

Abstract
Platelets play a critical role in both the initiation and progression of atherosclerosis, and even more so in the ensuing atherothrombotic complications. Low-dose aspirin remains the mainstay of antiplatelet therapy in high-risk patients by reducing the risk of myocardial ischemia (MI), stroke, or death due to cardiovascular disease (CVD). However, antiplatelet therapies lose their efficacy in people with diabetes mellitus (DM), increasing the risk of future atherothrombotic events. The molecular mechanisms that promote platelet hyperactivity remain unclear but could be due to glycation-induced conformational changes of platelet membranes resulting in impaired aspirin entry, or less-efficient acetylation/compensatory increase in COX-2 expression in newborn platelets. Emerging evidence from our laboratory and elsewhere suggest that enhanced platelet turnover (thrombopoiesis), particularly the production of immature reticulated platelets from the bone marrow, could contribute to atherosclerotic complications. We have identified a major role for neutrophil-derived S100A8/A9, a damage-associated molecular pattern, in driving reticulated thrombopoiesis by directly interacting with its receptors on Kupffer cells in the liver. In this review, we discuss the role of hepatic inflammation in driving reticulated platelet production and suggest potential targets to control their production, improve efficacy of current antiplatelet therapies, and reduce the risk of atherothrombotic complications.
Keywords: diabetes, immune system, cardiovascular, liver, hyperglycemia
Introduction:
Coronary artery disease (CAD) has been the leading cause of death worldwide for the last few decades except in the very low-income group population (World Health Organisation, 2017). Atherothrombosis and atherosclerosis are the pathologic basis for CAD and are widely appreciated to be multifactorial diseases with predominant factors of elevated lipid levels and chronic inflammation (Weber and Noels 2011; Woollard and Geissmann 2010). Emphasizing the role of lipids, statins have been the gold standard for treating CAD and a large number of randomized controlled statin trials have confirmed their beneficial effects in various patient groups of CAD (Pedersen 2016). However, in the last three decades evidence from experimental and clinical studies has also lent support to the inflammatory hypothesis in the pathogenesis of atherosclerotic disease in conjunction with or beyond elevated lipid levels (Reviewed by Libby 2012). The recent results of the CANTOS trial (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) showed clinical evidence that controlling vascular inflammation independently of lipid lowering could lower the rates of recurrent cardiovascular events in a large Phase 3 trial (Ridker, et al. 2017). Inflammatory signals likely cause plaque instability which could result in plaque rupture, fissuring, or erosion leading to a thrombotic response that causes myocardial infarction/ischemia (MI). However, strictly anti-inflammatory drugs have never been used alone to treat patients with CAD.
Elevated leukocyte count has been correlated with cardiovascular disease since the 1920s (Madjid, et al. 2004). The myeloid compartment of the white blood cells (WBCs), namely, monocytes (Choi, et al. 2017), neutrophils (Tan, et al. 2015; Wheeler, et al. 2004) and platelets (Huo, et al. 2003), are the predominant players in driving this association and have all been causally implicated in influencing CAD initiation or progression and impairing the regression of atheromatous plaques. In this context, it is not surprising to note that aspirin, by virtue of its inhibition of platelet aggregation by blocking thromboxane A2 formation, has been used for several decades as the gold standard in secondary prevention of CAD (Trialists’ Collaboration 1994). Here we review the common theme of myelopoiesis and more specifically, megakaryopoiesis, in driving the pathogenesis of CAD. We will examine the different pathways that promote the production of platelets, with an emphasis on reticulated platelets and the role of the liver in regulating megakaryopoiesis and thrombopoiesis.
Cardiovascular risk factor contribution to myelopoiesis
As stated above, monocyte levels are associated with CAD. However, different metabolic risk factors for CAD seem to promote the production of monocytes through varying mechanisms. While these risk factors act through independent pathways, it is important to note that these risk factors typically cluster together in metabolic diseases such as diabetes and metabolic syndrome to promote an exacerbated myelopoietic phenotype. The mechanisms of each of these risk factors in relation to myeloid-driven CAD are summarized in Figure 1 (Modified from Murphy and Tall 2016) and detailed below:
Figure 1: Cardiovascular risk factors promote myelopoiesis and accelerate atherosclerosis.
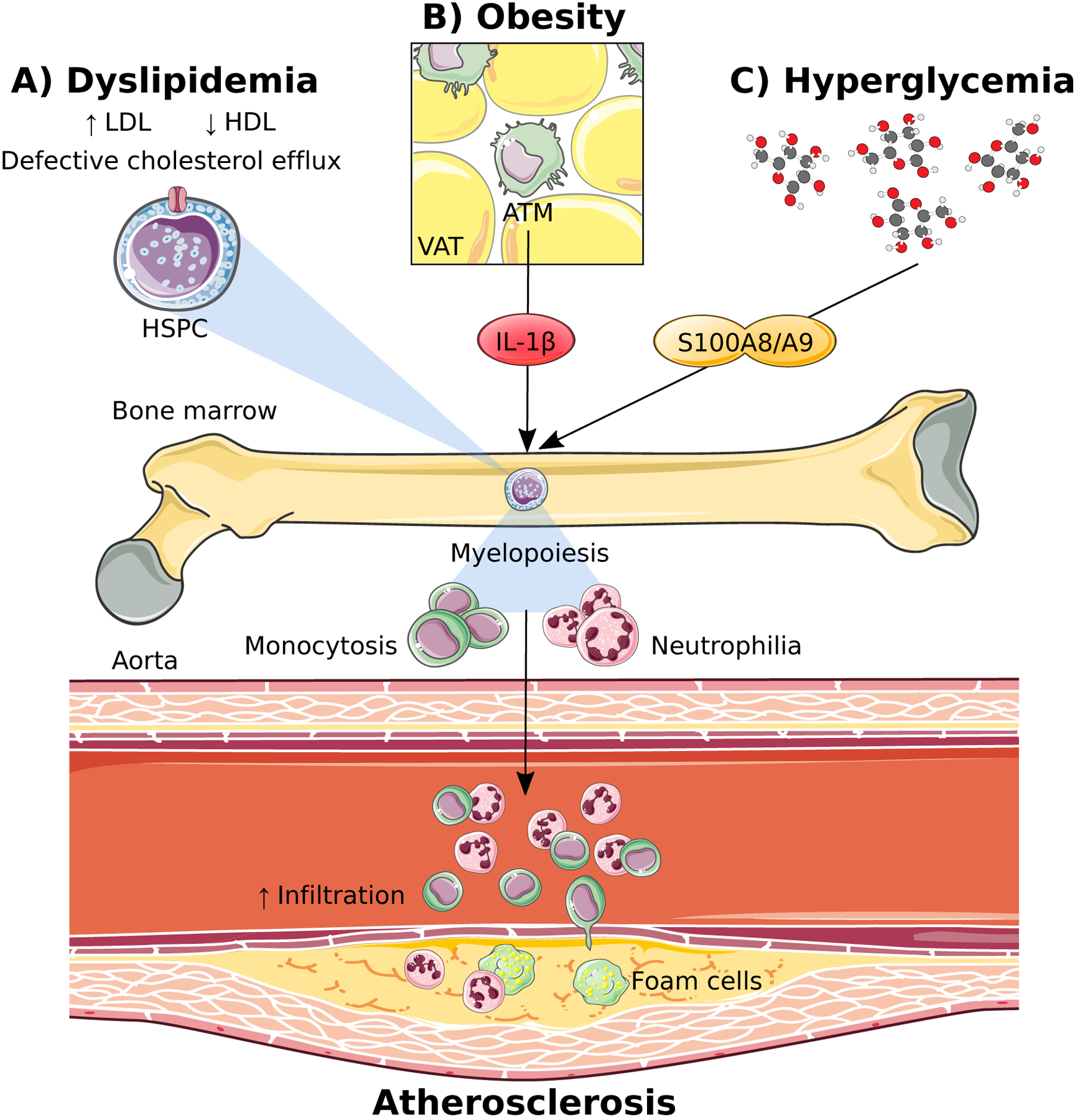
A) Dyslipidemia in the form of increased low-density lipoproteins (LDL) and decreased high-density lipoproteins (HDL) directly promotes haematopoietic stem and progenitor cell (HSPC) differentiation via impaired cholesterol efflux in these cells. B) Obesity indirectly promotes myelopoiesis by stimulating IL-1β production from adipose tissue macrophages (ATM) in the visceral adipose tissue (VAT). C) Hyperglycemia induces an upregulation in S100A8/A9 which travels to the bone marrow to stimulate myelopoiesis.
