as they are frequent and specifically seen in the neoplastic T‐cell clone. Discrete T‐cells such as CD4+CD10+ T‐cells and CD8+ T‐cells could be isolated using fluorescence‐activated cell sorting and subjected to single‐cell analysis for TCR gene rearrangement and somatic mutation analysis.
Unlike TET2 and DNMT3A mutations that occur at the haematopoietic stem cell level, both RHOA and IDH2 mutations are restricted to the neoplastic T‐cell clone in AITL and PTCL‐TFH 7, 16, 21, 22. In line with this, both the RHOA and IDH2 mutation loads were similar to each other, but significantly lower than the TET2 and DNMT3A mutation load (supplementary material, Figure S3). In mouse model, RhoA G17 V cooperates with TCR stimulation to promote TFH‐cell expansion, and lymphoma development in absence of Tet2 30, 31, 50. The IDH2 R172 mutation causes a high level of oncometabolite 2‐hydroxyglutarate in the neoplastic T cells, which inhibits multiple α‐KG–dependent dioxygenases involved in various cell functions, including histone methylation and the hypoxia response 21. The frequent concurrence of both RHOA and IDH2 mutations in AITL argues for their oncogenic synergy, and together they most likely play an important role in malignant transformation and also underpin the morphological and immunophenotypic presentations as discussed earlier.
By sequencing the rearranged TCR genes, we also demonstrated a significantly biased usage of certain TRB variable genes, particularly TRBV19*01 and TRBV27, in AITL and PTCL‐TFH. The high identify in the VDJ junctional sequence of TRBV19*01 rearrangement in two unrelated cases strongly argues for their similarity in antigen recognition. Although the antigen recognized by these TCRs and its impact on TCR signaling are unknown, a previous study demonstrated that among CD4+ T‐cells, those expressing TRBV19 or TRBV27 TCR were highly responsive to stimulation by staphylococcus enterotoxin B superantigen in vitro 51. Thus, it is pertinent to speculate that the intrinsic properties of TCR may render T cells to perpetual antigenic stimulation, chronic TCR signaling, hence clonal selection and expansion.
In summary, the present study shows evidence of bi‐ or oligoclonal T‐cell populations in a high proportion of AITL and PTCL‐TFH, and their derivation from a common TET2 mutant progenitor cell population in a representative case (Figure 6). We also demonstrate a significant biased TRB variable gene usage in these T‐cell lymphomas, suggesting an important role of TCR signaling in clonal selection and expansion for malignant transformation.
Author contributions statement
WQY and FW designed experiments, and collected and analysed data. LC carried out immunohistochemistry. SWZ, AC and MW designed the Fluidigm gene panel and provided technical assistance. JST and ZC carried out Illumina sequencing analysis and variant calling. HL, HB, YH, JWG, PW, HED, LRB, LF, GAF, ZG, ADA, SSC, MAK and WL contributed and reviewed cases. MQD, WY and FW wrote and prepared the manuscript. MQD designed and coordinated the study. All authors commented on the manuscript and approved its submission for publication.
Supporting information
Figure S1. Nature and distribution of mutations in TET2, DNMT3A, IDH2, RHOA, PLCG1, CCND3, and TNFRSF21 in AITL and PTCL‐TFH
Figure S2. Examples of somatic mutations identified by Fluidigm multiplex PCR and Illumina MiSeq sequencing in AITL
Figure S3. Comparison of mutation load among TET2, DNMT3A, RHOA, and IDH2 changes
Figure S4. Examples of TRB sequencing by BIOMED‐2 PCR and Illumina MiSeq sequencing
Table S1. PCR primers and conditions used in the study
Table S2. List of mutations identified in AITL, PTCL‐TFH, and PTCL‐NOS
Acknowledgements
The authors would like to thank Graeme Clark and Ezequiel Martin for their assistance with Illumina sequencing, Dianying Liao for preparation of cases, and all members of MQD's lab for their valuable suggestion and assistance in data analysis.The research in MQD's lab was supported by grants from Bloodwise (13 006, 15 019) UK, The Kay Kendall Leukaemia Fund (KKL582) UK, and The Pathological Society of Great Britain and Ireland, UK. WQY and FW were both supported by a research fellowship from the China Scholarship Council. The Human Research Tissue Bank is supported by the NIHR Cambridge Biomedical Research Centre.
Conflict of interest statement: M‐QD is an Associate Editor of The Journal of Pathology. No other conflicts of interest were declared.
References
- 1. Dogan A, Attygalle AD, Kyriakou C. Angioimmunoblastic T‐cell lymphoma. Br J Haematol 2003; 121:681–691. [DOI] [PubMed] [Google Scholar]
- 2. Dogan A, Gaulard P, Jaffe R, et al Angioimmunoblastic T‐cell lymphoma and other nodal lymphomas of T follicular helper cell origin In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Swerdlow SH, Campo E, Harris NL, et al (eds). WHO Press: Lyon, 2017; 407–412. [Google Scholar]
- 3. Gaulard P, de Leval L. Follicular helper T cells: implications in neoplastic hematopathology. Semin Diagn Pathol 2011; 28: 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahearne MJ, Allchin RL, Fox CP, et al Follicular helper T‐cells: expanding roles in T‐cell lymphoma and targets for treatment. Br J Haematol 2014; 166:326–335. [DOI] [PubMed] [Google Scholar]
- 5. Cairns RA, Iqbal J, Lemonnier F, et al IDH2 mutations are frequent in angioimmunoblastic T‐cell lymphoma. Blood 2012; 119:1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palomero T, Couronne L, Khiabanian H, et al Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 2014; 46:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sakata‐Yanagimoto M, Enami T, Yoshida K, et al Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:171–175. [DOI] [PubMed] [Google Scholar]
- 8. Yoo HY, Sung MK, Lee SH, et al A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:371–375. [DOI] [PubMed] [Google Scholar]
- 9. Odejide O, Weigert O, Lane AA, et al A targeted mutational landscape of angioimmunoblastic T‐cell lymphoma. Blood 2014; 123:1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang M, Zhang S, Chuang SS, et al Angioimmunoblastic T cell lymphoma: novel molecular insights by mutation profiling. Oncotarget 2017; 8:17763–17770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dobay MP, Lemonnier F, Missiaglia E, et al Integrative clinicopathological and molecular analyses of angioimmunoblastic T‐cell lymphoma and other nodal lymphomas of follicular helper T‐cell origin. Haematologica 2017; 102:e148–e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang C, McKeithan TW, Gong Q, et al IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T‐cell lymphoma. Blood 2015; 126:1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T‐cell lymphoma. N Engl J Med 2012; 366:95–96. [DOI] [PubMed] [Google Scholar]
- 14. Quivoron C, Couronne L, Della V, et al TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011; 20:25–38. [DOI] [PubMed] [Google Scholar]
- 15. Xie M, Lu C, Wang J, et al Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20:1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwartz FH, Cai Q, Fellmann E, et al TET2 mutations in B cells of patients affected by angioimmunoblastic T‐cell lymphoma. J Pathol 2017; 242:129–133. [DOI] [PubMed] [Google Scholar]
- 17. Vallois D, Dobay MP, Morin RD, et al Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T‐cell‐derived lymphomas. Blood 2016; 128:1490–1502. [DOI] [PubMed] [Google Scholar]
- 18. Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre‐leukemic stem cells in acute myeloid leukemia. Int J Hematol 2013; 98:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamidi T, Singh AK, Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015; 7:247–265. [DOI] [PubMed] [Google Scholar]
- 20. Yan H, Parsons DW, Jin G, et al IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lemonnier F, Cairns RA, Inoue S, et al The IDH2 R172K mutation associated with angioimmunoblastic T‐cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A 2016; 113:15084–15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen TB, Sakata‐Yanagimoto M, Asabe Y, et al Identification of cell‐type‐specific mutations in nodal T‐cell lymphomas. Blood Cancer J 2017; 7:e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cortes JR, Palomero T. The curious origins of angioimmunoblastic T‐cell lymphoma. Curr Opin Hematol 2016; 23:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fukumoto K, Nguyen TB, Chiba S, et al Review of the biologic and clinical significance of genetic mutations in angioimmunoblastic T‐cell lymphoma. Cancer Sci 2018; 109:490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kataoka K, Nagata Y, Kitanaka A, et al Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 2015; 47:1304–1315. [DOI] [PubMed] [Google Scholar]
- 26. Yoo HY, Kim P, Kim WS, et al Frequent CTLA4‐CD28 gene fusion in diverse types of T‐cell lymphoma. Haematologica 2016; 101:757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rohr J, Guo S, Huo J, et al Recurrent activating mutations of CD28 in peripheral T‐cell lymphomas. Leukemia 2016; 30:1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fujisawa M, Sakata‐Yanagimoto M, Nishizawa S, et al Activation of RHOA‐VAV1 signaling in angioimmunoblastic T‐cell lymphoma. Leukemia 2018; 32:694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scourzic L, Couronne L, Pedersen MT, et al DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 2016; 30:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zang S, Li J, Yang H, et al Mutations in 5‐methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest 2017; 127:2998–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cortes JR, Ambesi‐Impiombato A, Couronne L, et al RHOA G17V induces T follicular helper cell specification and promotes Lymphomagenesis. Cancer Cell 2018; 33:259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang M, Escudero‐Ibarz L, Moody S, et al Somatic mutation screening using archival formalin‐fixed, paraffin‐embedded tissues by Fluidigm multiplex PCR and Illumina sequencing. J Mol Diagn 2015; 17:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DePristo MA, Banks E, Poplin R, et al A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cucco F, Clipson A, Kennedy H, et al Mutation screening using formalin‐fixed paraffin‐embedded tissues: a stratified approach according to DNA quality. Lab Invest 2018; 98:1084–1092. [DOI] [PubMed] [Google Scholar]
- 35. Ye K, Schulz MH, Long Q, et al Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics 2009; 25:2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Dongen JJ, Langerak AW, Bruggemann M, et al Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T‐cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED‐2 concerted action BMH4‐CT98‐3936. Leukemia 2003; 17:2257–2317. [DOI] [PubMed] [Google Scholar]
- 37. Duez M, Giraud M, Herbert R, et al Vidjil: a web platform for analysis of high‐throughput repertoire sequencing. PLoS One 2016; 11:e0166126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weinstein JS, Lezon‐Geyda K, Maksimova Y, et al Global transcriptome analysis and enhancer landscape of human primary T follicular helper and T effector lymphocytes. Blood 2014; 124:3719–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Freeman JD, Warren RL, Webb JR, et al Profiling the T‐cell receptor beta‐chain repertoire by massively parallel sequencing. Genome Res 2009; 19:1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu L, Li Z, Cheng J, et al Crystal structure of TET2‐DNA complex: insight into TET‐mediated 5mC oxidation. Cell 2013; 155:1545–1555. [DOI] [PubMed] [Google Scholar]
- 41. Figueroa ME, Abdel‐Wahab O, Lu C, et al Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine‐286 prevents its rapid degradation via the ubiquitin‐proteasome pathway. Genes Dev 1997; 11:957–972. [DOI] [PubMed] [Google Scholar]
- 43. Rohde M, Bonn BR, Zimmermann M, et al Relevance of ID3‐TCF3‐CCND3 pathway mutations in pediatric aggressive B‐cell lymphoma treated according to the non‐Hodgkin Lymphoma Berlin‐Frankfurt‐Munster protocols. Haematologica 2017; 102:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmitz R, Young RM, Ceribelli M, et al Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012; 490:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ondrejka SL, Grzywacz B, Bodo J, et al Angioimmunoblastic T‐cell lymphomas with the RHOA p.Gly17Val mutation have classic clinical and pathologic features. Am J Surg Pathol 2016; 40:335–341. [DOI] [PubMed] [Google Scholar]
- 46. Nagao R, Kikuti YY, Carreras J, et al Clinicopathologic analysis of angioimmunoblastic T‐cell lymphoma with or without RHOA G17V mutation using formalin‐fixed paraffin‐embedded sections. Am J Surg Pathol 2016; 40:1041–1050. [DOI] [PubMed] [Google Scholar]
- 47. Steinhilber J, Mederake M, Bonzheim I, et al The pathological features of angioimmunoblastic T‐cell lymphomas with IDH2(R172) mutations. Mod Pathol 2019; 32:1123–1134. [DOI] [PubMed] [Google Scholar]
- 48. Tiacci E, Venanzi A, Ascani S, et al High‐risk clonal hematopoiesis as the origin of AITL and NPM1‐mutated AML. N Engl J Med 2018; 379:981–984. [DOI] [PubMed] [Google Scholar]
- 49. Lemonnier F, Sujobert P, Abraham J, et al Angioimmunoblastic T‐cell lymphoma and chronic myelomonocytic leukemia: a non‐fortuitous assocaition arising from precursor hematopoietic cells with acquired TET2 mutations. EAHP 2018; 2018:54. [Google Scholar]
- 50. Ng SY, Brown L, Stevenson K, et al RhoA G17V is sufficient to induce autoimmunity and promotes T‐cell lymphomagenesis in mice. Blood 2018; 132:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ozawa T, Tajiri K, Kishi H, et al Comprehensive analysis of the functional TCR repertoire at the single‐cell level. Biochem Biophys Res Commun 2008; 367:820–825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Nature and distribution of mutations in TET2, DNMT3A, IDH2, RHOA, PLCG1, CCND3, and TNFRSF21 in AITL and PTCL‐TFH
Figure S2. Examples of somatic mutations identified by Fluidigm multiplex PCR and Illumina MiSeq sequencing in AITL
Figure S3. Comparison of mutation load among TET2, DNMT3A, RHOA, and IDH2 changes
Figure S4. Examples of TRB sequencing by BIOMED‐2 PCR and Illumina MiSeq sequencing
Table S1. PCR primers and conditions used in the study
Table S2. List of mutations identified in AITL, PTCL‐TFH, and PTCL‐NOS