

Abstract
CDGSH iron–sulfur domain‐containing protein 2 (Cisd2), a protein that declines in an age‐dependent manner, mediates lifespan in mammals. Cisd2 deficiency causes accelerated aging and shortened lifespan, whereas persistent expression of Cisd2 promotes longevity in mice. Alzheimer's disease (AD) is the most prevalent form of senile dementia and is without an effective therapeutic strategy. We investigated whether Cisd2 upregulation is able to ameliorate amyloid β (Aβ) toxicity and prevent neuronal loss using an AD mouse model. Our study makes three major discoveries. First, using the AD mouse model (APP/PS1 double transgenic mice), the dosage of Cisd2 appears to modulate the severity of AD phenotypes. Cisd2 overexpression (∼two‐fold) significantly promoted survival and alleviated the pathological defects associated with AD. Conversely, Cisd2 deficiency accelerated AD pathogenesis. Secondly, Cisd2 overexpression protected against Aβ‐mediated mitochondrial damage and attenuated loss of neurons and neuronal progenitor cells. Finally, an increase in Cisd2 shifted the expression profiles of a panel of genes that are dysregulated by AD toward the patterns observed in wild‐type mice. These findings highlight Cisd2‐based therapies as a potential disease‐modifying strategy for AD. © 2019 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: Alzheimer's disease, Cisd2, mitochondria, track density imaging (TDI), transcriptomics
Introduction
With humans now having a longer lifespan, neurodegenerative disorders, cardiovascular diseases, and cancer become the most devastating illnesses currently affecting human populations. Alzheimer's disease (AD), a neurodegenerative disease and the most prevalent form of senile dementia, is associated with a deterioration in memory and a loss of cognitive function; it usually causes death in 3 to 9 years after diagnosis 1. AD is characterized by the presence of amyloid plaques, which mainly consist of amyloid β (Aβ) peptides, as well as the presence of neurofibrillary tangles that contain hyperphosphorylated tau protein. These features result in progressive neuronal cell death, a loss of synapses within the brain, memory impairment, and cognitive disturbances 2, 3. Loss of neurons in the entorhinal cortex, hippocampus, frontal cortex, parietal cortex, and temporal cortex of patients with AD has been documented 4. Previous reports have proposed various pathological mechanisms for AD, as well as various potential therapeutic targets that have been suggested for evaluation in clinical trials. However, the cause(s) of AD are not fully understood and there are no supplements or medications that have been shown to decrease the risk of AD.
Neurons as cells are highly energy‐dependent and rely on mitochondria to supply ATP for their many energy‐demanding neuronal functions, such as synaptic transmission, vesicle release, ion receptors, channel‐related signaling, and re‐uptake/recycling of neurotransmitters 5. Therefore, any defect that affects mitochondria‐related functions is likely to lead to neuronal death. Previous studies have revealed that oxidative stress occurs before Aβ plaque formation, which suggests that mitochondrial dysfunction and the accumulation of reactive oxygen species (ROS) play a pivotal role in AD pathogenesis 6, 7. Accumulation of Aβ in mitochondria leads to abnormal mitochondrial structure and increases the amounts of mitochondrial DNA, both of which are known to be compensatory responses to mitochondrial oxidative damage 8. Furthermore, Aβ has multiple adverse effects on the functioning and integrity of both presynaptic and postsynaptic terminals, including the induction of oxidative stress, impairment of calcium homeostasis, and perturbation of the functioning of the mitochondria and the endoplasmic reticulum (ER). These features mirror the primary alterations in synaptic/axonal functionality and the structural abnormalities present in patients with AD 9. Damage to the mitochondria often blocks the antioxidant mechanisms used to remove ROS and prevent oxidative stress 10; as a consequence, it is thought that the increase in oxidative stress induces tau phosphorylation and Aβ upregulation 11. Therefore, therapeutic agents that help to maintain mitochondrial integrity and functioning, as well as ones that help to eliminate Aβ‐induced ROS, should have neuroprotective properties against Aβ toxicity in the brains of patients with AD.
The major factor associated with the development of neurodegenerative diseases is aging. Our studies have revealed that CDGSH iron–sulfur domain‐containing protein 2 (Cisd2) plays a vital role in controlling mammalian lifespan 12, 13. The first molecular analysis of Cisd2 indicated that Cisd2 is one of the markers associated with early neuronal differentiation 14. Using Cisd2 knockout mice, we have demonstrated previously that Cisd2 deficiency shortened the lifespan of mice and this was accompanied by an accelerated aging phenotype. In addition, neuron and muscle degeneration were the primary defects observed using this mouse model 12. Conversely, a persistent expression of Cisd2 in mice extended their lifespan and prevents age‐dependent neuron and muscle degeneration. Furthermore, overexpression of Cisd2 appeared to protect mitochondria against age‐associated structural alterations and the functional decline associated with old age 13. Accordingly, it was of great interest to investigate with use of a mouse model whether an increase in Cisd2 expression was able to protect against the toxicity of Aβ and reduce the development of hippocampal neuronal damage, thereby attenuating AD pathogenesis by maintaining mitochondrial integrity and synaptic structure.
Materials and methods
Mouse models
The AD mouse model (JAX004462, kindly provided by Dr. Ding‐I Yang of National Yang Ming University) expresses a mutant chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1‐dE9), predominantly in the neurons of central nervous system (CNS); both transgenes are controlled by a mouse prion protein (MoPrP) promoter and are co‐integrated at the same chromosome location and thus co‐segregate 15. The AD mice were crossed with Cisd2 BAC transgenic mice to obtain AD mice carrying Cisd2 transgenes (AD;Cisd2TG). We used C57BL/6 as the genetic background for all mouse lines used as models. In addition, AD mice were also bred with Cisd2 knockout mice (KO) to generate AD mice with a Cisd2 deletion (AD;Cisd2KO). The genotypes of the mice were determined by PCR using tail genomic DNA. The mice were bred in a specific pathogen‐free facility. The animal protocol was approved by the Institutional Animal Care and Use Committee of the National Yang‐Ming University.
Cisd2 BAC transgenic mice
Cisd2 BAC transgenic mice, mouse Cisd2 genomic DNA obtained from a BAC clone (201 J1) was generated and reported previously 16. The Cisd2 BAC transgene was detected by Southern blot analysis using a probe designed on chloramphenicol resistance gene (CmR), specifically a 5.65 kb NcoI fragment. The expression levels of Cisd2 protein in line A301 and A302 were about two‐ to three‐fold higher than wild‐type (WT) mice.
Brain tissue sampling
Mice were anesthetized with an intraperitoneal injection of 2,2,2‐tribromoethanol in phosphate‐buffered saline (PBS) and perfused with ice‐cold 0.9% saline. Brains were then removed and divided in a sagittal manner. The left hemibrain was post‐fixed in phosphate‐buffered 4% paraformaldehyde and transferred to 30% sucrose to dehydrate the sample's cells. This hemisphere was used to produce frozen blocks using optimum cutting temperature (OCT) compound, whereas the right hemibrain was frozen and stored in liquid nitrogen for DNA, RNA, and protein extraction. To perform diffusion MRI analysis, mice were anesthetized and perfused, and then brains were removed and fixed in formalin (buffered, 10%). After MRI analysis, the whole brain was dehydrated and embedded in paraffin.
Full details of materials and methods for western blotting, transmission electron microscopy, immunofluorescence staining, thioflavin‐S amyloid labeling, Nissl staining (Cresyl violet staining), image analysis, oxygen consumption rate (OCR) measurement, diffusion MRI analysis, hippocampal RNA isolation, library preparation and RNA sequencing, and analysis of RNA sequencing are provided in supplementary material, Supplementary materials, and methods.
Statistics
Results are presented as means ± SD. Comparisons between two groups were carried out using Student's t‐test. Mouse survival rates were calculated using the Kaplan–Meier method, and the differences in the survival of the different groups of mice were determined using the log‐rank (Mantel–Cox) test. When analyzing statistical differences between different groups of mice, p < 0.05 was considered significant.
Results
Cisd2 upregulation in AD mice promotes their survival and protects against neuronal loss
To investigate whether Cisd2 overexpression can ameliorate the phenotypes associated with AD, we used a mouse AD model, namely APPswe and PS1‐dE9 double transgenic (APP/PS1) mice. This model shows accelerated plaque formation and logarithmic plaque deposition. This AD model is most useful when investigating the biochemical aspects of APP metabolism and the presence of intracellular damage compared to other AD models, which are most helpful when studying behavioral phenotypes 17, 18. We crossed the AD mice with our Cisd2 BAC transgenic mice 19, which carry two additional copies of the Cisd2 gene (see supplementary material, Figure S1A). We found that the level of Cisd2 protein was elevated by more than two‐fold in the hippocampus and cortex of our AD;Cisd2TG mice (AD mice carrying the Cisd2 transgene) compared with littermates carrying either the AD only or WT genotype (see supplementary material, Figure S1B). Our previous study revealed the mean lifespan of WT and Cisd2TG female mice to be 26.28 + 0.41 and 31.22 + 0.96 months, respectively 13. By 12 months of age, both the Cisd2TG and WT female mice have a 100% survival rate (Figure 1A). However, the AD female mice were found to have a significantly lower survival rate compared to WT controls. Intriguingly, Cisd2 overexpression significantly increased the survival rate of the AD;Cisd2TG female mice (Figure 1A). Less than 40% of the AD female mice survived to 4 months of age; however, on the other hand, more than 80% of the AD;Cisd2TG female mice survived to 4 months of age. By way of contrast, there was no overt effect of Cisd2 on the survival rate of AD male mice; this is probably because the survival rate of males from this AD model is not significantly decreased compared to WT males (see supplementary material, Figure S1C). Accordingly, from this point onward we focused mainly on the characterization of the female mice. Furthermore, our results showed that Cisd2 overexpression had no overt effect on the amyloid burden of these mice as revealed by western blotting (Figure 1B) and on thioflavin‐S staining of the extracellular Aβ plaques present in the cortex and hippocampus when the surviving AD and AD;Cisd2TG mice are compared at 12 months of age (Figure 1C,D).
Figure 1.
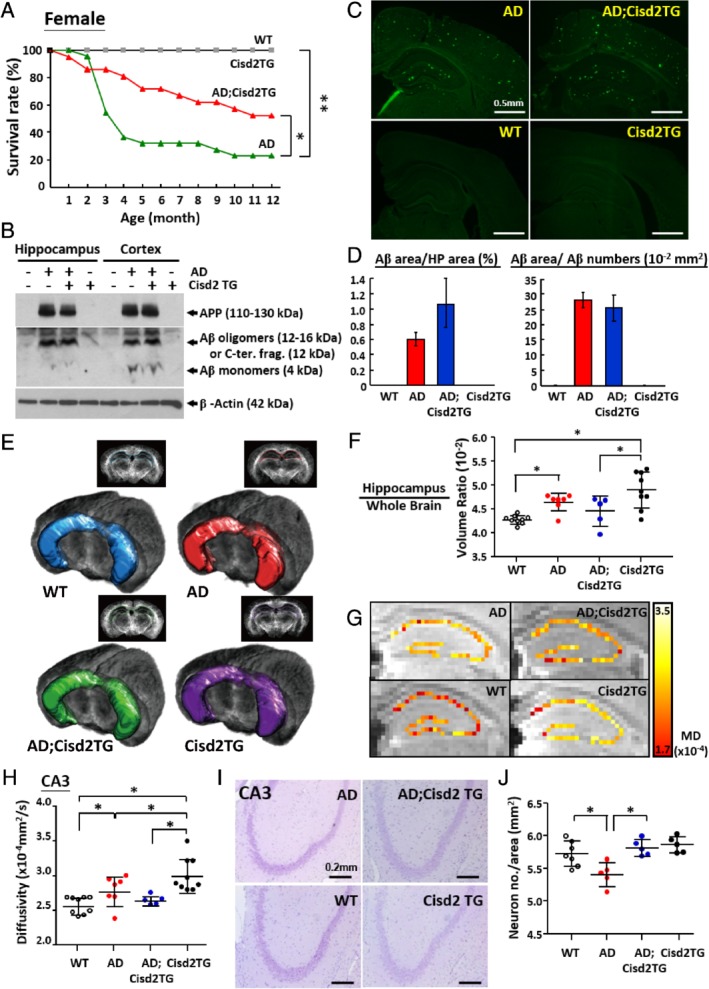
Overexpression of Cisd2 prevents premature death and reduces neuronal loss in female AD mice. (A) Premature lethality among female AD mice (APPswe and PS1‐dE9 double transgenic) can be recorded as early as 1 month of age, and >60% of female AD mice died by 4 months. Two‐fold overexpression of Cisd2 in female AD mice was able to partially rescue this premature mortality and increased their survival rate. The animal numbers of each group ranged from 12 to 36 mice. (B) Western blotting analysis revealed that AD and AD;Cisd2TG mice both exhibited similar levels of precursor APP and soluble Aβ species in their hippocampus and cortex. Total protein extracts from the hippocampus and cortex of 9‐month‐old mice were separated using 15% Tricine‐SDS PAGE and detected using 6E10 antibody. (C) Thioflavin‐S staining detects extracellular Aβ plaques in WT, AD, Cisd2TG, and AD;Cisd2TG female mice. (D) Quantification of Aβ plaque number in cortex and hippocampus. Total areas of Aβ were measured using MetaMorph by setting a threshold for fluorescent intensity, and then dividing by the counting area within the hippocampus. Total areas of Aβ were divided by the number of Aβ areas in order to measure the average size of the Aβ. No significant difference in the Aβ plaque burden and average size between AD and AD;Cisd2TG mice were found. (E) Target region of interest (ROI) definitions. The ROI shows the area of hippocampus and was rendered onto the mouse brain using 2D and 3D views for the WT, AD, Cisd2TG, and AD;Cisd2TG brains. (F) Ratio of hippocampus versus whole brain volume, which was measured using TDI images. (G) DTI in the hippocampus of the WT, AD, Cisd2TG, and AD;Cisd2TG mice. (H) Quantification of mean diffusivity in the cornu ammonis 3 (CA3). (I) Nissl staining of neurons in the hippocampus (CA3) of the WT, AD, Cisd2TG, and AD;Cisd2TG mice. (J) Calculation of neuron numbers in CA3. For (C–J) mice were 12‐months‐old. Mean ± SD. *p < 0.05, **p < 0.005.
Using MRI analysis, the results for track density imaging (TDI) showed that the ratio between hippocampus volume and total brain volume was significantly increased in AD female mice, whereas Cisd2 overexpression resulted in a slight decrease in this ratio (Figure 1E,F). Nevertheless, we were not able to detect any significant differences in the brain and hippocampus volumes among the various different groups (see supplementary material, Figure S1D,E). The diffusion parameters obtained from TDI revealed higher diffusivity in the CA3 region of hippocampus of the AD females, and, furthermore, Cisd2 overexpression appeared to reduce this diffusivity (Figure 1G,H); however, such a phenotypic effect was not detected in the CA1 and dentate gyrus (DG) regions of the AD;Cisd2TG mice (see supplementary material, Figure S1F,G). Higher diffusivity indicates that more neuronal cell loss has occurred in that brain region 20. Indeed, our pathological analysis revealed there was a significant decrease in neuron numbers in the CA3 region of hippocampus of the AD mice; intriguingly, Cisd2 overexpression protected against this neuronal loss and rescued this defect (Figure 1I,J; see supplementary material, Figure S1H). Immunofluorescence (IF) staining for neuronal markers further validated the neuroprotective effect of Cisd2. Results for NeuN (neuron‐specific nuclear protein) (Figure 2A,B) and MAP2 (a marker for neuronal cell body and dendrites) (see supplementary material, Figure S1I,J) staining showed neuronal loss in the hippocampus of AD females, and remarkably, Cisd2 overexpression appeared to significantly reduce neuronal loss and helped to maintain neuronal integrity. To understand the potential cause(s) of the increased hippocampus/brain ratio in AD mice, we analyzed the numbers of microglia and astrocytes by IF staining for the microglial marker, ionized calcium‐binding adapter molecule 1 (Iba1), and the astrocytic marker, glial fibrillary acidic protein (GFAP), respectively. The results revealed that Iba1 signals were significantly elevated in AD mice, whereas Cisd2TG and WT control mice had similarly (low) levels of Iba1 signals (Figure 2C,D; see supplementary material, Figure S1K,L). This suggests that the increase in hippocampus/brain ratio is likely to be attributable, at least in part, to an increase in the number of glial cells present. Collectively, these results indicated that Cisd2 was able to protect the hippocampus from Aβ‐induced neuronal loss, to attenuate microglia expansion, and to reduce the level of inflammation in AD mice.
Figure 2.
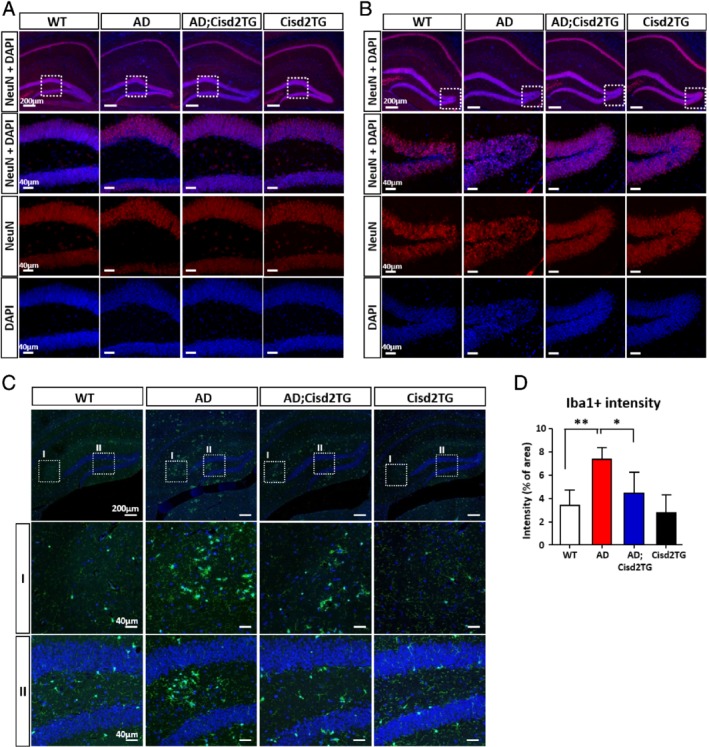
Overexpression of Cisd2 alleviates neuronal loss and inflammation in female AD mice. (A) (B) IF staining for NeuN (red) reveals a reduced number of neurons in the granule cell layer (A) and hilus (B) of the dentate gyrus (DG) in the hippocampus of AD mice. (C) IF staining for Iba1, a microglia marker, in the hippocampus of the WT, AD, Cisd2TG, and AD;Cisd2TG mice. (D) Density quantification of Iba1+ cells obtained from three sections per mouse and three mice for each group. All mice were 12‐months‐old. Mean ± SD. *p < 0.05, **p < 0.005.
Cisd2 deficiency accelerates Aβ‐mediated pathogenesis in the AD brain
Cisd2 levels decrease in an age‐dependent manner in the brains of mammals during natural aging 16. Because ‘aging’ is the major risk factor for AD, it is important to investigate whether Cisd2 deficiency accelerates AD pathogenesis and exacerbates the AD phenotype. To this end, we generated AD mice carrying a Cisd2KO background (AD;Cisd2KO) (see supplementary material, Figure S2A). At 2 months of age, although amyloid deposition was not yet detectable in the AD brain (see supplementary material, Figure S2B), the 2D and 3D views by TDI already revealed an enlarged brain volume in the AD mice; in particular, the hippocampus/brain ratio is significantly increased in the AD;Cisd2KO mice (Figure 3A–3C). No obvious difference in hippocampus diffusivity was observed among various groups (see supplementary material, Figure S2C–F). However, a pathological analysis revealed that neuron numbers in the CA3 region were significantly decreased in the Cisd2KO and AD;Cisd2KO mice (Figure 3D,E), and that more severe neuronal damage was observed in the AD;Cisd2KO mice compared to the AD and Cisd2KO mice, as revealed by staining of neuronal markers Map2 and NeuN, respectively (Figure 3E). Furthermore, the numbers of microglia (Iba1 positive) were significantly increased in the AD and Cisd2KO mice, and this was even more pronounced in the AD;Cisd2KO mice (Figure 3G,H). The numbers of astrocytes (GFAP positive) also seems to have increased in AD, Cisd2KO, and AD;Cisd2KO mice compared to WT mice (see supplementary material, Figure S2G,H). Together, these results reveal that Cisd2 deficiency not only damages neurons but also accelerate AD pathogenesis in mice.
Figure 3.
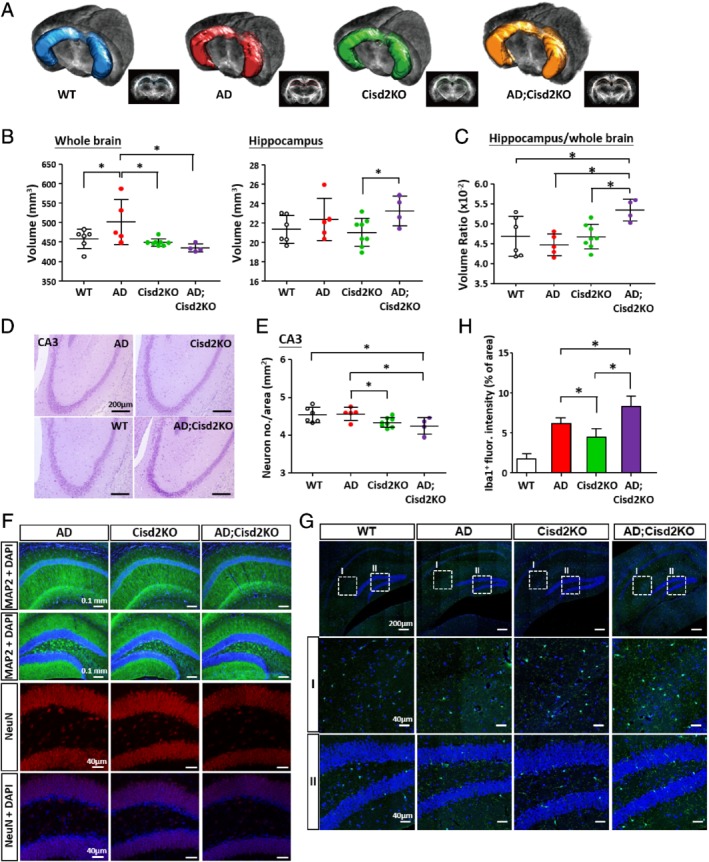
Cisd2 deficiency accelerates AD pathogenesis in mice. (A) Target region of interest (ROI) definitions. The ROI shows the area of hippocampus and was rendered on the mouse brain using 2D and 3D views. (B) Measurement of the volume of whole brain and hippocampus for the WT, AD, Cisd2KO, and AD;Cisd2KO female mice. The results were obtained from TDI images. (C) Volume ratio of hippocampus versus whole brain measured from TDI images. (D) Nissl staining of neurons in the CA3 of the hippocampus of WT, AD, Cisd2KO, AD;Cisd2KO female mice. (E) Quantification of neuron numbers in CA3. (F) Reduced immunoreactivity for MAP2 (green) in the CA1 and DG of the hippocampus was observed in the AD;Cisd2KO mice. Reduced immunoreactivity for NeuN (red) in the granule cell layer of DG of the hippocampus was also detected in the AD;Cisd2KO mice compared with the AD and Cisd2KO mice. (G) IF staining for Iba1, a microglia marker, in the hippocampus of the WT, AD, Cisd2KO, and AD;Cisd2KO mice. (H) Density quantification of Iba1+ cells. The results were obtained from three brain sections per mouse and three mice for each group. Mice were 2‐months‐old. Mean ± SD. *p < 0.05.
Cisd2 protects mitochondria and attenuates the loss of neuronal progenitor cells
To understand how Cisd2 overexpression ameliorates AD pathogenesis, we analyzed the neuronal progenitor cell population in the hippocampus. Notably, there were fewer proliferative cells (Ki‐67+ cells) and fewer neuronal progenitor cells (Dcx + or Tbr2+ cells) in the subgranular zone of the AD hippocampus, and Cisd2 overexpression seems to partially rescue this defect as well as significantly increase the number of neuronal progenitor cells present (Figure 4A,B). Ultrastructural examination of the hippocampus showed that the total number of synapses has no obvious difference among different groups, but Cisd2 did increase the relative ratio of excitatory versus inhibitory synapses in the AD;Cisd2TG mice (Figure 4C; see supplementary material, Figure S3A–E). The increase of the excitatory synapses suggests that there may be more neurons present that can generate new action potentials and transmit the information to another cell, thus helping to restore neuronal functionality 21. In addition, we monitored mitochondrial function and found that the OCR is significantly decreased in the AD mice, indicating a functional decline of mitochondria in terms of oxidative phosphorylation and the generation of ATP. Remarkably, Cisd2 overexpression rescues this mitochondrial defect in energy production (Figure 4D). In the hippocampus of AD mice, mitochondrial breakdown accompanied by autophagosome and autolysosome activity is a distinctive pathological feature; strikingly, in the AD;Cisd2TG mice, Cisd2 overexpression seems to revert these morphological abnormalities and move these mice toward a more normal state (Figure 4E). These results reveal that Cisd2 overexpression in AD mice can protect mitochondria from the Aβ‐induced structural defects and functional damage that affect ATP production, as well as attenuate the progressive loss of the neuronal progenitor cells.
Figure 4.
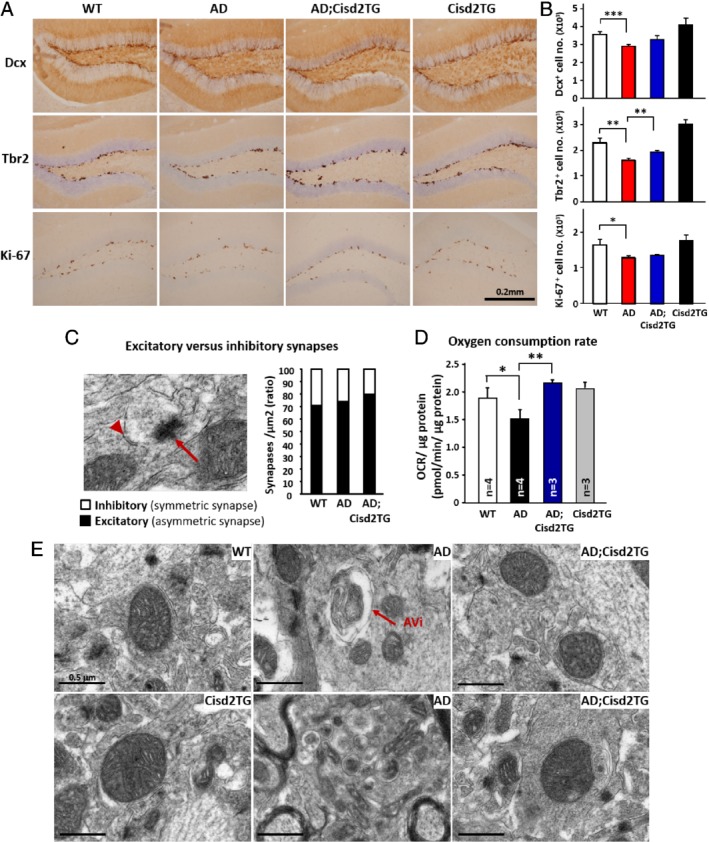
Elevated expression of Cisd2 attenuates the loss of neuronal progenitor cells and protects against Aβ‐induced mitochondrial defects. (A) Immunohistochemical staining of brain sections to label Doublecortin (Dcx), T‐box brain protein 2 (Tbr2), and antigen Ki‐67 (Ki67) in order to detect neuronal progenitor cells. (B) Quantification of cells positive for Dcx, Tbr2, and Ki‐67 in the DG region. (C) Excitatory (asymmetric) and inhibitory (symmetric) synapses revealed by TEM. Arrow indicates asymmetric synapses and arrowhead indicates symmetric synapses. Quantification results for the ratio of excitatory synapses to inhibitory synapses. (D) OCR measured using Seahorse equipment and hippocampus tissue samples obtained from the various groups of mice. (E) Ultrastructure of mitochondria in the hippocampus of the WT, AD, and AD;Cisd2 TG female mice. The mice used to create this figure were 12‐months‐old. Mean ± SD. *p < 0.05; **p < 0.005.
Cisd2 shifts the expression patterns of dysregulated genes in AD toward WT mice
To understand the molecular mechanism underlying the neuroprotective effect of Cisd2 in AD, we performed RNA sequencing using total RNA obtained from the hippocampus of WT, AD, and AD;Cisd2TG mice. Three sets of pair‐wise differential expression analysis were performed: these were between WT and AD mice (Set‐1), between AD;Cisd2TG and AD mice (Set‐2), and between WT and AD;Cisd2TG mice (Set‐3). A false discovery rate (FDR) less than 0.05 was set as significance threshold value. From these three sets of differentially expressed genes (DEGs), we select genes that were differentially expressed in Set‐1, that is, they showed a significant change in expression due to the AD genotype and that, in Set‐2, showed a significant change in expression due to Cisd2 overexpression, but were not differentially expressed in Set‐3. Thus their expression levels were not significantly different from their values in WT. A total of 154 genes were identified using these selection criteria (see supplementary material, Table S1). These findings indicate that Cisd2 overexpression may be able to reverse the expression pattern of a panel of dysregulated genes in the hippocampi of AD mice and move the expression pattern back toward one that is similar to that of WT mice (Figure 5; a high resolution figure with gene symbols is provided in supplementary material, Figure S4).
Figure 5.
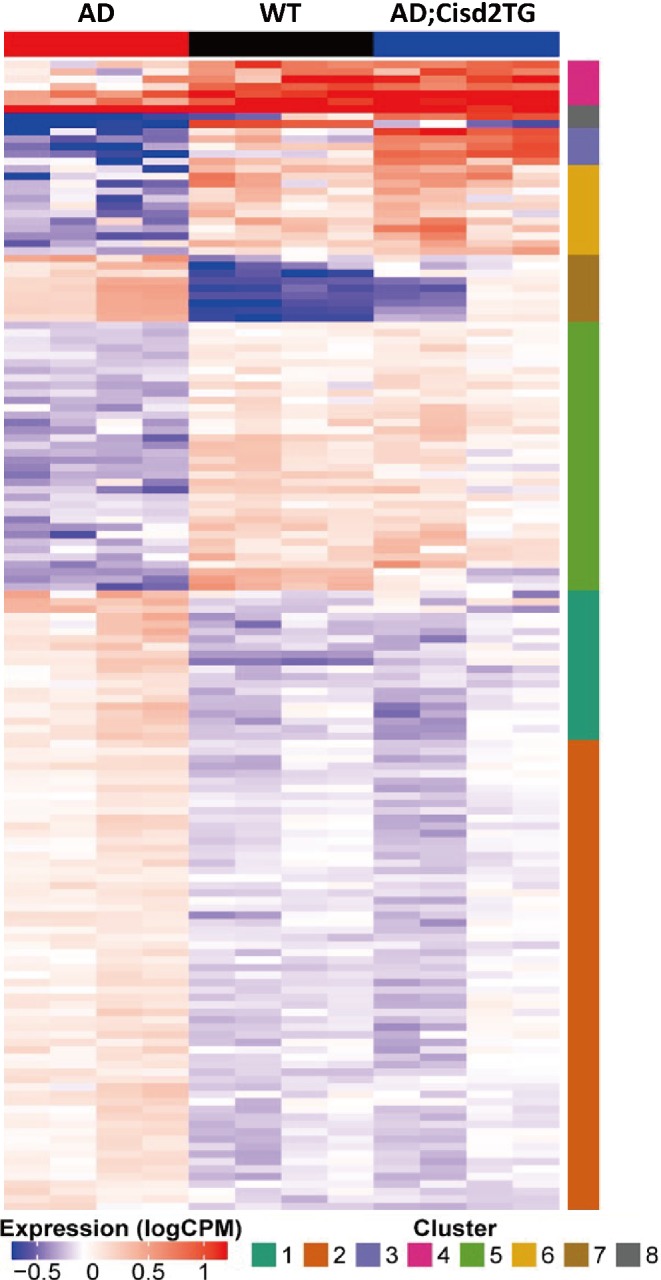
Cisd2 reverses the expression pattern of a panel of differentially expressed genes in the hippocampus of AD mice. The genes that showed significant difference in expression between the ‘WT and AD’, mice and between the AD;Cisd2TG and AD” mice, as well as having the same trend of expression changes were selected (FDR <0.05). A total of 154 genes were selected that showed high dissimilarity according to these criteria. The heat map reveals that Cisd2 overexpression in the AD;Cisd2TG mice shifts the expression patterns of the 154 genes toward the WT mice. Consensus clustering was performed, and this resulted in eight gene clusters. The intensities of gene expression are depicted in colors that range from blue (log2 count‐per‐million −1 and lower) to red (log2 count‐per‐million 1 and higher). A more detailed version of this figure listing the genes forming the clusters is provided as supplementary material, Figure S4.
The 154 DEGs can be classified into various different functional categories, namely, synapse‐related functions, mitochondrial functions, ion homeostasis, protein or organelle degradation and cell death, cellular and extracellular structure maintenance, cell cycle, transcriptional regulators, enzymes (except those found in mitochondria), and various others (see supplementary material, Table S1). In addition, when we compared our transcriptomic data with the GeneCards database, the expression levels of four AD‐associated genes –namely, aquaporin 1 (Aqp1), alpha‐2 macroglobulin (A2m), angiotensin‐converting enzyme (Ace), and transthyretin (Ttr) – were dramatically increased by Cisd2 overexpression in the AD;Cisd2TG mice. However, these four genes are not downregulated in AD mice (see supplementary material, Figure S5). Of interest, several of the selected DEGs are known to be associated with mitochondrial metabolism and synaptic neurotransmission (Figure 6). For example, Ras‐related protein Rab‐3C (Rab3c) and syntaxin 1B (Stx1b) are necessary for the regulation of synaptic vesicle transport and exocytosis 22, 23. Calcium voltage‐gated channel auxiliary subunit alpha2delta 1 (Cacna2d1), a voltage‐dependent calcium channel, is critical to excitation‐contraction coupling on both presynaptic and postsynaptic neuronal membranes 24. Glutamate decarboxylase 65 (Gad65), encoded by Gad2, undergoes activity‐dependent expression in axon terminals and is responsible for the synthesis and vesicle release of γ‐aminobutyric acid (GABA), an inhibitory neurotransmitter 25. Finally, glutamate receptor 1 (Gria1) is the prevailing excitatory neurotransmitter receptor and is involved mainly in synaptic plasticity 26. The GABA receptor (Gabra) is located post‐synapse and interacts with GABA to deliver the inhibitory signals 27. Thus, in summary, our transcriptomic analysis has revealed that Cisd2 overexpression seems to be able to reverse and rescue the expression patterns of a panel of genes dysregulated by AD, including genes that are crucial to the functioning of both mitochondria and synapses.
Figure 6.
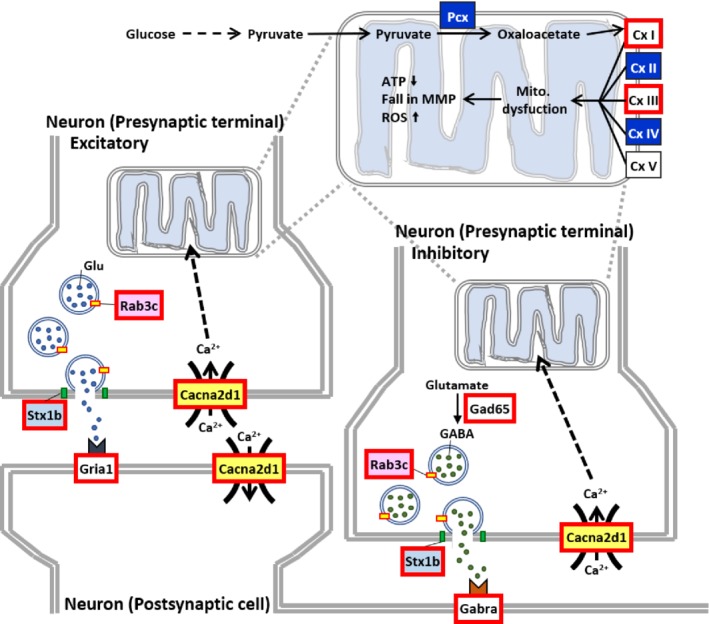
An overview of the possible effects of the genes dysregulated during AD that are can be reversed by Cisd2 overexpression and are involved in mitochondrial respiration and signal transduction during synaptic neurotransmission. Synapses demand a high energy supply from the mitochondria together with well‐controlled calcium homeostasis in order to permit the correct signal transduction from one neuron to another. In the hippocampus of AD mice, transcriptomic analysis revealed dysregulation of complex (Cx) genes (upregulation of Cx I and III and downregulation of Cx II and IV), which are essential components of electron transport chain that produces ATP. In addition, genes involved in the synapse neurotransmission are over‐activated (marked in red at the synapses); this might be a compensatory effect in response to neurodegeneration. It is notable that Cisd2 overexpression can reverse the abnormal expression pattern of these dysregulated genes and return them to a level similar to that found in WT mice.
Discussion
Implication of the findings
There are several new findings in this study. First, using this APP/PS1 mouse model, the dosage of Cisd2 present in the mice modulates the severity of the AD phenotypes. Second, Cisd2 protects against mitochondrial damage, protects against mitochondrial functional decline, and attenuates the loss of neurons and neuronal progenitor cells that occurs in the hippocampus of AD mice. Finally, an increase in Cisd2 reverses the abnormal expression pattern of a panel of genes that are dysregulated in AD; these are returned to an expression pattern similar to that of WT mice. The genes modulated by increased Cisd2 expression include ones that are involved in mitochondrial functioning within the presynaptic terminal of axons, in the release of neurotransmitters from synaptic vehicles, and in the passing of signals via receptor binding during neurotransmission. The above‐mentioned findings suggest that with higher Cisd2 expression there is a recovery of neuronal function and an attenuation of neuronal loss. These changes will ultimately ameliorate Aβ‐induced pathogenesis in the hippocampus of the AD mice. Alternatively, it is also possible that this shift in gene expression might be a secondary consequence of the neuroprotective effects of Cisd2, whereby it protects mitochondria from damage and thus maintains synaptic function.
Cisd2 upregulation attenuates AD progression
Neuroinflammation and the size of the hippocampus in the AD brain
Previous reports have shown that brain size, especially the size of hippocampus, decreased gradually as the disease progresses in AD patients and that this is also true for certain AD mouse models 28. However, in some of AD mouse models, the volume of the hippocampus and whole brain show no obvious differences 29, 30. Of interest, our TDI images showed that the ratio of the hippocampus versus whole brain was significantly increased in 12‐month‐old female AD mice. However, enhanced Cisd2 expression seemed to be able to rescue this phenotype and reduce the hippocampal volume to a size like that of the WT controls. Neuronal loss accompanied by neuroinflammation is a pathological hallmark of AD in human patients and mouse models. When amyloid oligomers begin to form in cells and extracellular spaces of the brain, the misfolded and aggregated proteins are recognized by microglia and astrocytes; this then triggers further immune responses, resulting in a vicious cycle 31, 32. Activated microglia are able to clean up debris and misfolded proteins; however, activated microglia are also harmful to neurons and participate in amyloid‐dependent synapse loss 33. In the present study, our results revealed that the hippocampus is enlarged in the AD mice and that this is accompanied by neuronal loss and severe neuroinflammation, as well as proliferation of microglia. One possible explanation for the enlarged hippocampus may be the increased number of microglia accumulating in the hippocampus of AD mice. Intriguingly, Cisd2 overexpression seems to attenuate neuronal loss and decrease the number of activated microglia, thereby reducing the size of the hippocampus.
Synaptic neurotransmission and mitochondrial function
Among human patients with neurodegenerative diseases, synapse loss is a critical feature during the early stages of the disease process 34. Substantial evidence indicates that, in AD, there is a decrease in the number of synapses, and that this occurs later than Aβ accumulation and is correlated with disease progression 35, 36. Dysregulation of several presynaptic and postsynaptic pathways may contribute to AD pathogenesis. Most interestingly, microglia play a direct role in neurodegeneration, especially of the synapse; this seems to occur via a modulation of the phagocytotic process 37. In our study, Cisd2 overexpression reversed the expression of several crucial genes that are known to be involved in synaptic neurotransmission (Figure 6); consequently, these changes may further improve the functioning of the synapse in the hippocampus of AD mice. In addition, previous studies have shown that an altered mitochondrial structure can lead to cerebral hypometabolism in AD‐affected brain regions 38, 39. Morphometric analysis in such circumstances has revealed that AD brain has significantly fewer mitochondria, and this is accompanied by increased ROS accumulation 38. Mitochondrial enzyme abnormalities underlie most of enzyme deficits affecting the AD brain, for example, cytochrome oxidase, the α‐ketoglutarate dehydrogenase complex, and the pyruvate dehydrogenase complex 40, 41. Numerous studies have demonstrated that increased Aβ accumulation contributes to structural and functional mitochondrial abnormalities, including effects on membrane potential, fusion/fission, transport, ROS production, and electron transfer chain 42. Intriguingly, elevated levels of Cisd2 protein appear to protect against mitochondrial breakdown, as revealed by ultrastructural examination using transmission electron microscopy (TEM), and attenuate Aβ‐mediated toxicity that affects mitochondrial function, as revealed by measuring the OCR of the electron transport chain.
Cisd2 overexpression leads to upregulation of four genes that have beneficial effects in relation to AD
Expression levels of the four genes, namely, Aqp1, A2m, Ace and Ttr, were found to be significantly increased in the hippocampus of AD mice when there was Cisd2 overexpression, although expression of these genes showed no significant difference when the AD and WT mice were compared. Aqp1 encodes an astrocytic water channel that can facilitate the maintenance of water homeostasis. Because Aβ accumulation interferes with the functioning of aquaporin 1, causing abnormalities in water homeostasis 43, an increased Aqp1 expression may have a beneficial effect on astrocytes at the site of Aβ depositions, which in turn may help them to degrade Aβ 44. Acute‐phase protein A2m, which functions as a pan‐protease inhibitor, is an important component of the innate immune system. In addition, A2m is able to bind to misfolded aggregation‐prone proteins 45. ACE had been reported to be able to facilitate the degradation of Aβ, to be able to enhance immune responses and to help to prevent cognitive decline in AD models 46. Finally, Ttr, which is an Aβ‐binding protein, has been reported to have proteolytic activity and can act as a neuroprotector in AD models 47. The elevated expression of these four genes in the AD;Cisd2TG mice is likely to help to protect against Aβ‐mediated damage and thus attenuate disease severity.
Diffusivity imaging may facilitate the monitoring of AD pathogenesis within the brain
TDI imaging allows high level resolution images to be captured and provides high anatomical contrast using a new MRI contrast mechanism. Previously, Calamante et al. have demonstrated that the TDI technique can provide meaningful and rich anatomical contrast results at a level similar to the information obtained using histological staining in the same brains 48. TDI combined with diffusion tensor imaging (DTI) has been used to characterize microstructural changes and differences in neuropathology 49. In this study, we substituted traditional structural imaging with TDI; our results revealed that TDI can indeed provide an improved spatial resolution and enhanced contrast between the grey matter and white matter. In addition, TDI can contour the hippocampal volume more precisely, and the diffusivity index of the TDI seems to represent the microstructure of neuronal tissue in the brain. Therefore, TDI should be able to offer, in the future, a noninvasive means for monitoring pathogenic progression in human patients with AD.
Finally, we have, by integrating different technology platforms across multiple disciplines, including brain imaging, mouse genetics, molecular pathology, and transcriptomics analysis, provided important evidence showing that Cisd2 overexpression is an effective way of attenuating Aβ‐mediated neuronal cell death and neuro‐inflammation; thus Cisd2 overexpression would seem to slow the progression of AD. Finally, we have provided evidence in this study to indicate that Cisd2‐based therapies hold great promise to serve as disease‐modifying strategies for AD.
Author contributions statement
YFC designed experiments, analyzed and interpreted results, and prepared the manuscript draft. TYC generated and characterized the mouse models at the beginning. IHL analyzed transcriptomic data, performed functional profiling, and drafted a portion of the manuscript. CGC and CPL designed, performed, and analyzed the diffuse MRI brain images. CHK designed and performed the TEM study. GJH analyzed the stem cell population and proliferation in brain. LKC and PNW participated in the experimental design for analyzing the AD phenotypes. TFT designed experiments, analyzed and interpreted results, and wrote the final manuscript. All authors read and approved the final version of this manuscript.
Data availability statement
The processed expression values and differential expression analysis results of 154 genes, which were mentioned in the manuscript, are provided in supplementary material, Table S2.
Supporting information
Supplementary materials and methods
Figure S1. Generation of Alzheimer's disease (AD) mice carrying two additional copies of the Cisd2 transgene and overexpressing Cisd2 in the brain
Figure S2. Generation and characterization of Alzheimer's disease (AD) mice carrying Cisd2KO background
Figure S3. Ultrastructural analysis of mitochondria in the hippocampus of Alzheimer's disease (AD) mice
Figure S4. The complete list of gene symbols in expression clusters of differentially expressed genes in the hippocampus of mice
Figure S5. Heatmap of genes associated with Alzheimer's disease
Table S1. The expression levels of genes, whose mRNA expression has an obvious change in Alzheimer's disease (AD) mice compared with wild‐type (WT) mice and their abnormal expression can be reversed by Cisd2 overexpression
Table S2. Processed expression values and differential expression analysis results of the 154 genes
Acknowledgements
We thank Dr. Ding‐I Yang and Dr. Irene H. Cheng (Institute of Brain Science, National Yang‐Ming University), Chia‐Sheng Chang, Yao‐Kuan Huang, and Hsin‐Yuan Chen for their technical assistance. We also thank the Clinical and Industrial Genomic Application Development Service Center of National Core Facility for Biopharmaceuticals, Taiwan (MOST 107‐2319‐B‐010‐002) for the RNA sequencing. We acknowledge support from the Ministry of Science and Technology (MOST 108‐2321‐B‐010‐013‐MY2 and MOST 107‐2634‐F‐010‐001 to TFT; MOST 108‐2321‐B‐010‐010‐MY2, MOST 108‐2634‐F‐010‐001 and MOST 108‐2321‐B‐010‐013‐MY2 to CPL;MOST105‐2320‐B‐038‐022‐MY3 to YFC) and support from the National Health Research Institutes (MG‐106‐PP‐17, MG‐107‐PP‐17 and MG‐108‐PP‐14).
No conflicts of interest were declared.
Contributor Information
Ching‐Po Lin, Email: cplin@ym.edu.tw.
Ting‐Fen Tsai, Email: tftsai@ym.edu.tw.
References
*Cited only in supplementary material.
- 1. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010; 362: 329–344. [DOI] [PubMed] [Google Scholar]
- 2. Vassar R, Bennett BD, Babu‐Khan S, et al Beta‐secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286: 735–741. [DOI] [PubMed] [Google Scholar]
- 3. Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 2005; 120: 545–555. [DOI] [PubMed] [Google Scholar]
- 4. West MJ, Coleman PD, Flood DG, et al Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 1994; 344: 769–772. [DOI] [PubMed] [Google Scholar]
- 5. Knott AB, Bossy‐Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci 2008; 1147: 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nunomura A, Perry G, Aliev G, et al Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001; 60: 759–767. [DOI] [PubMed] [Google Scholar]
- 7. Mattson MP. Pathways towards and away from Alzheimer's disease. Nature 2004; 430: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diana A, Simic G, Sinforiani E, et al Mitochondria morphology and DNA content upon sublethal exposure to beta‐amyloid(1–42) peptide. Coll Antropol 2008; 32(Suppl 1): 51–58. [PubMed] [Google Scholar]
- 9. Correia SC, Perry G, Moreira PI. Mitochondrial traffic jams in Alzheimer's disease ‐ pinpointing the roadblocks. Biochim Biophys Acta 2016; 1862: 1909–1917. [DOI] [PubMed] [Google Scholar]
- 10. Picone P, Nuzzo D, Caruana L, et al Mitochondrial dysfunction: different routes to Alzheimer's disease therapy. Oxid Med Cell Longev 2014; 2014: 780179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tamagno E, Parola M, Bardini P, et al Beta‐site APP cleaving enzyme up‐regulation induced by 4‐hydroxynonenal is mediated by stress‐activated protein kinases pathways. J Neurochem 2005; 92: 628–636. [DOI] [PubMed] [Google Scholar]
- 12. Chen YF, Kao CH, Chen YT, et al Cisd2 deficiency drives premature aging and causes mitochondria‐mediated defects in mice. Genes Dev 2009; 23: 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu CY, Chen YF, Wang CH, et al A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum Mol Genet 2012; 21: 3956–3968. [DOI] [PubMed] [Google Scholar]
- 14. Boucquey M, De Plaen E, Locker M, et al Noxp20 and Noxp70, two new markers of early neuronal differentiation, detected in teratocarcinoma‐derived neuroectodermic precursor cells. J Neurochem 2006; 99: 657–669. [DOI] [PubMed] [Google Scholar]
- 15. Jankowsky JL, Fadale DJ, Anderson J, et al Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet 2004; 13: 159–170. [DOI] [PubMed] [Google Scholar]
- 16. Chen YF, Wu CY, Kirby R, et al A role for the CISD2 gene in lifespan control and human disease. Ann N Y Acad Sci 2010; 1201: 58–64. [DOI] [PubMed] [Google Scholar]
- 17. Lee JE, Han PL. An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp Neurobiol 2013; 22: 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Webster SJ, Bachstetter AD, Van Eldik LJ. Comprehensive behavioral characterization of an APP/PS‐1 double knock‐in mouse model of Alzheimer's disease. Alzheimers Res Ther 2013; 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen ZQ, Chen YF, Chen JR, et al CISD2 haploinsufficiency disrupts calcium homeostasis, causes nonalcoholic fatty liver disease, and promotes hepatocellular carcinoma. Cell Rep 2017; 21: 2198–2211. [DOI] [PubMed] [Google Scholar]
- 20. Sykova E, Vorisek I, Antonova T, et al Changes in extracellular space size and geometry in APP23 transgenic mice: a model of Alzheimer's disease. Proc Natl Acad Sci U S A 2005; 102: 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 2007; 76: 823–847. [DOI] [PubMed] [Google Scholar]
- 22. Mishima T, Fujiwara T, Sanada M, et al Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PLoS One 2014; 9: e90004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Binotti B, Jahn R, Chua JJ. Functions of Rab proteins at presynaptic sites. Cell 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geisler S, Schopf CL, Obermair GJ. Emerging evidence for specific neuronal functions of auxiliary calcium channel alpha(2)delta subunits. Gen Physiol Biophys 2015; 34: 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Betley JN, Wright CV, Kawaguchi Y, et al Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell 2009; 139: 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu W, Bushong EA, Shih TP, et al The cell‐autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron 2013; 78: 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gamlin CR, Yu WQ, Wong ROL, et al Assembly and maintenance of GABAergic and Glycinergic circuits in the mammalian nervous system. Neural Dev 2018; 13: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Convit A, De Leon MJ, Tarshish C, et al Specific hippocampal volume reductions in individuals at risk for Alzheimer's disease. Neurobiol Aging 1997; 18: 131–138. [DOI] [PubMed] [Google Scholar]
- 29. Delatour B, Guegan M, Volk A, et al In vivo MRI and histological evaluation of brain atrophy in APP/PS1 transgenic mice. Neurobiol Aging 2006; 27: 835–847. [DOI] [PubMed] [Google Scholar]
- 30. Oberg J, Spenger C, Wang FH, et al Age related changes in brain metabolites observed by 1H MRS in APP/PS1 mice. Neurobiol Aging 2008; 29: 1423–1433. [DOI] [PubMed] [Google Scholar]
- 31. Rozemuller AJ, van Gool WA, Eikelenboom P. The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer's disease: therapeutic implications. Curr Drug Targets CNS Neurol Disord 2005; 4: 223–233. [DOI] [PubMed] [Google Scholar]
- 32. Heneka MT, Carson MJ, El Khoury J, et al Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015; 14: 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol 2018; 217: 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scheff SW, Price DA, Schmitt FA, et al Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 2006; 27: 1372–1384. [DOI] [PubMed] [Google Scholar]
- 35. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002; 298: 789–791. [DOI] [PubMed] [Google Scholar]
- 36. Shankar GM, Walsh DM. Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener 2009; 4: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rajendran L, Paolicelli RC. Microglia‐mediated synapse loss in Alzheimer's disease. J Neurosci 2018; 38: 2911–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirai K, Aliev G, Nunomura A, et al Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 2001; 21: 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moreira PI, Siedlak SL, Wang X, et al Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007; 3: 614–615. [DOI] [PubMed] [Google Scholar]
- 40. Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm (Vienna) 1998; 105: 855–870. [DOI] [PubMed] [Google Scholar]
- 41. Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging 2000; 21: 455–462. [DOI] [PubMed] [Google Scholar]
- 42. Calkins MJ, Manczak M, Mao P, et al Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet 2011; 20: 4515–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Misawa T, Arima K, Mizusawa H, et al Close association of water channel AQP1 with amyloid‐beta deposition in Alzheimer disease brains. Acta Neuropathol 2008; 116: 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoshi A, Yamamoto T, Shimizu K, et al Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. J Neuropathol Exp Neurol 2012; 71: 750–759. [DOI] [PubMed] [Google Scholar]
- 45. Varma VR, Varma S, An Y, et al Alpha‐2 macroglobulin in Alzheimer's disease: a marker of neuronal injury through the RCAN1 pathway. Mol Psychiatry 2017; 22: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bernstein KE, Koronyo Y, Salumbides BC, et al Angiotensin‐converting enzyme overexpression in myelomonocytes prevents Alzheimer's‐like cognitive decline. J Clin Invest 2014; 124: 1000–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Buxbaum JN, Ye Z, Reixach N, et al Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A 2008; 105: 2681–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Calamante F, Tournier JD, Kurniawan ND, et al Super‐resolution track‐density imaging studies of mouse brain: comparison to histology. Neuroimage 2012; 59: 286–296. [DOI] [PubMed] [Google Scholar]
- 49. Alexander AL, Lee JE, Lazar M, et al Diffusion tensor imaging of the brain. Neurotherapeutics 2007; 4: 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kao CH, Chen JK, Kuo JS, et al Visualization of the transport pathways of low density lipoproteins across the endothelial cells in the branched regions of rat arteries. Atherosclerosis 1995; 116: 27–41. [DOI] [PubMed] [Google Scholar]
- 51. Wolfe LS, Calabrese MF, Nath A, et al Protein‐induced photophysical changes to the amyloid indicator dye thioflavin T. Proc Natl Acad Sci USA 2010; 107: 16863–16868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tournier JD, Calamante F, Connelly A. Robust determination of the fibre orientation distribution in diffusion MRI: non‐negativity constrained super‐resolved spherical deconvolution. Neuroimage 2007; 35: 1459–1472. [DOI] [PubMed] [Google Scholar]
- 53. Tournier JD, Calamante F, Connelly A. Improved probabilistic streamlines tractography by 2nd order integration over fibre orientation distributions. Proc. 18th Annual Meeting of the Intl. Soc. Mag. Reson. Med. (ISMRM) 2010; 18: 1670. [Google Scholar]
- 54. Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual (3rd edn). Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Generation of Alzheimer's disease (AD) mice carrying two additional copies of the Cisd2 transgene and overexpressing Cisd2 in the brain
Figure S2. Generation and characterization of Alzheimer's disease (AD) mice carrying Cisd2KO background
Figure S3. Ultrastructural analysis of mitochondria in the hippocampus of Alzheimer's disease (AD) mice
Figure S4. The complete list of gene symbols in expression clusters of differentially expressed genes in the hippocampus of mice
Figure S5. Heatmap of genes associated with Alzheimer's disease
Table S1. The expression levels of genes, whose mRNA expression has an obvious change in Alzheimer's disease (AD) mice compared with wild‐type (WT) mice and their abnormal expression can be reversed by Cisd2 overexpression
Table S2. Processed expression values and differential expression analysis results of the 154 genes
Data Availability Statement
The processed expression values and differential expression analysis results of 154 genes, which were mentioned in the manuscript, are provided in supplementary material, Table S2.