1. Dyslipidemia:
Dyslipidemia refers to abnormal amounts of lipids in the blood and is a central component of both diabetes and metabolic syndrome, typically presenting as hyperlipidemia with an increase in triglycerides and low-density lipoprotein (LDL) cholesterol with a reduction in high density lipoprotein (HDL) cholesterol. In the context of dyslipidemia, lipid metabolism and cholesterol efflux are important regulators of myeloid cell production. The importance of lipids in regulating bone marrow (BM) hematopoietic stem and progenitor cell (HSPC) proliferation was originally discovered through the use of genetically deficient models of key cholesterol efflux genes, including ABCA1 and ABCG1, (ATP-binding cassette (ABC) transporters, belonging to a family of highly conserved cellular transmembrane proteins) as well as their cell-intrinsic ligand apolipoprotein E (apoE). Deletion of these genes resulted in cholesterol accumulation, enhanced proliferation and over production of monocytes and neutrophils (Murphy, et al. 2011; Yvan-Charvet, et al. 2010) (Figure 1A). Importantly, this phenotype could be reversed by increasing HDL levels to promote cholesterol efflux through either genetic or pharmacological approaches. It was also revealed that concurrently increasing LDL levels and reducing HDL levels using a genetic approach further influenced the proliferation of HSPCs resulting in enhanced monocytosis and accelerated atherosclerosis (Tolani, et al. 2013). Thus, regulating HSPC cholesterol content is important in maintaining normal hematopoiesis to prevent the overproduction of myeloid cells that exacerbate atherosclerosis in the presence of dyslipidemia.
2. Obesity:
Obesity is known as a low-grade inflammatory disorder with a heavy macrophage burden of the adipose tissue (Weisberg, et al. 2003). This discovery brought forward the hypothesis that signals emanating from this organ could have detrimental effects on the health of other organs around the body. This was recently demonstrated in the liver, where obese adipose tissue drove hepatic neutrophil recruitment (Bijnen, et al. 2017). Similarly, seeding of monocytes in the visceral adipose tissue (VAT) was shown to involve a regulatory feed-forward loop where inflamed VAT can signal to the bone marrow (BM) hematopoietic progenitor cells to proliferate, expand, and increase the production of myeloid cells (Nagareddy, et al. 2014) (Figure 1B). Specifically, we discovered that upregulation of S100A8/A9 in the VAT, signal adipose tissue macrophages (ATMs) to induce the expression of Pro-IL-1β and the Nlrp3 inflammasome. Activation of the Nlrp3 inflammasome by an second signal (yet to be clearly defined) results in the processing of Pro-IL-1β to form mature IL-1β which then travels to the BM to induce the proliferation of hematopoietic progenitor cells (common myeloid progenitors; CMPs and granulocyte macrophage progenitors; GMPs) resulting in monocytosis and neutrophilia (Nagareddy et al. 2014). The excess number of monocytes in the circulation give rise to more ATMs which in turn, trigger further adipose inflammation completing the loop and also promote insulin resistance. Given the positive outcome, albeit small, in reducing cardiovascular events in the CANTOS trial, alongside the remaining controversy over the explicit role of IL-1β in the general pathogenesis of atherosclerosis (Duewell, et al. 2010; Menu, et al. 2011; Ridker et al. 2017), it will be of specific interest to determine whether those of greatest benefit with IL-1β targeted therapies are the obese individuals, particularly those with a recent cardiovascular event. Additionally, but beyond the scope of this review, another sub-group of patients that could benefit from IL-1β targeted therapies are those with clonal hematopoiesis of intermediate potential (CHIP), particularly those with TET-2 mutations (Fuster, et al. 2017; Jaiswal, et al. 2017), or patients with IL-1β associated autoimmune disorders such as rheumatoid arthritis that is linked to accelerated CVD (Dragoljevic, et al. 2018).
3. Hyperglycemia:
It is well known that people with diabetes have elevated WBC counts (Ford 2002; Schmidt, et al. 1999; Vuckovic, et al. 2007; Woo, et al. 2011). Modeling this in mice, we discovered that hyperglycemia promotes myelopoiesis by increasing the release of S100A8/A9 from neutrophils which then interacts with the glucose-inducible receptor for advanced glycation end-products (RAGE) on CMPs, resulting in the production of inflammatory Ly6-Chi monocytes. The importance of this finding was that the increased production of Ly6-Chi monocytes contribute to persistent entry into the atherosclerotic lesion, impairing lesion regression as previously documented (even when cholesterol levels were effectively lowered) (Figure 1C) (Nagareddy, et al. 2013). Impaired lesion regression has also been observed in people with diabetes, even though LDL levels were lowered equally by statin treatment in comparison to people without diabetes(Hiro, et al. 2010), and modelled in mice (Parathath, et al. 2011). In the pre-clinical models, we found that this myelopoietic defect could be reversed by administration of an SGLT2 inhibitor, and given the recent success of this class of drugs in reducing cardiovascular events in people with diabetes (Nagareddy et al. 2013), it will be important to explore if the reduction is due to a dampening in the production of myeloid cells. Interestingly, dysregulated cholesterol metabolism has also been alluded to in the setting of diabetes (Mauldin, et al. 2008; Passarelli, et al. 2005), and silencing miR33, a negative regulator of ABCA1, can suppress myeloid cells in the setting of diabetes (Distel, et al. 2014). Therefore, it would also be interesting to determine if elevating HDL could reverse enhanced myelopoiesis in diabetes, with benefits that extend to the vasculature.
Platelets and atherosclerosis:
Apart from the intrinsic thrombotic tendency of platelets, platelet biogenesis plays an important role in atherogenesis due to two main reasons. Firstly, more activated platelets are found in the circulation of patients with CAD (Fitzgerald, et al. 1986; Schapiro, et al. 1990), hypercholesterolemia (Broijersen, et al. 1998) and diabetes (Grove, et al. 2011) which are prone to bind leukocytes, preferentially monocytes, to form platelet–leukocyte aggregates (Furman, et al. 1998; Rinder, et al. 1992) required for thrombus evolution. The enhanced platelet production also causes an increase in immature or reticulated platelets which are invariably linked to accelerated atherogenesis (Murphy, et al. 2013). Secondly, activated platelets can produce chemokines to exacerbate atherogenesis. For example, platelet CD40L (Henn, et al. 1998) and IL-1β (Hawrylowicz, et al. 1991), can activate endothelial cells, or platelets can deposit chemokines including platelet factor-4 (PF4/CXCL4) and RANTES (CCL5), which can trigger monocyte arrest on inflamed endothelium(von Hundelshausen, et al. 2001). It should be noted that antiplatelet therapy can reduce CVD risk in non-diabetic subjects (Trialists’ Collaboration 1994), however such therapies are less effective in the context of diabetes, potentially due to an increase in reticulated platelets which are somewhat resistant to standard antiplatelet therapies (Guthikonda, et al. 2007), possibly due to their ability to re-synthesize cyclooxygenase (COX)-1 and COX-2. This is because reticulated platelets, unlike matured platelets carry residual mRNA that may help replenish COX enzymes (Angénieux, et al. 2016).
Reticulated platelets and aspirin resistance
Antiplatelet drugs decrease platelet aggregation and prevent thrombus formation, even in the arterial circulation where anticoagulants are generally ineffective. The major classes of antiplatelet drugs are irreversible COX inhibitors such as aspirin and adenosine diphosphate (ADP) receptor inhibitors (e.g. clopidogrel). However, in the last couple of decades, inter-individual variability of platelet inhibition by antiplatelet therapies of aspirin and clopidogrel have been observed. As early as 2003, Gurbel et al. showed that the platelet inhibitory response from clopidogrel varied enormously and that patients undergoing elective coronary stenting were least protected when their ex vivo platelet reactivity was high to begin with (Gurbel, et al. 2003; Vlachojannis, et al. 2011). Similarly, inter-individual variability of platelet reactivity with aspirin has been termed aspirin resistance. Aspirin inhibits platelet activation by irreversible acetylation of platelet COX-1, thereby preventing the formation of thromboxane A2. Even though the half-life of aspirin is minutes, COX-1 modification by aspirin is sufficient for platelet inhibition for up to 24 hours as platelets are anucleate and, due to mRNA degradation, mature platelets lack sufficient mRNA required to replenish COX-1 protein (Angénieux et al. 2016; Patrono, et al. 2005). It has been estimated that with low dose aspirin, COX-1 activity is restored at a rate of 10% per day reflecting the 10-day lifespan of platelets, where 10% of the platelets are newly formed and not exposed to aspirin (Eikelboom and Warkentin 2014). Even though part of the inter-individual variability of platelet inhibition can be attributed to inherent variations in ex vivo measurements and polymorphisms of cytochrome (CYP) P450 2C19 (Shuldiner, et al. 2009), it is now well-accepted that new platelet generation plays a significant mechanistic role. Like increased reticulocyte (immature erythrocyte) count use as a marker for enhanced erythropoiesis, immature platelet counts (IPC) and reticulated platelet fractions (RPF) have emerged as a marker for increased thrombopoiesis. Immature or reticulated platelets are larger, metabolically more active, and have a higher thrombotic potential than mature platelets (Brown and Martin 1994). Although the terms “reticulated platelets” and “immature platelets” are used interchangeably, reticulated platelets are detected by flow cytometry with thiazole orange staining of the high ribonucleic acid (RNA) content in a reticulated pattern (hence the name “reticulated platelets”) (Kienast and Schmitz 1990), while “immature” platelets are detected on automated cell counters with an add-on module allowing platelets to be stained with a polyamine/oxazine combination (to detect RNA). Therefore, it should be noted that reticulated platelets and immature platelets do not represent necessarily identical populations even though there is a considerable overlap (Ibrahim, et al. 2016).
Reticulated platelet count has been strongly associated with aspirin resistance, especially with short lived antiplatelet therapies as they can re-synthesize COX-1/2 (Armstrong, et al. 2017; Guthikonda, et al. 2008; Guthikonda et al. 2007). However, IPC has emerged as the strongest independent platelet count–derived predictor of antiplatelet response (Stratz, et al. 2016). In a prospective study of 93 subjects with a median follow up of 31 months, IPC has also been associated with major adverse cardiovascular events (MACE), defined as a composite of death, MI, unplanned revascularization, or recurrent angina (Ibrahim, et al. 2014). RPF as an index of platelet turnover has also been shown to be an independent predictor of MACE (Freynhofer, et al. 2017). Thrombi generated in a perfusion chamber with whole blood from normal donors under shear conditions equivalent to a significant coronary artery stenosis, revealed that younger “reticulated” platelets had greater propensity for thrombus participation compared to older and mature platelets (McBane, et al. 2014).
Diabetes is associated with a higher number of clopidogrel non-responders and increased platelet reactivity on combined aspirin and clopidogrel treatment (Angiolillo, et al. 2005). Moreover, patients with DM but without any history of coronary event such as MI carry the same level of risk as a non-diabetic MI patient for subsequent acute coronary events (Schramm, et al. 2008). Significantly higher mean platelet volume (MPV) which is also a risk factor for MACE (Mayer, et al. 2014) was seen in diabetic patients compared to nondiabetic controls, suggesting a platelet dependent role in the macrovascular complications of diabetes (Hekimsoy, et al. 2004). Not surprisingly, diabetic patients with CAD have significantly higher MPV values compared to control subjects (Tavil, et al. 2010). Hence diabetic patients exhibit a prothrombotic milieu with reticulated and hyperreactive platelets, and a consistently impaired response to antiplatelet therapy (Reviewed by Neergaard-Petersen, et al. 2016).
Megakaryopoiesis and its regulation by the Liver
Platelets are produced through the myelopoietic pathway in the BM, specifically through the differentiation of hematopoietic stem cells through to megakaryocyte-erythroid progenitors (MEPs), then megakaryocyte progenitors (MkPs), and megakaryocytes before undergoing thrombopoiesis to form platelets. The primary cytokine regulator of megakaryopoiesis is thrombopoietin (TPO), which is constitutively produced by hepatocytes in the liver and to a lesser degree by proximal tubule cells in the kidney (Qian, et al. 1998; Sungaran, et al. 1997). TPO acts via its receptor, the cellular homologue of the myeloproliferative leukemia virus oncogene (c-MPL) on platelets and BM progenitor cells (de Sauvage, et al. 1994; Kuter, et al. 1994). Clearance of senescent desialyated platelets in the liver via the Ashwell-Morell receptor (AMR) induces STAT-3-mediated production of TPO in hepatocytes (Hoffmeister, et al. 2014). When circulating platelets are low, plasma TPO levels increase and promote thrombopoiesis by binding to MEPs, MkPs, and megakaryocytes. Mice lacking TPO or MPL are severely thrombocytopenic and deficient in megakaryocytes and their progenitor cells (Gurney, et al. 1994; Kaushansky 1995), a phenotype consistent with a role for TPO in maintaining appropriate megakaryocyte numbers in vivo. Platelet mass contains the highest concentration of cMpl receptor and MPL-receptor-mediated internalization and degradation through ubiquitination regulates systemic TPO levels (Saur, et al. 2010). After the platelets are released into the circulation, they act as a sink, binding to c-MPL and degrading TPO, which decreases platelet production (Saur et al. 2010). This feedback system effectively regulates TPO-driven platelet production in normal physiological conditions. The reciprocal relationship between platelet number (aka “MPL mass”) and circulating TPO levels may be an effective feedback loop during normal, but not in inflammatory conditions. For example, cytokines such as IL-6 and IL-1α can stimulate thrombopoiesis independent of platelet number or “MPL mass”, indicating that the number of platelets by themselves is not the sole determinant of circulating TPO levels and thus of thrombopoiesis (Broudy, et al. 1995; Gurney et al. 1994; Saur et al. 2010). During acute inflammation, TPO levels rise and a positive correlation is seen between the concentrations of circulating IL-6 and TPO (Hsu, et al. 1999; Ishiguro, et al. 2002). In fact, IL-6 has been shown to act directly on hepatocytes via the IL-6R to induce TPO expression, which is mediated by gp130-JAK1 activation of STAT3 independent to classical signaling via the hepatic AMR (Hoffmeister et al. 2014; Kaser, et al. 2001). Systemic TPO levels have been shown to be increased in diabetes and are associated with glycemia, insulinemia and adiposity (Maury, et al. 2010). Likewise, IL-6 is elevated in diabetes and metabolic syndrome and plays an important role in glucose metabolism, notably reducing hepatocyte insulin sensitivity (Kristiansen and Mandrup-Poulsen 2005; Pickup, et al. 1997). In the liver, IL-6 expression by Kupffer cells is increased in diabetes (Kraakman, et al. 2017) and this cytokine may play an important role in diabetes-induced thrombopoiesis. In contrast to the increased thrombopoiesis observed during inflammation, impaired liver function in patients with liver cirrhosis (LC) (Afdhal, et al. 2008) and in hepatitis B virus (HBV) related liver disease (Wang, et al. 2014a) is associated with 76% incidence of thrombocytopenia. The prevalence of cirrhosis as an etiology of thrombocytopenia of unknown origin in patients undergoing BM biopsies, is as high as 35% (Sheikh, et al. 2012). Liver cirrhosis patients typically present with decreased plasma TPO, decreased platelet production, as well as accelerated platelet destruction (Pradella, et al. 2011). Further, confirming the role of liver in thrombopoiesis, orthotopic liver transplantation in patients with LC induces platelet recovery through an increase in systemic TPO levels (Peck-Radosavljevic, et al. 2000).
Cardiovascular risk factor contribution to megakaryopoieisis
1. Dyslipidemia:
As is the case in diabetes, patients identified as hyperlipidemic also exhibit increased platelet activation (Podrez, et al. 2007; Vergeer, et al. 2011), higher platelet counts and increased MPV (Grove et al. 2011) indicating that dyslipidemia may play a direct role in platelet production and/or function and contribute to diabetes-associated thrombocytosis. For example, deficiency of ATP-binding cassette transporter B6 (ABCB6) and cholesterol transporter ABCG4, which promote cholesterol efflux to HDL in MkPs, can both lead to expansion and proliferation of MkPs (Murphy et al. 2013; Murphy, et al. 2014) (Figure 2A). ABCB6 is restricted to MEPs (Murphy et al. 2014) while ABCG4 has been shown to be highly expressed in BM MkPs (Murphy et al. 2013). Enhanced megakaryopoiesis in the absence of ABCB6 was accompanied by increased platelet counts, MPV, and platelet activity together with increased platelet–leukocyte aggregates and accelerated atherosclerosis (Murphy et al. 2014). In the absence of ABCG4, MkPs showed defective cholesterol efflux to HDL, increased cell surface expression of the TPO receptor, c-MPL, followed by enhanced proliferation and megakaryopoiesis (Nagareddy et al. 2013). Mechanistically, disruption of negative feedback regulation of c-MPL signaling by the E3 ligase c-CBL and the cholesterol-sensing LYN kinase were implicated. Interestingly, infusions of reconstituted HDL could reverse this phenotype in an ABCG4-dependent fashion. It is pertinent to note that people with myeloproliferative neoplasms like essential thrombocytosis and polycythemia vera, often die of atherothrombotic disease (Murphy and Tall 2016). However, the mechanism of enhanced thrombocytosis in these patients is due to cell-intrinsic mutations in MkPs and other BM progenitors resulting in uncontrolled proliferation (Maugeri, et al. 2011) which is different from megakaryopoiesis associated with CAD risk factors where the culprits are usually extrinsic. In the case of altered lipid levels, this extrinsic mediator is the abnormal cholesterol efflux in hematopoietic progenitors. In addition, while cholesterol handling of stem and progenitor cells in the BM plays an important role in regulating myelopoiesis and alterations in MkP cholesterol efflux leads to enhanced megakaryopoiesis, it is also possible that more global effects of dyslipidemia, particularly in the liver, may contribute to lipid-induced megakaryopoiesis.
Figure 2: Cardiovascular risk factors drive reticulated thrombopoiesis and accelerate atherosclerosis via enhanced megakaryopoiesis.
