

ABSTRACT
This study investigates how mesenchymal stem cell's (MSCs) proliferation and migration abilities are influenced by various platelet products (PP). Donor‐matched, clinical‐, and control laboratory‐standard PPs were generated and assessed based on their platelet and leukocyte concentrations. Bone marrow derived MSCs were exposed to these PP to quantify their effect on in vitro MSC proliferation and migration. An adapted colony forming unit fibroblast (CFU‐F) assay was carried out on bone marrow aspirate using clinical‐standard PP‐loaded electrospun poly(ϵ‐caprolactone) (PCL) membrane to mimic future clinical applications to contain bone defects. Clinical‐standard PP had lower platelet (2.5 fold, p < 0.0001) and higher leukocyte (14.1 fold, p < 0.0001) concentrations compared to laboratory‐standard PP. It induced suboptimal MSC proliferation compared to laboratory‐standard PP and fetal calf serum (FCS). All PP induced significantly more MSC migration than FCS up to 24 h. The removal of leukocytes from PP had no effect on MSC proliferation or migration. The PP‐loaded membranes successfully supported MSC colony formation. This study indicates that platelet concentrations in PP impact MSC proliferation more than the presence of leukocytes, whilst MSC migration in response to PP is not influenced by platelet or leukocyte numbers. Clinical‐standard PP could be applied alongside manufactured membranes in the future treatment of bone reconstruction. © 2019 The Authors. Journal of Orthopaedic Research® Published by Wiley Periodicals, Inc. on behalf of Orthopaedic Research Society. J Orthop Res 37:1329–1338, 2019.
Keywords: stem cells, platelet products, bone regeneration, fracture repair
Fracture nonunion persists as a prevalent complication, with the incidence in long bones reported to range between 5 and 10%.1, 2 Its etiopathogenesis remains multifactorial with a recent review identifying over 20 factors to be implicated in this process.3 To address impaired fracture healing, Giannoudis et al. proposed the diamond conceptual framework for bone repair highlighting that, for a successful healing response both mechanical stability and biological factors must be present.4 For biological stimulation, while autologous bone graft remains the gold standard, recently, other forms have gained popularity including bone marrow aspirates (BMA) and/or growth factors.5, 6 Since their introduction to the clinical setting, bone morphogenetic proteins (BMPs) have been extensively used for the treatment of nonunion fractures.7 There are numerous publications reporting on the clinical results of BMP‐2 and BMP‐7.8, 9, 10 However, since the withdrawal of BMP‐7 from the market and the license limitation of BMP‐2 to be used for the management of open tibial fractures, other inductive molecules gained popularity such as demineralized bone matrix (DBM),11 teriparatide,12 and platelet products (PP).13, 14
PPs are highly enriched sources of autologous growth factors and cytokines that act as biological stimulants to accelerate osteogenesis and bone repair.15, 16 PPs are usually applied as platelet rich plasma (PRP), where the cells remain intact, or more recently, as platelet lysate (PL) where only the growth‐factor containing plasma is used.17 Both forms have been found to be highly effective in the treatment of orthopaedic trauma for decades,18 and more recently for the treatment of fracture nonunion.19 Several studies into PP loaded membranes are already proving to be successful at promoting regeneration via the delivery of growth factors in cartilage and bone repair.20, 21 When working with PP loaded membranes, maximizing bone marrow derived MSC (BM‐MSC) proliferation, and migration is key. By ensuring these functions are enhanced, more viable cells are available to differentiate, as well as inducing more resident BM‐MSC homing to the site of injury. While some studies found that the presence of leukocytes in PP are advantageous due to their antimicrobial properties22 and high concentration of cytokines involved in bone repair such as VEGF,23, 24 there are also concerns related to leukocytes’ effect of inducing excessive inflammatory and necrotic pathways from the surrounding tissue.25, 26 Collectively, this provides a strong case for their depletion.
This study investigates the in vitro effect of platelets and leukocytes on the proliferation and migration of MSCs from BMA by studying a range of PP compositions including a clinical‐standard PRP (CPRP). A laboratory‐standard PRP made using a two‐spin centrifugation protocol to enrich platelets but deplete leukocytes was also used as control.18, 27
To investigate the specific importance of leukocytes, PRP was processed further by syringe‐filtration to remove leukocytes and produce a pure filtered platelet rich product (fPRP). These products were then lysed to ensure product consistency over the course of in vitro assays; thereafter referred to as clinical‐standard platelet lysate (CPL), platelet lysate (PL), and filtered PL (fPL). These PLs were then evaluated in vitro utilizing primary BM‐MSCs, including autologous BM‐MSC‐PP combinations, as well as loading onto a degradable electrospun membrane to achieve sustained localized delivery in a colony forming units‐fibroblast (CFU‐F) assay. The aim was to assess if high numbers of leukocytes would impede BM‐MSC proliferation and migration and whether their depletion from clinical PPs would be desirable for bone regenerative approaches.
METHODS
This study was carried out in adherence with the Helsinki Declaration under ethics code 06/Q1206/127 following approval from the local National Health Service Research & Development Department, Leeds East Research Ethics Committee to harvest these samples. Written informed consent was obtained from each participant.
Sample Collection and Processing of CPRP, PRP, and fPRP
Whole blood was collected from 11 healthy volunteers (8 males and 3 females, 22–58 yrs) in acid‐citrate dextrose solution A (ACD‐A). Each sample was divided and used to prepare CPRP and PRP in a single‐donor model. CPRP was made from whole blood using the BioCUE™ device (Zimmer Biomet) which was centrifuged at 1100 g for 15 min. After centrifugation, the device was agitated and the CPRP fraction was extracted using a syringe. PRP was generated using a two‐step centrifugation protocol28 whereby the patients’ whole blood was incubated at room temperature for 1 h before centrifugation at 400 g for 10 min. The platelet‐containing supernatant was extracted and centrifuged at 2700g for 10 min before resuspending the pellet in 1/5th of the residual supernatant.
fPRP was generated from PRP that was filtered through a white blood cell (WBC) syringe filter (Acrodisc) which entraps leukocytes and allows platelets to pass through for collection. The division of PRP for the production of fPRP meant both their final volumes were much smaller than CPRP limiting the number of assays they could be used in. Platelet and leukocyte concentrations of CPRP, PRP, fPRP, and whole blood (1 ml) were determined using an automatic haematology analyser (Sysmex).
Generating Lysate Products (CPL, PL, and fPL)
To generate the lysed products for in vitro assays, CPRP, PRP, and fPRP were processed through three freeze‐thaw cycles which involved freezing at −80 °C followed by thawing at 37 °C.29 To remove contaminating cell debris, CPL, PL, and fPL were centrifuged at 2700g for 10 min and the supernatants were extracted for cell stimulation.
Sample Collection and Processing of BM‐MSCs
Patients undergoing elective orthopaedic surgery with no underlying disease were recruited (n = 11, 6 males and 5 females, 17–69 yrs). 4 ml of BMA was collected from each donor's anterior iliac crest and was treated with ammonium chloride to lyse red blood cells. The remaining nucleated cells were plated into flasks at the cell seeding density of 5 × 105 cells/cm2. BM‐derived adherent cells were cultured in MSC expansion media (Miltenyi Biotec) for approximately 2 weeks until confluent.
Characterising BM‐MSCs
Cultured BM derived cells (passage two) were tested for the surface expression levels of MSC markers as defined by the International Society for Cell Therapy (ISCT).30 Following trypsinisation (Sigma Aldrich), cell cultures were stained with antibodies against positive markers of MSCs (CD90, CD73, and CD105) and negative hematopoietic‐lineage markers (CD34, CD14, HLA‐DR, CD19, CD45). All antibodies were used according to the manufacturer's recommendation (Miltenyi Biotec). Isotype controls (BD, Bio‐Rad) were used to gate for positive expression. The data were acquired on an Attune flow cytometer (ThermoFisher Scientific).
Trilineage Differentiation Potential of BM‐MSCs
BM derived cells were cultured up to passage four before being trypsinized and tested for multipotentiality as previously described.31 Cells were seeded with AdipoDiff Media (Miltenyi Biotec) or OsteoDiff Media (Miltenyi Biotec) for adipogenesis or osteogenesis, respectively, with bi‐weekly media changes. After 3 weeks, cell cultures were stained with oil red solution (adipogenesis)32 or Alizarin Red (osteogenesis). For chondrogenesis, cells were added to Eppendorf tubes to create a pellet, resuspended in ChondroDIFF Media (Miltenyi Biotec) and cultured for 21 days after which the pellets were stained with toluidine blue (Sigma).
Proliferation Assay
The XTT assay was used to quantify BM‐MSC proliferation in the presence of different PL products. Activity of mitochondrial dehydrogenase, and therefore cell number per well, is directly correlated to the amount of orange formazan formed, as monitored by the optical density (OD) at 450 nm. BM‐MSCs were seeded in triplicate in a 96‐well plate at a density of 500 cells/well with MSC expansion media and incubated for 24 h. The media was then replaced with basal Dulbecco's Modified Eagle's Media (DMEM; Sigma‐Aldrich) supplemented with 10% (v/v) of CPL, PL, fPL, or fetal calf serum (FCS) and incubated for 5 days with a half media change on day 3. Following exposure to the treatment media, cell proliferation was assessed on day 5 using an XTT cell proliferation kit (Roche) according to the manufacturer's instructions.
Migration Assay
The bottom wells contained basal DMEM (1% (v/v) Penicillin/Streptomycin and 2 I.U/ml sodium heparin solution) supplemented with 10% (v/v) of CPL, PL, fPL, or FCS. 10% was the selected concentration due to its frequent use in the literature.33, 34, 35 Migration was analyzed over 24 h using an IncuCyte® (Essen) to collect phase‐contrast images of the transwells as cells migrated along the chemotactic gradient. Images were captured every 30 min and a processing mask was created for each BM‐MSC culture to account for donor variation.
CFU‐F Assay With CPL‐Loaded Membrane
An adaptation of the CFU‐F assay was used to quantify how a CPL‐loaded membrane affected the proliferation and migration capacity of uncultured BM‐MSCs and to further validate the results of the assays using cultured BM‐MSCs. First, fresh BMA from three donors underwent red blood cell lysis after which a total of 5 × 105 cells were plated in 15 ml expansion media to adhere over 24 h. After initial attachment and a PBS wash the media was replaced with either 15 ml expansion media containing 2 I.U/ml sodium heparin solution (control) or 15 ml DMEM media (no serum) containing 2 I.U/ml sodium heparin solution (test). 1 cm2 square sections, 400 µm thick, of UV‐sterilized PCL that were made in‐house36 were soaked in CPL for 15 min until saturated before being placed in the centre of the test dishes. The dishes were incubated for a further 19 days with half‐media changes once a week. After 21 days, the dishes were washed in PBS, fixed in 10% (v/v) formalin (Sigma) and stained in 1% (w/v) methylene blue (Sigma). All colonies were imaged using a plate scanner (Epson) at 1200 dpi. Colony area, integrated density and number per dish was quantified using ImageJ whereby scanned images were converted to 8‐bit grayscale and a threshold mask was applied before particles were automatically analyzed and measured.
RESULTS
Characterising Cellular Content of PP
CPRP was generated using a BioCUE™ device whilst PRP was made following a previously optimised protocol18 and their cell populations were quantified using a haematology analyzer. While both PP significantly enriched platelets compared to their whole blood counterparts, PRP also had significantly higher numbers of platelets (15.9 × 105 PLT/µlL) (Fig. 1A) in comparison to CPRP (6.3 × 105 PLT/µl) as well as significantly lower leukocyte numbers (1.8 × 103 LEUK/µl) compared to CPRP (20.6 × 103 LEUK/µl) (Fig. 1B). Despite the significant difference in final volumes (p = 0.0078) (Fig. 1C), both products had comparable average fold decrease in volumes (10.0 ± 0.0 and 7.9 ± 0.5 CPRP and PRP, respectively). All CPRP replicates underwent identical reductions in volume causing their data points to overlap. The difference in the PP's cell populations is also visible in their appearance as CPRP is opaque with additional red blood cell contamination (Fig. 1D) while PRP had fewer contaminating red blood cells and was more translucent (Fig. 1E).
Figure 1.
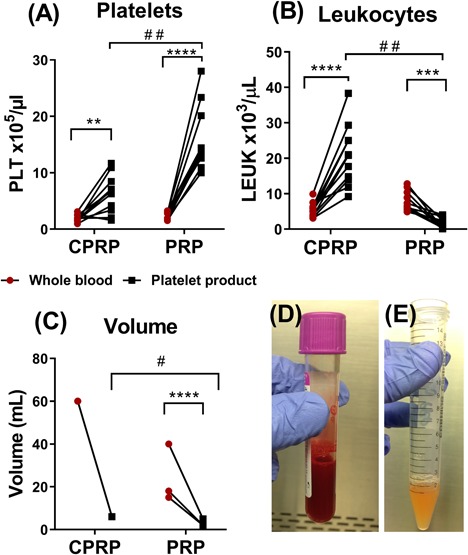
Characterising platelet rich plasma products. Using a haematology analyser, the change in platelets (A), leukocytes (B), and final volume (C) from whole blood to CPRP (n = 11) and PRP (n = 11) were quantified. The increase or decrease of each cell type indicates the change from whole blood (red) to the paired platelet product (black). A paired t‐test was used to compare change in cell type before and after processing (**p < 0.01, ***p < 0.001, and ****p < 0.0001) while a Mann‐Whitney test was used to compare CPRP and PRP (# = p < 0.001, # # = p < 0.0001). Statistics could not be conducted on CPRP's volume analysis (C) due to matching differences between each sample. The photographs show the CPRP fraction product (D) and the PRP product (E).
As previous studies have found that platelets and leukocytes are not stable at room temperature for the 5 days necessary for the proliferation and migration assays,37 the PP were lysed after production to ensure consistency of samples. Lysis of the products changed their nomenclature from CPRP and PRP to clinical platelet lysate (CPL) and platelet lysate (PL).
Characterizing BM‐MSCs
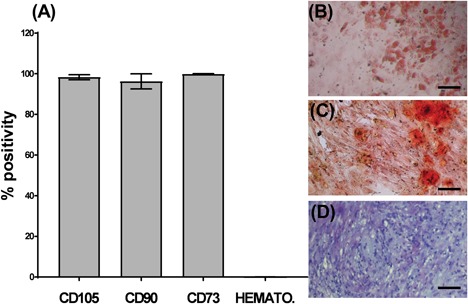
BMAs underwent red blood cell lysis, after which the remaining nucleated cells were plated into flasks, three randomly selected cultures were tested for MSC characterization according to the ISCT's definition.38 Cell surface marker expression was analyzed using flow cytometry which found cells were negative for CD14, CD19, CD34, CD45, HLA‐DR, and positive for CD73, CD90, and CD105 (Fig. 2A) consistent with the criteria for identifying MSCs. Their tri‐lineage differentiation capacity was confirmed following 3‐week culture in adipogenic, osteogenic, and chondrogenic stimulation medias. A representative culture is shown in Figure 2 which was stained with oil red o solution to visualize lipid droplets (Fig. 2B), alizarin red staining to visualize calcium depositions (Fig. 2C), and finally toluidine blue staining of the aggregated cell pellet indicative of glycosaminoglycan production. Nine of these cultures were used for subsequent PL proliferation and migration tests, each PL product was tested on at least three MSC cultures and the results were averaged.
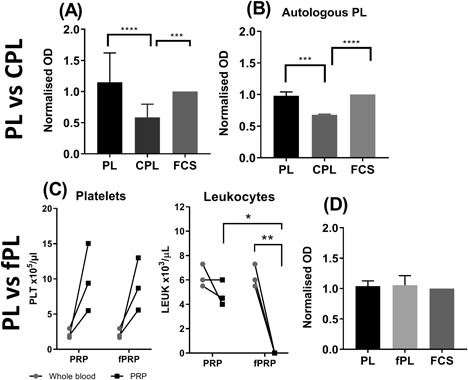
Figure 2.
Platelet product composition's impact on BM‐MSC proliferation. XTT assay quantifying BM‐MSC proliferation following four day exposure to media containing 10% platelet products or FCS as control. Proliferation was represented as OD normalized to FCS. (A) Cultured BM‐MSCs were exposed to 10% PL, CPL, and FCS. (B) Cultured BM‐MSCs were exposed to autologous 10% PL and CPL as well as 10% FCS. (C) The change in platelets and leukocytes from whole blood to PRP and fPRP (compared using a paired t‐test). (D) Cultured BM‐MSCs were exposed to 10% PL, fPL and FCS from three PL donors. One‐way ANOVA test was used to test significance between the platelet products effects on proliferation (3A, B, and D).(*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). Except for 3B, all experiments were performed on a minimum of three different PP donors and three BM‐MSC cultures. Error bars indicate variation between PP except for 3B where error bars indicate technical variation between replicates.
Platelet Product Composition's Impact on BM‐MSC Proliferation
Cultured cells were exposed to 10% PL, CPL, and control FCS containing media for 5 days, after which the cells were treated with XTT reagent causing a color change. BM‐MSC proliferation was expressed as OD normalized to FCS. BM‐MSCs treated with PL were found to proliferate significantly more than cells treated with CPL (p = 0.0001) and equal to cells treated with FCS (Fig. 3A). The same response was also seen from BM‐MSCs tested with autologous PL and CPL (Fig. 3B). To investigate whether CPL's suboptimal support of BM‐MSC proliferation was due to the high number of leukocytes, they were filtered out using a leukocyte syringe filter. The filtered PL's (fPL) platelet numbers were not significantly affected by the filtration process while leukocytes were shown to be significantly depleted in fPL (Fig. 3C). Removal of the leukocytes was found to not significantly improve proliferation as it was shown that both PL and fPL induced the same BM‐MSC proliferation as FCS (Fig. 3D).
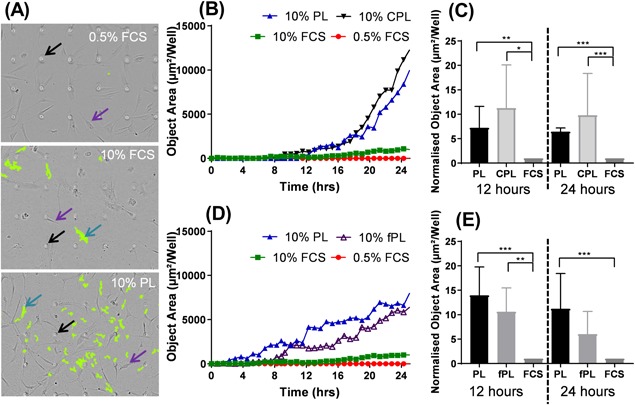
Figure 3.
Platelet product composition's impact on BM‐MSC migration. The IncuCyte® transwell assay quantifies BM‐MSCs moving toward a chemotactic gradient. (A) Representative images of wells of 0.5% FCS (top), 10% FCS (middle), and 10% PL (bottom). Arrows indicate pores (black), static cells on the top of the transwell (purple) and cells that have migrated (blue). The processing mask that quantifies the migrated cells is shown in green. (B) Representative time‐course response of the cells from one BM‐MSC culture that have migrated through the transwell towards 10% PL, 10% CPL 10% FCS, and 0.5% FCS. Data are shown as the area of the bottom of the well occupied by cells. (C) Average object area of the underside of the transwell occupied by BM‐MSCs and treated with 10% PL, 10% CPL, and 10% FCS was normalized to 10% FCS. (D) Representative time‐course response of the cells from one culture that have migrated through the transwell towards 10% PL, 10% fPL, 10% FCS, and 0.5% FCS. (E) Average object area of the underside of the transwell occupied by BM‐MSCs and treated with 10% PL, 10% fPL, and 10% FCS was normalized to 10% FCS. A one‐way ANOVA was carried out using the Kruskal‐Wallis test for normality (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Error bars indicate variation between PL donors. Experiments in C and E were performed on a minimum of three different BM‐MSC cultures.
Altogether, this data indicated that the leukocytes were not inhibitory for BM‐MSC proliferation and suggested that differing performances of PL and CPL observed were likely due to the different numbers of platelets present.
Platelet Product Composition's Impact on BM‐MSC Migration
To compare the chemotactic potential of the different PPs, the IncuCyte® transwell assay was used. Serum starved BM‐MSCs were seeded on top of the transwells and exposed to different PPs below. Over the course of 24 h, the top and bottom of the transwells were imaged continuously to track their migration. Representative images of the top of the transwells are shown in Figure 4A where non‐migrated cells are shown in focus (purple arrow), cells that have migrated through and identified by the software are highlighted in green for processing (blue arrow) and the pores are identified with black arrows. The migratory effects of PL and CPL were demonstrated as a time‐course assay using one representative culture (Fig. 4B) which showed that, both PL and CPL appear to induce far more migration than 10% FCS. To enable statistical analysis, three independent PL preparations were each tested on three BM‐MSC cultures and found that both PL and CPL induced significantly more migration than FCS at 12 h (p = 0.0068 and p = 0.0434, respectively) and at 24 h (p = 0.0006 and p = 0.0008) (Fig. 4C). Considering the higher levels of leukocytes in CPL, these findings suggest that the presence of leukocyte‐derived proteins is not a detriment to BM‐MSC migration and potentially contribute toward improved migration.
Figure 4.
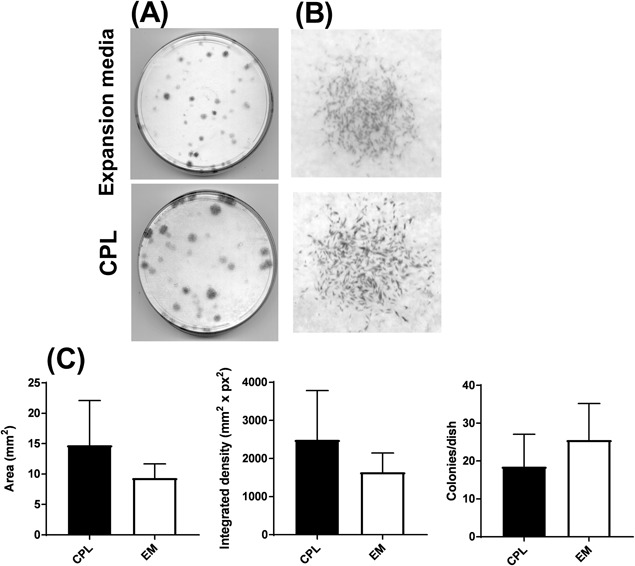
CPL‐loaded membrane supports cell proliferation in CFU‐F assay. (A) Representative CFU‐F dishes of BM‐MSCs grown in either standard expansion media (top dish) or serum‐free DMEM with the additional CPL‐loaded membrane as a source of released growth factors and cytokines (bottom dish). (B) Representative individual colonies of cells grown in expansion media (top) or CPL‐loaded membrane and DMEM (bottom). Three BMA donors were tested on a single CPL product. Images were collected using a photo scanner at 1200 dpi. (C) Comparison of average colony area, density, and total number between cells treated with a CPL‐loaded scaffold and expansion media (EM). An unpaired t‐test found no significant difference between CPL of EM.
To further investigate the effect of leukocyte‐derived proteins on BM‐MSC migration, PL and fPL's chemotactic effects were investigated. A representative culture is shown in Figure 4D which showed that, while the complete removal of leukocytes did appear to reduce BM‐MSC's migratory response this reduction was not statistically significant. In addition, PL and fPL both induced far more migration than 10% FCS. BM‐MSCs were found to be significantly more migratory toward PL and fPL than 10% FCS at 12 h (p = 0.0015 and 0.0198, respectively) and PL was also found to outperform 10% FCS at 24 h (p = 0.0024) (Fig. 4E). These findings suggest that while platelet and leukocyte numbers contribute toward BM‐MSC migration, it is likely that most of the cytokines that induce migration are present in the plasma component of PP—and so any additional platelet or leukocytes make no significant difference.
Loaded Membrane Supports Colony Formation From Native MSCs Present in BMA
To more closely mimic clinical applications involving a PP‐loaded biomaterial membrane as a MSC homing and containment device, experiments were performed using clinically approved PP. The aim of these experiments was to study if CPL released from a membrane could support colony formation from rare BM‐MSCs without their culture‐amplification. In standard CFU‐F assays, single rare BM‐MSCs give rise to individual colonies. The fresh BM‐MSCs used for this assay were grown in either standard MSC expansion media or basal serum‐free media with the addition of a CPL‐loaded membrane (Fig. 5A). Colony formation was observed in all test dishes, with similar morphology to control dishes containing MSC expansion media (Fig. 5B). Furthermore, although there were trends for higher colony numbers in MSC expansion media, and higher colony areas and densities in CPL‐loaded membrane dishes, the differences were not found to be statistically significant (Fig. 5C). This indicated that CPL‐loaded membranes were able to release growth factors that induced colony formation and supported rare BM‐MSC proliferation.
Figure 5.
Characterizing platelet rich plasma products.
DISCUSSION
While the clinical effectiveness of PP is generally accepted, due to the lack of quality control, the variation in manufacturing, processing, delivery, and its different applications (e.g., rotator cuff repair and osteoarthritis39, 40), the “optimal” composition is still hotly debated depending on its specific clinical application.
The main issue with lack of standardization is exemplified by one study which found that PRP had no significant effect on bone healing,41 however, a closer look at the platelet numbers used in the study found that some of the PRP tested had over 16‐fold more platelets than other PRP. Despite the vast differences in platelet numbers and omitted leukocyte numbers, these products were all classified under the same umbrella term “PRP.” This is commonplace across the literature, whereby, the absence of standardized nomenclature, quality control, as well as thorough analysis of cellular contents, makes it difficult to draw clear conclusions. According to Delong et al.'s classification system42 platelet number, activation method, and white blood cell number (PAW), CPRP is classified as P2‐A (which identifies the increase of platelets as “moderate” and the leukocytes as “enriched above baseline”) while PRP is classified as P3‐B (which identifies the platelet concentration as “highly enriched” above baseline and leukocytes depleted below baseline).
As well as variation in platelet concentrations, the lack of regulation also instils concern in the scientific community over the use of highly concentrated leukocytes, their associated pro‐inflammatory cytokines (specifically TNF‐α and IL‐1β), and the risk that they could counteract the platelet's beneficial effects and impede bone regeneration.26 TNF‐α and IL‐1β are known to induce inflammation causing a biphasic physiological response; whilst inflammation is necessary for healing,43 in excess it is thought to activate the NFκB pathway26 inhibiting osteogenesis and promoting osteoclastogenesis.44 However, more recently, these proinflammatory cytokines have been linked to increased osteogenesis,45 BM‐MSC migration,46 and proliferation47 indicative of the lack of consensus in the field.
This study addressed these concerns by better defining platelets and leukocyte's effects in the two key physiological processes of bone regeneration: BM‐MSC proliferation and BM‐MSC migration. With regards to proliferation, when compared to PL and FCS, CPL induced significantly less BM‐MSC proliferation than PL. This was also observed using donor‐matched BM‐MSCs and PP proving that the response from the cells was not due to the allogeneic nature of the PL. The cause of CPL's suboptimal ability to support BM‐MSC proliferation compared to PL is either due to its lower number of platelets or higher number of leukocytes—both of which have been reported to reduce proliferation in the literature.26, 42, 48, 49 To identify which cell type is the key player, leukocytes were filtered from PL while platelet numbers were kept the same to produce fPL. Direct comparison of PL and fPL showed that removing leukocytes did not improve proliferation. This suggests that in our experimental conditions, the cytokines released from leukocytes neither enhanced nor inhibited BM‐MSC proliferation and that platelets are most likely responsible for releasing the predominant growth factors involved in supporting BM‐MSC proliferation.
With regards to BM‐MSC migration, all three platelet products (PL, CPL, and fPL) induced significantly more migration than FCS, likely due to the greater concentrations of cytokines such as VEGF and SDF‐1.50, 51 Again, it was seen that removing leukocytes from PL did not further enhance BM‐MSC migration, but even seemed to reduce it. While there is no precedent for the use of platelet products in BM‐MSC migration studies, the current data supports previous literature of platelet products outperforming FCS.26, 34 The methods used are also unique in that, not only is the total migration shown, but also the increased rate of cell migration toward the platelet products.
Based on these findings it could be proposed that generating high quality platelet products should simply involve increasing platelet numbers as much as possible without regard for leukocytes—this could be easily achieved by decreasing the final volume to yield a more concentrated platelet product. However, the excessive enrichment of platelets faces the risk of paradoxically inhibiting cell proliferation, viability, and migration.42 A therapeutically effective range of platelet concentration is likely to be the case rather than a specific pure concentration. Our results indicate that platelet concentrations in the range of 6.3–15.9 × 105 PLTs/μl (with and without leukocytes) were effective in supporting BM‐MSC proliferation and migration without adverse effects on their attachment or morphology.
While several studies have already shown that PP increases the bone regeneration rate and prevention of non‐union fractures in animal models52, 53 and human subjects,14 as well as this work that aims to optimize PP's impact on BM‐MSCs, it is also important to consider how PP should be delivered to the site of injury. To address this, a membrane was loaded with CPL and its discharge was found to support BM‐MSC colony formation and BM‐MSC proliferation. Due to the manufacturing process of PL and fPL (specifically the division of samples), low volumes prevented their loading onto a membrane for CFU‐F analysis. In summary, this study supports the notion that the specific clinical application and desired outcome should be considered for defining best formulations of platelet products for bone regeneration. If cell proliferation is thought to be limiting regeneration, for example in elderly patients that have low numbers of autologous BM‐MSCs,54 then CPL will be sub‐optimal and PP with higher concentrations of platelets should be used. If however, the surgeon's priority is to induce BM‐MSC migration to the site of injury, for example to attract BM‐MSCs toward an unpopulated bone scaffold, then the current clinical standard CPL may be sufficient. As well, CPL was found to support colony formation when delivered using a membrane, with a trend of increased colony size and density than MSC expansion media; providing encouraging insight toward future delivery alternatives and streamlined surgeries.
AUTHOR CONTRIBUTIONS
All authors contributed extensively to the work presented in this paper. PVG, EJ, GT, SR, and KM were responsible for the study conception and design. KM generated the platelet products and performed the platelet characterization, proliferation, migration, and CFU‐F experimental work and analysis. JE conducted the flow cytometry experiments and analysis and HO conducted the tri‐lineage characterization experiments and analysis. PVG was responsible for patient recruitment and sample collection. Data analysis and interpretation were performed by KM, EJ, JE, and HO. Manuscript preparation and writing was completed by KM and EJ with additional critical review by PVG, JE, HO, GT, and SR. All authors have read and approved the final submitted manuscript
ACKNOWLEDGMENTS
Katrina Moisley and Heather Owston both hold the Centre of Doctoral Training in Tissue Engineering and Regenerative Medicine studentships funded by grant EP/L014823/1 from the Engineering and Physical Sciences Research Council. Jehan El‐Jawhari is funded by a start‐up grant S‐16‐132E from the AO foundation. The authors thank Dr Ala Altaie and Dr Karen Bieback for guidance, the staff at the Leeds General Infirmary Trauma Unit for collecting samples and staff at the Haematological Malignancy Diagnostic Service for assistance in characterizing the samples. We also gratefully acknowledge the technical expertise of Dr Thomas Baboolal and Dr. Richard Cuthbert.
Jehan J. El‐Jawhari and Heather Owston have contributed equally to this work.
Elena A. Jones and Peter V. Giannoudis are last author.
REFERENCES
- 1. Tzioupis C, Giannoudis P. 2007. Prevalence of long‐bone non‐unions. Injury 38:S3–S9. [DOI] [PubMed] [Google Scholar]
- 2. Calori GM, Mazza E, Colombo M, et al. 2011. Treatment of long bone non‐unions with polytherapy: indications and clinical results. Injury 42:587–590. [DOI] [PubMed] [Google Scholar]
- 3. Santolini E, West R, Giannoudis PV. 2015. Risk factors for long bone fracture non‐union: a stratification approach based on the level of the existing scientific evidence. Injury 46:S8–s19. [DOI] [PubMed] [Google Scholar]
- 4. Giannoudis PV, Einhorn T, Marsh D. 2007. Fracture healing: the diamond concept. Injury 4:3–6. [DOI] [PubMed] [Google Scholar]
- 5. Giannoudis PV, Faour O, Goff T, et al. 2011. Masquelet technique for the treatment of bone defects: tips‐tricks and future directions. Injury 42:591–598. [DOI] [PubMed] [Google Scholar]
- 6. Dimitriou R, Jones E, McGonagle D, et al. 2011. Bone regeneration: current concepts and future directions. BMC Med 9:66–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krishnakumar GS, Roffi A, Reale D, et al. 2017. Clinical application of bone morphogenetic proteins for bone healing: a systematic review. Int Orthop 41:1073–1083. [DOI] [PubMed] [Google Scholar]
- 8. Moghaddam‐Alvandi A, Zimmermann G, Büchler A, et al. 2012. Results of nonunion treatment with bone morphogenetic protein 7 (BMP‐7). Unfallchirurg 115:518–526. [DOI] [PubMed] [Google Scholar]
- 9. Lyon T, Scheele W, Bhandari M, et al. 2013. Efficacy and safety of recombinant human bone morphogenetic protein‐2/calcium phosphate matrix for closed tibial diaphyseal fracture: a double‐blind, randomized, controlled phase‐II/III trial. J Bone Joint Surg Am 95:2088–2096. [DOI] [PubMed] [Google Scholar]
- 10. Ristiniemi J, Flinkkilä T, Hyvönen P, et al. 2007. RhBMP‐7 accelerates the healing in distal tibial fractures treated by external fixation. J Bone Joint Surg Br 89:265–272. [DOI] [PubMed] [Google Scholar]
- 11. Babiker H, Ding M, Overgaard S. 2016. Demineralized bone matrix and human cancellous bone enhance fixation of porous‐coated titanium implants in sheep. J Tissue Eng Regen Med 10:245–251. [DOI] [PubMed] [Google Scholar]
- 12. Li XF, Xu DX, Chen YZ. 2017. Teriparatide as a nonoperative treatment for tibial and femoral fracture nonunion A case report. Medicine 96:e6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tall M. 2018. Treatment of aseptic tibial shaft non‐union without bone defect. Orthop Traumatol Surg Res 104:S63–S69. [DOI] [PubMed] [Google Scholar]
- 14. Galasso O, Mariconda M, Romano G, et al. 2008. Expandable intramedullary nailing and platelet rich plasma to treat long bone non‐unions. J Orthop Trauma 9:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martinotti S, Mariconda M, Romano G, et al. 2014. Platelet‐rich plasma induces mixed osteogenic/osteoclastogenic phenotype in osteosarcoma SaOS‐2 cells: role of TGF‐beta. Curr Pharm Biotechnol 15:120–126. [DOI] [PubMed] [Google Scholar]
- 16. Alsousou J, Thompson M, Hulley P, et al. 2009. The biology of platelet‐rich plasma and its application in trauma and orthopaedic surgery: a review of the literature. J Bone Joint Surg Br 91:987–996. [DOI] [PubMed] [Google Scholar]
- 17. Bieback K. 2013. Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transfus Med Hemother 40:326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Altaie A, Owston H, Jones E. 2016. Use of platelet lysate for bone regeneration − are we ready for clinical translation? World J Stem Cells 8:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buza JA, 3rd , Einhorn T. 2016. Bone healing in 2016. Clin Cases Miner Bone Metab 13:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moreira Teixeira LS, Leijten JC, Wennink JW, et al. 2012. The effect of platelet lysate supplementation of a dextran‐based hydrogel on cartilage formation. Biomaterials 33:3651–3661. [DOI] [PubMed] [Google Scholar]
- 21. Oliveira SM, Reis RL, Mano JF. 2015. Assembling human platelet lysate into multiscale 3D scaffolds for bone tissue engineering. ACS Biomater Sci Eng 1:2–6. [DOI] [PubMed] [Google Scholar]
- 22. Moojen DJ, Everts PA, Schure RM, et al. 2008. Antimicrobial activity of platelet‐leukocyte gel against Staphylococcus aureus. J Orthop Res 26:404–410. [DOI] [PubMed] [Google Scholar]
- 23. Boswell SG, Cole BJ, Sundman EA, et al. 2012. Platelet‐rich plasma: a milieu of bioactive factors. Arthroscopy 28:429–439. [DOI] [PubMed] [Google Scholar]
- 24. Robert Z, Jakubietz R, Jakubietz M, et al. 2001. Different preparation methods to obtain platelet components as a source of growth factors for local application. Transfusion 41:1217–1224. [DOI] [PubMed] [Google Scholar]
- 25. Xu Z, Yin W, Zhang Y, et al. 2017. Comparative evaluation of leukocyte‐ and platelet‐rich plasma and pure platelet‐rich plasma for cartilage regeneration. Sci Rep 7:43301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yin W, Qi X, Zhang Y, et al. 2016. Advantages of pure platelet‐rich plasma compared with leukocyte‐ and platelet‐rich plasma in promoting repair of bone defects. J Transl Med 14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shih DT, Burnouf T. 2015. Preparation, quality criteria, and properties of human blood platelet lysate supplements for ex vivo stem cell expansion. N Biotechnol 32:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dhurat R, Sukesh MS. 2014. Principles and methods of preparation of platelet‐rich plasma: a review and author's perspective. J Cutan Aesthet Surg 7:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krüger JP, Freymannx U, Vetterlein S, et al. 2013. Bioactive factors in platelet‐Rich plasma obtained by apheresis. Transfus Med Hemothery 40:432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dominici M, Le Blanc K, Mueller I, et al. 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 8:315–317. [DOI] [PubMed] [Google Scholar]
- 31. Jones EA, Kinsey SE, English A, et al. 2002. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum 46:3349–3360. [DOI] [PubMed] [Google Scholar]
- 32. Jones EA, Crawford A, English A, et al. 2008. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: detection and functional evaluation at the single‐cell level. Arthritis & Rheumatism 58:1731–1740. [DOI] [PubMed] [Google Scholar]
- 33. Mishra A, Tummala P, King A, et al. 2009. Buffered platelet‐rich plasma enhances mesenchymal stem cell proliferation and chondrogenic differentiation. Tissue Eng Part C Methods 15:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murphy MB, Blashki D, Buchanan RM, et al. 2012. Adult and umbilical cord blood‐derived platelet‐rich plasma for mesenchymal stem cell proliferation, chemotaxis, and cryo‐preservation. Biomaterials 33:5308–5316. [DOI] [PubMed] [Google Scholar]
- 35. Kocaoemer A, Kern S, Klüter H, et al. 2007. Human AB serum and thrombin‐activated platelet‐rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 25:1270–1278. [DOI] [PubMed] [Google Scholar]
- 36. Gharaei R, Tronci G, Davies RPW, et al. 2016. A structurally self‐assembled peptide nano‐architecture by one‐step electrospinning. J Mater Chem B 4:5475–5485. [DOI] [PubMed] [Google Scholar]
- 37. Zini G. 2013. Stability of complete blood count parameters with storage: toward defined specifications for different diagnostic applications. Int J Lab Hematol 36:111–113. [DOI] [PubMed] [Google Scholar]
- 38. Dominici M, Le Blanc K, Mueller I, et al. 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317. [DOI] [PubMed] [Google Scholar]
- 39. Sanchez M, Fiz N, Guadilla J, et al. 2014. Intraosseous infiltration of platelet‐rich plasma for severe knee osteoarthritis. Arthrosc Tech 3:e713–e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dai WL, Zhou AG, Zhang H, et al. 2017. Efficacy of platelet‐rich plasma in the treatment of knee osteoarthritis: a meta‐analysis of randomized controlled trials. Arthroscopy 33:659–670.e1. [DOI] [PubMed] [Google Scholar]
- 41. Joo MW, Chung SJ, Shin SH, et al. 2017. The effect of autologous platelet‐Rich plasma on bone regeneration by autologous mesenchymal stem cells loaded onto allogeneic cancellous bone granules. Cells Tissues Organs 203:327–338. [DOI] [PubMed] [Google Scholar]
- 42. DeLong JM, Russell RP, Mazzocca AD. 2012. Platelet‐rich plasma: the PAW classification system. Arthroscopy 28:998–1009. [DOI] [PubMed] [Google Scholar]
- 43. Glass GE, Chan JK, Freidin A, et al. 2011. TNF‐α promotes fracture repair by augmenting the recruitment and differentiation of muscle‐derived stromal cells. Proc Natl Acad Sci USA 108:1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lacey DC, Simmons PJ, Graves SE, et al. 2009. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: implications for bone repair during inflammation. Osteoarthritis and Cartilage 17:735–742. [DOI] [PubMed] [Google Scholar]
- 45. Croes M, Oner FC, Kruyt MC, et al. 2015. Proinflammatory mediators enhance the osteogenesis of human mesenchymal stem cells after lineage commitment. PLoS ONE 10:e0132781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomchuck SL, Zwezdaryk KJ, Coffelt SB, et al. 2008. Toll‐like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells 26:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. El‐Zayadi AA, Jones EA, Churchman SM, et al. 2017. Interleukin‐22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: a novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology 56:488–493. [DOI] [PubMed] [Google Scholar]
- 48. Doucet C, Ernou I, Zhang Y, et al. 2005. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell‐based therapy applications. J Cell Physiol 205:228–236. [DOI] [PubMed] [Google Scholar]
- 49. Muraglia A, Todeschi MR, Papait A, et al. 2015. Combined platelet and plasma derivatives enhance proliferation of stem/progenitor cells maintaining their differentiation potential. Cytotherapy 17:1793–1806. [DOI] [PubMed] [Google Scholar]
- 50. Crespo‐Diaz R, Behfar A, Butler GW, et al. 2011. Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplantation 20:797–812. [DOI] [PubMed] [Google Scholar]
- 51. Cuthbert RJ, Churchman SM, Tan HB, et al. 2013. Induced periosteum a complex cellular scaffold for the treatment of large bone defects. Bone 57:484–492. [DOI] [PubMed] [Google Scholar]
- 52. Hakimi M, Jungbluth P, Sager M, et al. 2010. Combined use of platelet‐rich plasma and autologous bone grafts in the treatment of long bone defects in mini‐pigs. Injury 41:717–723. [DOI] [PubMed] [Google Scholar]
- 53. Kanthan SR, Kavitha G, Addi S, et al. 2011. Platelet‐rich plasma (PRP) enhances bone healing in non‐united critical‐sized defects: a preliminary study involving rabbit models. Injury 42:782–789. [DOI] [PubMed] [Google Scholar]
- 54. Block TJ, Marinkovic M, Tran ON, et al. 2017. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell‐based therapies. Stem Cell Res Ther. 8:239. [DOI] [PMC free article] [PubMed] [Google Scholar]