

Abstract
Trained immunity is a process in which innate immune cells undergo functional reprogramming in response to pathogens or damage‐associated molecules leading to an enhanced non‐specific immune response to subsequent stimulation. While this capacity to respond more strongly to stimuli is beneficial for host defense, in some circumstances it can lead to maladaptive programming and chronic inflammation. Gout is characterized by persistent low‐grade inflammation and is associated with an increased number of comorbidities. Hyperuricemia is the main risk factor for gout and is linked to the development of comorbidities. Several experimental studies have shown that urate can mechanistically alter the inflammatory capacity of myeloid cells, while observational studies have indicated an association of hyperuricemia to a wide spectrum of common adult inflammatory diseases. In this review, we argue that hyperuricemia is a main culprit in the development of the long‐term systemic inflammation seen in gout. We revisit existing evidence for urate‐induced transcriptional and epigenetic reprogramming that could lead to an altered functional state of circulating monocytes consisting in enhanced responsiveness and maladaptive immune responses. By discussing specific functional adaptations of monocytes and macrophages induced by soluble urate or monosodium urate crystals and their contribution to inflammation in vitro and in vivo, we further enforce that urate is a metabolite that can induce innate immune memory and we discuss future research and possible new therapeutic approaches for gout and its comorbidities.
Keywords: gout, hyperuricemia, inflammation, innate immune memory
1. INTRODUCTION
Gout is a highly prevalent, debilitating, autoinflammatory arthropathy having hyperuricemia as a prerequisite for disease development.1 When the serum urate level exceeds the solubility threshold, it undergoes a phase change in the presence of sodium and precipitates into monosodium urate (MSU) crystals.2 Gout manifests as acute episodes of painful arthritis due to the deposition of MSU crystals in the joints and surrounding tissues, commonly affecting the big toe, although it can involve other joints as well. 3 The natural course of an acute gouty attack generally lasts between 7 and 10 days, followed by an inter‐critical period in which there is a complete resolution of signs and symptoms.4 While some patients exhibit no recurrence after the first attack, in most patients the disease takes its natural course leading to advanced gout, characterized by chronic inflammation, frequent attacks, tophus formation, and joint destruction.5
Despite the progress made in understanding the mechanisms behind MSU‐elicited inflammation, open questions remain regarding why some patients present with one single attack, while others develop chronic gout. Additionally, it is not fully understood what precipitates the attacks and why most patients with hyperuricemia and some patients with MSU crystal deposition never develop gout at all.
Gout is no longer viewed as an isolated pathology, and the elucidation of the underlying mechanisms in gout could hold relevance for associated diseases. An increasing number of studies link gout with comorbidities such as hypertension, cardiovascular disease, renal disease, diabetes mellitus, metabolic syndrome, hyperlipidemia, increased incidence of cancer, and premature aging.6, 7, 8, 9, 10
In this review, we focus on the hypothesis that urate induces immune programming and maladaptive inflammatory responses that could play a role in the development of gout and its comorbidities. We describe recent work investigating the effects of urate on the inflammatory responses of the host and discuss its importance in health and disease, as well as its role in opening new prospective treatment targets and approaches.
2. THE INNATE IMMUNE SYSTEM AND INNATE IMMUNE MEMORY
Pattern recognition receptors (PRRs) expressed by antigen‐presenting cells (APCs) are known to confer a certain degree of specificity in the recognition of microorganisms and danger signals by the innate immune cells.11 Collectively termed alarmins, PAMPs (pathogen‐associated molecular patterns) and DAMPs (danger‐associated molecular patterns) serve as ligands for PRRs activating APC cells to present antigens and induce the production of cytokines.12 PRRs are either membrane‐bound or intracellular and include Toll‐like receptors (TLRs), RIG‐I‐like receptors (RLRs), and Nod‐like receptors (NLRs).13 Some of the more prototypical PAMPs are represented by bacterial lipopolysaccharides (LPSs), which are found on the cell membranes of Gram‐negative bacteria and can be specifically recognized by TLR4.14 Others include peptidoglycans, flagellin, lipoteichoic acid from Gram‐positive bacteria, nucleic acids such as double‐stranded RNA (dsRNA) associated with viruses, and unmethylated CpG motifs that mimic bacterial DNA.15
DAMPs represent molecules that are normally found inside the cells and released in the extracellular space upon injury, stress, or necrosis.16 More recently, it has been shown that any modified or dysfunctional molecule, such as oxidized or denatured molecules, can act as DAMPs.17, 18, 19 Some of the known danger signals include nucleic acids, heat shock proteins (Hsps), adenosine triphosphate (ATP), urate, hyaluronic acid, oxidized low‐density lipoproteins (oxLDL), cytokines (such as interferon alpha, interleukin‐1α, interleukin‐1β, HMGB1), and others.16, 20, 21, 22
Trained immunity is a term coined by Netea et al23 to describe the ability of the innate immune system to mount innate memory by altering the response after an encounter with a PAMP or DAMP, leading to an enhanced and non‐specific later response.23, 24, 25 This memory of the innate immune cells, independent of the adaptive immune system, is mediated by epigenetic reprogramming and immunometabolism.26, 27 The reprogramming of the innate immune cells was observed to last from weeks to months, although the life span of these cells in blood is much shorter than the duration of trained immunity.26, 28 It is now accepted that innate immune memory can be developed by hematopoietic stem and progenitor cells, providing one explanation for the long‐term effect of trained immunity.29, 30
The mechanisms by which trained immune cells can mount an enhanced immune response are shown to be dependent on epigenetic modifications and cellular metabolism reprogramming.30 Kleinnijenhuis et al showed that increased inflammatory response after Bacillus Calmette‐Guérin (BCG) vaccination is associated with chromatin marks on histone H3, such as lysine trimethylation (H3K4me3) in monocytes.26 Epigenomic data of monocytes trained with β‐glucan show distinct histone signatures compared to non‐treated monocyte‐derived macrophages.31 Transcriptomics data from monocytes trained with β‐glucan show a metabolic shift toward glycolysis characterized by high glucose consumption, lactate production, and a high intracellular ratio of nicotinamide adenine dinucleotide (NAD+/NADH).32 Glutaminolysis represents another metabolic reconfiguration observed in immune training of cells with β‐glucan, and inhibition of this metabolic shift leads to reduced induction of trained immunity in vivo. 33 Similarly altered immune states with enhanced inflammatory cytokine production associated with epigenetic modifications have not only been observed in settings using microbial stimulations, but also in response to sterile ligands such as oxLDL or hypercholesterolemia in the context of atherosclerosis.34, 35
3. URATE‐INDUCED IMMUNE PROGRAMMING
PAMPs and DAMPs use the same recognition machinery to mount innate immune responses, and innate immune memory is seen in both microbial and sterile stimulations. This suggests that abnormal levels of endogenous molecules or metabolic stimuli can lead to elevated inflammatory responses via resetting the transcriptional program of innate immune cells. In this section, we discuss urate as a metabolic stimulus that, at higher concentrations, can lead to innate immune activation and potentially long‐term inflammatory consequences for disease.
3.1. Urate physiology
In humans and higher primates, urate is the final oxidation product of purine catabolism. At the physiological pH of 7.4, urate is found in the ionized form of urate.36 It is mainly synthesized by the liver, but it can also be produced by other tissues that possess xanthine oxidase activity, such as the intestines, muscles, kidneys, and vascular endothelium.37 Due to the pseudogenization of the uricase gene during the evolution of hominoid primates, humans among other mammals lost uricase activity, rendering them unable to further oxidize urate to the more water‐soluble compound allantoin.38 Thus, serum urate (sUA) levels are three to ten times higher in humans than in uricase‐preserving organisms. 37, 38 About two‐thirds of urate is excreted in the kidneys, out of which 90% is reabsorbed.39 This evolutionary preserving mechanism implies a beneficial role for urate, such as its antioxidant capacity.40, 41 Despite accounting for almost half of the plasma total antioxidant capacity, there is still no apparent biological importance of urate, since in xanthinuria (a genetic condition in which patients lack xanthine oxidase, being unable to produce urate) there are yet no known consequences, except for the development of xanthine stones.42 Intriguingly, clinical and epidemiological studies show a detrimental role for low levels of urate regarding neurodegenerative diseases and high urate levels are associated with neuroprotection against Parkinson disease, multiple sclerosis, Alzheimer disease, and amyotrophic lateral sclerosis.40, 43, 44, 45 However, whether these effects can be attributed to urate's antioxidant capacity is still much debated, as these studies mainly show immune activation as potential mechanism for urate‐induced neuroprotection.46, 47
3.2. Urate acts as a DAMP
Tissues are continuously surveilled by the immune system for antigens and potential dangers to the host. During necrosis (cellular stress or damage that represent manifestations of an underlying pathological process), cells release danger signals, including urate which alerts the immune system and promotes immunity.48
DAMPs can stimulate the dendritic cells to mature.11 It has been shown that the long use of alum salts as vaccine adjuvants is due to their ability to increase urate production.49 When urate is added to cultures of primary bone marrow–derived dendritic cells, it increases the expression of CD86, providing T cells with the necessary co‐stimulatory signals to initiate a T‐cell response. Even more so, depletion of urate inhibits the immune response associated with dying cells.48 In line with these findings, other studies confirm the role of urate as an adjuvant in the anti‐tumor immunity induced by dendritic cell vaccines.50, 51
Under normal circumstances, cells contain high levels of urate and, when cellular damage or necrosis occurs, they produce even more owing to purine degradation and release of intracellular urate.48, 52 It is hypothesized that due to high levels of sodium present in the extracellular fluid, the supra‐saturation of urate upon cellular death triggers nucleation of MSU crystals, which are classically thought to be immunologically active, capable of acting as a DAMP.52 Indeed, in any setup which studies supra‐saturated urate effects, crystal‐dependent mechanisms cannot be excluded when assessing the immunological activities of urate. The release of urate by dying cells contributes to the host defense but can also lead to sterile inflammation and pathologic consequences.
3.2.1. Evidence of crystal‐dependent urate‐driven inflammation
Martinon et al described that MSU crystal–driven inflammation in vitro is dependent on the assembly of the NLRP3 inflammasome, both in THP‐1 cells and in human monocytes.53 In the context of MSU crystal–induced production of active IL‐1β, two observations need mentioning. First, the cells need to be primed so that transcription of pro‐IL‐1β is active, and second, in vitro MSU crystals need TLR synergism to induce active IL‐1β production and release. 54, 55
IL‐1 is the prototypical pro‐inflammatory cytokine, virtually affecting all cells and organs.56 IL‐1α and IL‐1β are both active forms of IL‐1, encoded by distinct genes, but binding to the same receptor (IL‐1R1) and having similar immunological effects.57 Unlike IL‐1α, for IL‐1β to exert its biological function, it needs to be cleaved from its precursor form, pro‐IL1β. The production of active IL‐1β is a two‐step process, requiring cells to be first stimulated to induce the transcription of IL‐1β.56 PAMPs and DAMPs can act through PRRs to induce the expression of pro‐IL‐1β, referred to as a priming step, partly dependent on NF‐κB activation.58
Transcriptional control
In vitro, MSU crystals have been shown to stimulate macrophages to produce IL‐1β in a MyD88‐dependent pathway (Figure 2).59 MyD88 is an intracellular protein that functions as an adapter for signal transduction, linking Toll‐like receptor (TLR) family members and IL‐1 receptor (IL‐1R) to IL‐1R‐associated kinase (IRAK), leading to the activation of nuclear factor kappa B (NF‐κB).60 The transcription factor NF‐κB upregulates a large number of inflammatory genes, including those encoding IL‐1β, TNF‐α, IL‐6, IL‐12p40, and cyclooxygenase.61
Figure 2.
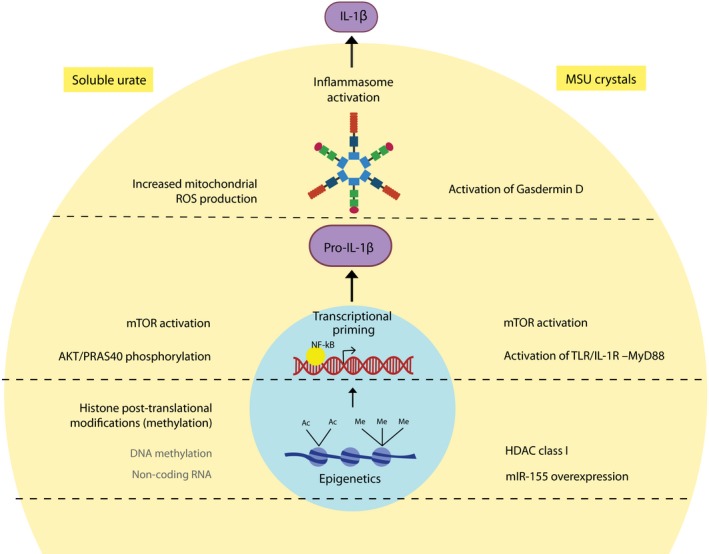
Mechanisms of soluble urate– and monosodium urate crystal–induced inflammation. Soluble urate and MSU crystals promote inflammation at several different levels. Inflammasome activation: MSU crystals activate the NLRP3 inflammasome and are accompanied by activation of gasdermin D. Soluble urate can also activate the NLRP3 inflammasome and seems to be dependent on the production of mitochondrial ROS. Transcriptional control: MSU crystals induce mTOR activation and inhibition of PTEN (an mTOR inhibitor) which further induce IL‐1β and inhibition of mTOR leads to lower IL‐1β production. MSU crystals act through MyD‐88, an intracellular adapter protein that leads to activation of NF‐κB and IL‐1β expression. Soluble urate priming enhanced PRAS40 phosphorylation which activates mTOR through Akt signaling leading to IL‐1β expression. Epigenetic regulations: MSU crystal stimulation of innate immune cells is associated with class I HDAC activation accompanied by SOCS1 downregulation, NF‐κB activation, and enhanced IL‐1β transcription. MSU crystals also induce the expression of miR‐155 which by inhibiting SHIP‐1 leads to phosphorylation of Akt with subsequent activation of NF‐κB and IL‐1β transcription. Soluble urate priming is regulated epigenetically by histone methylation, perhaps also by involvement of DNA methylation and non‐coding RNAs, with inhibition of histone methyltransferase opposing the effects of urate. MSU: monosodium urate; HDAC: histone deacetylase; SOCS1: suppressor of cytokine signaling 1; NF‐κB: nuclear factor kappa B; IL: interleukin; miR: microRNA; Akt: protein kinase B; SHIP‐1: Src homology 2 (SH2) domain‐containing inositol polyphosphate 5‐phosphatase 1; PTEN: phosphatase and tensin homolog; mTOR: mammalian target of rapamycin; MyD‐88: myeloid differentiation primary response 88; PRAS40: proline‐rich Akt substrate of 40 kDa; NLRP3: NACHT, LRR, and PYD domain‐containing protein 3; ROS: reactive oxygen species
Inflammasome activation
The primed cell must encounter a second stimulus to induce the processing and secretion of the biologically active IL‐1β. Pro‐IL‐1β is cleaved by the inflammatory protease caspase‐1, located inside the cell.62 Caspase‐1 is normally found in its inactive zymogen form (pro‐caspase‐1) and is autoactivated through oligomerization upon inflammasome activation.63 Inflammasomes are multi‐protein intracellular pattern recognition receptors composed of a C‐terminal leucine‐rich repeat capable of recognizing PAMPs and DAMPs, a central nucleotide binding domain (NACHT), and N‐terminal pyrin domain (PYD).64 The PYD domain recruits the adapter molecule apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC). Pro‐caspase‐1 is recruited to ASC by homotypic interaction of caspase recruitment domains resulting in caspase‐1 activation. Once activated, caspase‐1 will cleave pro‐IL‐1β into its mature active form readily available to be released by the cell and initiate IL‐1 signaling (Figure 1).65 Inhibition of actin and tubulin polymerization by colchicine (a common drug used in the treatment of gout) blocked NLRP3 activation and subsequent processing of IL‐1β induced by MSU crystals, providing a mechanism for MSU‐dependent inflammasome activation via microtubule polymerization and ASC recruitment at the site of inflammasome assembly.66, 67
Figure 1.
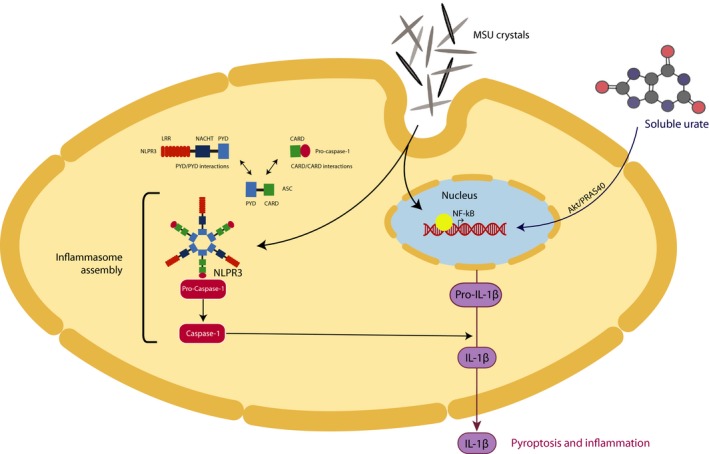
The two‐step hit model of inflammasome activation and IL‐1β production and release. The initial priming step of inflammasome activation can be mediated by both MSU crystals and soluble urate resulting in translocation of NF‐κB into the nucleus to initiate transcription of pro‐IL‐1β. Soluble urate enhances phosphorylation of PRAS40 which acts at the intersection of Akt and mTOR signaling, further leading to transcriptional regulation of cytokine genes. MSU crystals can induce NF‐κB activation through mTOR signaling. Once pro‐IL‐1β expression is induced, MSU crystals can induce NLRP3 inflammasome assembly (second step). Pro‐caspase‐1 is self‐activated by proteolytic cleavage into the enzymatically activate caspase‐1. Activate caspase‐1 cleaves pro‐IL‐1β into the biological active form IL‐1β which can be readily secreted into the extracellular milieu, mediating inflammation and pyroptosis. MSU: monosodium urate; NF‐κB: nuclear factor kappa B; IL: interleukin; PRAS40: proline‐rich Akt substrate of 40 kDa; Akt: protein kinase B; mTOR: mammalian target of rapamycin; NLRP3: NACHT, LRR, and PYD domain‐containing protein 3
Release of mature IL‐1β is associated with caspase‐1‐dependent pyroptosis.68 Pyroptosis is a type of inflammatory cell death mediated by gasdermin D (GSDMD) processing to release N‐terminal GSDMD fragment which, subsequently, contributes to pore structure formation at the plasma membrane.69, 70 This results in the release into the extracellular milieu of DAMPs such as DNA, ATP, ASC oligomers, or intracellular cytokines that further recruit immune cells and perpetuate the inflammatory response and tissue damage.71, 72 MSU crystal–induced activation of GSDMD has been recently described (Figure 2).73 Although GSDMD is rapidly activated in response to MSU crystals, it was shown to be dispensable for MSU crystal–mediated IL‐1β release and cell death both in vitro and in vivo.73 Interestingly, necrosulfonamide (NSA), a GSDMD inhibitor, can hinder IL‐1β release in response to MSU crystals, suggesting a role for NSA in blocking inflammasome activation pathway upstream of GSDMD.73, 74
Pyroptosis ultimately functions as a defense mechanism against infection by clearing out pathogens. When innate immune cells sense danger signals, they become activated, produce inflammatory cytokines, and can undergo pyroptosis, augmenting inflammation, thus contributing to the development of adaptive responses.75 If danger signals persist, inflammation will continue to amplify leading to metabolic disorders, autoinflammatory diseases, and chronic inflammation.76 The priming step of inflammasome activation in response to MSU crystals in vivo is still debated, since in vitro studies show the need for a two‐step activation of the NLRP3 inflammasome.54, 55
Production of IL‐1β in peripheral blood mononuclear cells (PBMCs) isolated from patients with gout also needs co‐stimulation, showing that MSU alone is an inefficient stimulus for IL‐1β secretion.55 When incubated with TLR2 ligands, MSU crystals synergize to induce higher cytokine production in cells of gout patients compared to healthy controls.55 No difference in mRNA transcripts levels of IL‐1β was observed after stimulation of PBMCs of gout patients with MSU crystals, enforcing lack of effect of MSU crystals on transcriptional priming but rather on the post‐translational processing of IL‐1.55, 77
3.2.2. Evidence of soluble urate–driven inflammation
In contrast to the findings described for MSU crystals, the evidence on soluble urate points toward a priming effect to induce pro‐inflammatory cytokine production.78, 79
Transcriptional control
In an experimental setting mimicking hyperuricemia in vitro, monocytes from healthy donors were exposed to soluble urate and subsequently stimulated with MSU crystals in the presence or absence of TLR2 or TLR4 ligands. Soluble urate failed to induce IL‐1β secretion on its own, but dose‐dependently increased IL‐1β cytokine production in the presence of TLR ligands, without microscopic evidence for MSU crystallization.78 Beside inducing IL‐1β transcription, soluble urate specifically downregulated IL‐1Ra production, both at the level of gene transcription and cytokine secretion, effect that was consistent even at lower urate doses, in the range of clinical hyperuricemia.78 When the same setting was applied to PBMCs from gout patients, it revealed that urate priming enhanced IL‐1β production to a lesser degree in cells from gout patients compared to healthy controls and that these patients had higher basal IL‐1β transcript levels.78 Monocytes exposed to soluble urate showed enhanced phosphorylation of proline‐rich AKT substrate 40 kD (Pras40) and a decrease in C3 II‐positive autophagosome formation (Figure 2).79 Pras40 is a substrate of Akt and a component of mammalian target of rapamycin complex 1 (mTORC1). Phosphorylation of PRAS40 strongly induces mTOR, which in turn inhibits autophagy.80 Autophagic modulators, 3‐methyladenine (3MA) and wortmannin that act as PI3K inhibitors, inhibited autophagy in a similar manner, recapitulating the cytokine pattern seen induced by urate priming.79, 81 The enhanced production of IL‐1β and decrease of IL‐1Ra make the Akt‐PRAS40 autophagy pathway a likely candidate for urate signaling in inflammation.
Another report showed upregulation of mRNA levels of inflammasome‐related genes, such as IL‐1β and NLRP3 in a model of murine obstructive nephropathy, dependent on urate production.82 Upregulation of IL1B transcription was also reported in bone marrow–derived macrophages in response to normouricemic concentrations of soluble urate.82 These suggest that soluble urate can alter the transcriptional program of the cell and modulate cytokine production in the absence of apparent urate crystal formation.
Inflammasome activation
In line with the findings described for MSU crystals, Braga et al showed that soluble urate also activates the NLRP3 inflammasome and induces the production of IL‐1β.53, 82 These effects were reversed upon addition of allopurinol, a urate lowering compound.82
A few mechanisms were proposed to mediate inflammasome activation, involving reactive oxygen species and phagosomal proteases.75 The hypothesis of a redox‐regulated NLRP3 activation occurred after the observation that, in alveolar macrophages, ATP induces caspase‐1 activation and subsequent IL‐1β production which was reversible upon co‐incubation with diphenyleneiodonium (DPI), an inhibitor of NADPH oxidase.83 Moreover, the discovery of a disulfide bond in the crystal structure of the NLRP3 protein suggested that the NLRP3 inflammasome might be sensitive to redox regulation.83, 84 In line with this, another report demonstrated that IL‐1β release in response to MSU crystals was suppressed by knockdown of the p22phox subunit of NADPH oxidase in THP1 cells.85 Indeed, Braga et al also showed that the NLRP3 inflammasome activation as a result of soluble urate exposure is dependent on mitochondrial ROS production.82
However, the redox‐dependent inflammasome activation by urate might be owing to experimental settings and cell origin, without a clear mechanism being established yet. In a setting of urate priming of PBMCs from patients with chronic granulomatous disease (CGD), a genetic disorder characterized by a deficiency in phagocyte NADPH oxidase 2 activity, and inability to efficiently produce reactive oxygen compounds, the pro‐inflammatory shift in cytokine pattern induced by soluble urate could still be reproduced.79, 86 This argues against the dependence of urate priming on NADPH oxidase–derived ROS; nevertheless, it does not exclude a redox regulation of the NLRP3 via alternative sources of reactive oxygen species (ROS). Zhou et al showed that mitochondrial‐derived ROS can activate the NLRP3 inflammasome and that autophagy opposed this effect by targeting dysfunctional mitochondria.87 Supporting the mitochondrial‐derived ROS hypothesis, another report showed an increase in mitochondrial ROS production following stimulation of macrophages with soluble urate (Figure 2).82 Although a clear mechanism has not been established yet, further research is needed to validate the effects of soluble urate on ROS production and production of IL‐1β in vitro and in vivo.
3.3. Urate induces epigenetic regulation
Epigenetic modifications represent functional changes of the genome that affect gene activity and expression without involvement of the underlying nucleotide sequence.88 DNA methylation, histone post‐translational modifications, or non‐coding RNA‐dependent processes determine transcriptional accessibility in different cell states.88 Core promoters generally have low basal activity, but can be activated by enhancers or can be further suppressed by chromatin remodeling.89 Active promoters are characterized by an open chromatin state that facilitates binding of activating transcription factors. Enhancers and active promoters generally associate acetylation of histone subunit H3 at lysine residues 9 and/or 27 (H3K9ac, H3K27ac) and methylation of histone subunit H3 lysine 4 (H3K4me), while methylation at histone subunit H3 lysine 9 or 27 (H3K9me, H3K27me) is usually repressive.90, 91, 92 Histone lysine acetylation is regulated by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs), while histone methylation at lysine residues is under the control of lysine methyltransferases (KMT) and lysine demethylases (KDM).93 Other effectors of trained immunity are represented by micro(mi)RNAs that function as regulators of gene expression.94, 95 Because of their relatively long half‐life, they can persist after the removal of the initial stimulus, making them likely candidates to mediate the innate immune memory.96 Although the main marks of epigenetic regulation have been characterized, the stimuli that influence these dynamic processes are under continuous investigation. The reprogramming of the innate immune system under variation of metabolite levels is an emerging concept, due to a role of metabolites acting as cofactors for epigenetic enzyme activities.32
The higher cytokine production in response to TLR agonists seen in gout patients78 and after urate exposure in vitro suggests a long‐term effect of urate exposure, consistent with innate immune memory. As discussed above, from studies on trained immunity it is known that innate immune cells alter their functional state and enhance their responsiveness to secondary stimuli via epigenetic modifications and metabolic reprogramming. In this section, we review the currently available evidence of epigenetic regulation as a critical determinant of urate‐induced innate immune responses.
3.3.1. Urate crystal–dependent inflammation and epigenetic regulation
Inflammatory responses precipitated by MSU crystals appear to be dependent on epigenetic modifiers. A recent study showed that romidepsin, a histone deacetylase (HDAC) 1/2 inhibitor, decreased cytokine production in PBMCs in response to MSU crystal stimulation (Figure 2).97 Inhibition of class I HDAC upregulated the expression of SOCS1 and further decreased the phosphorylation of STAT1 and STAT3.97, 98 The inhibitory effect of romidepsin on cytokine production was also observed at the level of IL‐1β transcription in response to MSU crystals and saturated fatty acid palmitate (C16.0) known to signal via TLR2, consistently with the two‐step model of inflammasome activation and processing of IL‐1β.56, 97 Although the mRNA levels of IL‐1β were decreased in response to romidepsin, the transcripts of inflammasome components were not significantly affected.97 SOCS1 modulates the immune response by targeting inflammatory molecules for proteasomal degradation, and when bortezomib, a proteasome inhibitor was added, it reversed the effect of romidepsin.97, 99, 100 Taken together, these results show that HDAC inhibition by romidepsin controls inflammation by increasing the expression of SOCS1, targeting inflammatory molecules for proteasomal degradation and preventing the transcription of IL‐1β.97, 99
Similarly, butyrate, a short‐chain fatty acid which has class I HDAC inhibition properties, can modulate inflammation by decreasing NF‐κB activation.101, 102 Butyrate was shown to suppress MSU‐ and C16‐induced IL‐1β production in a dose‐dependent manner, effect that was also exerted at transcriptional levels.103 The suppressive effect of butyrate was also seen ex vivo in cells from gout patients, with a strong decrease in cytokine production regardless of urate levels, treatment, or phase of the disease.103
MicroRNA‐dependent processes have also been studied in MSU‐induced models of inflammation. PBMCs and synovial fluid mononuclear cells (SFMCs) from patients with gout exhibit higher levels of miR‐155. Furthermore, MSU crystal stimulation was shown to strongly induce miR‐155 (Figure 2).104 Overexpression of miR‐155 decreased SH‐2 containing inositol 5' polyphosphatase 1 (SHIP‐1) and promoted IL‐1β production in response to MSU. SHIP‐1 acts to control inflammation, and its inhibition led to phosphorylation of protein kinase B (Akt) with probable subsequent activation of NF‐κB.104, 105, 106 By activating NF‐κB and inducing transcription of IL‐1β, miR‐155 acted as a priming signal for IL‐1β processing in response to MSU crystal stimulation.104
While MSU crystal–driven inflammation is mostly studied in experimental models mimicking the acute response in gout inflammation, the data presented in this section suggest that epigenetic mechanisms could also be at play in MSU‐induced responses. Gout is known to be a disease where the deposition of urate crystals is continuous, while the flares are intermittent.5 Moreover, the higher the urate load, the more frequent the acute attacks are known to occur.107 It is tempting to speculate that MSU crystals could also prime circulating or resident cells for elevated inflammatory responses via innate immune memory‐like mechanisms. Indeed, one study did show that MSU crystals were able to enhance the phagocytosis and killing capacity of murine macrophages leading to a more enduring response to BCG vaccination in mice.108
3.3.2. Soluble urate–dependent inflammation and epigenetic regulation
During priming with soluble urate, a persistent increase in IL‐1β gene transcription accompanied by increased responsiveness to secondary stimulation is described.78 This enhanced IL‐1β production is thought to be regulated epigenetically, as the exposure of PBMCs to a histone methyltransferase inhibitor, (5’‐deoxy‐5’methylthio‐adenosine, MTA), abolished the effects of urate on cytokine production (Figure 2).78 Furthermore, in vitro, modulation of cytokine production by urate was maintained after extended resting times between priming and restimulation. In an in vivo murine model of acute gout using intra‐articular knee injections of MSU crystals and palmitic acid after exposure for 1 day to urate and potassium oxonate (uricase inhibitor), methyltransferase inhibition was validated to oppose the effects of urate‐induced inflammation (T Crisan, LAB Joosten, poster communication, European Journal of Human Genetics, July 2019). In vivo intravenous administration of urate in healthy human volunteers was consistent with higher levels of plasma IL‐6 after acute lipid ingestion, whereas potently lowering of serum urate with rasburicase did not have a significant effect on IL‐6 plasma values after lipid oral tolerance test.109 This further shows that elevated urate levels can enhance inflammatory responses to subsequent challenges.
The transcriptional rewiring of urate stimulated cells is suggestive of innate immune memory developed in response to endogenous sterile stimuli, leading to a scenario in which soluble urate acts as a stimulus that drives epigenetic reprogramming in circulating or tissue‐resident cells, thereby causing persistent inflammatory effects in response to continuous exposure to urate. We propose a model in which cells exposed to high urate levels are more prone to develop a strong pro‐inflammatory response when exposed to specific triggers due to innate immune memory, epigenetic changes in exposed cells, or persistence of exposure to high urate levels. The epigenetic landscape driven by soluble urate in myeloid cells is a field of study that requires further research in order to describe epigenetic signatures and possible reversible targets for the therapy of the inflammatory consequences of hyperuricemia.
3.4. The AKT‐mTOR pathway as an effector of urate‐induced immune priming
Upon stimulation, innate immune cells undergo differentiation from quiescence to activation.110 In the case of trained immunity, this associates epigenetic changes that are accompanied by extensive metabolic rewiring adapted to cellular nutrient and metabolic demands. A central player that has been shown to modulate the immunometabolic adaptation in innate immune memory is the PI3K‐Akt‐mTOR pathway.111 Classically activated macrophages (CAMs) reprogram their metabolism from oxidative phosphorylation (OXPHOS) to glycolysis, as measured by a decreased oxygen consumption rate and an increase in tricarboxylic acid cycle (TCA) intermediates, phenomenon described as aerobic glycolysis.112 A role for HIF‐1α has emerged as the primary regulator of glycolysis in CAMs.111 Consistent with this, a shift from OXPHOS to glycolysis dependent on Akt‐mTOR‐HIF‐1α has been described in trained immunity induced by β‐glucan.32 Accumulation of TCA metabolites during training of monocytes links immunometabolism with epigenetic changes. Macrophages activated in response to LPS induce succinate accumulation which stabilizes hypoxia‐inducible factor α (HIF‐1α) leading to increased IL‐1β production during inflammation.113 By acting as a cofactor, succinate is an important determinant for the activity of epigenetic enzymes. Succinate has been shown to inhibit JMJD3, also known as lysine‐specific demethylase 6 (KDM6) leading to enhanced H3K27 trimethylation of particular loci, such as regions on genes associated with the anti‐inflammatory M2 phenotype.114 Inhibition of glycolysis by 2‐deoxyglucose (2‐DG), of mTOR with rapamycin and metformin, blocks the formation of histone marks and inhibits cytokine production.33
Evidence of similar mechanistic data has also been described in the context of MSU crystal– or soluble urate‐induced inflammation. One study showed that stimulation of PBMCs and human monocytes with MSU crystals enhances mTOR activity (Figure 2).115 Intracellular pro‐IL‐1β levels were shown to be increased in response to MSU crystals, without an elevation in cytokine mRNA levels, which is consistent with evidence showing that MSU acts as a second signal for inflammasome activation. Although mTOR activity was enhanced, pro‐IL‐1β protein synthesis was selectively regulated by p38 MAPK signaling and not on mTOR.115 However, another report showed that gout patients exhibit higher expression of genes involved in mTOR pathway and lower expression of PTEN, an mTOR inhibitor, compared to healthy controls.116 The group showed that MSU crystals induce mTOR pathway gene expression in monocytes from healthy donors, and this was also reflected at protein level, as well as induction of IL‐1β. They also showed that positively selected CD14+ monocytes undergo pyroptosis upon MSU crystal encounter. The effects of MSU crystals were reversed upon induction of autophagy and mTOR inhibition by metformin or rapamycin, which reduced cell death and lowered inflammasome activation and inflammation. In line with these data, in a retrospective cohort analysis, patients who received metformin as comedication were shown to have less frequent gout attacks and patients who were treated with colchicine expressed lower levels of mTOR activation.116 Metformin exerts its anti‐inflammatory effects by activating AMPK (5′ adenosine monophosphate‐activated protein kinase) which is a negative regulator of NF‐κB via the PI3K (phosphoinositide 3‐kinase)–Akt1 pathway.117 It has also been shown that inhibition of mTOR with rapamycin in macrophages induces autophagy and targets pro‐IL‐1β for degradation, resulting in a decrease of NLRP3 inflammasome activation.118 As presented in previous section, in urate priming of human monocytes, the phosphorylation of PRAS40 inducing mTOR activation with subsequent decrease in autophagosome formation was described, while the pharmacological inhibition of PI3K recapitulated the effects of soluble urate exposure.79, 80 Collectively, these data enforce that similar pathways to the ones described in trained macrophages are involved in soluble urate– and crystalline urate–induced inflammatory responses. Immunometabolism to our knowledge has not been studied in the context of hyperuricemia or gout. Potential immunometabolism deregulations in hyperuricemia represent an interesting subject of further study as this could shed light on mechanisms of AKT‐mTOR induction as well as intracellular metabolites that further could act as cofactors for epigenetic modifiers.
4. GOUT AS A MODEL OF MALADAPTIVE IMMUNE PROGRAMMING
While the heightened capacity of innate immune memory‐bearing cells to respond more strongly to stimuli is beneficial to host defense, long‐term immune programming can have systemic consequences that lead to chronic inflammation and tissue damage. Even more so, when trained immunity is induced by endogenous ligands, they can change the landscape of the immune response in a perpetuating cycle of activation and hyper‐responsiveness.
Gout is no longer viewed solely as an articular disease and, in its most comprehensive definition, it is an inflammatory and metabolic disorder as the manifestations of gout extend beyond the local inflammatory consequence of MSU crystal deposition.119 Hyperuricemic patients exhibit higher incidence of comorbidities such as cardiovascular disease, type 2 diabetes, metabolic syndrome, chronic kidney disease, cancer, and premature aging (Figure 3).6, 7, 8, 9, 10 Whether urate elevation is involved in the pathogenesis of comorbidities seen in gout is still a debated issue, but an increasing number of epidemiological studies have found hyperuricemia as an independent risk factor for gout comorbidities: A meta‐analysis including cohorts of over a million subjects shows serum urate levels to increase cardiovascular mortality risk in a dose‐dependent manner, and experimental animal models show positive correlation between urate levels, arterial hypertension, and insulin resistance, with reversal of manifestation upon lowering and inhibition of urate.6, 7, 8, 120, 121
Figure 3.
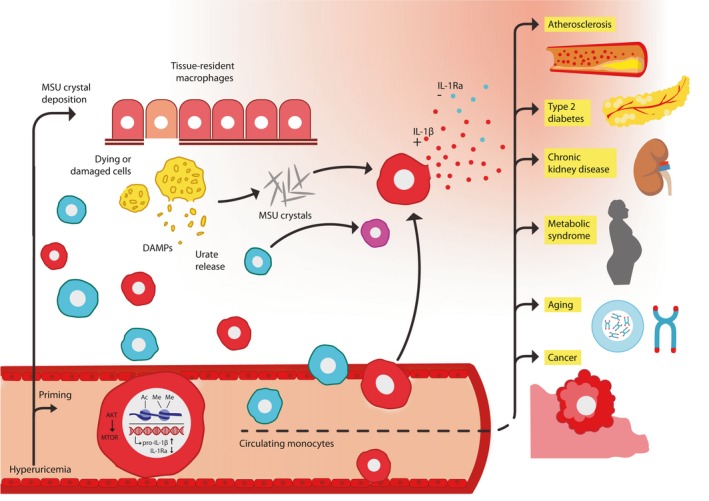
Suggested model for the interactions of soluble urate– and monosodium urate crystal–driven inflammation in gout and comorbidities. Soluble urate primes circulating monocytes acting through upregulation of the Pras40‐Akt‐mTOR pathway with upregulation of IL‐1β and specific downregulation of IL‐1Ra. Supra‐saturated serum urate leads to crystallization of urate into MSU and deposition of crystals in tissues which drives resident macrophage activation, tissue damage, and release of DAMPs and more urate. During a gout attack, circulating monocytes arrive at the site of tissue inflammation where, upon contact with MSU crystals, they become activated. Monocytes already primed by serum urate can readily secrete IL‐1β after encountering DAMPs and MSU crystals, which activate the inflammasome and processing of pro‐IL‐1β. This leads to enhanced IL‐1β secretion and pyroptosis in the context of low IL‐1Ra antagonism. Non‐primed circulating monocytes could potentially also become primed upon MSU crystal encounter. Upon tissue damage and pyroptosis, more urate is released which can further prime new monocytes, perpetuating inflammation and tissue damage during a gout flare. In the circulation, primed monocytes can readily secrete IL‐1β in response to second activation by stimuli, shifting the immune response toward pro‐inflammation with long‐term predisposition for comorbidities. Akt: protein kinase B; mTOR: mammalian target of rapamycin; IL: interleukin; MSU: monosodium urate; DAMPs: damage‐associated molecular patterns
The common link in all these diseases is the presence of sterile inflammation, with overlapping mechanisms involved in their pathogenesis. The NLRP3 pathway, associated with MSU‐induced inflammation, has also been described in atherosclerosis and metabolic syndrome.122, 123 In patients with arterial hypertension, elevated circulating levels of IL‐1β have been reported and linked to possible inflammatory and atherogenic consequences of hypertension.124, 125 A causative role for urate in the development of these comorbidities has not been established, but associations between urate levels and inflammatory markers have also been described in asymptomatic hyperuricemic individuals.126 Therefore, it can be hypothesized that urate could contribute to systemic inflammation and act as a co‐signal for the initiation of innate immune responses.
Chronic inflammation is associated with increased cellular senescence, which strongly correlates with the risk of cardiovascular disease.127, 128, 129, 130 Patients with gout have increased replicative senescence, and telomere length is an independent risk factor for cardiovascular disease in these patients.131 Gout patients have an increased risk of cancer development, and it has been suggested that hyperuricemia may play a protective role due to the antioxidant properties of urate.41, 132 A prospective study on gout and cancer risk shows that the incidence of cancers is elevated in these patients and that hyperuricemia plays no role in the prevention of carcinogenesis.133 Gout is a disorder of purine metabolism, and it is known that high cellular turnover can lead to hyperuricemia and tumorigenesis.134, 135 High serum urate levels are independently correlated with an increased risk of cancer.136, 137 Data from a meta‐analysis study show gout as an independent risk factor for the incidence of total cancers, but there is strong evidence for a role of serum urate in cancer biology, with elevated levels strongly associated with increased premature mortality in relation to cancer in both men and women. 138, 139
Although it was believed that tissue‐resident macrophages are responsible for the initiating and driving of the early inflammatory response in gout, kinetic studies on murine gout models have shown that circulating monocytes are the initial responders and are recruited to the site of MSU crystal deposition.140, 141 One of the monocyte chemoattractants involved in monocyte recruitment is chemokine (C‐C motif) ligand 2 (CCL2), and animal models studies have shown CCL2 as a local chemoattractant of monocytes in gout.142, 143, 144 CCL2 levels have been shown to positively correlate with serum urate levels in both gout patients and asymptomatic hyperuricemic individuals.145 Soluble urate can induce CCL2 production in vascular smooth muscle cells, and this might be one source of elevated CCL2 production seen in hyperuricemia with implications in cardiovascular diseases. 146, 147, 148 Elevated serum urate and CCL2 levels increase circulating CD14+ monocyte, even in the absence of active inflammation, thus priming monocytes to rapidly respond to inflammatory stimuli.145
Interestingly, when investigating IL‐1β levels in patients with gout ex vivo, there is evidence for an in vivo priming effect of IL‐1β induction.78 Positively selected monocytes from patients with gout present higher basal IL‐1β mRNA levels compared to healthy controls.78 The IL‐1 system is a potent mediator of acute and chronic inflammation, and it has been shown that IL‐1β can induce its own production.149 One of the ways by which this system is prevented from inducing exacerbated local and systemic inflammation with subsequent tissue damage is by the action of the IL‐1 receptor antagonist (IL‐1Ra).150 As described in the previous sections, the specific downregulation of IL‐1Ra in vitro in PBMCs and monocytes exposed to soluble urate is a unique finding that can enhance susceptibility to inflammation. The interleukin‐1 receptor antagonist is a secreted, soluble protein that specifically inhibits the activities of IL‐1α and IL‐1β. Another two isoforms of IL‐1Ra exist intracellularly and are released upon tissue damage, subsiding inflammation induced by cellular injury.150 Genetically engineered mice to lack IL‐1Ra were shown to spontaneously develop inflammatory poly‐arthropathy and overexpress genes encoding pro‐inflammatory cytokines, even before the onset of clinical manifestations.151 Loss‐of‐function mutations in the gene encoding for IL‐1Ra result in phenotypes of sterile osteomyelitis, periostitis, and skin inflammation.152
In conclusion, we propose a concept for soluble urate–induced facilitation of inflammation in which urate, potentially through altered metabolism and epigenetic modifications, primes monocytes to enhance their pro‐inflammatory response. By inducing CCL2, urate also alters the number of circulating monocytes, thus setting the landscape for chronic low‐grade inflammation. Upon secondary stimulation, already primed monocytes can be readily recruited at the site of inflammation and facilitate the production of pro‐inflammatory cytokines, amplifying the immune response. Ultimately, monocyte priming could act as a defense mechanism against invading pathogens. However, in conditions of hyperuricemia or tissue damage, urate induces an exacerbated response and maintains a loop of inflammation in which urate released from dying cells primes new monocytes and precipitates into MSU crystals leading to more inflammation and tissue damage. Moreover, by specifically downregulating IL‐1Ra, urate renders cells unable to oppose IL‐1 effects and the IL‐1 autoinduction loop, which maintains innate activation and tissue destruction (Figure 3).56
5. CONCLUDING REMARKS
In this review, we present data of the main mechanisms supporting the concept of urate‐induced immune programming. Ex vivo monocytes and macrophages from patients with gout as well as in vitro stimulations of monocytes with urate lead to functional changes consistent with a trained immunity phenotype. The increased cytokine production, changes in cellular metabolism, and epigenetic reconfiguration are hallmarks of trained immunity that have been observed in the context of crystalline or soluble urate inflammation models. Urate‐induced immune programming can initiate gout development and maintenance of chronic inflammation as well as partake in the development of comorbidities commonly associated with gout progression. Evidence of urate‐induced long‐term functional alterations is rapidly accumulating, but further research is needed to determine the metabolic adaptations of innate immune cells in response to urate. In trained immunity, metabolic reprogramming leads to epigenetic rewiring consistent with an enhanced inflammatory response. We have shown that by inhibiting specific metabolic pathways or by inhibiting epigenetic effectors, the inflammation and tissue damage induced by urate can be counteracted. Therefore, by elucidating the mechanisms of urate in the induction of trained immunity and progression of disease, we can describe new pathways for immunotherapy intervention in autoinflammatory diseases and in diseases in which trained immunity becomes maladaptive.
CONFLICT OF INTEREST
The authors declare they have no competing interests.
ACKNOWLEDGEMENTS
This work was supported by a Competitiveness Operational Programme grant of the Romanian Ministry of European Funds (HINT, P_37_762, MySMIS 103587)
Cabău G, Crișan TO, Klück V, Popp RA, Joosten LAB. Urate‐induced immune programming: Consequences for gouty arthritis and hyperuricemia. Immunol Rev. 2020;294:92–105. 10.1111/imr.12833
This article is part of a series of reviews covering Inflammatory Arthritis appearing in Volume 294 of Immunological Reviews.
REFERENCES
- 1. Punzi L, Scanu A, Ramonda R, Oliviero F. Gout as autoinflammatory disease: New mechanisms for more appropriated treatment targets. Autoimmun Rev. 2012;12(1):66‐71. [DOI] [PubMed] [Google Scholar]
- 2. Martillo MA, Nazzal L, Crittenden DB. The crystallization of monosodium urate. Curr Rheumatol Rep. 2014;16(2):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richette P, Bardin T, Médicale UFR. Gout. 2010.
- 4. Dalbeth N, Merriman TR, Stamp LK. Gout. The Lancet. 2016;388(10055):2039‐2052. 10.1016/s0140-6736(16)00346-9 [DOI] [PubMed] [Google Scholar]
- 5. Ragab G, Elshahaly M, Bardin T. Gout: An old disease in new perspective – A review. J Adv Res. 2017;8(5):495‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pillinger MH, Goldfarb DS, Keenan RT. Gout and its comorbidities. Bull NYU Hosp Jt Dis. 2010;68(3):199‐203. [PubMed] [Google Scholar]
- 7. Krishnan E. Inflammation, oxidative stress and lipids: the risk triad for atherosclerosis in gout. Rheumatology. 2010;49(7):1229‐1238. [DOI] [PubMed] [Google Scholar]
- 8. Ichikawa N, Taniguchi A, Urano W, Nakajima A, Yamanaka H. Comorbidities in patients with gout. Nucleosides, Nucleotides Nucleic Acids. 2011;30(12):1045‐1050. [DOI] [PubMed] [Google Scholar]
- 9. Feldman N, Rotter‐Maskowitz A, Okun E. DAMPs as mediators of sterile inflammation in aging‐related pathologies. Ageing Res Rev. 2015;24:29‐39. [DOI] [PubMed] [Google Scholar]
- 10. Eisenbacher JL, Schrezenmeier H, Jahrsdörfer B, et al. S100A4 and uric acid promote mesenchymal stromal cell induction of IL‐10 + /IDO + lymphocytes. J Immunol. 2014;192(12):6102‐6110. [DOI] [PubMed] [Google Scholar]
- 11. Medzhitov R, Janeway CJ. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173(1):89‐97. [DOI] [PubMed] [Google Scholar]
- 12. Medzhitov R. Approaching the asymptote: 20 years later. Immunity. 2009;30(6):766‐775. [DOI] [PubMed] [Google Scholar]
- 13. Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG‐I‐like receptors. Immunol Rev. 2009;227(1):54‐65. [DOI] [PubMed] [Google Scholar]
- 14. Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahla RS, Reddy MC, Vijaya Raghava Prasad D, Kumar H. Sweeten PAMPs: Role of sugar complexed PAMPs in innate immunity and vaccine biology. Front Immunol. 2013;4:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301‐305. [DOI] [PubMed] [Google Scholar]
- 17. Matzinger P. Extended family. Annu Rev Immunol. 1994;12:991‐1045. [DOI] [PubMed] [Google Scholar]
- 18. Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol. 2012;3:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seong SY, Matzinger P. Hydrophobicity: an ancient damage‐associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469‐478. [DOI] [PubMed] [Google Scholar]
- 20. Jounai N, Kobiyama K, Takeshita F, Ishii KJ. Recognition of damage‐associated molecular patterns related to nucleic acids during inflammation and vaccination. Front Cell Infect Microbiol. 2013;4:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): The prototypical endogenous danger molecule. Mol Med. 2015;21:S6‐S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller YI, Choi S‐H, Wiesner P, et al. Oxidation‐specific epitopes are danger‐associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Netea MG, Quintin J, Van Der Meer JWM. Trained immunity: A memory for innate host defense. Cell Host Microbe. 2011;9(5):355‐361. [DOI] [PubMed] [Google Scholar]
- 24. Bowdish DME, Loffredo MS, Mukhopadhyay S, Mantovani A, Gordon S. Macrophage receptors implicated in the “adaptive” form of innate immunity. Microbes Infect. 2007;9(14–15):1680‐1687. [DOI] [PubMed] [Google Scholar]
- 25. Boraschi D, Italiani P. Innate immune memory: Time for adopting a correct terminology. Front Immunol. 2018;9:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette‐Guérin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012;109(43):17537‐17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Netea MG, Latz E, Mills KHG, O’Neill LAJ. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol. 2015;16(7):675‐679. [DOI] [PubMed] [Google Scholar]
- 28. Yona S, Viukov S, Guilliams M, Misharin A. Tissue macrophages under homeostasis. Immunity. 2013;38(1):79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yáñez A, Hassanzadeh‐Kiabi N, Ng MY, et al. Detection of a TLR2 agonist by hematopoietic stem and progenitor cells impacts the function of the macrophages they produce. Eur J Immunol. 2013;43(8):2114‐2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dominguez‐Andres J, Netea MG. Long‐term reprogramming of the innate immune system. J Leukoc Biol. 2019;105(2):329‐338. [DOI] [PubMed] [Google Scholar]
- 31. Saeed S, Quintin J, Kerstens HHD, et al. Epigenetic programming of monocyte‐to‐macrophage differentiation and trained innate immunity. Science. 2014;345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumar V, Giamarellos‐bourboulis EJ, Martens JHA, et al. mTOR/HIF1α‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arts RJW, Novakovic B, ter Horst R, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24(6):807‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bekkering S, Arts RJW, Novakovic B, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018;172(1–2):135‐146. [DOI] [PubMed] [Google Scholar]
- 35. Bekkering S, Stiekema LCA, Bernelot Moens S, et al. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. 2019;30(1):1‐2. [DOI] [PubMed] [Google Scholar]
- 36. Mandel NS, Mandel GS. Monosodium urate monohydrate, the gout culprit. J Am Chem Soc. 1976;98(8):2319‐2323. [DOI] [PubMed] [Google Scholar]
- 37. El Ridi R, Tallima H. Physiological functions and pathogenic potential of uric acid: a review. J Adv Res. 2017;8(5):487‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang BSW. Ancient insights into uric acid metabolism in primates. Proc Natl Acad Sci USA. 2014;111(10):3657‐3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8‐14. [DOI] [PubMed] [Google Scholar]
- 40. Álvarez‐Lario B, Macarrón‐Vicente J. Uric acid and evolution. Rheumatology. 2010;49(11):2010‐2015. [DOI] [PubMed] [Google Scholar]
- 41. Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant‐ and radical‐caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA. 1981;78(11):6858‐6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frayha RA, Salti IS, Arnaout A, Khatchadurian A, Uthman SM. Hereditary xanthinuria. Report on three patients and short review of the literature. Nephron. 1977;19(6):328‐332. [DOI] [PubMed] [Google Scholar]
- 43. Drulović J, Dujmović I, Stojsavljević N, et al. Uric acid levels in sera from patients with multiple sclerosis. J Neurol. 2001;248(2):121‐126. [DOI] [PubMed] [Google Scholar]
- 44. Schwarzschild MA, Schwid SR, Marek K, et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol. 2008;65(6):716‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kutzing MK, Firestein BL. Altered uric acid levels and disease states. J Pharmacol Exp Ther. 2008;324(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 46. Cipriani S, Desjardins CA, Burdett TC, Xu Y, Xu K, Schwarzschild MA. Protection of dopaminergic cells by urate requires its accumulation in astrocytes. J Neurochem. 2012;123(1):172‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hooper DC, Spitsin S, Kean RB, et al. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1998;95(2):675‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi Y, Evans JE, Rock KL. Apoptotic_cell_MSU_Nat2003. Nature. 2003;425(6957):516‐521. [DOI] [PubMed] [Google Scholar]
- 49. Kool M, Soullié T, van Nimwegen M, et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205(4):869‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y, Ma X, Su C, et al. Uric acid enhances the antitumor immunity of dendritic cell‐based vaccine. Sci Rep. 2015;5:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu DE, Moore AM, Thomsen LL, Brindle KM. Uric acid promotes tumor immune rejection. Cancer Res. 2004;64(15):5059‐5062. [DOI] [PubMed] [Google Scholar]
- 52. Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. 2010;120(6):1939‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237‐241. [DOI] [PubMed] [Google Scholar]
- 54. Giamarellos‐Bourboulis EJ, Mouktaroudi M, Bodar E, et al. Crystals of monosodium urate monohydrate enhance lipopolysaccharide‐induced release of interleukin 1 βby mononuclear cells through a caspase 1‐mediated process. Ann Rheum Dis. 2009;68(2):273‐278. [DOI] [PubMed] [Google Scholar]
- 55. Mylona EE, Mouktaroudi M, Crisan TO, et al. Enhanced interleukin‐1β production of PBMCs from patients with gout after stimulation with Toll‐like receptor‐2 ligands and urate crystals. Arthritis Res Ther. 2012;14(4):R158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dinarello CA. Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol. 2009;27(1):519‐550. [DOI] [PubMed] [Google Scholar]
- 57. Veerapandian B. Structure and function of interleukin‐1, based on crystallographic and modeling studies. Biophys J. 1992;62(1):112‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license nlrp3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen CJ, Shi Y, Hearn A, et al. MyD88‐dependent IL‐1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116(8):2262‐2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deguine J, Barton GM. MyD88: A central player in innate immune signaling. F1000Prime . Rep. 2014;6:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. 2014;5:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thornberry NA, Molineaux SM. Interleukin‐1βconverting enzyme: A novel cysteine protease required for IL‐1β production and implicated in programmed cell death. Protein Sci. 1995;4(1):3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase‐1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem. 2009;284(10):6546‐6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schroder K, Tschopp J. Leading edge the inflammasomes. Cell. 2010;140:821‐832. [DOI] [PubMed] [Google Scholar]
- 65. Brough D, Rothwell NJ. Caspase‐1‐dependent processing of pro‐interleukin‐1β is cytosolic and precedes cell death. J Cell Sci. 2007;120(5):772‐781. [DOI] [PubMed] [Google Scholar]
- 66. McCarty DJ. Urate crystals, inflammation, and colchicine. Arthritis Rheum. 2008;58(2):20‐24. [DOI] [PubMed] [Google Scholar]
- 67. Misawa T, Takahama M, Kozaki T, et al. Microtubule‐driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454‐460. [DOI] [PubMed] [Google Scholar]
- 68. Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase‐1‐dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105(11):4312‐4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sborgi L, Rühl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35(16):1766‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kuang S, Zheng J, Yang H, et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci USA. 2017;114(40):10642‐10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baroja‐Mazo A, Martín‐Sánchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15(8):738‐748. [DOI] [PubMed] [Google Scholar]
- 72. Franklin BS, Bossaller L, De Nardo D , et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol. 2014;15(8):727‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rashidi M, Simpson DS, Hempel A, et al. The pyroptotic cell death effector gasdermin D is activated by gout‐associated uric acid crystals but is dispensable for cell death and IL‐1β release. J Immunol. 2019;203(3):736‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore‐forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3(26):eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. He Y, Hara H, Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016;41(12):1012‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Davis BK, Wen H, Ting JP‐Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29(1):707‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jansen TL, Berendsen D, Crisan TO, Cleophas MCP, Janssen MCH, Joosten LAB. New gout test: Enhanced ex vivo cytokine production from PBMCS in common gout patients and a gout patient with Kearns‐Sayre syndrome. Clin Rheumatol. 2014;33(9):1341‐1346. [DOI] [PubMed] [Google Scholar]
- 78. Crișan TO, Cleophas MCP, Oosting M, et al. Soluble uric acid primes TLR‐induced proinflammatory cytokine production by human primary cells via inhibition of IL‐1Ra. Ann Rheum Dis. 2016;75(4):755‐762. [DOI] [PubMed] [Google Scholar]
- 79. Crişan TO, Cleophas MCP, Novakovic B, et al. Uric acid priming in human monocytes is driven by the AKT‐PRAS40 autophagy pathway. Proc Natl Acad Sci USA. 2017;114(21):5485‐5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wiza C, Nascimento EBM, Ouwens DM. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol ‐ Endocrinol Metab. 2012;302(12):1453‐1461. [DOI] [PubMed] [Google Scholar]
- 81. Wu Y‐T, Tan H‐L, Shui G, et al. Dual role of 3‐methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3‐kinase. J Biol Chem. 2010;285(14):10850‐10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Braga TT, Forni MF, Correa‐Costa M, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. 2017;7(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species‐dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282(5):2871‐2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of nlrp3 inflammasomes: ROS as trigger or effector? Antioxidants Redox Signal. 2015;22(13):1111‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dostert C, Pétrilli V, Van Bruggen R , Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Segal AW. The NADPH oxidase and chronic granulomatous disease. Mol Med Today. 1996;2(3):129‐135. [DOI] [PubMed] [Google Scholar]
- 87. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221‐226. [DOI] [PubMed] [Google Scholar]
- 88. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057‐1068. [DOI] [PubMed] [Google Scholar]
- 89. Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613‐626. [DOI] [PubMed] [Google Scholar]
- 90. Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. 2007;39(3):311-318. [DOI] [PubMed] [Google Scholar]
- 91. Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell‐type‐specific gene expression. Nature. 2009;459(7243):108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074‐1080. [DOI] [PubMed] [Google Scholar]
- 94. Chen CZ, Schaffert S, Fragoso R, Loh C. Regulation of immune responses and tolerance: The microRNA perspective. Immunol Rev. 2013;253(1):112‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597‐610. [DOI] [PubMed] [Google Scholar]
- 97. Cleophas MCP, Crişan TO, Klück V, et al. Romidepsin suppresses monosodium urate crystal‐induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res Ther. 2019;21(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xiong H, Du W, Zhang Y‐J, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter‐associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol Carcinog. 2012;51(2):174‐184. [DOI] [PubMed] [Google Scholar]
- 99. Mansell A, Smith R, Doyle SL, et al. Suppressor of cytokine signaling 1 negatively regulates Toll‐like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7(2):148‐155. [DOI] [PubMed] [Google Scholar]
- 100. Chen D, Frezza M, Schmitt S, Kanwar J, P. Dou Q. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Place RF, Noonan EJ, Giardina C. HDAC inhibition prevents NF‐κB activation by suppressing proteasome activity: down‐regulation of proteasome subunit expression stabilizes IκBα. Biochem Pharmacol. 2005;70(3):394‐406. [DOI] [PubMed] [Google Scholar]
- 102. Segain JP, Raingeard de la Blétière D, Bourreille A, et al. Butyrate inhibits inflammatory responses through NFkappa B inhibition: implications for Crohn's disease. Gut. 2000;47(3):397‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cleophas MCP, Crişan TO, Lemmers H, et al. Suppression of monosodium urate crystal‐induced cytokine production by butyrate is mediated by the inhibition of class I histone deacetylases. Ann Rheum Dis. 2016;75(3):593‐600. [DOI] [PubMed] [Google Scholar]
- 104. Jin H, Kim T‐J, Choi J‐H, et al. MicroRNA‐155 as a proinflammatory regulator via SHIP‐1 down‐regulation in acute gouty arthritis. Arthritis Res Ther. 2014;16(2):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rauh MJ, Sly LM, Kalesnikoff J, et al. The role of SHIP1 in macrophage programming and activation. Biochem Soc Trans. 2004;32(5):785‐788. [DOI] [PubMed] [Google Scholar]
- 106. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR‐155. Proc Natl Acad Sci USA. 2009;106(17):7113‐7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shiozawa A, Szabo SM, Bolzani A, Cheung A, Choi HK. Serum uric acid and the risk of incident and recurrent gout: A systematic review. J Rheumatol. 2017;44(3):388‐396. [DOI] [PubMed] [Google Scholar]
- 108. Taus F, Santucci MB, Greco E, et al. Monosodium urate crystals promote innate anti‐mycobacterial immunity and improve BCG efficacy as a vaccine against tuberculosis. PLoS ONE. 2015;10(5):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tanaka T, Milaneschi Y, Zhang Y, Becker KG, Zukley L, Ferrucci L. A double blind placebo controlled randomized trial of the effect of acute uric acid changes on inflammatory markers in humans: a pilot study. PLoS ONE. 2017;12(8):1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mitroulis I, Kalafati L, Hajishengallis G, Chavakis T. Myelopoiesis in the Context of Innate Immunity. J Innate Immun. 2018;10(5–6):365‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol. 2014;32:609‐634. 10.1146/annurev-immunol-032713-120236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rodríguez‐Prados J‐C, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605‐614. [DOI] [PubMed] [Google Scholar]
- 113. Tannahill G, Curtis A, Adamik J, et al. Succinate is a danger signal that induces IL‐1β via HIF‐1α. Nature. 2013;496(7444):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. pluripotency of embryonic stem cells. Nature. 2015;518(7539):413‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chung YH, Kim DH, Lee WW. Monosodium urate crystal‐induced pro‐interleukin‐1β production is post‐transcriptionally regulated via the p38 signaling pathway in human monocytes. Sci Rep. 2016;6:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vazirpanah N, Ottria A, van der Linden M, et al. MTOR inhibition by metformin impacts monosodium urate crystal‐induced inflammation and cell death in gout: A prelude to a new add‐on therapy? Ann Rheum Dis. 2019;78(5):663‐671. [DOI] [PubMed] [Google Scholar]
- 117. Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine‐induced nuclear factor κB activation via AMP‐activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47(6):1183‐1188. [DOI] [PubMed] [Google Scholar]
- 118. Harris J, Hartman M, Roche C, et al. Autophagy controls IL‐1β secretion by targeting Pro‐IL‐1β for degradation. J Biol Chem. 2011;286(11):9587‐9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Terkeltaub R. What makes gouty inflammation so variable? BMC Med. 2017;15(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose‐induced metabolic syndrome. Am J Physiol ‐ Ren Physiol. 2006;290(3):625‐631. [DOI] [PubMed] [Google Scholar]
- 121. Rahimi‐Sakak F, Maroofi M, Rahmani J, Bellissimo N, Hekmatdoost A. Serum uric acid and risk of cardiovascular mortality: a systematic review and dose‐response meta‐analysis of cohort studies of over a million participants. BMC Cardiovasc Disord. 2019;19(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Vandanmagsar B, Youm Y‐H, Ravussin A, et al. The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med. 2011;17(2):179‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin‐1β in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129(3):300‐308. [DOI] [PubMed] [Google Scholar]
- 125. Peeters ACTM, Netea MG, Janssen MCH, Kullberg BJ, Van Der Meer JWM, Thien T. Pro‐inflammatory cytokines in patients with essential hypertension. Eur J Clin Invest. 2001;31(1):31‐36. [DOI] [PubMed] [Google Scholar]
- 126. Ruggiero C, Cherubini A, Ble A, et al. Uric acid and inflammatory markers. Eur Heart J. 2006;27(10):1174‐1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhang J, Rane G, Dai X, et al. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res Rev. 2016;25:55‐69. [DOI] [PubMed] [Google Scholar]
- 128. Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta‐ Analysis. BMJ. 2014;349:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jurk D, Wilson C, Passos JF, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Minamino T, Miyauchi H, Yoshida T, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomeres in endothelial dysfunction. J Cardiol. 2003;41(1):39‐40. [PubMed] [Google Scholar]
- 131. Vazirpanah N, Kienhorst LBE, Van Lochem E, et al. Patients with gout have short telomeres compared with healthy participants: Association of telomere length with flare frequency and cardiovascular disease in gout. Ann Rheum Dis. 2017;76(7):1309‐1315. [DOI] [PubMed] [Google Scholar]
- 132. Wang W, Xu D, Wang B, et al. Increased risk of cancer in relation to gout: a review of three prospective cohort studies with 50,358 subjects. Mediators Inflamm. 2015;2015:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Boffetta P, Nordenvall C, Nyren O, Ye W. A prospective study of gout and cancer. Eur J Cancer Prev. 2009;18(2):127‐132. [DOI] [PubMed] [Google Scholar]
- 134. Robinson PC, Horsburgh S. Gout: Joints and beyond, epidemiology, clinical features, treatment and co‐morbidities. Maturitas. 2014;78(4):245‐251. [DOI] [PubMed] [Google Scholar]
- 135. Kuo CF, Luo SF, See LC, Chou IJ, Fang YF, Yu KH. Increased risk of cancer among gout patients: A nationwide population study. Jt Bone Spine. 2012;79(4):375‐378. [DOI] [PubMed] [Google Scholar]
- 136. Yan S, Zhang P, Xu W, et al. Serum uric acid increases risk of cancer incidence and mortality: a systematic review and meta‐analysis. Mediators Inflamm. 2015;2015:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med. 2012;1(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Petersson B, Trell E. Raised serum urate concentration as risk factor for premature mortality in middle aged men: relation to death from cancer. Br Med J (Clin Res Ed). 1983;287(6384):7‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Levine W, Dyer AR, Shekelle RB, Schoenberger JA, Stamler J. Serum uric acid and 11.5‐year mortality of middle‐aged women: findings of the Chicago heart association detection project in industry. J Clin Epidemiol. 1989;42(3):257‐267. [DOI] [PubMed] [Google Scholar]
- 140. Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal‐induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60(1):281‐289. [DOI] [PubMed] [Google Scholar]
- 141. Schiltz C, Lioté F, Prudhommeaux F, et al. Monosodium urate monohydrate crystal‐induced inflammation in vivo: quantitative histomorphometric analysis of cellular events. Arthritis Rheum. 2002;46(6):1643‐1650. [DOI] [PubMed] [Google Scholar]
- 142. Rollins BJ. Monocyte chemoattractant protein 1: A potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today. 1996;2(5):198‐204. [DOI] [PubMed] [Google Scholar]
- 143. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7(3):311‐317. [DOI] [PubMed] [Google Scholar]
- 144. Matsukawa A, Miyazaki S, Maeda T, et al. Production and regulation of monocyte chemoattractant protein‐1 in lipopolysaccharide‐ or monosodium urate crystal‐induced arthritis in rabbits: roles of tumor necrosis factor alpha, interleukin‐1, and interleukin‐8. Lab Invest. 1998;78(8):973‐985. [PubMed] [Google Scholar]
- 145. Grainger R, McLaughlin RJ, Harrison AA, Harper JL. Hyperuricaemia elevates circulating CCL2 levels and primes monocyte trafficking in subjects with inter‐critical gout. Rheumatol (United Kingdom). 2013;52(6):1018‐1021. [DOI] [PubMed] [Google Scholar]
- 146. Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54(8):2688‐2696. [DOI] [PubMed] [Google Scholar]
- 147. Kanellis J, Watanabe S, Li JH, et al. Uric acid stimulates monocyte chemoattractant protein‐1 production in vascular smooth muscle cells via mitogen‐activated protein kinase and cyclooxygenase‐2. Hypertension. 2003;41(6):1287‐1293. [DOI] [PubMed] [Google Scholar]
- 148. Kang D‐H, Han L, Ouyang X, et al. Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005;25(5):425‐433. [DOI] [PubMed] [Google Scholar]
- 149. Dinarello CA, Ikejima T, Warner SJ, et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139(6):1902‐1910. [PubMed] [Google Scholar]
- 150. Dinarello CA. IL‐1: Discoveries, controversies and future directions. Eur J Immunol. 2010;40(3):599‐606. [DOI] [PubMed] [Google Scholar]
- 151. Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin I receptor antagonist‐deficient mice. J Exp Med. 2000;191(2):313‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin‐1‐receptor antagonist. N Engl J Med. 2009;360(23):2426‐2437. [DOI] [PMC free article] [PubMed] [Google Scholar]