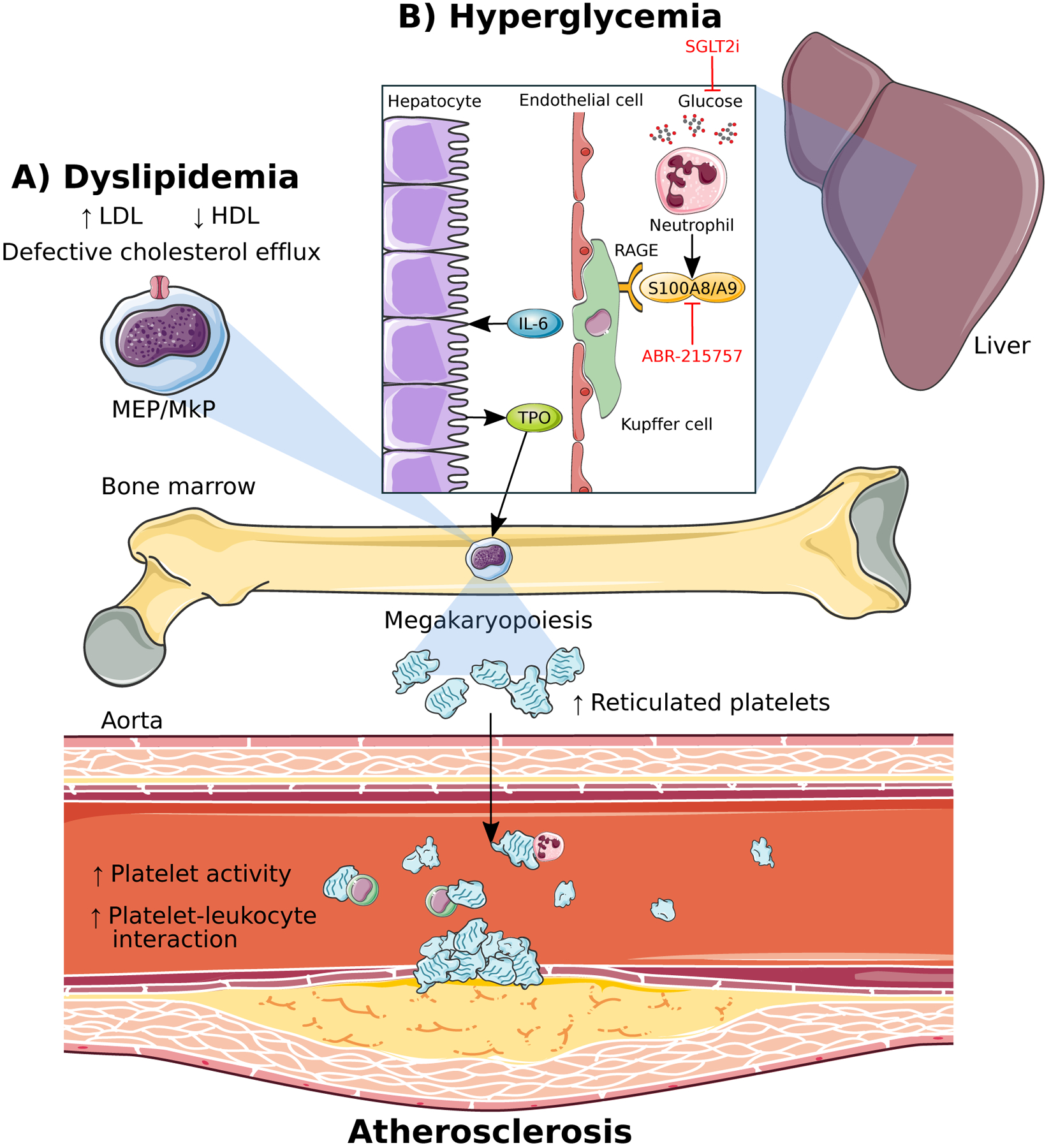
A) Dyslipidemia in the form of form of increased low-density lipoproteins (LDL) and decreased high-density lipoproteins (HDL) along with defective cholesterol efflux promotes proliferation and differentiation of megakaryocyte-erythroid progenitors (MEPs) and megakaryocyte progenitors (MkPs). B) Hyperglycemia-induced S100A8/A9 from neutrophils stimulates megakaryopoiesis and promotes atherosclerosis via enhanced liver inflammation. Binding of S100A8/A9 to the receptor for advanced glycation end-products (RAGE) on Kuppfer cells in the liver stimulates IL-6 production leading to TPO production from hepatocytes which travels to the bone marrow to promote megakaryopoiesis. Reticulated thrombocytosis resulting from enhanced megakaryopoiesis in both dyslipidemia and hyperglycemia leads to increased platelet activity, platelet-leukocyte interactions and accelerated atherosclerosis. Lowering blood glucose using the sodium-glucose cotransporter 2 inhibitor (SGLT2i), dapagliflozin, as well as blocking S100A8/A9 binding to RAGE, prevents hyperglycemia-induced thrombocytosis and atherosclerosis.
2. Hyperglycemia:
The residual risk of CVD in diabetes despite lipid lowering, as described earlier, implies that there are major gaps in our understanding of the underlying mechanisms responsible for increased CVD in diabetes. In our recent study, we unraveled a novel link between hyperglycemia and enhanced platelet production and reactivity (Kraakman et al. 2017). We found that diabetic mice had significantly higher production of reticulated platelets through overall expansion of BM progenitor populations; specifically, CMPs, MEPs and MkPs (Figure 2B). This appeared to be driven by TPO originating from the liver, as IL-6-driven liver inflammation was evident. Transplantation of IL-6 knockout BM followed by injection of STZ led to normalized levels of reticulated platelets and BM MkPs without any decrease in liver Kupffer cell numbers. This implied that IL-6 downstream of Kupffer cells is the key mediator of diabetes-associated thrombocytosis through induction of TPO (Kraakman et al. 2017). In a cohort of diabetic patients, increased plasma levels of TPO were also observed, similar to our pre-clinical model (Grove et al. 2011; Kraakman et al. 2017). Through a series of bone marrow transplant (BMT) studies using BM from different genetic deficient models, we found that neutrophil-derived S100A8/A9, binding to the RAGE on the Kupffer cells from diabetic mice caused increases in Kupffer cell abundance and IL-6 expression, consistent with the increased inflammatory tone of the liver. Furthermore, S100a9–/– mice, or hematopoietic deletion of S100A9 through BMTs also protected mice from diabetes-induced thrombocytosis confirming that the source of the ligands were hematopoietic cells. Independent of hyperglycemia, S100A8/A9 is also known to contribute to the biological response by promoting leukocyte recruitment in vascular injury and regulate thrombosis (Croce, et al. 2009; Wang, et al. 2014b).
Neutrophils are the predominant source of the serum S100A8/A9 (Nagareddy et al. 2013) and neutrophil depletion using an anti–Ly6-G antibody in diabetic mice caused significant lowering of circulating platelets and BM MEPs. Increased serum S100A8/A9 are biomarkers for chronic inflammatory disorders and localize to sites of infection or sterile injury (Pruenster, et al. 2016). Does this imply that neutrophils are sensors of hyperglycemia? Indeed, neutrophil depletion studies decrease the levels of S100A8/A9. It is interesting to note that leukocytes have differential glycolytic and oxidative metabolic profiles where lymphocytes primarily rely on oxidative phosphorylation to metabolise glucose, while neutrophils are almost entirely glycolytic with platelets and monocytes being relatively intermediate (Kramer, et al. 2014). S100A8/A9 levels were increased in a cohort of individuals with type 1 diabetes (T1D) with complications (Nagareddy et al. 2013) and similar to results in mice, T2DM patients had a significant increase in the abundance of reticulated platelets that correlates with glycated haemoglobin as well as increased plasma S100A8/A9 levels. Our studies have identified two potential therapeutic strategies, 1) mice treated with sodium-glucose cotransporter 2 inhibitor (SGLT2i), dapagliflozin, lowered blood glucose concomitantly with reduced number of reticulated platelets, 2) STZ-treated mice ameliorated their diabetes-induced athero-progression with S100A9 inhibitor ABR-215757. Not surprisingly, in the EMPA-REG OUTCOME trial a sodium-glucose cotransporter 2 inhibitor, empagliflozin, reduced cardiovascular morbidity and mortality in patients with type 2 diabetes mellitus and established cardiovascular disease (Wanner, et al. 2018). It would be interesting to see the effect of S100A8/A9 targeting small-molecule inhibitor ABR-215757 (Paquinimod) in diabetic patients, which has already been shown to have fewer side effects and encouraging pharmacokinetics in a dose-ranging study with cohorts of SLE patients (Bengtsson, et al. 2012).
