

Abstract
Objective
Pulmonary arterial hypertension (PAH), one of the major complications of systemic sclerosis (SSc), is a rare disease with unknown etiopathogenesis and noncurative treatments. As mice deficient in P‐selectin glycoprotein ligand 1 (PSGL‐1) develop a spontaneous SSc‐like syndrome, we undertook this study to analyze whether they develop PAH and to examine the molecular mechanisms involved.
Methods
Doppler echocardiography was used to estimate pulmonary pressure, immunohistochemistry was used to assess vascular remodeling, and myography of dissected pulmonary artery rings was used to analyze vascular reactivity. Angiotensin II (Ang II) levels were quantified by enzyme‐linked immunosorbent assay, and Western blotting was used to measure Ang II type 1 receptor (AT1R), AT2R, endothelial cell nitric oxide synthase (eNOS), and phosphorylated eNOS expression in lung lysates. Flow cytometry allowed us to determine cytokine production by immune cells and NO production by endothelial cells. In all cases, there were 4–8 mice per experimental group.
Results
PSGL‐1−/− mice showed lung vessel wall remodeling and a reduced mean ± SD expression of pulmonary AT2R (expression ratio [relative to β‐actin] in female mice age >18 months: wild‐type mice 0.799 ± 0.508 versus knockout mice 0.346 ± 0.229). With aging, female PSGL‐1−/− mice had impaired up‐regulation of estrogen receptor α (ERα) and developed lung vascular endothelial dysfunction coinciding with an increase in mean ± SEM pulmonary Ang II levels (wild‐type 48.70 ± 5.13 pg/gm lung tissue versus knockout 78.02 ± 28.09 pg/gm lung tissue) and a decrease in eNOS phosphorylation, leading to reduced endothelial NO production. These events led to a reduction in the pulmonary artery acceleration time:ejection time ratio in 33% of aged female PSGL‐1−/− mice, indicating pulmonary hypertension. Importantly, we found expanded populations of interferon‐γ–producing PSGL‐1−/− T cells and B cells and a reduced presence of regulatory T cells.
Conclusion
The absence of PSGL‐1 induces a reduction in Treg cells, NO production, and ERα expression and causes an increase in Ang II in the lungs of female mice, favoring the development of PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare and progressive disease that mainly affects women. PAH is characterized by hypertrophic distal pulmonary vascular remodeling resulting from endothelial dysfunction, dysregulated vascular smooth muscle cell proliferation, and inflammation, which together promote medial thickening of pulmonary arteries and luminal obliteration 1. These pathologic events increase pulmonary vascular resistance and pulmonary artery pressure (PAP), leading to an increased hemodynamic load on the right ventricle (RV). The RV adapts with a compensatory increase in wall thickness and contractility 2, 3. PAH develops in 7–12% of patients with systemic sclerosis (SSc), constituting a leading cause of death 4, 5, 6. Indeed, SSc is a major cause of connective tissue disease (CTD)–associated PAH 4.
Several molecular mechanisms have been implicated in the control of pulmonary pressure and are dysregulated in PAH. Pulmonary artery endothelial cells (ECs) from patients with idiopathic PAH produce reduced amounts of nitric oxide (NO) 4. Angiotensin II (Ang II) plays a major role in the control of blood pressure and vascular tone in peripheral blood vessels 7, 8, 9. In this context, the binding of Ang II to Ang II receptor 1 (AT1R) induces vasoconstriction, while binding to AT2R triggers vasodilation 7. Thus, elevated levels of renin, angiotensin‐converting enzyme (ACE), Ang II, and AT1R have been observed in experimental models as well as in patients with pulmonary hypertension (PH) 10, 11, 12.
P‐selectin glycoprotein ligand 1 is a leukocyte receptor responsible for the initial contacts between white blood cells and endothelium. PSGL‐1 interacts with P‐, E‐, and L‐selectin, allowing leukocyte tethering and rolling before extravasation to the inflammatory foci 13. The PSGL‐1–P‐selectin interaction triggers a tolerogenic program in human monocyte‐derived dendritic cells, which drive Treg cell generation 14. Accordingly, disease exacerbation has been described in PSGL‐1–deficient (PSGL‐1−/−) mice in different experimental inflammatory models 15, 16, 17, 18, 19. More importantly, PSGL‐1−/− mice progressively develop an autoimmune syndrome which shares multiple features with human SSc, such as autoantibody production, dermal fibrosis, and vascular damage 15.
Given that PSGL‐1−/− mice develop an autoimmune syndrome similar to SSc, and that there are not good mouse models for SSc associated with PAH (SSc‐PAH), we questioned whether, as a part of the scleroderma‐like syndrome, these mice develop PH. Interestingly, Doppler echocardiography is now considered a validated noninvasive method to assess the systolic pressure in the pulmonary artery and right ventricle 20, 21. The reduction in the ratio of pulmonary artery acceleration time (PAAT) to ejection time (ET) is associated with high PAP in humans and in mice 20, 21, 22, 23. In the present study, we analyzed the lungs and heart of PSGL‐1−/− mice, finding pulmonary small vessel remodeling and increased PAP in female mice, and we examined the possible molecular events implicated in this phenotype.
MATERIALS AND METHODS
Animals
C57BL/6 PSGL‐1−/− mice were kindly provided by Dr. M. K. Wild and Dr. D. Vestweber (Max Planck Institute for Molecular Biomedicine, Münster, Germany). Wild‐type (WT) C57BL/6 mice were obtained from The Jackson Laboratory and were backcrossed with PSGL‐1−/− mice. Mice were kept in pathogen‐free conditions at the Animal Facility of the School of Medicine, Universidad Autónoma de Madrid and the Animal Facility of the Centro Nacional de Investigaciones Cardiovasculares. Mice were killed by cervical dislocation, and internal organs were extracted for analysis. All experiments and breeding were performed in accordance with national and institutional guidelines for animal care (EU Directive 2010/63/EU for animal experiments). The experimental procedures were approved by the Director General de Medio Ambiente of Madrid (ref. PROEX 69/14 and PROEX 162/15).
Immunohistochemistry and vessel wall thickness calculation
Immunohistochemistry analysis using antibody against murine α‐smooth muscle actin (α‐SMA) was performed in paraffin‐embedded lung sections. Small blood vessel (diameter <50 μm) wall thickness was calculated by measuring the internal and total vessel diameter of anti–α‐SMA–stained lungs and calculating the vessel wall area occupied by the vessel wall using ImageJ (National Institutes of Health). Vessels were clustered according to their diameter, and the mean wall thickness area was calculated.
Transthoracic Doppler echocardiography and Fulton index calculation
All transthoracic echocardiography measurements were obtained with an echocardiography system (VEVO 2100; Visualsonics) and an 18–38 ‐Hz ultrasound probe. Chest hair was removed with hypoallergenic depilatory cream, and animals were anesthetized with a continuous influx of 2% isoflurane with an oxygen flow rate of 1.5 liter/minute. Pulsed wave Doppler mode was used to measure the PAAT and ET of blood flow in the pulmonary artery at the level of the pulmonary valve. The PAAT:ET ratio was then calculated and used as an indirect measure of systolic pulmonary artery blood pressure. M‐mode in transversal and longitudinal axis was used to measure the systolic and diastolic left ventricular internal diameters (LVIDs) and the longitudinal length of the LV. LV end systolic volume (LVESV) and LV end diastolic volume (LVEDV) were estimated as follows: LVESV = (7/[2.4 + systolic LVID]) × systolic LVID3; LVEDV = (7/[2.4 + diastolic LVID]) × diastolic LVID3. The ejection fraction was then calculated as the ratio (LVEDV − LVESV):LVEDV. Diastolic function was assessed using analyses of transmitral blood flow by pulsed wave Doppler. E wave (early ventricular filling) and A wave (late ventricular filling caused by atrial contraction) velocity were measured, and the E/A ratio was calculated.
Systemic pressure measurements
Systemic arterial pressure was measured using a BP‐2000 system (Visitech Systems), and data were analyzed with BP‐2000 Analysis Software. Eighteen‐month‐old female WT and PSGL‐1−/− mice were subjected to pressure measurement for a week for protocol habituation. Next, consecutive measurements were obtained over 5 days, and the mean systolic pressure value was calculated.
Vascular reactivity
Murine pulmonary arteries were carefully dissected free of surrounding tissue and cut into rings (1.8–2 mm in length). Vessel segments were mounted on a wire myograph in Krebs physiologic solution. Buffer solutions were continuously bubbled with 21% O2, 5% CO2, and 74% N2 (PO2¼ 17–19 kPa) 24, and stretched to a transmural pressure equivalent to 30 mm Hg. Contractility was recorded with an isometric force transducer and a displacement device coupled with a digitalization and data acquisition system (PowerLab). To confirm smooth muscle viability, arteries were first stimulated by raising the K+ concentration of the buffer to 80 mmoles/liter. Thereafter, concentration‐response curves to acetylcholine (10−9–10−5 moles/liter) and sodium nitroprusside (10−11–10−5 moles/liter) were performed using cumulative addition to analyze the endothelium‐dependent and endothelium‐independent vasodilatation, respectively. For the reactive oxygen species (ROS) scavenging experiments, the concentration of superoxide dismutase (SOD)–polyethylene glycol (Sigma) used was 50 units/ml.
Enzyme‐linked immunosorbent assay (ELISA)
The left lung was mechanically disrupted in 1× phosphate buffered saline (PBS). After 4 freeze/defreeze cycles to break cell membranes, samples were centrifuged at 5,000g for 5 minutes at 4ºC, and supernatants were recovered. Ang II concentrations were measured using an Ang II ELISA kit (CSB‐E04495 ml; Cusabio).
Western blotting
The right lung was frozen, pulverized, and diluted in radioimmunoprecipitation assay buffer (1% Triton X‐100, 0.24M sodium deoxycholate, 0.35M sodium dodecyl sulfate in 1× Tris buffered saline) with protease and phosphatase inhibitors. Lung lysates were used for Western blot assays. The following primary antibodies were used: rabbit anti‐AT1R and anti‐AT2R (1:1,000; Novus Biologicals), rabbit anti–β‐actin (1:5,000; Sigma), rabbit antivinculin (1:2,000; Sigma), mouse anti‐eNOS (1:1,000; BD Pharmingen), mouse anti–phosphorylated eNOS Ser1176 (1:1,000; BD Pharmingen), mouse anti–estrogen receptor α (ERα) (1:1,000; R&D), rabbit anti‐ERβ (1:1,000; ThermoFisher Scientific), and mouse anti‐GAPDH (1:1,000; Biolegend).
Bound antibodies were visualized by chemiluminescence with a Luminata Forte Western HRP Substrate (Merck KGaA) using either a horseradish peroxidase–conjugated goat anti‐rabbit or goat anti‐mouse IgG secondary antibody. Band intensity was analyzed using ImageJ, and results were normalized to the expression of β‐actin, GAPDH, or vinculin, as loading controls.
Flow cytometry
Lungs were weighed, minced into ~1‐mm2 pieces, and digested for 1 hour with 1 mg/ml collagenase A (Sigma), 2.5 mg/ml Dispase II (Roche), and 40 μg/ml DNase (Sigma) in RPMI 1640 medium. Cell aggregates and undigested pieces of tissue were eliminated using a 70‐μm cell strainer (BD Falcon). Cells were then washed with 25 ml of PBS, 0.5% bovine serum albumin, 5 mM EDTA, concentrated in 700 μl, and filtered through a 30‐μm cell strainer (BD Pharmingen). After incubation with 1:200 Fc Block (BD Pharmingen), cells were stained with the cocktail of surface antibodies for 15 minutes at 4ºC. Subsequently, cells were permeabilized with 2 ml of fluorescence‐activated cell sorting (FACS) Lysing Solution (BD Pharmingen) for 15 minutes, washed, and stained for 30 minutes at 4ºC with a cocktail of antibodies directed against intracellular cytokines. For intranuclear FoxP3 staining, a fluorescein isothiocyanate (FITC)–conjugated anti‐mouse/rat FoxP3 staining set was used according to the instructions of the manufacturer (eBioscience). Flow cytometry was performed using a FACSCanto II and FACS Diva Software (BD Pharmingen).
Cell gating strategy and flow cytometry reagents
T cells and B cells were gated as CD45.2+CD3+CD19−/B220− and CD45.2+CD19+CD3− or CD45.2+B220+CD3−, respectively. Neutrophils were gated as CD45+Ly−6G+ and Ly−6C+ (see Supplementary Figure 1, on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract). Alveolar macrophages were gated as CD45.2+CD11c+Siglec F+, interstitial macrophages were gated as CD45.2+MHC‐II+Siglec F−CD11c−CD11b+, and dendritic cells were gated as CD45.2+Siglec F−MHC‐II+CD11c+ (Supplementary Figure 1). The expression of interleukin‐10 (IL‐10), IL‐17, and interferon‐γ (IFNγ) was analyzed in these subsets. Lung ECs were identified as CD45.2−CD31+. Antibodies used for the identification of the aforementioned cell populations were as follows: phycoerythrin (PE)–Cy7–conjugated CD11c (1:50; eBioscience), PE–Cy7–conjugated CD3ε (1:200; eBioscience), allophycocyanin (APC)–conjugated CD31 (1:200; BD Pharmingen), FITC‐ and BV421‐conjugated CD45.2 (1:200; BD Pharmingen), APC–Cy7–conjugated IL‐17A (1:200; BD Pharmingen), APC‐conjugated IFNγ (1:50; Miltenyi Biotec), VioBlue‐conjugated CD19 (1:50; Miltenyi Biotec), APC–Vio770–conjugated B220 (1:50; Miltenyi Biotec), PerCP–Cy5.5–conjugated IL‐10 (1:100; BioLegend), Gr‐1– and APC‐conjugated Ly‐6G/Ly‐6C (1:100; BD Pharmingen), APC‐conjugated CD11b (1:50; Miltenyi Biotec), PerCP–Vio 700–conjugated major histocompatibility complex class II (MHC‐II) (1:100; Miltenyi Biotec), and PE‐conjugated Siglec F (1:100; BD Pharmingen). CountBright absolute counting beads (Invitrogen) were used for the quantification of absolute cell numbers.
Intracellular NO evaluation
After blocking and surface molecule staining (CD45 and CD31), cells were washed and incubated with the NO‐sensing fluorescent probe diaminorhodamine‐4M acetoxymethyl ester (DAR‐4M AM) (5 μm; Sigma) in PBS for 30 minutes at 37ºC. Finally, cells were fixed and analyzed with a FACSCanto II cytometer. Cells that were not incubated with the probe were used as negative controls for fluorescence. Two groups of cells could be distinguished according to the fluorescence levels at Em 580 nm. The fold change in the DAR‐4M AM mean fluorescence intensity (MFI) for ECs from WT mice and PSGL‐1−/− mice was calculated as the ratio of the MFI obtained in each mouse in relation to the average MFI for all WT mice analyzed.
Statistical analysis
Statistical significance between 2 groups was calculated using Student's 2‐tailed t‐test for parametric variables and the Mann‐Whitney U test for nonparametric variables. Statistical significance between 3 groups was calculated using one‐way analysis of variance (ANOVA) with the Bonferroni post hoc test. For dose‐dependent relaxation studies, statistical significance was calculated using two‐way ANOVA. P values less than 0.05 were considered significant. All statistical analyses were performed using SPSS, version 15.0 (IBM).
RESULTS
Altered echocardiographic parameters consistent with PH in PSGL‐1−/− mice
Given the elevated rate of death in PSGL‐1−/− mice after reaching 1 year of age 15, echocardiography was used to measure the PAAT:ET ratio (Figure 1A), an indirect evaluation of pulmonary pressure. Since preliminary data suggested differences in the PAAT:ET ratio between WT and PSGL‐1−/− mice, follow‐up transthoracic Doppler echocardiography was performed on WT and PSGL‐1−/− littermates between 1.5 and 18 months of age. A recurrent tendency toward a reduced PAAT:ET ratio was observed in female PSGL‐1−/− mice from 3 months of age (Figure 1B). Considering that a group of 3 female PSGL‐1−/− mice (33% of all female PSGL‐1−/− mice) died between 15 and 18 months of age, we differentiated between the dead and survivor groups. The group of PSGL‐1−/− mice that died prematurely showed increased PAP, detected by a reduced PAAT:ET ratio, compared to WT mice and to the surviving group of PSGL‐1−/− mice (Figure 1B). PSGL‐1−/− mice that survived maintained a PAAT:ET ratio below that observed in WT mice. In contrast, both male WT and male PSGL‐1−/− mice exhibited similar PAAT:ET ratios throughout the experiment (Figure 1C).
Figure 1.
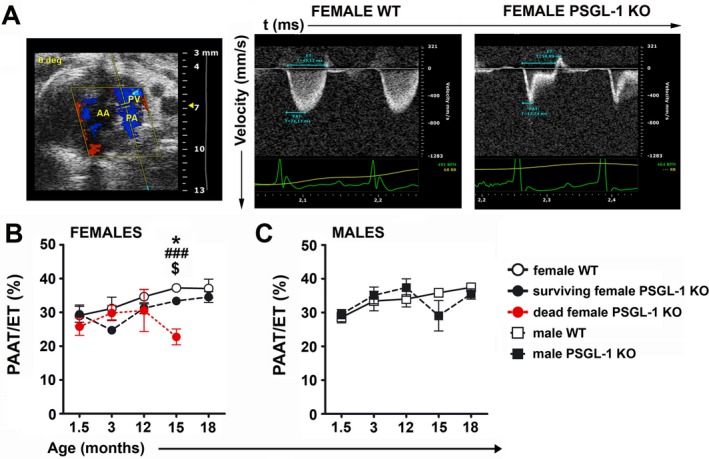
Development of pulmonary hypertension with aging in female PSGL‐1−/− mice. A, B‐mode showing the echocardiographic plane used for Doppler pulmonary flow acquisition (left), and representative Doppler pulmonary artery flow of female wild‐type (WT) mice (middle) and PSGL‐1–knockout (KO) mice (right). B, Longitudinal study of the pulmonary artery acceleration time:ejection time (PAAT:ET) ratio between 1.5 and 18 months of age in female WT mice (n = 4), surviving female PSGL‐1−/− mice (n = 6), and female PSGL‐1−/− mice that died prematurely (n = 3). C, Longitudinal study of the PAAT:ET ratio between 1.5 and 18 months of age in male WT mice (n = 6) and male PSGL‐1−/− mice (n = 6). Results are representative of 3 replicate experiments. Values are the mean ± SD. * = P < 0.05, WT versus surviving PSGL‐1−/− mice; ### = P < 0.005, WT versus dead PSGL‐1−/− mice; $ = P < 0.05, surviving PSGL‐1−/− versus dead PSGL‐1−/− mice, all by one‐way analysis of variance with Bonferroni post hoc test. AA = ascending aorta; PV = pulmonary valve; PA = pulmonary artery.
No differences in the ejection fraction and the E/A ratio were found between the 3 groups (Supplementary Figures 2A and B, http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract). In addition, systolic arterial pressure and the PAAT:ET ratio were measured in another cohort of aged female WT and PSGL‐1−/− mice, and WT and KO mice showed similar systolic arterial pressure (Supplementary Figure 2C).
Remodeling of pulmonary small vessels in PSGL‐1−/− mice
The presence of pulmonary vascular remodeling that could explain the increase in pulmonary pressure was analyzed. Immunohistochemical staining, using an antibody against α‐SMA, of lung sections from female WT and PSGL‐1−/− mice revealed a thicker medial wall of small vessels in PSGL‐1−/− mouse lungs (Figure 2A). Quantification demonstrated a significant increase of the relative wall area in almost all groups of PSGL‐1−/− mouse vessels, independent of age (Figure 2B).
Figure 2.
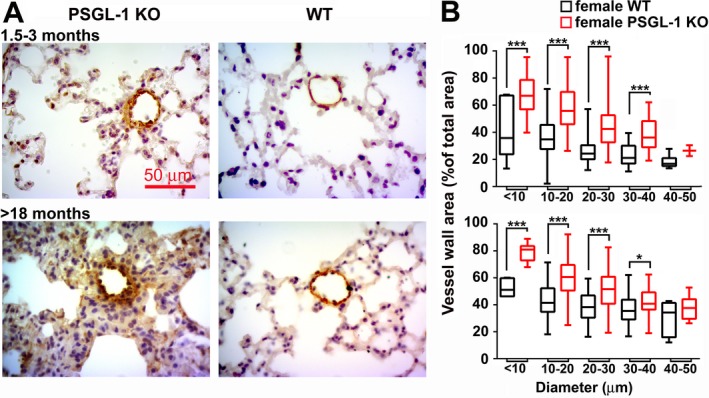
Vascular remodeling in pulmonary small vessels from female PSGL‐1−/− mice. A, Representative photomicrographs of anti–α–smooth muscle actin–immunostained lung sections from mice ages 1.5–3 months and >18 months. B, Percentage of vessel wall area in <50‐μm–diameter pulmonary blood vessels in mice ages 1.5–3 months (top) and >18 months (bottom). Fewer than 150 vessels were analyzed for each age group (n = 5–7 per group). Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th and 90th percentiles. * = P < 0.05; *** = P < 0.005, by Student's 2‐tailed t‐test. See Figure 1 for definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract.
Decreased endothelial NO‐dependent relaxing response in pulmonary arteries of aged female PSGL‐1−/− mice
Vascular reactivity in pulmonary and mesenteric arterial rings was assessed using wire myography. When compared to female WT littermates, pulmonary arteries isolated from female PSGL‐1−/− mice showed increased vasoconstriction in response to 80 mM KCl (Figure 3A). The vasodilating response to acetylcholine was impaired in PSGL‐1−/− mouse arterial rings (Figure 3B); however, the addition of an external NO donor (sodium nitroprusside) was sufficient to fully relax both WT and PSGL‐1−/− mouse arterial rings (Figure 3C). Conversely, vascular reactivity did not differ between mesenteric arterial rings of PSGL‐1−/− and WT littermates (Supplementary Figures 3A and B, http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract), which suggests that the endothelial dysfunction is restricted to the pulmonary circulation. The inhibition of ROS production by addition of SOD did not restore the relaxation capability of PSGL‐1−/− mouse lung arteries, thus ruling out the notion of NO scavenging by ROS (Supplementary Figure 3C).
Figure 3.
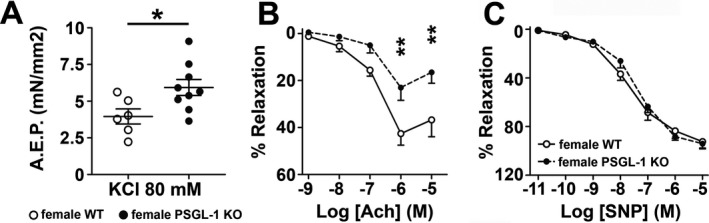
Vascular response to vasoconstrictor and vasodilator agents. A, Contractile response to KCl in pulmonary arterial rings obtained from female mice >18 months of age. Symbols represent individual mice; bars show the mean ± SEM. B and C, Vasodilating response to acetylcholine (Ach) (B) and sodium nitroprusside (SNP) (C) in pulmonary arterial rings obtained from female mice >18 months of age. Values are the mean ± SEM. * = P < 0.05; ** = P < 0.01, by Student's 2‐tailed t‐test (A) and by two‐way analysis of variance (B). AEP = active effective pressure (see Figure 1 for other definitions).
Reduced pulmonary endothelial NO production and eNOS phosphorylation in aged female PSGL‐1−/− mice
To understand the molecular mechanisms responsible for the impaired EC‐dependent relaxation of pulmonary arteries, NO production by ECs was assessed. The percentage of NO‐producing lung ECs was reduced in aged female PSGL‐1−/− mice (Figures 4A–C), although this was significant only within the highest NO‐producing EC subset (Figure 4C). Additionally, the MFI for the NO‐sensing probe DAR‐4M AM was lower in lung ECs from aged PSGL‐1−/− mice than in those from aged WT mice (Figure 4D). These changes were not observed in 3‐month‐old female mice (Supplementary Figures 4A–C, http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract). Remarkably, although eNOS protein expression was measured in lung lysates and no differences were found between female WT and PSGL‐1−/− mice (Figure 4E), reduced levels of eNOS phosphorylation at the Ser1176 activator site were found in the lung lysates of aged female PSGL‐1−/− mice (Figure 4F).
Figure 4.
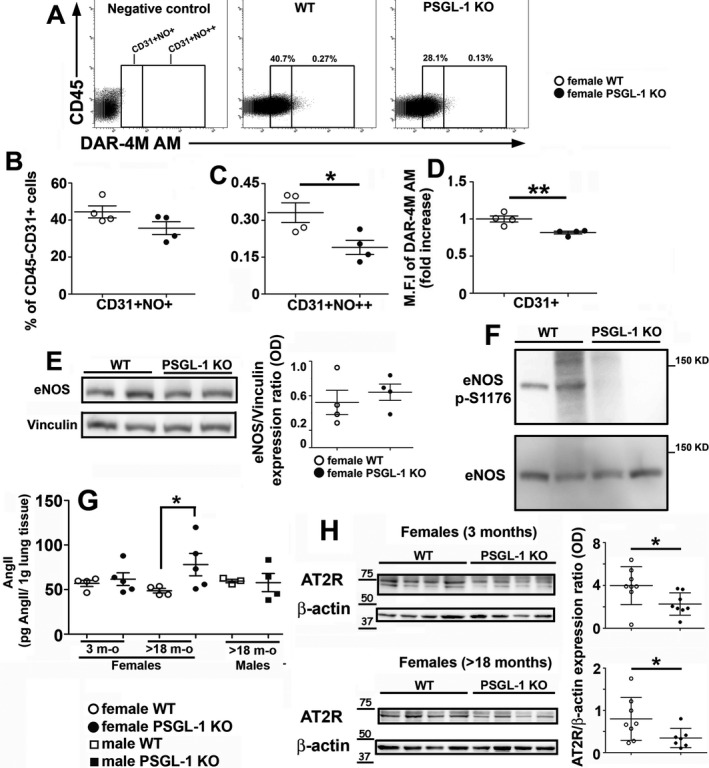
Quantification of nitric oxide (NO) production and assessment of endothelial cell nitric oxide synthase (eNOS) phosphorylation. A, Dot plots of diaminorhodamine‐4M acetoxymethyl ester (DAR‐4M AM) signal in female mice >18 months of age. Highly positive population is shown to the right of the internal vertical line. B and C, Percentage of lung endothelial cells producing moderate (B) or high (C) amounts of NO measured in WT and PSGL‐1−/− female mice >18 months of age. D, Fold change of mean fluorescence intensity (MFI) for the NO‐sensing probe DAR‐4M AM measured in lung endothelial cells of female WT and PSGL‐1−/− mice >18 months of age (n = 4–6 per group). E, Western blot showing eNOS expression in the lungs of female mice >18 months of age (left) and densitometric quantification (right). Vinculin was used as a loading control. F, Western blot showing phosphorylated eNOS expression in the lungs of female mice >18 months of age. G, Angiotensin II (Ang II) concentration in lung lysates from mice (n = 5 per group). H, Immunoblots showing Ang II type 2 receptor (AT2R) expression in the lungs of mice (n = 7–8 per group) and densitometric quantification. β‐actin was used as a loading control. In B–E, G, and H, symbols represent individual mice; bars show the mean ± SEM (B–E and G) or the mean ± SD (H). * = P < 0.05; ** = P < 0.01, by Mann‐Whitney U test. See Figure 1 for other definitions.
Increased pulmonary levels of Ang II and reduced expression of AT2R in aged female PSGL‐1−/− mice
No differences were found in the pulmonary concentration of Ang II between young female WT and PSGL‐1−/− mice; however, the mean ± SEM Ang II concentration was significantly higher in aged female PSGL‐1−/− mice (78.02 ± 28.09 pg/gm lung tissue) than in aged female WT mice (48.70 ± 5.13 pg/gm lung tissue) (Figure 4G). In contrast, Ang II levels in aged males were similar between the 2 genotypes (Figure 4G).
Regarding Ang II receptors, the mean ± SD expression of AT2R was lower in both young and aged female PSGL‐1−/− mice than in WT mice (expression ratio [relative to β‐actin] in young WT 3.983 ± 1.765 versus young KO 2.266 ± 1.045; in aged WT 0.799 ± 0.508 versus aged KO 0.346 ± 0.229) (Figure 4H), whereas no differences in expression were found for AT1R in any case (Supplementary Figure 4D, http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract).
Increased IFNγ‐producing T cells, B cells, and macrophages and reduced Treg cells in the lungs of aged female PSGL‐1−/− mice
Because PSGL‐1 is a leukocyte receptor, the pulmonary immune system was analyzed in female WT and PSGL‐1−/− mice. The total number of CD45+ cells was increased in the bronchoalveolar lavage (BAL) fluid and lung tissue of young and aged female PSGL‐1−/− mice (Figures 5A and B). Deeper analysis showed that the BAL fluid in young female mice had higher numbers of alveolar macrophages and neutrophils, and aged female mice had an increased number of neutrophils (Figure 5A). In the lung tissue, female PSGL‐1−/− mice showed increased numbers of neutrophils and T cells at a young age and an increased number of neutrophils when elderly (Figure 5B). Because IFNγ has been associated with vascular dysfunction 25, 26, the presence of IFNγ‐producing cells was analyzed in the lung tissue. Interestingly, IFNγ‐producing interstitial macrophages and T cell populations were increased in the lung tissue of young female PSGL‐1−/− mice, and the IFNγ‐producing T cell subset was increased in aged female PSGL‐1−/− mice (Figure 5C and Supplementary Figure 5, http://onlinelibrary.wiley.com/doi/10.1002/art.41100/abstract). Notably, the analysis of B cell and T cell populations in the lung tissue showed that aged female PSGL‐1−/− mice had reduced percentages of IL‐10–producing T cells and increased IFNγ‐producing T cells and B cells (Figure 5D and Supplementary Figure 5), while the percentage of IL‐17–producing populations was similar in WT and KO mice (Supplementary Figures 5A and B). Importantly, the percentage of Treg cells was highly reduced in the T cell population, which increased the Th1/Treg cell balance in the lung tissue of aged female PSGL‐1−/− mice (Figure 5D and Supplementary Figure 5A).
Figure 5.
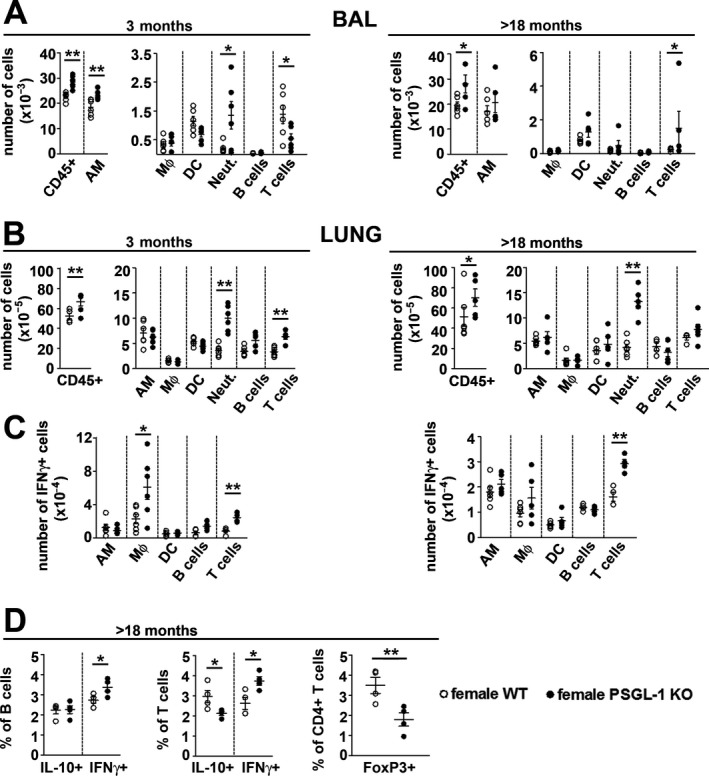
Immune system analysis of the lungs of female wild‐type (WT) and PSGL‐1−/− mice. A, Absolute numbers of immune cell populations isolated from the bronchoalveolar lavage (BAL) fluid of female WT and PSGL‐1−/− mice. B, Absolute numbers of immune cell populations found in the whole lungs of female WT and PSGL‐1−/− mice. C, Absolute numbers of interferon‐γ (IFNγ)–positive cells obtained from the whole lungs of female WT and PSGL‐1−/− mice. D, Percentage of interleukin‐10 (IL‐10)– and IFNγ‐producing cells in the B cell and T cell populations and the frequency of Treg cells (FoxP3+) in the CD4+ T cell subset in lungs isolated from mice (n = 4–6 per group). Symbols represent individual mice; bars show the mean ± SEM. * = P < 0.05; ** = P < 0.01, by Student’s 2‐tailed t‐test with Bonferroni post hoc test. AM = alveolar macrophages; MФ = interstitial macrophages; DC = dendritic cells; Neut. = neutrophils.
Reduced ERα expression in aged female PSGL‐1−/− mice
To elucidate the molecular mechanisms responsible for the sex bias implicated in the development of pulmonary hypertension in female PSGL‐1−/− mice, the expression of estrogen receptors was quantified in lung lysates. Aging induced a reduction in the expression of ERβ in a genotype‐ and sex‐independent manner, with no differences between WT and KO mice (Figures 6A–C). In contrast, ERα expression increased with aging in female WT mice but not in female PSGL‐1−/− mice, while aging did not affect the expression of ERα in males, and no differences between male WT and PSGL‐1−/− mice were found (Figures 6D–F).
Figure 6.
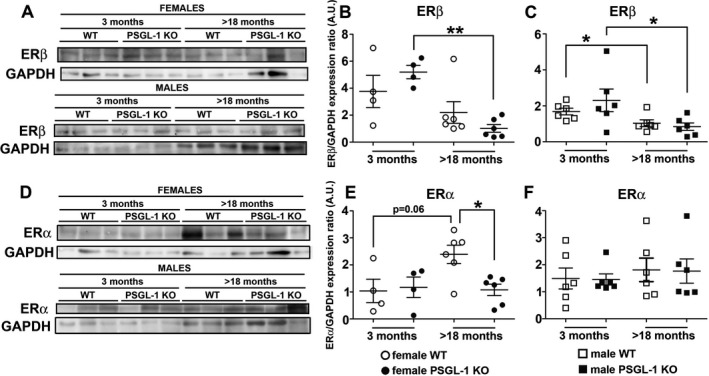
Analysis of estrogen receptor β (ERβ) and ERα expression in the lungs of mice. A–C, Western blot (A) and quantification of the expression of ERβ in the lungs of female (B) and male (C) wild‐type (WT) and PSGL‐1−/− mice. D–F, Representative Western blot (D) and quantification of the expression of ERα in the lungs of female (E) and male (F) WT and PSGL‐1−/− mice. Symbols represent individual mice; bars show the mean ± SEM. * = P < 0.05; ** = P < 0.01, by Mann‐Whitney U‐test.
DISCUSSION
PAH is a particularly severe life‐threatening complication of some CTDs that leads to RV remodeling, right heart failure, and premature death 27. Although many efforts have been made in searching for an efficient treatment for PAH, no curative therapy is available. Our work shows that female mice deficient in PSGL‐1 exhibit reduced expression of pulmonary AT2R and thickened small vessel walls. With aging, female PSGL‐1−/− mice show increased pulmonary levels of Ang II, which coincides with decreased eNOS phosphorylation and reduced NO production by lung ECs and leads to RV dysfunction and death. Interestingly, aged female PSGL‐1−/− mice have impaired ERα up‐regulation.
The increase in relative wall vessel area in lung vasculature has been described as a histologic marker of PAH in both animal models and patients 1, 4, 27 and is thought to be one of the first events that occurs in the development of PAH 2, 12. The vascular remodeling observed in PSGL‐1−/− mice might cause an increase in the lung vascular resistance to blood flow, the main consequence of which would be the elevation of pressure in the pulmonary artery.
Transthoracic Doppler echocardiography is a noninvasive diagnostic tool for patients with suspected PAH, providing information not only about diagnosis but also about the causes or consequences of PAH 27, 28. This method has been validated for estimating PAP in PAH patients and in rat and mouse models 20, 21, 22, 23, 29 and is becoming increasingly more relevant in murine models of heart diseases 30. Using this echocardiography modality, and in accordance with the pulmonary vessel remodeling, we found a reduced PAAT:ET ratio in the pulmonary arteries of 3‐month‐old female PSGL‐1−/− mice, suggesting that PSGL‐1−/− mice are susceptible to PH at young age. Importantly, female PSGL‐1−/− mice had preserved ejection fraction and E/A ratio, indicating that the incremental increase in PAP was not a consequence of an alteration in LV systolic/diastolic function or an elevated systemic arterial pressure, which suggests primary PAH.
Reduced expression of AT2R, together with increased Ang II concentration, reduced eNOS phosphorylation at Ser1176, and NO production by ECs, was found in the lungs of aged female PSGL‐1−/− mice. This might explain the higher contractility and reduced relaxation capability of lung arteries at this age, which ultimately leads to an elevated flow resistance and then to increased pulmonary pressure and a reduced PAAT:ET ratio. In this context, pentraxin 3, a soluble ligand of P‐selectin that interferes with the PSGL‐1–P‐selectin interaction 31, 32, induced endothelial dysfunction and increased blood pressure 32, suggesting that PSGL‐1 binding to P‐selectin may be involved in the maintenance of correct endothelial function and integrity.
Molecular systems involved in the control of vascular tone, such as endothelin 1, Ang II, and NO, have been described as being altered in several animal models and in patients with PAH 4, 7, 27. In our model, we found elevated lung concentrations of Ang II in aged female PSGL‐1−/− mice. Thus, hypoxic and monocrotaline‐treated rats showed increased Ang II and AT1R levels 11, 33, 34, and patients with PAH showed higher levels of pulmonary Ang II due to increased activity of ACE, as well as increased expression and signaling of AT1R, resulting in augmented vascular smooth muscle cell (VSMC) proliferation 35. Inhibitors of the renin–angiotensin–aldosterone system (RAAS) have been shown to have an effect in reducing PAP and other PAH signs in some animal models and in pilot studies with small patient cohorts, highlighting the role of RAAS in the pathology of PAH. For example, treatment with losartan (AT1R antagonist) or captopril (ACE inhibitor) was able to reduce mean PAP, RV hypertrophy, and lung VR in rats exposed to hypobaric hypoxia for 14 days 35, 36. Although no differences in ACE expression were found in the lungs of female WT and PSGL‐1−/− mice, the increased levels of pulmonary Ang II found in aged female PSGL‐1−/− mice might be explained by enhanced ACE activity, which has previously been reported in patients with PAH and in animal models 35, 37.
NO is a critical regulator of vascular homeostasis, not only by modulating vascular tone but also by controlling VSMC proliferation and migration and leukocyte adhesion to endothelium 27, 38. Accordingly, reduced NO production by PSGL‐1−/− mouse lung ECs, together with a reduction of eNOS phosphorylated on Ser1176 in lung lysates, suggests an impairment of eNOS activation 39. Interestingly, the reduction of NO is not due to NO sequestering by ROS. It has been reported that an increase in Ang II could lead to eNOS uncoupling, which results in reduced NO production 25.
Several studies have highlighted the relevant role of inflammation and the immune system in endothelial dysfunction 25, 26, 40. Importantly, the Treg cell population is reduced and the balance between effector cells in each leukocyte subset is altered in aged PSGL‐1−/− mice, as previously described in the colon and skin of these mice (15,16). In this context, it was observed that vascular dysfunction in aortic ECs can be mediated by inflammatory monocytes and natural killer cells producing IFNγ and IL‐12 in an Ang II–dependent manner 25, 26. A similar mechanism could be operating in the pulmonary vasculature of female PSGL‐1−/− mice, in which aging increases IFNγ production in B cells and T cells as well as pulmonary Ang II levels that can account for the endothelial dysfunction with the ensuing reduction in the endothelial NO. Moreover, we found a reduction in the lung Treg cell population. In this regard, hypoxic mice treated with Treg cells showed reduced RV systolic pressure and Fulton index scores, accompanied by a reduction of the expression of proinflammatory cytokines 41. Furthermore, patients with CTD‐associated PAH showed reduced numbers of circulating Treg cells, illustrating the importance of this T cell subset in the pathogenesis of PAH 42.
The mechanisms that mediate sex bias in pulmonary hypertension have not yet been described. It has been suggested that17β estradiol exerts vasoprotective actions through both ERα and ERβ 43. In ECs, ERα promotes eNOS phosphorylation in a phosphatidylinositol 3‐kinase– and Akt‐dependent manner 44, 45, 46, 47, 48. In addition, treatment with Ang II promotes vascular adhesion and migration of leukocytes, which can be abolished through treatment with 17β estradiol 49, 50 and exacerbated by incubation with l‐N G‐nitroarginine methyl ester 51. Moreover, coculture with Treg cells induces the up‐regulation of ERα and ERβ in human cardiac microvascular ECs and increases culture supernatant concentrations of plasma prostacyclin and IL‐10 52. In female PSGL‐1−/− mice, the reduction in the percentage of Treg cells could explain why the pulmonary levels of ERα were not increased with aging, declining at the same time as the phosphorylation of eNOS and NO production. Our data also indicate that the increase in ERα expression along with aging may prevent the increase of Ang II in female WT mice, which has recently been described in the context of hepatic ischemia‐reperfusion injury 53. Notably, ERα expression in males is not regulated by aging, suggesting that it is not as crucial in males as in females to control Ang II elevation and to increase NO production. These observations may explain why female PSGL‐1−/− mice develop PAH. Since PSGL‐1 might participate in the control of ERα expression with aging, it would be of great interest to understand the molecular mechanisms implicated.
Our findings demonstrate that, with aging, female PSGL‐1−/− mice develop PH as part of an SSc‐like autoimmune syndrome. Our study highlights the importance of leukocyte–endothelium interactions for the maintenance of vascular homeostasis in lungs and protection against PAH. Leukocytic deficiency of PSGL‐1, which interacts with P‐selectin and E‐selectin on the surface of activated endothelium, reduces the presence of Treg cells in the lungs and the expression of ERα in females and also promotes endothelial dysfunction characterized by reduced vasodilation response due to impaired NO production. This impairment in endothelial function leads to vascular remodeling, PH, and, ultimately, premature death in mice.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Urzainqui had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
González‐Tajuelo, Urzainqui.
Acquisition of data
González‐Tajuelo, de la Fuente‐Fernández, Morales‐Cano, Muñoz‐Callejas, González‐Sánchez, Silván, Serrador, Cadenas, Barreira, Espartero‐Santos, Jiménez‐Borreguero, Urzainqui.
Analysis and interpretation of data
González‐Tajuelo, Serrador, Gamallo, Vicente‐Rabaneda, Castañeda, Pérez‐Vizcaíno, Cogolludo, Jiménez‐Borreguero, Urzainqui.
Supporting information
ACKNOWLEDGMENTS
We thank the UAM and CNIC animal facilities for animal breeding and care. We are indebted to Ana Vanesa Alonso and Lorena Flores (Vascular Imaging Unit at CNIC) for performing echocardiography examinations. We also thank the Cytometry Unit and Statistical and Methodological Support Unit of the Hospital de la Princesa for technical support. We thank Dr. Kenneth McCreath for manuscript editing.
Supported by the Spanish Ministry of Economy and Competitiveness (grants SAF2015‐69396‐R, SAF2011‐28150, and SAF2014‐55399) and the Spanish Ministry of Health and Instituto de Salud Carlos III (cofinanced by Fondos FEDER; grants AC17‐00027, FIS‐PI14‐01698, FIS‐PI17‐01819, and FIS‐PI12‐01578, Red de Investigación de Enfermedades Reumáticas [RIER] RD12/0009/0017). The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Ministerio de Economía, Industria y Competitividad, and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence (SEV‐2015‐0505).
1Rafael González‐Tajuelo, PhD, María de la Fuente‐Fernández, MS, Antonio Muñoz‐Callejas, MS, Elena González‐Sánchez, PhD, Javier Silván, MS, Marina Espartero‐Santos, BS, Carlos Gamallo, PhD, MD, Esther F. Vicente‐Rabaneda, PhD, MD, Ana Urzainqui, PhD: Fundación de Investigación Biomédica‐Hospital de la Princesa, IIS‐Princesa, Servicio de Inmunlogía, Madrid, Spain; 2Daniel Morales‐Cano, PhD, Bianca Barreira, BS, Francisco Pérez‐Vizcaíno, PhD, Ángel Cogolludo, PhD: University Complutense of Madrid School of Medicine and Ciber Enfermedades Respiratorias, Madrid, Spain; 3Juan Manuel Serrador, PhD: Centro de Biología Molecular Severo Ochoa (CBMSO) and Instituto de Física Teórica CSIC/Universidad Autónoma de Madrid (UAM), Madrid, Spain; 4Susana Cadenas, PhD: Fundación de Investigación Biomédica‐Hospital de la Princesa, IIS‐Princesa, and CBMSO, CSIC‐UAM, Madrid Spain; 5Santos Castañeda, PhD, MD: Fundación de Investigación Biomédica‐Hospital de la Princesa, IIS‐Princesa, and Catedra UAM‐ROCHE, Madrid, Spain; 6Luis Jesús Jiménez‐Borreguero, MD: Hospital de la Princesa and Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain.
Ms de la Fuente‐Fernández and Dr. Morales‐Cano contributed equally to this work.
No potential conflicts of interest relevant to this article were reported.
REFERENCES
- 1. Farber H, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004;351:1655–65. [DOI] [PubMed] [Google Scholar]
- 2. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci 2016;17:E761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vonk Noordegraaf A, Westerhof B, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017;69:236–43. [DOI] [PubMed] [Google Scholar]
- 4. Montani D, Günther S, Dorfmüller P, Perros F, Girerd B, Garcia G, et al. Pulmonary arterial hypertension [review]. Orphanet J Rare Dis 2013;8:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Derrett‐Smith EC, Dooley A, Gilbane AJ, Trinder SL, Khan K, Baliga R, et al. Endothelial injury in a transforming growth factor β–dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum 2013;65:2928–39. [DOI] [PubMed] [Google Scholar]
- 6. Rhee RL, Gabler NB, Praestgaard A, Merkel PA, Kawut SM. Adverse events in connective tissue disease–associated pulmonary arterial hypertension. Arthritis Rheumatol 2015;67:2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maron BA, Leopold JA. The role of the renin‐angiotensin‐aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference Series) [review]. Pulm Circ 2014;4:200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaddam RR, Chambers S, Bhatia M. ACE and ACE2 in inflammation: a tale of two enzymes. Inflamm Allergy Drug Targets 2014;13:224–34. [DOI] [PubMed] [Google Scholar]
- 9. Liu SS, Wang HY, Tang JM, Zhou XM. Hypoxia‐induced collagen synthesis of human lung fibroblasts by activating the angiotensin system. Int J Mol Sci 2013;14:24029–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruce E, Shenoy V, Rathinasabapathy A, Espejo A, Horowitz A, Oswalt A, et al. Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br J Pharmacol 2015;172:2219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan YM, Luo L, Guo Z, Yang M, Ye RS, Luo C. Activation of renin–angiotensin–aldosterone system (RAAS) in the lung of smoking‐induced pulmonary arterial hypertension (PAH) rats. J Renin Angiotensin Aldosterone Syst 2015;16:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maron BA, Loscalzo J. Pulmonary hypertension: pathophysiology and signaling pathways In: Humbert M, Evgenov OV, Stasch JP. editors. Pharmacotherapy of pulmonary hypertension. Heidelberg: Springer, 2013. p. 31–58. [DOI] [PubMed] [Google Scholar]
- 13. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011;118:6743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Urzainqui A, Martínez del Hoyo G, Lamana A, de la Fuente H, Barreiro O, Olazabal IM, et al. Functional role of P‐selectin glycoprotein ligand 1/P‐selectin interaction in the generation of tolerogenic dendritic cells. J Immunol 2007;179:7457–65. [DOI] [PubMed] [Google Scholar]
- 15. Pérez‐Frías A, González‐Tajuelo R, Núñez‐Andrade N, Tejedor R, García‐Blanco MJ, Vicente‐Rabaneda E, et al. Development of an autoimmune syndrome affecting the skin and internal organs in P‐selectin glycoprotein ligand 1 leukocyte receptor–deficient mice. Arthritis Rheumatol 2014;66:3178–89. [DOI] [PubMed] [Google Scholar]
- 16. Nuñez‐Andrade N, Lamana A, Sancho D, Gisbert JP, Gonzalez‐Amaro R, Sanchez‐Madrid F, et al. P‐selectin glycoprotein ligand‐1 modulates immune inflammatory responses in the enteric lamina propria. J Pathol 2011;224:212–21. [DOI] [PubMed] [Google Scholar]
- 17. He X, Schoeb TR, Panoskaltsis‐Mortari A, Zinn KR, Kesterson RA, Zhang J, et al. Deficiency of P‐selectin or P‐selectin glycoprotein ligand‐1 leads to accelerated development of glomerulonephritis and increased expression of CC chemokine ligand 2 in lupus‐prone mice. J Immunol 2006;177:8748–56. [DOI] [PubMed] [Google Scholar]
- 18. Rivera‐Nieves J, Burcin T, Olson T, Morris MA, McDuffie M, Cominelli F, et al. Critical role of endothelial P‐selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med 2006;203:907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Angiari S, Rossi B, Piccio L, Zinselmeyer BH, Budui S, Zenaro E, et al. Regulatory T cells suppress the late phase of the immune response in lymph nodes through P‐selectin glycoprotein ligand‐1. J Immunol 2013;191:5489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thibault H, Kurtz B, Raher M, Shaik RS, Waxman A, Derumeaux G, et al. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 2010;3:157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yared K, Noseworthy P, Weyman A, McCabe E, Picard MH, Baggish AL. Pulmonary artery acceleration time provides an accurate estimate of systolic pulmonary arterial pressure during transthoracic echocardiography. J Am Soc Echocardiogr 2011;24:687–92. [DOI] [PubMed] [Google Scholar]
- 22. Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of high‐resolution echocardiography and magnetic resonance imaging vs. high‐fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2010;299:401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tania NP, Maarsingh H, Bos ST, Mattiotti A, Prakash S, Timens W, et al. Endothelial follistatin‐like‐1 regulates the postnatal development of the pulmonary vasculature by modulating BMP/Smad signaling. Pulm Circ 2017;7:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cogolludo A, Frazziano G, Briones A, Cobeño L, Moreno L, Lodi F, et al. The dietary flavonoid quercetin activates BKCa currents in coronary arteries via production of H2O2. Role in vasodilatation. Cardiovasc Res 2007;73:424–31. [DOI] [PubMed] [Google Scholar]
- 25. Kossmann S, Hu H, Steven S, Schönfelder T, Fraccarollo D, Mikhed Y, et al. Inflammatory monocytes determine endothelial nitric‐oxide synthase uncoupling and nitro‐oxidative stress induced by angiotensin II. J Biol Chem 2014;289:27540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II–induced vascular dysfunction depends on interferon‐γ–driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 2013;33:1313–9. [DOI] [PubMed] [Google Scholar]
- 27. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012;122:4306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bleeker GB, Steendijk P, Holman ER, Yu CM, Breithardt OA, Kaandorp TA, et al. Assessing right ventricular function: the role of echocardiography and complementary technologies. Heart 2006;92:i19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brittain E, Penner NL, West J, Hemnes A. Echocardiographic assessment of the right heart in mice. J Vis Exp 2013;81:50912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen G, Li Y, Tian J, Zhang L, Jean‐Charles P, Gobara N, et al. Application of echocardiography on transgenic mice with cardiomyopathies. Biochem Res Int 2012;2012:7151972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol 2010;11:328–34. [DOI] [PubMed] [Google Scholar]
- 32. Carrizzo A, Lenzi P, Procaccini C, Damato A, Biagioni F, Ambrosio M, et al. Pentraxin 3 induces vascular endothelial dysfunction through a P‐selectin/matrix metalloproteinase‐1 pathway. Circulation 2015;131:1495–505. [DOI] [PubMed] [Google Scholar]
- 33. Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med 2014;190:808–17. [DOI] [PubMed] [Google Scholar]
- 34. Li G, Liu Y, Zhu Y, Liu A, Xu Y, Li X, et al. ACE2 activation confers endothelial protection and attenuates neointimal lesions in prevention of severe pulmonary arterial hypertension in rats. Lung 2013;191:327–36. [DOI] [PubMed] [Google Scholar]
- 35. De Man FS, Tu L, Handoko ML, Rain S, Ruiter G, François C, et al. Dysregulated renin–angiotensin–aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrell N, Morris K, Stenmark KR. Role of angiotensin‐converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol 1995;269:1186–94. [DOI] [PubMed] [Google Scholar]
- 37. Shenoy V, Qi Y, Katovich MJ, Raizada MK. ACE2, a promising therapeutic target for pulmonary hypertension. Curr Opin Pharmacol 2011;11:150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martinelli R, Gegg M, Longbottom R, Adamson P, Turowski P, Greenwood J. ICAM‐1–mediated endothelial nitric oxide synthase activation via calcium and AMP‐activated protein kinase is required for transendothelial lymphocyte migration. Mol Biol Cell 2009;20:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci 2015;129:83–94. [DOI] [PubMed] [Google Scholar]
- 40. Stenmark K, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 2009;297:L1013–32. [DOI] [PubMed] [Google Scholar]
- 41. Chu Y, Xiangli X, Xiao W. Regulatory T cells protect against hypoxia‐induced pulmonary arterial hypertension in mice. Mol Med Rep 2015;11:3181–7. [DOI] [PubMed] [Google Scholar]
- 42. Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, et al. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators Inflamm 2014;2014:247372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Austin ED, Lahm T, West J, Tofovic SP, Johansen AK, MacLean MR, et al. Gender, sex hormones and pulmonary hypertension. Pulm Circ 2013;3:294–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lantin‐Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW. Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol 1997;273:L119–26. [DOI] [PubMed] [Google Scholar]
- 45. Chambliss K, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev 2002;23:665–86. [DOI] [PubMed] [Google Scholar]
- 46. Simoncini T, Hafezi‐Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol‐3‐OH kinase [letter]. Nature 2000;407:538–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt‐dependent activation of endothelial nitric‐oxide synthase in vascular endothelial cells. J Biol Chem 2001;276:3459–67. [DOI] [PubMed] [Google Scholar]
- 48. Stirone C, Boroujerdi A, Duckles SP, Krause DN. Estrogen receptor activation of phosphoinositide‐3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: rapid and long‐term effects. Mol Pharmacol 2005;67:105–13. [DOI] [PubMed] [Google Scholar]
- 49. Álvarez Á, Cerdá‐Nicolás M, Nabah YN, Mata M, Issekutz AC, Panés J, et al. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood 2004;104:402–8. [DOI] [PubMed] [Google Scholar]
- 50. Alvarez A, Hermenegildo C, Issekutz A, Esplugues J, Sanz MJ. Estrogens inhibit angiotensin II‐induced leukocyte‐endothelial cell interactions in vivo via rapid endothelial nitric oxide synthase and cyclooxygenase activation. Circ Res 2002;91:1142–50. [DOI] [PubMed] [Google Scholar]
- 51. Nabah YN, Mateo T, Cerdá‐Nicolás M, Alvarez A, Martinez M, Issekutz AC, et al. L‐NAME induces direct arteriolar leukocyte adhesion, which is mainly mediated by angiotensin‐II. Microcirculation 2005;12:443–53. [DOI] [PubMed] [Google Scholar]
- 52. Tamosiuniene R, Manouvakhova O, Mesange P, Saito T, Qian J, Sanyal M, et al. Dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res 2018;122:1689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li W, Li D, Sun L, Li Z, Yu L, Wu S. The protective effects of estrogen on hepatic ischemia‐reperfusion injury in rats by downregulating the Ang II/AT1R pathway. Biochem Biophys Res Commun 2018;503:2543–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials