

Abstract
Aim
To compare the safety and efficacy of U500‐R delivered by a novel, specifically designed U500‐R insulin pump with U‐500R delivered by multiple daily injections (MDI).
Methods
The phase 3 VIVID study randomized people with type 2 diabetes to U‐500R by continuous subcutaneous insulin infusion (CSII) or MDI. Participants (aged 18–85 years) had HbA1c ≥7.5% and ≤12.0% and a total daily dose of insulin >200 and ≤600 U/day. After a 2‐week transition to three times daily injections of U‐500R, participants were treated for 24 weeks with U‐500R by CSII or MDI. Treatment arms were compared using mixed model repeated measures analysis.
Results
The study randomized 420 participants (CSII: 209, MDI: 211) with 365 completers. Mean changes from baseline were: HbA1c, −1.27% (−13.9 mmol/mol) with CSII and −0.85% (−9.3 mmol/mol) with MDI (difference − 0.42% [−4.6 mmol/mol], P <0.001); fasting plasma glucose, −33.9 mg/dL (−1.9 mmol/L) with CSII and 1.7 mg/dL (0.09 mmol/L) with MDI (difference − 35.6 mg/dL [−2.0 mmol/L], P <0.001); total daily dose, 2.8 U with CSII and 51.3 U with MDI (P < 0.001). Weight changes and rates of documented symptomatic and severe hypoglycaemia were similar between groups; the CSII group had a higher rate of nocturnal hypoglycaemia.
Conclusions
In type 2 diabetes requiring high doses of insulin, both methods of U‐500R delivery lowered HbA1c. However, the CSII group attained greater HbA1c reduction with significantly less insulin. Individualized dose titration will be important to balance glycaemic control with hypoglycaemia risk.
Keywords: clinical trial, continuous subcutaneous insulin infusion, insulin resistance, insulin therapy, randomized trial, type 2 diabetes.
1. INTRODUCTION
The clinical use of recombinant human regular U‐500 insulin (Humulin R U‐500; U‐500R) has recently increased with the worsening epidemic of obesity and type 2 diabetes.1 High‐dose insulin‐resistant subjects (requiring >200 U/day of U‐100 insulin) have been treated successfully with U‐500R by injection, resulting in improved glycaemic control (HbA1c reductions from 1% to >3%), with no increase in severe hypoglycaemia.2
U‐500R is typically administered by utilizing multiple daily injections (MDIs). A U‐500R pharmacokinetic (PK) and pharmacodynamic (PD) simulation modelling study indicated that the steady‐state basal PK/PD effect from both twice daily (BID) and three times daily (TID) dosing supports the use of U‐500R as insulin monotherapy.3 A subsequent clinical trial showed that both BID and TID dosing, when transitioning people uncontrolled on high dose U‐100 insulin, reduced HbA1c by >1% after 24 weeks with low overall rates of documented symptomatic hypoglycaemia <50 mg/dL.4 The long mean duration of action of U‐500R (21 hours, range 13–24 hours) compared with the same dose of U‐100 regular insulin, albeit with a similar area under the curve, allows U‐500R to provide basal insulin coverage.5, 6 U‐500R also provides postprandial glucose lowering as the mean onset of action is within 15 minutes of injection, and it has a comparable time to peak action at relevant doses.5, 6 These properties make it possible to use U‐500R as insulin monotherapy.
Despite the growing pharmacotherapeutic options for type 2 diabetes, insulin therapy is a longstanding proven therapeutic agent for glycaemic control and U‐500 insulin continues to be a clinically relevant treatment option. The extended application of U‐500R as insulin monotherapy via continuous subcutaneous insulin infusion (CSII) is driven by the need for novel alternative regimens for high insulin‐requiring people with type 2 diabetes.7 An estimated 25% of U‐500R insulin use is by CSII already, using pumps designed for U‐100 insulin.8 The safety and efficacy of U‐500R delivered by CSII has not been determined.6 Use of U‐500 insulin in a U‐100 pump requires mathematical conversion and could be confusing; for example, 10 U of bolus insulin displayed on a U‐100 pump would in fact deliver 50 U, or 5‐fold more, if using U‐500 insulin. Previous studies of U‐500R in CSII therapy lacked controls, were mostly retrospective, or included a small number of subjects.2, 9 Therefore, the use of U‐500R by CSII needed to be studied in a larger, prospective, randomized clinical trial.
The Omnipod DASH U‐500 Insulin Management System (Insulet Corporation, Acton, MA, USA) is a novel, specifically designed U‐500R pump, scaled for delivering U‐500 insulin without the need for dose conversion. The Evaluating U‐500R Infusion Versus Injection in Type 2 Diabetes Mellitus (VIVID) study is the first prospective, randomized clinical trial to compare U‐500R by CSII (using an investigational version of the Omnipod DASH U‐500 System) with U‐500R by MDI, in people with type 2 diabetes requiring a high dose of insulin.
2. RESEARCH DESIGN AND METHODS
2.1. Participants
The VIVID study was a randomized, controlled, multicentre, open‐label, parallel design, 26‐week, phase 3 study of U‐500R delivered by CSII compared with U‐500R delivered by MDI in participants with type 2 diabetes who require high doses of insulin. Participants were aged 18–85 years, had a body mass index of 25–50 kg/m2, an HbA1c of ≥7.5% and ≤12.0%, and a total daily dose (TDD) of insulin >200 U, but ≤600 U of U‐500 insulin by injection or non‐U‐500 insulin by injection or CSII (U‐100 rapid‐acting insulin analogue only) (Table S1). This population represents people who will probably be recommended to start an insulin pump with U‐500R insulin. Participants were excluded for liver disease, stage 4 or higher kidney disease, class III or IV cardiac disease, or a history of more than one episode of severe hypoglycaemia requiring the assistance of another person within the last 6 months. Concomitant glucose‐lowering therapy with metformin, dipeptidyl peptidase‐4 (DPP‐4) inhibitors, glucagon‐like peptide‐1 receptor agonists (GLP‐1RAs), sodium glucose co‐transporter‐2 inhibitors (SGLT2is) and/or pioglitazone (doses ≤30 mg/day) was permitted. Participants not using GLP‐1RAs or SGLT2is were enrolled into the primary cohort (Group A), as these medications were not yet widely used with insulin at the time when the study was being designed, while those using GLP‐1RAs or SGLT2is comprised Group B. Group A plus Group B constituted the all randomized population.
All participants were required to give informed consent for participation in the study and prior to any study‐specific procedures. The protocol was approved by local ethical review boards and was conducted according to International Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. The trial is registered at http://clinicaltrials.gov as NCT02561078.
2.2. Objectives
The primary objective of the VIVID study was to show in Group A that the change in HbA1c of the U‐500R CSII group was non‐inferior to that of the U‐500R MDI group from baseline to week 26 (non‐inferiority margin 0.4%). The key secondary objectives, which will be presented for the all randomized population without adjustment for multiplicity, were to show that U‐500R delivered by CSII was superior to U‐500R by MDI in the following variables: change from baseline of fasting plasma glucose (FPG); change from baseline of HbA1c; and the proportion of participants achieving HbA1c target values of <7% and <7.5%.
Additionally, between treatment group comparisons were performed in the all randomized population on: the proportions of participants achieving other HbA1c targets (≤6.5% and <8.0%); change in seven‐point self‐monitored blood glucose (SMBG) profile, including mean SMBG for each time point measurement; change in TDD; rate and incidence of hypoglycaemia; and change in body weight.
The categories of hypoglycaemia analysed in the study were documented: symptomatic hypoglycaemia (signs/symptoms with a blood glucose level of ≤70 mg/dL), nocturnal hypoglycaemia (any hypoglycaemic event, documented or severe, occurring between bedtime and waking), and severe hypoglycaemia (an event such that the patient required the assistance of another person). An additional post hoc analysis of hypoglycaemia was conducted with documented hypoglycaemia defined by the clinically relevant blood glucose level of <54 mg/dL.10
2.3. Procedures
Prior to randomization (week 0) participants continued prestudy insulins with dose adjustments at the investigator's discretion. Assignment to MDI or CSII treatment arms was determined by a computer‐generated random sequence using an interactive web response system, with stratification by HbA1c ≥8.5% or <8.5%, non‐users versus users of GLP‐1RAs or SGLT2is, and U‐500R at entry versus other insulins. After randomization, both groups underwent a 2‐week transition to U‐500R by MDI, whereupon participants who had been randomized to CSII switched from MDI to the investigational U‐500 system. Treatment was continued for 26 weeks (Figure S1). All participants were instructed to record seven‐point SMBG profiles on any two non‐consecutive days in the 2 weeks prior to visits at weeks 0, 14 and 26; profiles were to include a weekend day, if possible. A central laboratory was used for analysis of FPG at weeks 0, 14 and 26, and for analysis of HbA1c at weeks 0, 8, 14, 20 and 26.
2.4. Transition period and MDI dosing
During the transition period to U‐500R MDI (weeks 0–2), participants injected U‐500R TID with a 40:30:30 dose proportion before breakfast, lunch and dinner. Participants used a vial and U‐100 syringe as neither the U‐500 Kwik‐Pen nor U‐500 calibrated syringe were available at study start. Participants previously taking U‐500R continued their prestudy TDD; for those entering the trial using other insulins, the initial U‐500R dose was based on entry HbA1c and SMBG from screening to week 0 (Table S5).
2.5. Device
The Omnipod DASH U‐500 Insulin Management System is a novel, specifically designed U‐500R CSII device (Insulet Corporation) that allows customizable basal and bolus insulin delivery for the management of diabetes in persons requiring insulin. The investigational U‐500 system consisted of an insulin pump and controller based on the U‐100 Omnipod Insulin Management System with software changes to accommodate U‐500 insulin. The duration of insulin action was increased from a maximum of 8 hours for the U‐100 Omnipod System to a maximum of 12 hours to allow the investigator flexibility in personalizing the insulin on board (IOB) calculation for people using U‐500, given that U‐500 insulin has a longer duration of action.5 Other settings were calibrated for U‐500; for example, the maximum bolus was 150 units, maximum basal rate was 150 U/h, and the minimum dose increment was 0.25 U.
2.6. CSII dosing
For participants assigned to CSII, the TDD was divided into 50:50 bolus and basal. Basal was split into two rates: 06:00 am to 09:00 pm at the calculated basal rate and 09:00 pm to 06:00 am at 10% reduction in basal rate. Up to four basal rates were allowed. As an additional precaution against nocturnal hypoglycaemia, participants were encouraged to have a bedtime snack without an insulin bolus.
CSII participants used the pump bolus calculator to determine their bolus doses. The investigator assigned one of three bolus options: fixed bolus (40:30:30 proportion for breakfast, lunch and dinner), carbohydrate (CHO) counting, or meal size per participant estimate (programmed as small = 30 g CHO; medium = 60 g CHO and large = 90 g CHO). For the latter two options, the suggested starting insulin:CHO ratio was programmed between 1:3 and 1:5 U/g, customized by the investigator. This allowed use of the IOB and correction portion (including negative adjustment) for the mealtime U‐500R dose calculation. The duration of insulin action setting was programmed at 6 hours.
2.7. Dose titration
For both study arms, titrations were based on an insulin algorithm with a premeal and bedtime glucose target range of 80–140 mg/dL. At a minimum, the titrations were performed weekly until week 4, biweekly until week 14, and triweekly until study end (week 26).
2.8. Statistical analyses
The data were analysed for a modified intent‐to‐treat population, defined as all randomized participants who took at least one dose of study drug. Summary statistics for continuous measurements, including sample size, mean, median, SD, minimum and maximum, were provided for measurements at baseline, each post‐treatment visit, and change from baseline to each visit.
A mixed model repeated measures (MMRM) analysis was used for continuous outcomes with repeated postbaseline measurements to compare treatment arms, unless otherwise noted. A sensitivity analysis was also performed for the primary objective (Group A population) by the copy reference multiple imputation method (CRMIM), followed by ANCOVA at endpoint. Least squares (LS) mean (SE) by treatment group and visit (if applicable), LS mean difference between treatment groups, 95% confidence limits of the treatment differences, and the P‐value for the treatment comparison are given. For the change from baseline, P‐values for the within‐treatment changes are also given. All tests of treatment effects were conducted at a two‐sided alpha of 0.05.
3. RESULTS
3.1. Participants
A total of 632 participants were screened; 420 were randomly assigned to treatment (CSII: 209, MDI: 211). Of these, 340 participants were in Group A (CSII: 170, MDI: 170). The study was conducted between October 20, 2015 and May 9, 2017 at 55 sites in the USA and Puerto Rico (Table S4). A total of 365 participants completed the study (179 [85.6%] CSII; 186 [88.2%] MDI); see Figure S2 for the complete participant disposition. The treatment groups were similar in baseline demographic and clinical characteristics except for baseline systolic blood pressure (mean: MDI = 135 mmHg; CSII = 131 mmHg; P = 0.042) (Table 1). Previous insulin use among participants is given in Table S1. Other glucose‐lowering medications are given in Table S2.
Table 1.
Demographics and baseline characteristics (all randomized population)
| U‐500R CSII N = 209 | U‐500R MDI N = 211 | |
|---|---|---|
| Sex, male | 104 (49.8) | 117 (55.5) |
| Age, years | 57.6 ± 10.3 | 56.7 ± 10.1 |
| Race | ||
| White | 160 (76.6) | 160 (75.8) |
| Black/African American | 19 (9.1) | 14 (6.6) |
| Multiple | 25 (12.0) | 26 (12.3) |
| Other/missing | 5 (2.4) | 11 (5.2) |
| Hispanic or Latino | 39 (18.7) | 49 (23.2) |
| BMI, kg/m2 | 39.3 ± 5.6 | 40.1 ± 5.8 |
| HbA1c, % | 8.75 ± 1.03 | 8.77 ± 1.08 |
| HbA1c, mmol/mol | 72.1 ± 11.3 | 72.3 ± 11.8 |
| Duration of diabetes, years | 17.4 ± 7.6 | 16.9 ± 7.7 |
| FPG, mg/dL | 165 ± 60 | 165 ± 59 |
| FPG, mmol/L | 9.2 ± 3.3 | 9.2 ± 3.3 |
| Systolic BP, mmHg | 131 ± 17 | 135 ± 18 |
| Diastolic BP, mmHg | 75 ± 9.6 | 77 ± 10.0 |
| TDD insulin, U | 288 ± 108 | 291 ± 93 |
| TDD insulin, U/kg | 2.56 ± 0.91 | 2.54 ± 0.89 |
| U‐500R use at entry | 59 (28.2) | 59 (28.0) |
| Glucose‐lowering medications, alone or in combination | N = 118 | N = 125 |
| DPP‐4 | 10 (8.5) | 10 (8.0) |
| SGLT2i | 25 (21.2) | 24 (19.2) |
| GLP‐1RA | 17 (14.4) | 21 (16.8) |
| Metformin | 99 (83.9) | 103 (82.4) |
| Pioglitazone | 4 (3.4) | 5 (4.0) |
| Bolus dosing option (CSII group) | ||
| Fixed dose | 169 (80.9) | N/A |
| Meal size | 22 (10.5) | N/A |
| CHO counting | 12 (5.7) | N/A |
| Missing | 6 (2.9) | N/A |
Abbreviations: BMI, body mass index; BP, blood pressure; CHO, carbohydrate; CSII, continuous subcutaneous insulin infusion; FPG, fasting plasma glucose; DPP‐4, dipeptidylpeptidase‐4 inhibitor; GLP‐1RA, glucagon‐like peptide‐1 receptor agonist; MDI, multiple daily injection; SGLT2i, sodium glucose co‐transporter‐2 inhibitor.
Data are n (%) or mean ± SD.
3.2. Objectives
The study met the primary objective. For Group A, U‐500R by CSII was non‐inferior in HbA1c change from baseline to week 26 compared with U‐500R by MDI (LS mean [SE]) (−1.30% [0.082] vs. ‐0.86% [0.081]) (−14.2 [0.9] vs. ‐9.4 [0.9] mmol/mol) (difference − 0.44% [−4.8 mmol/mol]; 95% CI: −0.67, −0.22 [−7.3, −2.4]; P < 0.001). The sensitivity analysis involving CRMIM and ANCOVA confirmed the results of the MMRM analysis (Figure S3).
For the all randomized population (Groups A and B), both treatment groups improved their glycaemic control compared with baseline. HbA1c change from baseline to week 26 with U500‐R by CSII (LS mean [SE]) was −1.27% (0.072) (−13.9 [0.8] mmol/mol) and by MDI was −0.85% (0.070) (−9.3 [0.8] mmol/mol) (LS mean difference − 0.42% [−4.6 mmol/mol]; 95% CI: −0.62%, −0.22% [−6.8, −2.4]; P < 0.001) (Figure 1A). The percentage of participants reaching the HbA1c target value of <7% was 28.7% with U500‐R by CSII compared with 18.4% by MDI (odds ratio [OR] 1.97 [95% CI: 1.14, 3.39]; P = 0.015). The percentage of participants reaching the HbA1c target value of <7.5% was 52.6% with U500‐R by CSII compared with 38.6% by MDI (OR 1.94 [95% CI: 1.23, 3.07]; P = 0.005).
Figure 1.
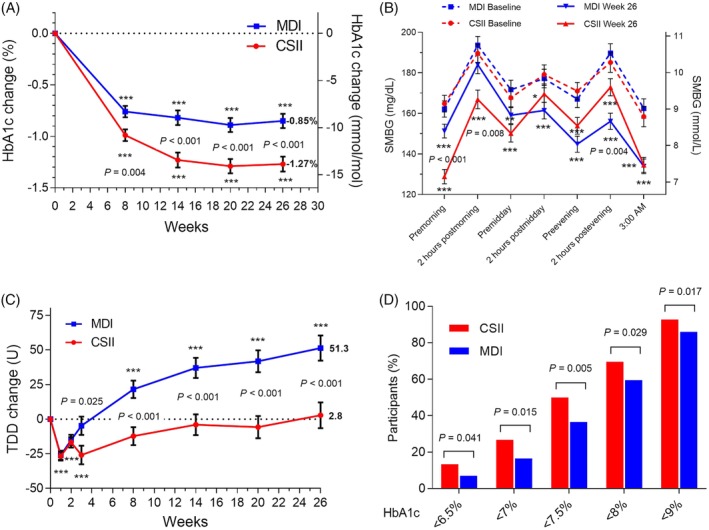
Selected outcome measures, all randomized participants by treatment (continuous subcutaneous insulin infusion [CSII], n = 209; multiple daily injections [MDI], n = 211). (A) Change from baseline in HbA1c. Data are LS mean ± SE; ***P <0.001 for change from baseline; P‐values are given for significant differences between treatments. (B) Seven‐point self‐monitored blood glucose (SMBG) profile at baseline and week 26. Only participants with a non‐missing baseline value and at least one non‐missing postbaseline SMBG value were included in analysis (CSII, n = 163; MDI, n = 174). *P <0.05, **P <0.01, ***P <0.001 for change from baseline; P‐values are given for significant differences between treatments. (C) Change from baseline in total daily insulin dose (TDD). Data are LS mean ± SE; ***P <0.001 for change from baseline; P‐values are given for significant differences between treatments. (D) Percentage of participants reaching HbA1c targets at 26 weeks by treatment; P‐values are given for significant differences between treatments
FPG (LS mean [SE]) at week 26 was 143 (5.03) mg/dL (7.9 [0.3] mmol/L) in the CSII group and 179 (4.90) mg/dL (9.9 [0.3] mmol/L) in the MDI group (P < 0.001). FPG change from baseline to week 26 with U500‐R by CSII was −33.9 (5.03) mg/dL (−1.9 [0.3] mmol/L) and with U‐500R by MDI was 1.7 (4.90) mg/dL (0.09 [0.3] mmol/L) (difference − 35.6 mg/dL [−2.0 mmol/L]; 95% CI: −49.4, −21.7 [−2.7, −1.2]; P < 0.001). The FPG results in the MDI group are similar to the FPG results from our previous study comparing BID with TID U‐500R by MDI in a similar population (Table S3).4
SMBG profiles at baseline and week 26 are shown in Figure 1B. Significant decreases from baseline were observed in both treatment groups. The CSII group had significantly lower premorning meal and 2‐hour postmorning meal blood glucose compared with the MDI group, while the MDI group had significantly lower 2‐hour postevening meal blood glucose.
The TDD (LS mean [SE]) at week 26 was 285 (9.33) U (2.42 [0.090] U/kg) for the CSII group and 333 (9.14) U (2.90 [0.088] U/kg) for the MDI group. This represented a significant increase in the MDI group from baseline of 51.3 (9.14) U (0.42 [0.09] U/kg), while the TDD of the CSII group was unchanged (+2.8 [9.3] U; −0.05 [0.09] U/kg) (LS mean difference − 48.4 U; 95% CI: −74.1, −22.8 [−0.48; −0.73, −0.23]; P < 0.001; Figure 1C). The CSII group had significantly greater proportions of participants achieving specific HbA1c target values at week 26 (Figure 1D). Body weight change from baseline to week 26 was not significantly different between groups (LS mean [SE]: 4.2 kg [0.4] in the CSII group and 3.4 kg [0.3] in the MDI group) (Figure S4).
3.3. Safety
There were no significant differences in incidences or rates (weeks 0–26) of documented symptomatic hypoglycaemia or severe hypoglycaemia between treatment groups (Table 2). The CSII group had a higher nocturnal hypoglycaemia rate at both blood glucose cut‐offs (<54 and ≤70 mg/dL). However, nocturnal hypoglycaemia incidence was similar between groups (Table 2) with most participants having 0 to 5 (median: CSII = 4, MDI = 4) events during this 26‐week study (Figure S5). The rate of nocturnal hypoglycaemia (documented symptomatic or severe) in the CSII group was highest at week 8, and decreased steadily from week 8 until study end (Figure S6).
Table 2.
Incidence and rate of hypoglycaemia
| CSII N = 209 | MDI N = 211 | Ratio CSII/MDI | 95% CI | P‐value | |
|---|---|---|---|---|---|
| Documented symptomatic ≤70 mg/dL | |||||
| Incidence, n (%) | 183 (87.6) | 193 (91.9) | 0.61 | (0.32, 1.16) | 0.13 |
| 1‐year event rate | 37.0 ± 3.69 | 33.9 ± 3.01 | 1.09 | (0.88, 1.35) | 0.42 |
| Documented symptomatic <54 mg/dL | |||||
| Incidence, n (%) | 152 (72.7) | 155 (73.8) | 0.95 | (0.61, 1.47) | 0.81 |
| 1‐year event rate | 14.4 ± 1.76 | 13.8 ± 1.43 | 1.04 | (0.80, 1.35) | 0.76 |
| Nocturnal ≤70 mg/dL | |||||
| Incidence, n (%) | 169 (80.9) | 167 (79.5) | 1.09 | (0.67, 1.79) | 0.72 |
| 1‐year event rate | 19.6 ± 2.25 | 11.8 ± 1.13 | 1.66 | (1.29, 2.13) | <0.001 |
| Nocturnal <54 mg/dL | |||||
| Incidence, n (%) | 123 (58.9) | 126 (60.0) | 0.95 | (0.64, 1.41) | 0.80 |
| 1‐year event rate | 8.18 ± 1.10 | 5.39 ± 0.66 | 1.52 | (1.11, 2.07) | 0.008 |
| Severe hypoglycaemia | |||||
| Incidence, n (%) | 11 (5.26) | 5 (2.38) | 2.28 | (0.78, 6.72) | 0.13 |
| 1‐year event rate | 0.11 ± 0.04 | 0.05 ± 0.03 | 2.45 | (0.85, 7.06) | 0.096 |
Abbreviations: CSII, continuous subcutaneous insulin infusion; MDI, multiple daily injection.
Data are LS mean ± SE or n (%).
There were 17 severe hypoglycaemia events in 16 participants: 12 events in 11 participants in the CSII group and five events in five participants in the MDI group (P = 0.13). All participants with severe hypoglycaemic events fully recovered.
A total of 57 participants (13.6%) experienced at least one serious adverse event (SAE) after randomization; 34 (16.3%) in the CSII group and 23 (10.9%) in the MDI group. Severe hypoglycaemia was designated as an SAE as per the protocol. Hypoglycaemia was the most common SAE in both groups (n [%]; CSII 10 [4.8], MDI 5 [2.4]). There were no SAEs related to the study device. Three deaths occurred during the study (CSII, two [heart failure, cardiac arrest]; MDI, one [urosepsis]); none of these were judged by the investigator to be related to study drug, device or procedure.
Treatment‐emergent adverse events (TEAEs) possibly related to study drug were reported by 29 participants (13.9%) in the CSII group and 22 participants (10.4%) in the MDI group (P = 0.30). TEAEs possibly related to study drug reported by five or more participants in the study were hypoglycaemia (CSII: 9 [4.3%]; MDI: 5 [2.4%]; P = 0.29) and weight increased (CSII: 7 [3.3%]; MDI: 11 [5.2%]; P = 0.47).
TEAEs possibly related to study device were reported by 13 (6.2%) participants in the CSII group. One patient reported pain, pruritus and irritation associated with the pump injection site as AEs; no product complaints contained SAEs. Hyperglycaemia during non‐routine infusion site changes was reported by five (2.4%) participants in the CSII group.
4. DISCUSSION
The VIVID study is the first randomized, controlled trial to robustly evaluate the safety and efficacy of using U‐500R by CSII compared with MDI in people with type 2 diabetes requiring high‐dose insulin. Overall, both CSII and MDI methods of U‐500R delivery resulted in reduced HbA1c (Figure 1A) and seven‐point SMBG (Figure 1B) from baseline, attesting to the glucose‐lowering efficacy of U‐500R in people needing high‐dose insulin. The two delivery methods of U‐500R resulted in similar weight gain and no significant differences in severe or clinically relevant hypoglycaemia, except for higher nocturnal hypoglycaemia rates in the CSII group.
The CSII group, using a dedicated device designed for delivery of U‐500 insulin, which allowed for customization of insulin delivery, had a greater reduction from baseline in HbA1c at week 26 compared with the MDI group (difference − 0.42%), and a substantial reduction from baseline in FPG (difference >34 mg/dL [1.9 mmol/L]), while FPG in the MDI group did not change significantly. This was achieved with little change in TDD in the CSII group (mean increase of 2.8 U), while the MDI group increased mean TDD by 51.3 U at study end. Weight gain, an expected consequence of intensive insulin treatment,11 was seen in both groups with no significant difference between treatments (CSII, 4.2 kg; MDI, 3.4 kg).
The 24‐hour SMBG profiles from both treatment groups showed significant reduction in blood glucose from baseline at all time points (Figure 1B). Both groups had the same glucose target, and investigators were expected to titrate according to the algorithm. However, the pattern of blood glucose reductions relative to baseline was different between the two treatment groups, which reflects the method of insulin delivery; CSII lowered blood glucose more in the morning while MDI lowered it more in the afternoon and evening. In this treat‐to‐target study, the MDI doses were titrated according to the three daily premeal SMBG values, rather than the premorning value alone (Table S5), which might explain the greater SMBG change as the day progressed. The significant reductions in postevening and 03:00 am SMBG levels with MDI, for example, suggest that the pre‐evening meal U‐500R dose was probably titrated. The CSII basal doses were titrated according to the premorning meal, the pre‐evening meal, and the 03:00 am SMBG values; the CSII bolus doses were titrated according to the premidday meal, pre‐evening meal and bedtime SMBG values (Table S5). The significant reduction in the premorning glucose level with CSII could be attributed to a better ability to fine‐tune overnight basal insulin. These data support the need for individualized dose adjustments and can inform prescribers on how to implement and optimize U‐500R use via CSII or MDI in practice.
The incidence of hypoglycaemia (documented symptomatic, nocturnal and clinically relevant) was similar between treatments. There were no differences in rates of documented symptomatic or severe hypoglycaemia between treatments. The rate of severe hypoglycaemia was low in this cohort, 0.12 events/person/year for the CSII group and 0.05 events/person/year in the MDI group, and was comparable with published rates in insulin‐treated type 2 diabetes populations.12, 13, 14
Although the median rate of nocturnal hypoglycaemia events was similar between groups (median events/participant/year was 4 for both MDI and CSII groups, Figure S5), more patients on CSII were in the outlier tail, indicating that the difference in rate was driven by such patients, who may need closer attention and individualization of treatment. Steps taken in the protocol to reduce nocturnal hypoglycaemia in the CSII group included a 10% reduction in the night‐time basal insulin rate compared with the daytime, and the recommendation to take a bedtime snack without an insulin bolus.
To gain a better understanding of nocturnal hypoglycaemia as it relates to changes in insulin dose, we examined the rate of nocturnal (documented symptomatic or severe) hypoglycaemia and TDD during the course of the study (ie, excluding events without an accompanying blood glucose reading or if signs/symptoms were absent, unknown or missing). The rate of nocturnal (documented symptomatic or severe) hypoglycaemia in the CSII group increased between weeks 2 to 4 (transition from U‐500R TID to CSII) and was higher compared with MDI from weeks 8 to 14, then steadily decreased through to study end (Figure S6). The TDD of the CSII group started to plateau at 14 weeks (Figure 1C), around the time that this nocturnal hypoglycaemia rate with CSII was decreasing. This suggests attainment of an insulin dose that allowed mitigation of nocturnal hypoglycaemia without compromising glycaemic control. Further, the nocturnal hypoglycaemia rates had converged by week 23, and there were no significant differences between MDI and CSII groups at the end of the study (Figure S6). These findings suggest that once increased hypoglycaemia rates were noted during the active titration phase with U‐500R by either MDI or CSII, the investigators adapted and adjusted insulin doses accordingly to reduce hypoglycaemia over time.
With the obesity epidemic contributing to the prevalence of insulin resistance and rising insulin requirements among insulin‐treated persons, the findings in this study provide timely confirmation about the efficacy and safety profile of U500‐R MDI while also showing that U‐500R CSII is a potential alternative delivery method. Having these choices of delivery for U‐500R could help overcome clinical challenges in this population related to injecting frequently or with large volumes to meet their insulin needs.
In conclusion, U‐500R by CSII using a U‐500 dedicated pump, with individualization of therapy for optimal safety and efficacy, could be a viable treatment option, in addition to U‐500R MDI, for glucose lowering in people who require high doses of insulin.
CONFLICT OF INTEREST
T. L. is an employee and stockholder of Insulet Corporation. L. L. I., L. F., S. Z. and J. J. are employees and stockholders of Eli Lilly and Company. J. M. is a former employee and stockholder of Eli Lilly and Company, and serves as a consultant for Thermalin, Inc. G. G. has research funding from Medtronic and serves on the speakers’ bureau for Sanofi, Novo Nordisk, Eli Lilly, and Boehringer Ingelheim. H. Z. is an employee of Verily Life Sciences. A. B. has research funding from Sanofi, United BioSource Corp., Dexcom, Teijin American Inc., Astra Zeneca, Bayer Health Care Pharmaceuticals Inc., Boehringer Ingelheim, Bristol‐Meyers Squibb Research and Development, Eli Lilly and Company, Gan & Lee Pharmaceuticals, Jaeb Center for Health Research, KOWA Research Institute, Inc., Medtronic MiniMed, Merck Research Laboratories, Mylan GmbH, Novo Nordisk, and Theracos Sub, LLC, and serves on the speakers’ bureau for AstraZeneca and Sanofi.
AUTHOR CONTRIBUTIONS
G. G., J. J., H. Z. and J. M. designed the study. G. G. and A. B. collected the data. L. F. and S. Z. analyzed the data. L. I., J. J. and T. L. drafted the paper. All authors critically reviewed the paper and approved the manuscript for publication.
FUNDING INFORMATION
The study was funded by Eli Lilly and Company and Insulet Corporation.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
We thank the participants, their caregivers and the investigators for their participation in this study. Caryl J. Antalis, PhD, Eli Lilly and Company, provided writing and editorial support. The data in this paper were presented in part in a poster and an oral presentation at the 78th Scientific Sessions of the American Diabetes Association, June 22‐26, 2018.
The study was funded by Eli Lilly and Company and Insulet Corporation.
Grunberger G, Bhargava A, Ly T, et al. Human regular U‐500 insulin via continuous subcutaneous insulin infusion versus multiple daily injections in adults with type 2 diabetes: The VIVID study. Diabetes Obes Metab. 2020;22:434–441. 10.1111/dom.13947
Funding information Insulet Corporation; Eli Lilly and Company; American Diabetes Association; Eli Lilly and Company
REFERENCES
- 1. Cochran EK, Valentine V, Samaan KH, Corey IB, Jackson JA. Practice tips and tools for the successful use of U‐500 regular human insulin: the diabetes educator is key. Diabetes Educ. 2014;40:153‐165. [DOI] [PubMed] [Google Scholar]
- 2. Reutrakul S, Wroblewski K, Brown RL. Clinical use of U‐500 regular insulin: review and meta‐analysis. J Diabetes Sci Technol. 2012;6:412‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de la Pena A, Ma X, Reddy S, et al. Application of PK/PD modeling and simulation to dosing regimen optimization of high‐dose human regular U‐500 insulin. J Diabetes Sci Technol. 2014;8:821‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hood RC, Arakaki RF, Wysham C, Li YG, Settles JA, Jackson JA. Two treatment approaches for human regular U‐500 insulin in patients with type 2 diabetes not achieving adequate glycymic control on high‐dose U‐100 insulin therapy with or without oral agents: a randomized, titration‐to‐target clinical trial. Endocr Pract. 2015;21:782‐793. [DOI] [PubMed] [Google Scholar]
- 5. de la Pena A, Riddle M, Morrow LA, et al. Pharmacokinetics and pharmacodynamics of high‐dose human regular U‐500 insulin versus human regular U‐100 insulin in healthy obese subjects. Diabetes Care. 2011;34:2496‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Highlights of prescribing information for HUMULIN R U‐500: 2018. https://pi.lilly.com/us/humulin-r-u500-pi.pdf. Accessed July 25, 2019.
- 7. Knee TS, Seidensticker DF, Walton JL, Solberg LM, Lasseter DH. A novel use of U‐500 insulin for continuous subcutaneous insulin infusion in patients with insulin resistance: a case series. Endocr Pract. 2003;9:181‐186. [DOI] [PubMed] [Google Scholar]
- 8. Eby EL, Zagar AJ, Wang P, et al. Healthcare costs and adherence associated with human regular U‐500 versus high‐dose U‐100 insulin in patients with diabetes. Endocr Pract. 2014;20:663‐670. [DOI] [PubMed] [Google Scholar]
- 9. Martin C, Perez‐Molinar D, Shah M, Billington C. U500 disposable patch insulin pump: results and discussion of a VA pilot study. J Endocrine Soc. 2018;2:1275‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. International Hypoglycaemia Study Group . Glucose concentrations of less than 3.0 mmol/L (54 mg/dL) should be reported in clinical trials: a joint position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2017;40:155‐157. [DOI] [PubMed] [Google Scholar]
- 11. Purnell JQ, Zinman B, Brunzell JD, DCCT/EDIC Research Group . The effect of excess weight gain with intensive diabetes mellitus treatment on cardiovascular disease risk factors and atherosclerosis in type 1 diabetes mellitus: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study (DCCT/EDIC) study. Circulation. 2013;127:180‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herman WH, Ilag LL, Johnson SL, et al. A clinical trial of continuous subcutaneous insulin infusion versus multiple daily injections in older adults with type 2 diabetes. Diabetes Care. 2005;28:1568‐1573. [DOI] [PubMed] [Google Scholar]
- 13. Edridge CL, Dunkley AJ, Bodicoat DH, et al. Prevalence and incidence of hypoglycaemia in 532,542 people with type 2 diabetes on oral therapies and insulin: a systematic review and meta‐analysis of population based studies. PLoS One. 2015;10:e0126427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khunti K, Alsifri S, Aronson R, et al. Rates and predictors of hypoglycaemia in 27 585 people from 24 countries with insulin‐treated type 1 and type 2 diabetes: the global HAT study. Diabetes Obes Metab. 2016;18:907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information