Conclusions:
Diabetes is associated with enhanced atherosclerosis and atherothrombosis which is resistant to antiplatelet therapies. Liver inflammation in diabetes as well as impaired cholesterol efflux in the platelet-lineage progenitor cells promotes megakaryopoiesis and leads to reticulated thrombopoiesis, generating new platelets uninhibited by antiplatelet drugs, increasing platelet activity, and accelerating atherosclerosis. S100A8/A9 from neutrophils induced by hyperglycemia promotes this liver inflammation, resulting in the IL-6-mediated production of the megakaryopoiesis-inducing factor, TPO, in the liver. S100A8/A9 also both directly and indirectly promotes the production of monocytes and neutrophils through enhanced myelopoiesis in the context of hyperglycemia and obesity. Moreover, dyslipidemia and impaired cholesterol efflux in the stem and progenitor cells upstream of monocytes and neutrophils also contributes to enhanced myelopoiesis and accelerates atherosclerosis. Given the cumulative effects of these risk factors underlying CVD in promoting thrombocytosis and in generating reticulated platelets resistant to antiplatelet drugs such as aspirin, it is unsurprising that current therapies for thrombocytosis often fail to achieve effective platelet inhibition. This is particularly evident in patients with diabetes, resulting in persistent high platelet reactivity with is associated with an increased risk of cardiovascular events. Targeting platelet production along with mature platelet reactivity may provide a more effective and reliable approach to reducing thrombocytosis associated with metabolic disease. Moreover, while the potential additive or synergistic effects of inhibiting S100A8/A9 alongside lipid lowering to inhibit thrombocytosis have yet to be explored, these data suggest that combined targeting of these pathways could be beneficial in lowering platelet activity as well as monocyte and neutrophil levels in patients with metabolic diseases and have the overall potential to reduce CVD in these ‘at-risk’ patients.
Funding
PRN was supported by grants from the NIH (R01HL1379 & R00HL1225). AJM was supported by a Career Development Fellowship from the NHMRC (APP1085752), a Future Leader Fellowship from the National Heart Foundation (100440), and a Centenary Award from CSL.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as influencing the objectivity of this review.
References
- Afdhal N, McHutchison J, Brown R, Jacobson I, Manns M, Poordad F, Weksler B & Esteban R 2008. Thrombocytopenia associated with chronic liver disease. J Hepatol 48 1000–1007. [DOI] [PubMed] [Google Scholar]
- Angénieux C, Maître B, Eckly A, Lanza F, Gachet C & de la Salle H 2016. Time-dependent decay of mRNA and ribosomal RNA during platelet aging and its correlation with translation activity. PloS one 11 e0148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramirez C, Sabate M, Jimenez-Quevedo P, Hernandez R, Moreno R, Escaned J, Alfonso F, et al. 2005. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes 54 2430–2435. [DOI] [PubMed] [Google Scholar]
- Armstrong PC, Hoefer T, Knowles RB, Tucker AT, Hayman MA, Ferreira PM, Chan MV & Warner TD 2017. Newly Formed Reticulated Platelets Undermine Pharmacokinetically Short-Lived Antiplatelet Therapies. Arterioscler Thromb Vasc Biol 37 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson AA, Sturfelt G, Lood C, Ronnblom L, van Vollenhoven RF, Axelsson B, Sparre B, Tuvesson H, Ohman MW & Leanderson T 2012. Pharmacokinetics, tolerability, and preliminary efficacy of paquinimod (ABR-215757), a new quinoline-3-carboxamide derivative: studies in lupus-prone mice and a multicenter, randomized, double-blind, placebo-controlled, repeat-dose, dose-ranging study in patients with systemic lupus erythematosus. Arthritis Rheum 64 1579–1588. [DOI] [PubMed] [Google Scholar]
- Bijnen M, Josefs T, Cuijpers I, Maalsen CJ, van de Gaar J, Vroomen M, Wijnands E, Rensen SS, Greve JWM & Hofker MH 2017. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut gutjnl-2016-313654. [DOI] [PubMed] [Google Scholar]
- Broijersen A, Hamsten A, Eriksson M, Angelin B & Hjemdahl P 1998. Platelet activity in vivo in hyperlipoproteinemia--importance of combined hyperlipidemia. Thromb Haemost 79 268–275. [PubMed] [Google Scholar]
- Broudy VC, Lin NL & Kaushansky K 1995. Thrombopoietin (c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood 85 1719–1726. [PubMed] [Google Scholar]
- Brown AS & Martin JF 1994. The megakaryocyte platelet system and vascular disease. Eur J Clin Invest 24 Suppl 1 9–15. [DOI] [PubMed] [Google Scholar]
- Choi SH, Kim JH, Lim S, Lim JY, Kim KW, Park KS & Jang HC 2017. Monocyte count as a predictor of cardiovascular mortality in older Korean people. Age Ageing 46 433–438. [DOI] [PubMed] [Google Scholar]
- Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N & Libby P 2009. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 120 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, Darbonne WC, Henzel WJ, Wong SC, Kuang WJ, et al. 1994. Stimulation of megakaryocytopoiesis and thrombopoiesis by the c-Mpl ligand. Nature 369 533–538. [DOI] [PubMed] [Google Scholar]
- Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ & Fisher EA 2014. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res 115 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoljevic D, Kraakman MJ, Nagareddy PR, Ngo D, Shihata W, Kammoun HL, Whillas A, Lee MKS, Al-Sharea A & Pernes G 2018. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. European Heart Journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. 2010. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelboom JW & Warkentin TE 2014. Immature platelet count: part of the cardiologist’s complete blood count? J Am Coll Cardiol 64 2130–2132. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DJ, Roy L, Catella F & FitzGerald GA 1986. Platelet activation in unstable coronary disease. N Engl J Med 315 983–989. [DOI] [PubMed] [Google Scholar]
- Ford ES 2002. Leukocyte count, erythrocyte sedimentation rate, and diabetes incidence in a national sample of US adults. Am J Epidemiol 155 57–64. [DOI] [PubMed] [Google Scholar]
- Freynhofer MK, Iliev L, Bruno V, Rohla M, Egger F, Weiss TW, Hubl W, Willheim M, Wojta J & Huber K 2017. Platelet turnover predicts outcome after coronary intervention. Thromb Haemost 117 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, Hechtman HB & Michelson AD 1998. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol 31 352–358. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. 2017. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove EL, Hvas AM, Mortensen SB, Larsen SB & Kristensen SD 2011. Effect of platelet turnover on whole blood platelet aggregation in patients with coronary artery disease. J Thromb Haemost 9 185–191. [DOI] [PubMed] [Google Scholar]
- Gurbel PA, Bliden KP, Hiatt BL & O’Connor CM 2003. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation 107 2908–2913. [DOI] [PubMed] [Google Scholar]
- Gurney AL, Carver-Moore K, de Sauvage FJ & Moore MW 1994. Thrombocytopenia in c-mpl-deficient mice. Science 265 1445–1447. [DOI] [PubMed] [Google Scholar]
- Guthikonda S, Alviar CL, Vaduganathan M, Arikan M, Tellez A, DeLao T, Granada JF, Dong JF, Kleiman NS & Lev EI 2008. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol 52 743–749. [DOI] [PubMed] [Google Scholar]
- Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong JF & Kleiman NS 2007. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost 5 490–496. [DOI] [PubMed] [Google Scholar]
- Hawrylowicz CM, Howells GL & Feldmann M 1991. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J Exp Med 174 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimsoy Z, Payzin B, Ornek T & Kandogan G 2004. Mean platelet volume in Type 2 diabetic patients. J Diabetes Complications 18 173–176. [DOI] [PubMed] [Google Scholar]
- Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G & Kroczek RA 1998. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 391 591–594. [DOI] [PubMed] [Google Scholar]
- Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, et al. 2010. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome--serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial). Circ J 74 1165–1174. [DOI] [PubMed] [Google Scholar]
- Hoffmeister KM, Falet H & Grozovsky R 2014. Platelets Regulate Thrombopoietin Production: The Missing Link. Blood 124 SCI-52-SCI-52. [Google Scholar]
- Hsu HC, Tsai WH, Jiang ML, Ho CH, Hsu ML, Ho CK & Wang SY 1999. Circulating levels of thrombopoietic and inflammatory cytokines in patients with clonal and reactive thrombocytosis. J Lab Clin Med 134 392–397. [DOI] [PubMed] [Google Scholar]
- Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C & Ley K 2003. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 9 61–67. [DOI] [PubMed] [Google Scholar]
- Ibrahim H, Nadipalli S, Usmani S, DeLao T, Green L & Kleiman NS 2016. Detection and quantification of circulating immature platelets: agreement between flow cytometric and automated detection. J Thromb Thrombolysis 42 77–83. [DOI] [PubMed] [Google Scholar]
- Ibrahim H, Schutt RC, Hannawi B, DeLao T, Barker CM & Kleiman NS 2014. Association of immature platelets with adverse cardiovascular outcomes. J Am Coll Cardiol 64 2122–2129. [DOI] [PubMed] [Google Scholar]
- Ishiguro A, Suzuki Y, Mito M, Shimbo T, Matsubara K, Kato T & Miyazaki H 2002. Elevation of serum thrombopoietin precedes thrombocytosis in acute infections. Br J Haematol 116 612–618. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. 2017. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, Theurl I, Widder W, Molnar C, Ludwiczek O, et al. 2001. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood 98 2720–2725. [DOI] [PubMed] [Google Scholar]
- Kaushansky K 1995. Thrombopoietin: the primary regulator of platelet production. Blood 86 419–431. [PubMed] [Google Scholar]
- Kienast J & Schmitz G 1990. Flow cytometric analysis of thiazole orange uptake by platelets: a diagnostic aid in the evaluation of thrombocytopenic disorders. Blood 75 116–121. [PubMed] [Google Scholar]
- Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E, Basu D, Heywood S, Kammoun HL, Flynn M, et al. 2017. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest 127 2133–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PA, Ravi S, Chacko B, Johnson MS & Darley-Usmar VM 2014. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biol 2 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen OP & Mandrup-Poulsen T 2005. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes 54 S114–S124. [DOI] [PubMed] [Google Scholar]
- Kuter DJ, Beeler DL & Rosenberg RD 1994. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Natl Acad Sci U S A 91 11104–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P 2012. Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 32 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madjid M, Awan I, Willerson JT & Casscells SW 2004. Leukocyte count and coronary heart disease: implications for risk assessment. J Am Coll Cardiol 44 1945–1956. [DOI] [PubMed] [Google Scholar]
- Maugeri N, Malato S, Femia EA, Pugliano M, Campana L, Lunghi F, Rovere-Querini P, Lussana F, Podda G, Cattaneo M, et al. 2011. Clearance of circulating activated platelets in polycythemia vera and essential thrombocythemia. Blood 118 3359–3366. [DOI] [PubMed] [Google Scholar]
- Mauldin JP, Nagelin MH, Wojcik AJ, Srinivasan S, Skaflen MD, Ayers CR, McNamara CA & Hedrick CC 2008. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation 117 2785–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury E, Brichard SM, Pataky Z, Carpentier A, Golay A & Bobbioni‐Harsch E 2010. Effect of Obesity on Growth‐related Oncogene Factor‐α, Thrombopoietin, and Tissue Inhibitor Metalloproteinase‐1 Serum Levels. Obesity 18 1503–1509. [DOI] [PubMed] [Google Scholar]
- Mayer FJ, Hoke M, Schillinger M, Minar E, Arbesu I, Koppensteiner R & Mannhalter C 2014. Mean platelet volume predicts outcome in patients with asymptomatic carotid artery disease. Eur J Clin Invest 44 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBane RD 2nd, Gonzalez C, Hodge DO & Wysokinski WE 2014. Propensity for young reticulated platelet recruitment into arterial thrombi. J Thromb Thrombolysis 37 148–154. [DOI] [PubMed] [Google Scholar]
- Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L & Tschopp J 2011. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis 2 e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, et al. 2011. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest 121 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, Wang Y, Shaw JA, Levine RL, Ni H, et al. 2013. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med 19 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Sarrazy V, Wang N, Bijl N, Abramowicz S, Westerterp M, Welch CB, Schuetz JD & Yvan-Charvet L 2014. Deficiency of ATP-binding cassette transporter B6 in megakaryocyte progenitors accelerates atherosclerosis in mice. Arterioscler Thromb Vasc Biol 34 751–758. [DOI] [PubMed] [Google Scholar]
- Murphy AJ & Tall AR 2016. Disordered haematopoiesis and athero-thrombosis. Eur Heart J 37 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. 2014. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19 821–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, et al. 2013. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 17 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neergaard-Petersen S, Hvas AM, Kristensen SD & Grove EL 2016. Platelets and Antiplatelet Therapy in Patients with Coronary Artery Disease and Diabetes. Semin Thromb Hemost 42 234–241. [DOI] [PubMed] [Google Scholar]
- Organisation WH 2017. Cardiovascular Diseases.
- Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ & Fisher EA 2011. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes 60 1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarelli M, Tang C, McDonald TO, O’Brien KD, Gerrity RG, Heinecke JW & Oram JF 2005. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 54 2198–2205. [DOI] [PubMed] [Google Scholar]
- Patrono C, Garcia Rodriguez LA, Landolfi R & Baigent C 2005. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 353 2373–2383. [DOI] [PubMed] [Google Scholar]
- Peck-Radosavljevic M, Wichlas M, Zacherl J, Stiegler G, Stohlawetz P, Fuchsjager M, Kreil A, Metz-Schimmerl S, Panzer S, Steininger R, et al. 2000. Thrombopoietin induces rapid resolution of thrombocytopenia after orthotopic liver transplantation through increased platelet production. Blood 95 795–801. [PubMed] [Google Scholar]
- Pedersen TR 2016. The Success Story of LDL Cholesterol Lowering. Circ Res 118 721–731. [DOI] [PubMed] [Google Scholar]
- Pickup J, Mattock M, Chusney G & Burt D 1997. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40 1286. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, et al. 2007. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med 13 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradella P, Bonetto S, Turchetto S, Uxa L, Comar C, Zorat F, De Angelis V & Pozzato G 2011. Platelet production and destruction in liver cirrhosis. J Hepatol 54 894–900. [DOI] [PubMed] [Google Scholar]
- Pruenster M, Vogl T, Roth J & Sperandio M 2016. S100A8/A9: From basic science to clinical application. Pharmacol Ther 167 120–131. [DOI] [PubMed] [Google Scholar]
- Qian S, Fu F, Li W, Chen Q & de Sauvage FJ 1998. Primary role of the liver in thrombopoietin production shown by tissue-specific knockout. Blood 92 2189–2191. [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. 2017. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377 1119–1131. [DOI] [PubMed] [Google Scholar]
- Rinder CS, Bonan JL, Rinder HM, Mathew J, Hines R & Smith BR 1992. Cardiopulmonary bypass induces leukocyte-platelet adhesion. Blood 79 1201–1205. [PubMed] [Google Scholar]
- Saur SJ, Sangkhae V, Geddis AE, Kaushansky K & Hitchcock IS 2010. Ubiquitination and degradation of the thrombopoietin receptor c-Mpl. Blood 115 1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro JM, Arber N & Sidi Y 1990. Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med 323 1707. [DOI] [PubMed] [Google Scholar]
- Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP & Heiss G 1999. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet 353 1649–1652. [DOI] [PubMed] [Google Scholar]
- Schramm TK, Gislason GH, Kober L, Rasmussen S, Rasmussen JN, Abildstrom SZ, Hansen ML, Folke F, Buch P, Madsen M, et al. 2008. Diabetes patients requiring glucose-lowering therapy and nondiabetics with a prior myocardial infarction carry the same cardiovascular risk: a population study of 3.3 million people. Circulation 117 1945–1954. [DOI] [PubMed] [Google Scholar]
- Sheikh MY, Raoufi R, Atla PR, Riaz M, Oberer C & Moffett MJ 2012. Prevalence of cirrhosis in patients with thrombocytopenia who receive bone marrow biopsy. Saudi J Gastroenterol 18 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB, Damcott CM, Pakyz R, Tantry US, Gibson Q, et al. 2009. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratz C, Bomicke T, Younas I, Kittel A, Amann M, Valina CM, Nuhrenberg T, Trenk D, Neumann FJ & Hochholzer W 2016. Comparison of Immature Platelet Count to Established Predictors of Platelet Reactivity During Thienopyridine Therapy. J Am Coll Cardiol 68 286–293. [DOI] [PubMed] [Google Scholar]
- Sungaran R, Markovic B & Chong BH 1997. Localization and regulation of thrombopoietin mRNa expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood 89 101–107. [PubMed] [Google Scholar]
- Tan TP, Arekapudi A, Metha J, Prasad A & Venkatraghavan L 2015. Neutrophil-lymphocyte ratio as predictor of mortality and morbidity in cardiovascular surgery: a systematic review. ANZ J Surg 85 414–419. [DOI] [PubMed] [Google Scholar]
- Tavil Y, Sen N, Yazici H, Turfan M, Hizal F, Cengel A & Abaci A 2010. Coronary heart disease is associated with mean platelet volume in type 2 diabetic patients. Platelets 21 368–372. [DOI] [PubMed] [Google Scholar]
- Tolani S, Pagler TA, Murphy AJ, Bochem AE, Abramowicz S, Welch C, Nagareddy PR, Holleran S, Hovingh GK, Kuivenhoven JA, et al. 2013. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis 229 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trialists’ Collaboration A 1994. Collaborative overview ofrandomised trials ofantiplatelet therapy-Ill: Reductionin venous thrombosis andpulmonary embolism by antplatelet prophylaxis among surgical and medical. Bmj 308 235. [PMC free article] [PubMed] [Google Scholar]
- Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, Hoekstra M, Sierts JA, Dallinga-Thie GM, Motazacker MM, et al. 2011. Genetic variant of the scavenger receptor BI in humans. N Engl J Med 364 136–145. [DOI] [PubMed] [Google Scholar]
- Vlachojannis GJ, Dimitropoulos G & Alexopoulos D 2011. Clopidogrel resistance: current aspects and future directions. Hellenic J Cardiol 52 236–245. [PubMed] [Google Scholar]
- von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K & Weber C 2001. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 103 1772–1777. [DOI] [PubMed] [Google Scholar]
- Vuckovic S, Withers G, Harris M, Khalil D, Gardiner D, Flesch I, Tepes S, Greer R, Cowley D, Cotterill A, et al. 2007. Decreased blood dendritic cell counts in type 1 diabetic children. Clin Immunol 123 281–288. [DOI] [PubMed] [Google Scholar]
- Wang X, Jiang W, Li F, Hua F, Zhan Y, Li Y, Ji L, Zou S, Min Z, Song D, et al. 2014a. Abnormal platelet kinetics are detected before the occurrence of thrombocytopaenia in HBV-related liver disease. Liver Int 34 535–543. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fang C, Gao H, Bilodeau ML, Zhang Z, Croce K, Liu S, Morooka T, Sakuma M & Nakajima K 2014b. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. The Journal of clinical investigation 124 2160–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George J, Woerle HJ, Broedl UC, von Eynatten M, Zinman B, et al. 2018. Empagliflozin and Clinical Outcomes in Patients With Type 2 Diabetes Mellitus, Established Cardiovascular Disease, and Chronic Kidney Disease. Circulation 137 119–129. [DOI] [PubMed] [Google Scholar]
- Weber C & Noels H 2011. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17 1410–1422. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL & Ferrante AW Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JG, Mussolino ME, Gillum RF & Danesh J 2004. Associations between differential leucocyte count and incident coronary heart disease: 1764 incident cases from seven prospective studies of 30,374 individuals. Eur Heart J 25 1287–1292. [DOI] [PubMed] [Google Scholar]
- Woo SJ, Ahn SJ, Ahn J, Park KH & Lee K 2011. Elevated systemic neutrophil count in diabetic retinopathy and diabetes: a hospital-based cross-sectional study of 30,793 Korean subjects. Invest Ophthalmol Vis Sci 52 7697–7703. [DOI] [PubMed] [Google Scholar]
- Woollard KJ & Geissmann F 2010. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 7 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, et al. 2010. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328 1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]