

Abstract
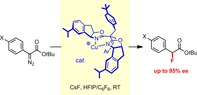
The Cu‐catalyzed reaction of substituted α‐diazoesters with fluoride gives α‐fluoroesters with ee values of up to 95 %, provided that chiral indane‐derived bis(oxazoline) ligands are used that carry bulky benzyl substituents at the bridge and moderately bulky isopropyl groups on their core. The apparently homogeneous solution of CsF in C6F6/hexafluoroisopropanol (HFIP) is the best reaction medium, but CsF in the biphasic mixture CH2Cl2/HFIP also provides good results. DFT studies suggest that fluoride initially attacks the Cu‐ rather than the C‐atom of the transient donor/acceptor carbene intermediate. This unusual step is followed by 1,2‐fluoride shift; for this migratory insertion to occur, the carbene must rotate about the Cu−C bond to ensure orbital overlap. The directionality of this rotatory movement within the C 2‐symmetric binding site determines the sense of induction. This model is in excellent accord with the absolute configuration of the resulting product as determined by X‐ray diffraction using single crystals of this a priori wax‐like material grown by capillary crystallization.
Keywords: asymmetric catalysis, bisoxazoline ligands, carbene complexes, copper, organofluorine compounds
Let's work together: Even though bisoxazolines are privileged ligands in asymmetric catalysis, standard representatives give poor ee values in the copper catalyzed asymmetric fluorination of α‐diazoesters. The outcome was greatly improved, however, upon gem‐benzylation of the bridge, proper substitution of the core, and optimization of the medium. The reaction follows an unusual course in that the incoming fluoride first hits the metal rather than the C‐atom of the transient copper carbene complex.
Introduction
The incorporation of fluorine into active pharmaceutical ingredients (API′s) or agrochemicals often entails substantial advantages in chemical and/or biological terms.1 (Chiral) α‐fluoro carbonyl compounds represent a privileged motif and are therefore prominently featured in API′s;1, 2 at the same time, they constitute valuable building blocks for further elaboration. Most methods for their synthesis invoke the carbonyl compound as (masked) enolate or enamine; the price to pay is the need for an electrophilic fluorine source as the reaction partner.3, 4, 5 The alternative approach of using an “umpoled” substrate in combination with an ordinary fluoride salt is currently less developed, despite potential chemical virtues and practical advantages.6, 7, 8
A recent report on the fluorination of highly electrophilic copper carbenes generated in situ provides an interesting foray in this direction (Scheme 1).9, 10 Specifically, it was shown that α‐diazoesters such as 1 a, on treatment with catalytic amounts of [Cu(MeCN)4]PF6 (or [Cu(OTf)]2⋅PhMe) and L1 as the preferred ligand, react with excess KF in a biphasic mixture comprised of hexafluoroisopropanol (HFIP) and 1,2‐dichloroethane (DCE) at 40 °C to give the corresponding α‐fluoroester 2 a in good yield. Despite the excellent track record of chiral bis(oxazolines) (BOX) and related ligands in asymmetric catalysis,11, 12 however, only poor enantioselectivity (≤31 % ee) was reached. Use of a monodentate chiral phosphoramidite in lieu of L1 entailed higher optical purity (86 % ee) at the expense of the chemical yield, which dropped to only 12 %.9
Scheme 1.
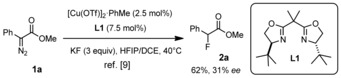
Lead finding of a copper catalyzed formation of α‐fluoroesters.
Results and Discussion
Reaction optimization and scope
As part of our studies into structure, bonding and reactivity of organotransition metal carbene complexes,13, 14, 15, 16, 17, 18 we sought to improve on these lead findings. We were fully apprehensive that the small size of the fluoride ion constitutes a formidable and inherent challenge for asymmetric catalysis; moreover, any uncatalyzed and hence racemic background reaction had to be prevented.19 To this end, it was deemed imperative to find milder reaction conditions and to carry out a broad ligand screening. The first goal was readily achieved in that the use of CsF instead of KF allowed the reaction to proceed at ambient temperature in the biphasic mixture CH2Cl2/HFIP as a ligand‐accelerated process.20 Of arguably higher relevance—even beyond the present context—is the observation that mixtures of HFIP and hexafluorobenzene form an apparently homogeneous phase capable of dissolving CsF, in which the α‐fluorination proceeds particularly cleanly.
Not unexpectedly, the search for effective chiral ligands proved challenging, not least because clear‐cut structure/selectivity relationships were difficult to deduce from the acquired data (for the full list, see the Supporting Information). As one consistent trend, however, it was noticed that standard BOX ligands led to clean reactions. The poor chiral induction notwithstanding (cf. Scheme 1 and the Supporting Information),9 the ligand optimization exercise remained largely focused on this privileged scaffold.11, 12 Our efforts were guided by the notion that efficient steering of the small incoming fluoride anion likely mandates a tight chiral binding site. We conjectured that this goal might be reached by adjustment of the following three parameters: (i) implementation of substituents at the methylene bridge to exert remote control,21, 22 (ii) extension of the lateral “walls” of the C 2‐symmetric ligand scaffold beyond the tert‐butyl substituents in L1, and (iii) increase of the size of the electrophilic partner by using bulkier α‐diazoesters.
Variation of any of these factors alone did not grant success; gratifyingly though, they were found to synergize. While the commercially available indane‐based bis(oxazoline) L2 gave methyl ester 2 a with only 31 % ee (Figure 1) and the corresponding tert‐butyl ester 2 b with equally disappointing 35 % ee (Table 1, entries 1, 2), gem‐disubstitution of the methylene bridge entailed some improvement. This effect has ample precedent in the literature and is usually ascribed to a buttressing effect, the fine‐tuning of the ligand's bite‐angle, and suppression of enolization at this site.11, 12 Although these factors are likely relevant in the present context too, additional aspects seem to play an important role. Specifically, introduction of a cycloheptyl or cyclopropyl group improved the ee to 45 % and 54 % for product 2 b, respectively (entries 3, 4). Equally good or even better results were obtained with ligands such as L5–L9 carrying different benzyl substituents at this position, which furnished 2 b with ee values of up to 75 % (entries 5–9);23 moreover, the size of the ester group in the diazo compound had a marked effect (cf. entries 11 and 12).
Figure 1.
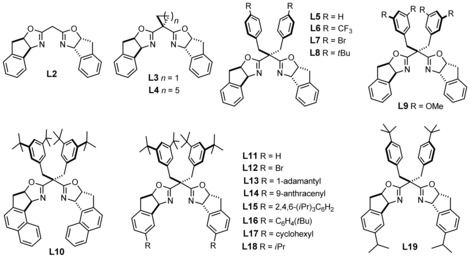
Subset of the tested BOX‐ligands; for the full list, see the Supporting Information.
Table 1.
Ligand screening and optimization of the reaction conditions
|
| ||||
|---|---|---|---|---|
|
Entry |
R |
Medium[a] |
Ligand |
ee [%][b] (yield) |
|
1 |
Me |
A |
L2 |
31 |
|
2 |
tBu |
A |
L2 |
35 |
|
3 |
tBu |
A |
L3 |
54 |
|
4 |
tBu |
A |
L4 |
45 |
|
5 |
tBu |
A |
L5 |
66 |
|
6 |
tBu |
A |
L6 |
61 |
|
7 |
tBu |
A |
L7 |
59 |
|
8 |
tBu |
A |
L8 |
75 |
|
9 |
tBu |
A |
L9 |
71 |
|
10 |
tBu |
A |
L10 |
16 |
|
11 |
Me |
A |
L11 |
52 |
|
12 |
tBu |
A |
L11 |
73 |
|
13 |
tBu |
A |
L12 |
76 |
|
14 |
tBu |
A |
L13 |
66 |
|
15 |
tBu |
A |
L14 |
71 |
|
16 |
tBu |
A |
L15 |
52 |
|
17 |
tBu |
A |
L16 |
72 |
|
18 |
tBu |
A |
L17 |
70 |
|
19 |
tBu |
A |
L18 |
81 |
|
20 |
tBu |
A |
L19 |
85 (72)[c] |
|
21 |
tBu |
A |
L19 |
87 (65)[c,d] |
|
22 |
tBu |
B |
L19 |
89 (87)[c] |
[a] A: HFIP (10 equiv), CH2Cl2; B: HFIP (10 equiv), C6F6. [b] ee of the isolated pure compound; for details, see the Supporting Information. [c] Isolated yield of analytically pure product. [d] Using [Cu(OtBu)]4 instead of [Cu(OTf)]2⋅PhMe
Next, we considered placement of substituents at strategic positions of the indane core or an extension of the scaffold by annulation of additional aryl rings. Such modifications are rare,24, 25 but it was hoped that they might entail a narrower trajectory for the incoming fluoride anion. Screening of a number of variants showed that bis(oxazoline) L19 carrying bulky p‐tert‐butyl substituted benzyl moieties at the bridge and an only moderately big isopropyl substituent on the indane nucleus gave the best result (for the full data set, see the Supporting Information). This particular ligand furnished α‐fluorophenylacetate 2 b in 87 % yield with 89 % ee when the reaction was carried out in HFIP/C6F6 as the optimal medium (entry 22). More electron‐deficient α‐diazocarbonyl derivatives performed even better, especially those carrying the electron‐withdrawing group at the para‐position of the aryl ring (Figure 2); the corresponding products were isolated in high yields with optical purities of up to 95 % ee. Comparison of the results obtained for products 4 and 5 illustrates the influence of the substitution pattern on the outcome; in this context it is noteworthy that attempted formation of the corresponding ortho‐chlorinated analogue failed due to decomposition of the diazo ester precursor.
Figure 2.
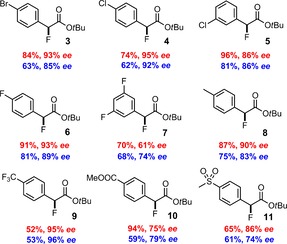
Chiral α‐fluoroester derivatives prepared by copper‐catalyzed diazoester decomposition. Reaction conditions: [Cu(OTf)]2⋅PhMe (2.5 mol %), L19 (5 mol %), CsF (3.5 equiv), RT, 12 h, HFIP (10 equiv)/CH2Cl2 (blue) or HFIP (10 equiv)/C6F6 (red).
Configurational assignment via capillary crystallization
Since none of the resulting α‐fluorinated esters shown in Figure 2 had previously been prepared in the literature in optically active form, we faced the need to rigorously establish the absolute configuration prior to any mechanistic discussion.26 To this end, a sample of 2 b (89 % ee) as the parent compound of this series was subjected to preparative HPLC on a chiral stationary phase to remove the remaining minor enantiomer. Low temperature differential scanning calorimetry showed that 2 b (ee ≥99 %) features a broad melting range close to ambient temperature (ca. +3 to +22 °C) and a remarkably strong hysteresis upon cooling (see the Supporting Information). Therefore crystallization was expected to be non‐trivial.
The challenge was met by capillary crystallization: the neat “liquid” sample was filled into a capillary, which was tightly fused before being chilled in a stream of cold air; the resulting polycrystalline material was then locally warmed to slightly above melting temperature. This cooling/warming cycle led to single crystals suitable for X‐ray diffraction. The capillary was transferred quickly to the diffractometer; the collected data set afforded a statistically significant absolute structure parameter and allowed the absolute configuration to be determined;27 to the best of our knowledge is this the first example of its kind28 and bodes well for configurational assignments of other chiral liquid samples in the future. As shown in Figure 3, the major isomer of the α‐fluoroester 2 b ( =+65.9) formed in the copper catalyzed reaction with ligand L19 is (S)‐configured. All other compounds shown in Figure 2 were assigned by analogy, based on the fact that they are invariably dextrorotatory.
Figure 3.
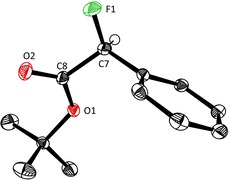
Structure of compound 2 b in the solid state which allowed the absolute configuration of the α‐fluorinated ester derivative to be determined; all hydrogen atoms—except the one on the (S)‐configured chiral center C7—are omitted for clarity.
Mechanistic discussion
Despite numerous attempts, we failed to grow single crystals of the copper precatalyst carrying the optimal ligand L19, most likely because of the greasy lateral substituents. Therefore, we resorted to solving the structure of the related complex derived from L3, which leads to the same sense of induction in the fluorination reaction but to a lower ee of 54 % (Table 1, entry 3). In contrast to the copious information on BOX complexes of CuII and other (Lewis‐acidic) transition metals,29 surprisingly few crystal structures of (chiral) [BOX⋅CuI]X complexes are known in the literature;30, 31, 32, 33 actually, some of them are intricate dimeric or oligomeric arrays, which likely have to disassemble before any catalytic reaction can take place.
In contrast, complex [L3⋅Cu(MeCN)]BF4 is a monomeric entity (Figure 4); the coordination geometry about the Cu‐center is distorted trigonal. The N1‐Cu1‐N2 bite‐angle of 95.9(1)° is wider than that of the few comparable cases in the literature,31 which likely reflects the influence of the gem‐disubstitution at the bridge as well as the steric demand of the indane core. Although the crystal structure provides only a static picture, it is noteworthy that the complex is not strictly C 2‐symmetric in the solid state; specifically, the N3‐Cu1‐N1 (121.8(1)°) and the N3‐Cu1‐N2 (141.5°) angles are notably uneven, as are the distances Cu1−N1 (2.004(2) Å) and Cu1−N2 (1.952(2) Å).31, 34
Figure 4.
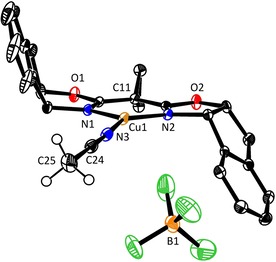
Structure of complex [L3⋅Cu(MeCN)]BF4 in the solid state; hydrogen atoms on the ligand backbone are omitted for clarity.
NMR investigations provided some insights into the role of the substituents at the bridge between the oxazoline rings.34 NOESY spectra of complex [L11⋅Cu(MeCN)]BF4 in CDCl3 show numerous cross peaks between the protons of the substituted benzyl groups and those of the indane ring (see Figure 5). The benzyl substituents are hence “forward” oriented in at least one averaged conformer on the NMR timescale; as such, they (temporarily) reside in vicinity of the incipient carbene, above and below the catalytically active Cu‐center. The crystal structure of a related BOX⋅Cu complex with pendant p‐(tert‐butyl)benzyl substituents corroborates the notion that these seemingly remote substituents actually shield the upper and lower face of the coordination plane.31, 35
Figure 5.
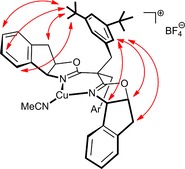
Graphical representation of relevant NOE contacts in [L11⋅Cu(MeCN)]BF4, which indicate that the substituted benzyl groups are, on average, “forward” oriented on the NMR time scale.
If one takes the ligated acetonitrile as a dummy for the carbene to be formed upon reaction of the Cu‐center with the α‐diazoester, attack onto the Re face of the electrophilic C‐atom seems most plausible at first sight, which would entail formation of (R)‐2 b (compare Figures 5 and 6). Since (S)‐2 b, however, has been shown to be the major product, a more involved scenario must be operative. Therefore, a computational study was performed to gain a better understanding. Density functionals based on the generalized gradient approximation (GGA) were chosen for the good balance between accuracy and performance. The BP86 functional was found to reproduce the experimental structure of the copper complex [L3⋅Cu(MeCN)]BF4 particularly well and was therefore used throughout (for the computational methods, see the Supporting Information).
Figure 6.
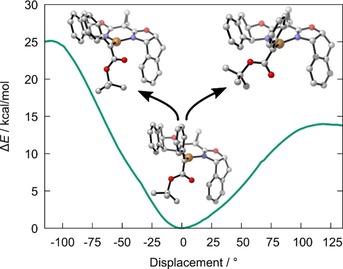
Computed ground state structure of the donor/acceptor copper carbene complex [L3⋅Cu=CPh(COOtBu)]BF4 formed from [L3⋅Cu(MeCN)]BF4 and the diazoester 1 b; illustration of the uneven barriers for clockwise and counterclockwise rotation about the Cu−C bond.
The plane defined by the trigonal donor/acceptor carbene center in [L3⋅Cu=CPh(COOtBu)]BF4 is orthogonal to the {N2Cu} plane of the chiral catalyst (Figure 6);36 this arrangement is geometrically favorable and, at the same time, arguably maximizes back‐bonding from the filled metal d‐orbital that is most destabilized by the N,N‐donor ligand into the empty carbene p‐orbital. As one might expect for a C 2‐symmetric ligand environment,34 the barriers for clockwise and counter‐clockwise rotation about the Cu−C bond are notably different. The appreciable barrier heights of ≈14 and ≈25 kcal mol−1, respectively, imply that the carbene moiety does not freely rotate at ambient temperature at which the reaction with fluoride does occur, but rather oscillates about the conformation of lowest energy. Importantly, this swinging motion is directional and the amplitudes are uneven, in that clockwise movement will prevail by large margins as it avoids a clash between the carbene's substituents and the rigid indane scaffold.37
In exploring possible trajectories for the attack of fluoride onto this complex, we were surprised to find that the Cu‐center rather than the electrophilic carbene C‐atom constitutes the prime site of attack; no transition state was found for direct C−F bond formation, despite considerable search efforts.38 The incoming nucleophile approaches the metal center from above or below the coordination plane to form the copper fluoride complexes F@Cutop and F@Cubot, respectively. It is striking that top‐side attack is barrierless with a submerged transition state TSF→Cu top,39 provided that a continuum solvent environment was used in the computations (Figure 7). In stark contrast, attack from the bottom face shows an activation barrier of no less than 12.3 kcal mol−1 (see the Supporting Information). Qualitatively, this differential is rather intuitive and very well in line with the conclusions previously drawn from the solid‐state structure of electrophilic donor/acceptor dirhodium carbenes:13 Any incoming nucleophile, especially when charged, will avoid dipole repulsion with the lone‐pairs of the ester O‐atoms40 and hence preferentially follow a Bürgi–Dunitz trajectory alongside the aryl substituent of the donor/acceptor carbene.13, 41
Figure 7.
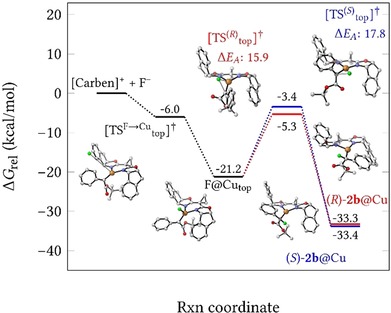
Reaction profile for nucleophilic attack of fluoride from the top‐face of the Cu‐carbene complex [L3⋅Cu=CPh(COOtBu)]BF4, followed by isomerization of F@Cutop into (S)‐ or (R)‐2 b; for the free energy profile of the bottom‐face approach, see the Supporting Information.
Since the top–face approach wins out by far, only the fate of the resulting complex F@Cutop needs to be considered at this point. Orbital alignment is necessary before the fluoride ligand can migrate from copper to carbon,42, 43 which, in turn, mandates that the carbene unit rotates about the Cu−C bond. The transition state for clockwise rotation is 1.9 kcal mol−1 lower in energy (Figure 7); it leads to complex (R)‐2 b@Cu, which ultimately delivers (S)‐2 b upon protonation (note that the “inversion” is due to the formalism of the CIP rules). Although this computational result overestimates the level of induction, it correctly predicts the sense of induction of the copper catalyzed α‐fluorination reaction.
Conclusions
The first highly enantioselective metal catalyzed addition of fluoride to α‐diazoesters is described, featuring ee values of up to 95 %. To properly assess this result, one has to consider the inherent challenges for asymmetric catalysis posed by this particular nucleophile, which had basically thwarted an earlier attempt at rendering this transformation asymmetric.9 Interestingly, the reaction most likely proceeds by initial attack of fluoride onto the Cu‐ rather than the C‐atom of the electrophilic metal carbene intermediate; a subsequent 1,2‐fluoride shift then leads to product formation. For this migration to happen, the carbene must rotate about the Cu−C bond to ensure the necessary orbital overlap; the directionality of this rotatory motion, enacted by the C 2‐symmetric ligand environment, is the major enantiodetermining factor. Further work on the structure and reactivity of transition metal carbene complexes and the stereoselective preparation of organofluorine derivatives44 is underway in our laboratory and will be reported in due course.
Experimental Section
All experimental details can be found in the Supporting Information. The material includes compound characterization data, additional crystallographic information, the full list of ligands investigated, HPLC traces, supporting computational data, and copies of spectra of new compounds.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Generous financial support by the Max‐Planck‐Society, the Alexander‐von‐Humboldt Foundation (return scholarship to P.J.), and the Xunta de Galicia and European Union (European Social Fund, predoctoral fellowship to I.P.) is gratefully acknowledged. We thank the analytical departments of our Institute for excellent support.
M. Buchsteiner, L. Martinez-Rodriguez, P. Jerabek, I. Pozo, M. Patzer, N. Nöthling, C. W. Lehmann, A. Fürstner, Chem. Eur. J. 2020, 26, 2509.
References
- 1.
- 1a. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 1b. Wang J., Sánchez-Roselló M., Acena J. L., del Pozo C., Sorochinsky A. E., Fustero S., Soloshonok V. A., Liu H., Chem. Rev. 2014, 114, 2432–2506; [DOI] [PubMed] [Google Scholar]
- 1c. Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A., J. Med. Chem. 2015, 58, 8315–8359; [DOI] [PubMed] [Google Scholar]
- 1d. Zhou Y., Wang J., Gu Z., Wang S., Zhu W., Acena J. L., Soloshonok V. A., Izawa K., Liu H., Chem. Rev. 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- 2.For an instructive case, see: Lee K. L., Ambler C. M., Anderson D. R., Boscoe B. P., Bree A. G., Brodfuehrer J. I., Chang J. S., Choi C., Chung S., Curran K. J., Day J. E., Dehnhardt C. M., Dower K., Drozda S. E., Frisbie R. K., Gavrin L. K., Goldberg J. A., Han S., Hegen M., Hepworth D., Hope H.-R., Kamtekar S., Kilty I. C., Lee A., Lin L.-L., Lovering F. E., Lowe M. D., Mathias J. P., Morgan H. M., Murphy E. A., Papaioannou N., Patny A., Pierce B. S., Rao V. R., Saiah E., Samardjiev I. J., Samas B. M., Shen M. W. H., Shin J. H., Soutter H. H., Strohbach J. W., Symanowicz P. T., Thomason J. R., Trzupek J. D., Vargas R., Vincent F., Yan J., Zapf C. W., Wright S. W., J. Med. Chem. 2017, 60, 5521–5542. [DOI] [PubMed] [Google Scholar]
- 3. Liang T., Neumann C. N., Ritter T., Angew. Chem. Int. Ed. 2013, 52, 8214–8264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8372–8423. [Google Scholar]
- 4.For leading references on stoichiometric methods, see the following and literature cited therein:
- 4a. Davis F. A., Kasu P. V. N., Tetrahedron Lett. 1998, 39, 6135–6138; [Google Scholar]
- 4b. Alvarado J., Herrmann A. T., Zakarian A., J. Org. Chem. 2014, 79, 6206–6220. [DOI] [PubMed] [Google Scholar]
- 5.For leading references on catalytic procedures, see the following and literature cited therein:
- 5a. Beeson T. D., MacMillan D. W. C., J. Am. Chem. Soc. 2005, 127, 8826–8828; [DOI] [PubMed] [Google Scholar]
- 5b. Suzuki T., Hamashima Y., Sodeoka M., Angew. Chem. Int. Ed. 2007, 46, 5435–5439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5531–5535; [Google Scholar]
- 5c. Paull D. H., Scerba M. T., Alden-Danforth E., Widger L.-R., Lectka T., J. Am. Chem. Soc. 2008, 130, 17260–17261; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Wheeler P., Vora H. U., Rovis T., Chem. Sci. 2013, 4, 1674–1679; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Peng J., Du D. M., RSC Adv. 2014, 4, 2061–2067; [Google Scholar]
- 5f. Li F., Wu Z., Wang J., Angew. Chem. Int. Ed. 2015, 54, 656–659; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 666–669; [Google Scholar]
- 5g. Dong X., Yang W., Hu W., Sun J., Angew. Chem. Int. Ed. 2015, 54, 660–663; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 670–673; [Google Scholar]
- 5h. Wang L., Jiang X., Chen J., Huang Y., Angew. Chem. Int. Ed. 2019, 58, 7410–7414; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7488–7492. [Google Scholar]
- 6.
- 6a. Hollingworth C., Gouverneur V., Chem. Commun. 2012, 48, 2929–2942; [DOI] [PubMed] [Google Scholar]
- 6b. Wu J., Tetrahedron Lett. 2014, 55, 4289–4294. [Google Scholar]
- 7. Adler P., Teskey C. J., Kaiser D., Holy M., Sitte H. H., Maulide N., Nat. Chem. 2019, 11, 329–334. [DOI] [PubMed] [Google Scholar]
- 8.Formation of scalemic α-aryl-α-fluoro esters from mandelate or arylglycine derivatives is challenging and/or requires special reagents, see:
- 8a. Bresciani S., O'Hagan D., Tetrahedron Lett. 2010, 51, 5795–5797; [Google Scholar]
- 8b. Hamman S., Barrelle N., Tetaz F., Beguin C. G., J. Fluorine Chem. 1987, 37, 85–94. [Google Scholar]
- 9. Gray E. E., Nielsen M. K., Choquette K. A., Kalow J. A., Graham T. J. A., Doyle A. G., J. Am. Chem. Soc. 2016, 138, 10802–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For racemic “vinylogous” fluorination of diazo derivatives, see:
- 10a. Qin C., Davies H. M. L., Org. Lett. 2013, 15, 6152–6154; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Thompson S., Lee S. J., Jackson I. M., Ichiishi N., Brooks A. F., Sanford M. S., Scott P. J. H., Synthesis 2019, 51, 4401–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfaltz A., Acc. Chem. Res. 1993, 26, 339–345. [Google Scholar]
- 12.
- 12a. Johnson J. S., Evans D. A., Acc. Chem. Res. 2000, 33, 325–335; [DOI] [PubMed] [Google Scholar]
- 12b. McManus H. A., Guiry P. J., Chem. Rev. 2004, 104, 4151–4202; [DOI] [PubMed] [Google Scholar]
- 12c. Desimoni G., Faita G., Jorgensen K. A., Chem. Rev. 2006, 106, 3561–3651; [DOI] [PubMed] [Google Scholar]
- 12d. Desimoni G., Faita G., Jorgensen K. A., Chem. Rev. 2011, 111, PR284–PR437. [Google Scholar]
- 13.
- 13a. Werlé C., Goddard R., Fürstner A., Angew. Chem. Int. Ed. 2015, 54, 15452–15456; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15672–15676; [Google Scholar]
- 13b. Werlé C., Goddard R., Philipps P., Farès C., Fürstner A., J. Am. Chem. Soc. 2016, 138, 3797–3805; [DOI] [PubMed] [Google Scholar]
- 13c. Werlé C., Goddard R., Philipps P., Farès C., Fürstner A., Angew. Chem. Int. Ed. 2016, 55, 10760–10765; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10918–10923. [Google Scholar]
- 14.
- 14a. Collins L. R., van Gastel M., Neese F., Fürstner A., J. Am. Chem. Soc. 2018, 140, 13042–13055; [DOI] [PubMed] [Google Scholar]
- 14b. Collins L. R., Auris S., Goddard R., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 3557–3561; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3595–3599. [Google Scholar]
- 15. Tindall D. J., Werlé C., Goddard R., Philipps P., Farès C., Fürstner A., J. Am. Chem. Soc. 2018, 140, 1884–1893. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Fürstner A., J. Am. Chem. Soc. 2019, 141, 11–24; [DOI] [PubMed] [Google Scholar]
- 16b. Guthertz A., Leutzsch M., Wolf L. M., Gupta P., Rummelt S. M., Goddard R., Farès C., Thiel W., Fürstner A., J. Am. Chem. Soc. 2018, 140, 3156–3169; [DOI] [PubMed] [Google Scholar]
- 16c. Biberger T., Gordon C. P., Leutzsch M., Peil S., Guthertz A., Copéret C., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 8845–8850; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8937–8942; [Google Scholar]
- 16d. Peil S., Guthertz A., Biberger T., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 8851–8856; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8851–8856. [Google Scholar]
- 17. Tskhovrebov A. G., Goddard R., Fürstner A., Angew. Chem. Int. Ed. 2018, 57, 8089–8094; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8221–8226. [Google Scholar]
- 18.
- 18a. Seidel G., Fürstner A., Angew. Chem. Int. Ed. 2014, 53, 4807–4811; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4907–4911; [Google Scholar]
- 18b. Seidel G., Gabor B., Goddard R., Heggen B., Thiel W., Angew. Chem. Int. Ed. 2014, 53, 879–882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 898–901; [Google Scholar]
- 18c. Seidel G., Mynott R., Fürstner A., Angew. Chem. Int. Ed. 2009, 48, 2510–2513; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2548–2551; [Google Scholar]
- 18d. Tskhovrebov A. G., Lingnau J. B., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 8834–8838; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8926–8930. [Google Scholar]
- 19.For metal-free reactions of α-diazocarbonyl compounds with various fluoride sources see:
- 19a. Olah G. A., Welch J., Synthesis 1974, 896–898; [Google Scholar]
- 19b. Setti E. L., Mascaretti O. A., J. Chem. Soc. Perkin Trans. 1 1988, 2059–2060; [Google Scholar]
- 19c. Ohno M., Itoh M., Ohashi T., Eguchi S., Synthesis 1993, 793–796; [Google Scholar]
- 19d. Pasceri R., Bartrum H. E., Hayes C. J., Moody C. J., Chem. Commun. 2012, 48, 12077–12079. [DOI] [PubMed] [Google Scholar]
- 20.In the absence of ligand, reaction of 1 b with CsF (3 equiv) and [Cu(OTf)]2⋅PhMe (5 mol %) in CH2Cl2/HFIP led to ≤1 % conversion after 4 h reaction time at ambient temperature.
- 21.
- 21a. Evans D. A., Woerpel K. A., Hinman M. M., Faul M. M., J. Am. Chem. Soc. 1991, 113, 726–728; [Google Scholar]
- 21b. Corey E. J., Imai N., Zhang H.-Y., J. Am. Chem. Soc. 1991, 113, 728–729. [Google Scholar]
- 22.For an excellent review, see: Liao S., Sun X.-L., Tang Y., Acc. Chem. Res. 2014, 47, 2260–2272. [DOI] [PubMed] [Google Scholar]
- 23.However, the use of ligands with π-extended benzylic groups was counterproductive, see the Supporting Information.
- 24. Liu C., Yi J.-C., Zheng Z.-B., Tang Y., Dai L.-X., You S.-L., Angew. Chem. Int. Ed. 2016, 55, 751–754; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 761–764. [Google Scholar]
- 25. Rendina V. L., Goetz S. A., Neitzel A. E., Kaplan H., Kingsbury J. S., Tetrahedron Lett. 2012, 53, 15–18. [Google Scholar]
- 26.Parenthetically, we note that the configurational assignment of α-fluoro-α-phenylacetic acid methyl ester, which is often used as a reference point, was based on either plausibility arguments or on three-step derivatization of an only modestly enriched sample, see ref. [8b] and the following: Miyamoto K., Tsuchiya S., Ohta H., J. Fluorine Chem. 1992, 59, 225–232. [Google Scholar]
- 27. Flack H. D., Acta Crystallogr. A 1983, 39, 876–881. [Google Scholar]
- 28.For a review on low-temperature crystallization, see:
- 28a. Veith M., Frank W., Chem. Rev. 1988, 88, 81–92; for a recent example, see: [Google Scholar]
- 28b. Seidel R. W., Goddard R., Nöthling N., Lehmann C. W., CrystEngComm 2019, 21, 3295–3303. [Google Scholar]
- 29. Rasappan R., Laventine D., Reiser O., Coord. Chem. Rev. 2008, 252, 702–714. [Google Scholar]
- 30.
- 30a. Evans D. A., Woerpel K. A., Scott M. J., Angew. Chem. Int. Ed. Engl. 1992, 31, 430–432; [Google Scholar]; Angew. Chem. 1992, 104, 439–441; see also: [Google Scholar]
- 30b. Bellemin-Laponnaz S., Gade L. H., Chem. Commun. 2002, 1286–1287. [DOI] [PubMed] [Google Scholar]
- 31. Deng C., Wang L.-J., Zhu J., Tang Y., Angew. Chem. Int. Ed. 2012, 51, 11620–11623; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11788–11791. [Google Scholar]
- 32.
- 32a. Walli A., Dechert S., Bauer M., Demeshko S., Meyer F., Eur. J. Inorg. Chem. 2014, 4660–4676; [Google Scholar]
- 32b. Goswami V. E., Walli A., Forster M., Dechert S., Demeshko S., Holthausen M. C., Meyer F., Chem. Sci. 2017, 8, 3031–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Srinivas H. D., Maity P., Yap G. P. A., Watson M. P., J. Org. Chem. 2015, 80, 4003–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The spectra of [L11⋅Cu(MeCN)]BF4 in CDCl3 indicate C 2 symmetry on the NMR timescale.
- 35.For a similar situation in [BOX⋅CuII] complexes, see:
- 35a. Xiong H., Xu H., Liao S., Xie Z., Tang Y., J. Am. Chem. Soc. 2013, 135, 7851–7854; [DOI] [PubMed] [Google Scholar]
- 35b. Li Y., Zhou K., Wen Z., Cao S., Shen X., Lei M., Gong L., J. Am. Chem. Soc. 2018, 140, 15850–15858. [DOI] [PubMed] [Google Scholar]
- 36.This geometrical feature is well in line with the X-ray structures of terminal copper carbenes, see:
- 36a. Dai X., Warren T. H., J. Am. Chem. Soc. 2004, 126, 10085–10094; [DOI] [PubMed] [Google Scholar]
- 36b. Straub B. F., Hofmann P., Angew. Chem. Int. Ed. 2001, 40, 1288–1290; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1328–1330; [Google Scholar]
- 36c. Hofmann P., Shishkov I. V., Rominger F., Inorg. Chem. 2008, 47, 11755–11762; [DOI] [PubMed] [Google Scholar]
- 36d. Shishkov I. V., Rominger F., Hofmann P., Organometallics 2009, 28, 1049–1059. [Google Scholar]
- 37.
- 37a.The directionality of the rotatory motion is also the essence of the Pfaltz model explaining the course of Cu-catalyzed cyclopropanations; however, the incoming olefin directly interacts with the carbene-C atom rather than the Cu center, see: Fritschi H., Leutenegger U., Pfaltz A., Helv. Chim. Acta 1988, 71, 1553–1565; [Google Scholar]
- 37b.for a later computational study, see: Rasmussen T., Jensen J. F., Ostergaard N., Tanner D., Ziegler T., Norrby P.-O., Chem. Eur. J. 2002, 8, 177–184. [DOI] [PubMed] [Google Scholar]
- 38.For a cationic Fischer Ir–carbene complex, which is attacked by fluoride at the metal rather than carbon atom, see: O'Connor J. M., Pu L., Woolard S., Chadha R. K., J. Am. Chem. Soc. 1990, 112, 6731–6732. [Google Scholar]
- 39.This phenomenon is particularly well precedented for reactions in which one partner is small and highly reactive; it is thought to indicate stabilizing effects near the transition state, resulting in a strong non-Arrhenius behavior, cf.: Donahue N. M., Chem. Rev. 2003, 103, 4593–4604.14664625 [Google Scholar]
- 40.In the ground state, the ester is orthogonal to the carbene to minimize orbital overlap and avoid further withdrawal of electron density from this highly electrophilic site; the barrier for rotation about the C1−C3 bond is ≈8 kcal mol−1, see the Supporting Information.
- 41.For crystallographic evidence for the validity of the Bürgi–Dunitz trajectory in metal carbene chemistry, see: Peil S., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 18476–18481; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18647–18652. [Google Scholar]
- 42.Related migratory insertion reactions were proposed for different metals, including copper, cf.:
- 42a. Xiao Q., Zhang Y., Wang J., Acc. Chem. Res. 2013, 46, 236–247; [DOI] [PubMed] [Google Scholar]
- 42b. Zhao X., Zhang Y., Wang J., Chem. Commun. 2012, 48, 10162–10173; [DOI] [PubMed] [Google Scholar]
- 42c. Barluenga J., Valdés C., Angew. Chem. Int. Ed. 2011, 50, 7486–7500; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7626–7640; [Google Scholar]
- 42d. Franssen N. M. G., Walters A. J. C., Reek J. N. H., de Bruin B., Catal. Sci. Technol. 2011, 1, 153–165; see also: [Google Scholar]
- 42e. Cairncross A., Sheppard W. A., J. Am. Chem. Soc. 1968, 90, 2186–2187. [Google Scholar]
- 43.For Cu-mediated trifluoromethylation and trifluoromethylthiolation of diazo esters, which seem to proceed by a similar mechanism, see:
- 43a. Hu M., Ni C., Hu J., J. Am. Chem. Soc. 2012, 134, 15257–15260; [DOI] [PubMed] [Google Scholar]
- 43b. Wang X., Zhou Y., Ji G., Wu G., Li M., Zhang Y., Wang J., Eur. J. Org. Chem. 2014, 3093–3096; [Google Scholar]
- 43c. Lefebvre Q., Fava E., Nikolaienko P., Rueping M., Chem. Commun. 2014, 50, 6617–6619. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Sommer H., Fürstner A., Chem. Eur. J. 2017, 23, 558–562; [DOI] [PubMed] [Google Scholar]
- 44b. Mo X., Letort A., Rosca D.-A., Higashida K., Fürstner A., Chem. Eur. J. 2018, 24, 9667–9674; [DOI] [PubMed] [Google Scholar]
- 44c. Meng Z., Souillart L., Monks B., Huwyler N., Herrmann J., Müller R., Fürstner A., J. Org. Chem. 2018, 83, 6977–6994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary