

Abstract
Pathogenic somatic missense mutations within the DNA polymerase epsilon (POLE) exonuclease domain define the important subtype of ultramutated tumours (‘POLE‐ultramutated’) within the novel molecular classification of endometrial carcinoma (EC). However, clinical implementation of this classifier requires systematic evaluation of the pathogenicity of POLE mutations. To address this, we examined base changes, tumour mutational burden (TMB), DNA microsatellite instability (MSI) status, POLE variant frequency, and the results from six in silico tools on 82 ECs with whole‐exome sequencing from The Cancer Genome Atlas (TCGA). Of these, 41 had one of five known pathogenic POLE exonuclease domain mutations (EDM) and showed characteristic genomic alterations: C>A substitution > 20%, T>G substitutions > 4%, C>G substitutions < 0.6%, indels < 5%, TMB > 100 mut/Mb. A scoring system to assess these alterations (POLE‐score) was developed; based on their scores, 7/18 (39%) additional tumours with EDM were classified as POLE‐ultramutated ECs, and the six POLE mutations present in these tumours were considered pathogenic. Only 1/23 (4%) tumours with non‐EDM showed these genomic alterations, indicating that a large majority of mutations outside the exonuclease domain are not pathogenic. The infrequent combination of MSI‐H with POLE EDM led us to investigate the clinical significance of this association. Tumours with pathogenic POLE EDM co‐existent with MSI‐H showed genomic alterations characteristic of POLE‐ultramutated ECs. In a pooled analysis of 3361 ECs, 13 ECs with DNA mismatch repair deficiency (MMRd)/MSI‐H and a pathogenic POLE EDM had a 5‐year recurrence‐free survival (RFS) of 92.3%, comparable to previously reported POLE‐ultramutated ECs. Additionally, 14 cases with non‐pathogenic POLE EDM and MMRd/MSI‐H had a 5‐year RFS of 76.2%, similar to MMRd/MSI‐H, POLE wild‐type ECs, suggesting that these should be categorised as MMRd, rather than POLE‐ultramutated ECs for prognostication. This work provides guidance on classification of ECs with POLE mutations, facilitating implementation of POLE testing in routine clinical care. © 2019 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: POLE, molecular classification, endometrial cancer
Introduction
Pathogenic somatic mutations in the exonuclease domain of the replicative DNA polymerase epsilon (POLE) define a subgroup of endometrial cancers (ECs) with ultramutation (frequently ≥ 100 mutations/Mb), characteristic mutation signature (COSMIC signature 10) 1, enhanced immune response 2, 3, and excellent clinical outcome 4, 5, 6, 7. ‘POLE ultramutated’ EC (POLEmut EC) has therefore been proposed as a distinct clinical entity that can be diagnosed in the presence of a pathogenic POLE exonuclease domain mutation (EDM) 8. For the five most common POLE mutations (P286R, V411L, S297F, A456P, and S459F), pathogenicity (in this sense meaning causal for tumour ultramutation) has been confirmed 4, 5, 6, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25; however, the classification of other, less frequent POLE variants is currently challenging. This is becoming an urgent problem, as POLE sequencing for molecular EC classification is rapidly entering clinical practice.
Previous work has shown that ECs with a pathogenic POLE EDM typically display characteristic genomic alterations, with a high prevalence of C>A substitutions, frequently exceeding 20%; a low proportion of small insertion and deletion mutations (indels); and an extremely high tumour mutational burden (TMB; > 100 mut/Mb) 12, 26. In the pivotal 2013 EC study from The Cancer Genome Atlas (TCGA), all 17 tumours classified as ultramutated had a POLE EDM, including recurrent P286R and V411L substitutions (eight and five cases, respectively), and one case each of S297F, A456P, M444K, and L424I substitutions 7. Interestingly, 10 of 231 non‐ultramutated ECs in this study also had a POLE mutation either within or outside the exonuclease domain. Following the TCGA report, further studies have confirmed the prevalence of the five pathogenic mutations listed above and identified additional variants of uncertain pathogenicity. The parameters by which to evaluate the latter are ill defined, and thus classification of such cases is challenging, particularly in the absence of whole‐exome or whole‐genome sequencing (WES/WGS). In order to facilitate the classification of ECs in clinical practice, we aimed to develop a scoring system to estimate the pathogenicity of novel POLE mutations based on the presence or absence of genomic alterations associated with known pathogenic POLE mutations. We also sought to provide pragmatic guidelines for the interpretation of POLE variants in cases analysed by targeted POLE sequencing where such comprehensive genomic data are unavailable, being mindful that the designation of a tumour as POLE‐ultramutated EC may lead to withholding treatment, given the very favourable prognosis of this EC molecular subtype, so that a conservative approach to diagnosis is warranted.
Materials and methods
Data extraction TCGA EC cohort
To analyse the base change proportions of the TCGA cohort of ECs (n = 530), we downloaded the MAF files [using Mutect for point somatic mutation call as well as small insertions and deletions (indels)] from Genome Data Commons (https://portal.gdc.cancer.gov/; accessed 27 February 2019). We used somatic called coding variants [single nucleotide substitutions (SNV), including synonymous mutations, and indels] as mutation count. To calculate tumour mutational burden (TMB), we used 38 Mb as the estimate of the exome size. Microsatellite status, as defined by the Bethesda Protocol classification 27, was obtained from the Genome Data Analysis Center (GDAC) database (https://gdac.broadinstitute.org/; accessed 30 October 2018).
COSMIC signatures from all 530 TCGA ECs were obtained from mSignatureDB (http://tardis.cgu.edu.tw/msignaturedb/; accessed 22 October 2019) 28, 29.
Recurrence of somatic POLE mutations in EC and pan‐cancer
We searched for each somatic POLE mutation in the complete TCGA (Genome Data Commons) catalogues and COSMIC (https://cancer.sanger.ac.uk/cosmic, accessed 10 January 2019), annotating their recurrence on all cancer types (pan‐cancer) and exclusively within ECs (supplementary material, Table S1). Recurrent mutations were defined as those present in two or more cancer samples in the COSMIC and TCGA databases combined (cases present in both databases were counted only once). A mutation was considered non‐recurrent if it was found only once.
In silico prediction tools
To evaluate the functional status of somatic POLE mutations, we used six widely‐used in silico tools: SIFT 30, PROVEAN 31, PolyPhen‐2 32, PANTHER 33, SNAP2 34, and the meta predictor REVEL 35. SIFT is a multi‐step algorithm using sequence‐based predictive features to predict the effect of single‐nucleotide polymorphisms (SNPs) 30. PROVEAN extends this approach, additionally incorporating analysis of in‐frame insertions, deletions, and multiple substitutions 31. PolyPhen‐2 implements sequence‐based and structure‐based predictive features and compares wild‐type and mutant allele through a decision tree 32; ‘possibly damaging’ results were interpreted as benign. PANTHER is based on protein sequence, using a metric based on evolutionary conservation on direct ancestors of the organism 33; ‘possibly damaging’ and ‘probably benign’ results were interpreted as benign. SNAP2 is a neural network‐based classifier that uses sequence and structural‐based data as inputs 34. REVEL is an ensemble method based on 13 individual tools 35; scores below 0.5 were considered benign.
Somatic POLE mutations reported in ECs and not detected in TCGA cases
A review of the literature was undertaken, to the end of 2018, to identify ECs in which POLE had been sequenced and the mutations published 6, 7, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20, 21, 22, 24, 25. All literature contributing entries into the COSMIC database (POLE + endometrium) were reviewed; in addition, searches in PubMed and Web of Science were undertaken with the keywords ‘POLE + Endometrial + Carcinoma’ and ‘POLE + Endometrial + Cancer’, noting that ‘POLϵ’ is interpreted as ‘POLE’ in these resources.
DNA mismatch repair‐deficient/microsatellite‐unstable, POLE exonuclease domain‐mutated endometrial cancer cohort
Tumours with concomitant mismatch repair deficiency (MMRd) and somatic POLE EDM, and clinical follow‐up were identified from a pooled cohort of 2988 molecularly profiled ECs across ten participating institutes (a detailed description can be found in León‐Castillo et al 36). Informed consent and ethical approvals were obtained according to local protocols in each participating centre. These tumours were combined with five tumours with concomitant microsatellite instability and POLE EDM from the 2013 TCGA EC cohort 7 for survival analysis.
Statistical analysis
Nominal variables were compared by χ2 statistics or Fisher's exact test and ordinal variables using the Mann–Whitney test. All statistical tests were two‐sided and statistical significance was accepted at p < 0.05. We generated Kaplan–Meier curves for recurrence‐free survival (RFS) and overall survival (OS), and differences were tested by the log‐rank test. The median follow‐up was estimated by the reverse Kaplan–Meier method.
Results
Genomic characteristics of endometrial cancers with somatic POLE mutations in the complete TCGA cohort
To elucidate which genomic alterations best define pathogenic somatic POLE mutations (which we use in this context to mean very likely causal for tumour ultramutation), we used data from 530 ECs profiled by TCGA, including those reported in the 2013 publication 7. This included 82 tumours with a somatic POLE mutation, of which 59 (72%) were located within the exonuclease domain and 23 (28%) outside the exonuclease domain. The 59 exonuclease domain mutations comprised 21 unique variants, the five most common of which (P286R, 21 cases; V411L, 13 cases; S297F, 3 cases; A456P, 2 cases; and S459F, 2 cases) were classified as pathogenic based on previous reports 7, 8, 26 and designated as ‘hotspot’ POLE mutations for the purpose of this study (Table 1).
Table 1.
POLE variants in TCGA EC
| Protein change | No. of cases | Nucleotide substitution | Exon | MSI‐H cases (%) | Mutation recurrence in EC | Mutation recurrence pan‐cancer | No. of ‘benign’ results by in silico tools | POLE‐score | EDM | Signature 10 contribution |
|---|---|---|---|---|---|---|---|---|---|---|
| P286R | 21 | c.857C>G | 9 | 1 (4.8) | Recurrent | Recurrent | 0 | 5–6 | Y | 0.225–0.978 |
| V411L | 13 | c.1231G>T/C | 13 | 1 (7.7) | Recurrent | Recurrent | 1 | 4–6 | Y | 0.000–0.751 |
| S297F | 3 | c.890C>T | 9 | 2 (66.7) | Recurrent | Recurrent | 0 | 5–6 | Y | 0.123–0.611 |
| S459F | 2 | c.1376C>T | 14 | 0 (0) | Recurrent | Recurrent | 1 | 5–6 | Y | 0.940–0.955 |
| A456P | 2 | c.1366G>C | 14 | 0 (0) | Recurrent | Recurrent | 0 | 5–6 | Y | 0.277–0.837 |
| F367S | 2 | c.1100T>C | 11 | 2 (100) | Recurrent | Recurrent | 0 | 6 | Y | 0.095–0.100 |
| L424I | 2 | c.1270C>A | 13 | 2 (100) | Recurrent | Recurrent | 1 | 5 or 3 | Y | 0.000–0.000 |
| M295R | 1 | c.884T>G | 9 | 1 (100) | Recurrent | Recurrent | 0 | 6 | Y | 0.785 |
| P436R | 1 | c.1307C>G | 13 | 0 (0) | Recurrent | Recurrent | 0 | 6 | Y | 0.230 |
| M444K | 1 | c.1331T>A | 13 | 0 (0) | Recurrent | Recurrent | 0 | 5 | Y | 1.000 |
| R705W | 1 | c.2113C>T | 19 | 0 (0) | Novel | Novel | 1 | 5 | N | 0.821 |
| D368Y | 1 | c.1102G>T | 11 | 1 (100) | Novel | Recurrent | 0 | 4 | Y | 0.042 |
| M1754V | 1 | c.5260A>G | 39 | 1 (100) | Novel | Novel | 5 | 3 | N | 0.000 |
| K1070N | 1 | c.3210G>T | 26 | 1 (100) | Novel | Novel | 1 | 3 | N | 0.000 |
| L424V | 1 | c.1270C>G | 13 | 0 (0) | Recurrent | Recurrent | 0 | 3 | Y | 0.529 |
| A428T | 1 | c.1282G>A | 13 | 0 (0) | Novel | Novel | 5 | 3 | Y | 0.000 |
| R742H | 1 | c.2225G>A | 20 | 1 (100) | Novel | Recurrent | 1 | 3 | N | 0.018 |
| Q1335* | 1 | c.4003C>T | 30 | 1 (100) | Novel | Novel | NA | 3 | N | 0.000 |
| T278M | 1 | c.833C>T | 9 | 1 (100) | Recurrent | Recurrent | 0 | 3 | Y | 0.000 |
| A465V | 1 | c.1394C>T | 14 | 1 (100) | Recurrent | Recurrent | 0 | 3 | Y | 0.000 |
| S461L | 1 | c.1382C>T | 14 | 1 (100) | Novel | Novel | 0 | 2 | Y | 0.000 |
| R114* | 1 | c.340C>T | 5 | 1 (100) | Recurrent | Recurrent | NA | 2 | N | 0.000 |
| F990C | 1 | c.2969T>G | 25 | 0 (0) | Novel | Novel | 0 | 1 | N | 0.000 |
| W1824C | 1 | c.5472G>T | 40 | 0 (0) | Novel | Novel | 0 | 1 | N | 0.000 |
| E396G | 1 | c.1187A>G | 12 | 1 (100) | Recurrent | Recurrent | 2 | 1 | Y | 0.000 |
| A1140T | 1 | c.3418G>A | 28 | 1 (100) | Novel | Recurrent | 5 | 1 | N | 0.000 |
| Y1889C | 1 | c.5666A > G | 41 | 1 (100) | Novel | Novel | 0 | 1 | N | 0.000 |
| A781S | 1 | c.2341G>T | 21 | 1 (100) | Novel | Novel | 6 | 1 | N | 0.000 |
| R34C | 1 | c.100C>T | 2 | 0 (0) | Recurrent | Recurrent | 1 | 1 | N | 0.000 |
| E1461V | 1 | c.4382A>T | 34 | 1 (100) | Novel | Novel | 5 | 1 | N | 0.000 |
| R976S | 1 | c.2926C > A | 25 | 0 (0) | Novel | Novel | 1 | 0 | N | 0.000 |
| V2025M | 1 | c.6073G>A | 44 | 1 (100) | Novel | Novel | 6 | 0 | N | 0.000 |
| A566T | 1 | c.1696G>A | 16 | 1 (100) | Novel | Novel | 2 | 0 | N | 0.000 |
| R1386Q | 1 | c.4157G>A | 33 | 0 (0) | Novel | Novel | 2 | 0 | N | 0.022 |
| D368* | 1 | c.1101dupT | 11 | 1 (100) | Novel | Novel | NA | 0 | Y | 0.011 |
| R1321K | 1 | c.3962G>A | 31 | 1 (100) | Novel | Novel | 5 | 0 | N | 0.000 |
| Q1049H | 1 | c.3147G>T | 26 | 1 (100) | Novel | Novel | 2 | 0 | N | 0.000 |
| R764M | 1 | c.2291G>T | 20 | 1 (100) | Novel | Novel | 0 | 0 | N | 0.000 |
| E1698D | 1 | c.5094G > T | 38 | 1 (100) | Novel | Novel | 1 | 0 | N | 0.000 |
| A1010T | 1 | c.3028G>A | 25 | 1 (100) | Novel | Novel | 1 | 0 | N | 0.000 |
| C402R | 1 | c.1204T>C | 12 | 1 (100) | Novel | Novel | 3 | 0 | Y | 0.000 |
| T906I | 1 | c.2717C>T | 24 | 1 (100) | Novel | Novel | 0 | 0 | N | 0.000 |
| Q352H | 1 | c.1056G>T | 11 | 1 (100) | Novel | Novel | 4 | 0 | Y | 0.000 |
| Q453R | 1 | c.1358A>G | 13 | 1 (100) | Novel | Novel | 3 | 0 | Y | 0.000 |
NA, not assessable. Pathogenic mutations in the exonuclease domain are in bold. Y = yes; N = no.
The location of each POLE mutation (exonuclease domain versus non‐exonuclease domain), its recurrent or non‐recurrent status in EC in the TCGA and COSMIC databases, and genomic characteristics are shown for all cases in Figure 1. As previously reported 1, 7, 12, 26, the five hotspot POLE mutations were reliably associated with elevated TMB (median = 268 mut/Mb), which exceeded 100 mut/Mb (typically used to define ultramutation) in most tumours (33/41). Interestingly, TMB varied between different hotspot mutations (range 37.5–791.9 mut/Mb), and among tumours with identical hotspot mutations (e.g. P286R: 41.9–550.1 mut/Mb). POLE hotspot‐mutant ECs typically displayed a high proportion of C>A substitutions (median 32.5, > 20% in 37/41 tumours) and T>G substitutions (median 12.8%), whereas the proportion of C>G substitutions (median 0.3%) and indels (median 0.5%) was small. For comparison, 321 microsatellite‐stable (MSS), POLE wild‐type ECs all had TMB < 100 mut/Mb (median 2.1), a lower C>A proportion (median 13.5%) and T>G proportion (median 3.9%), and a higher C>G proportion (median 8.9%) and indel proportion (median 7.4%) (Table 2). We defined tumours with POLE hotspot mutations as a set of ‘true positives’, for subsequent identification of genomic alterations associated with pathogenic POLE mutations (Table 2).
Figure 1.
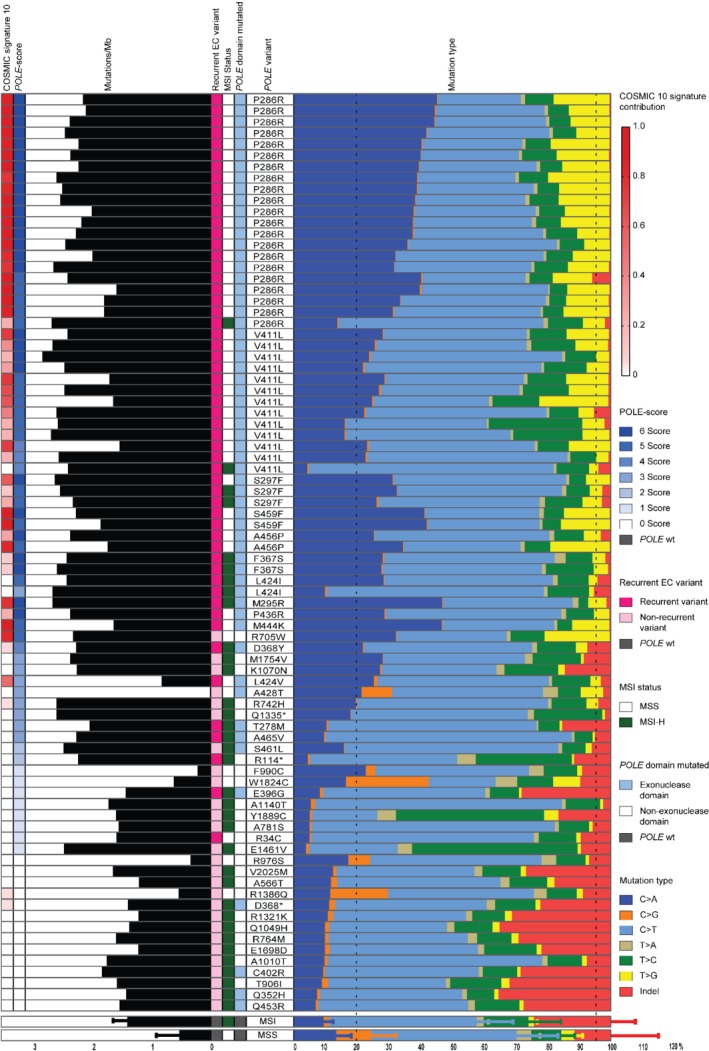
Mutational features of EC with POLE variants in the TCGA. The colour scheme for the mutation type is on the right of the histogram. Cases are grouped by mutations, with the most frequent POLE mutations in first place. The COSMIC 10 signature contribution, the points obtained in the POLE pathogenicity score (POLE‐score), the recurrence of the variant in EC, microsatellite instability (MSI) status, and POLE domain mutated are colour‐coded (legend on the right of the histogram). Below are the cases without POLE mutations; two rows depict the median plus standard deviation of the base change proportions and tumour mutation burden (TMB) of MSI‐H and MSS ECs without a POLE mutation in the TCGA.
Table 2.
Tumour mutation burden and SNV/indel by POLE mutation location and tumour MSI status in TCGA endometrial cancers
| ECs with hotspot POLE mutations | ECs with non‐hotspot POLE EDMs | ECs with POLE non‐EDMs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | MSS | MSI | Total | MSS | MSI | Total | MSS | MSI | MSI‐POLEwt ECs | MSS‐POLEwt ECs | |
| n = 41 | n = 37 | n = 4 | n = 18 | 4,0 | n = 14 | n = 23 | n = 6 | n = 17 | n = 127 | n = 321 | |
| Tumour mutational burden | |||||||||||
| Median (range) | 268.0 (37.5–791.9) | 262.8 (37.5–791.9) | 339.0 (237.7–550.1) | 164.4 (1.1–530.4) | 27.3 (1.1–262.9) | 207.1 (26.9–530.4) | 42.8 (1.7–452.9) | 4 (1.7–236.2) | 48.5 (17.4–452.9) | 21.5 (0.0–150.5) | 2.1 (0.3–59.0) |
| ≥ 100 mut/Mb (%) | 33 (80.5) | 29 (78.4) | 4 (100) | 10 (55.6) | 1 (25) | 9 (64.3) | 7 (30.4) | 1 (16.7) | 6 (35.3) | 1 (0.8) | 0 (0) |
| Percentage of C:G>A:T | |||||||||||
| Median (range) | 32.5 (4.3–45.2) | 33.0 (16.0–45.2) | 20.0 (4.3–32.5) | 20.2 (6.9–46.9) | 27.0 (21.4–46.7) | 10.8 (6.9–46.9) | 10.8 (3.9–32.3) | 16.9 (5.0–32.3) | 9.9 (3.9–28.1) | 9.1 (0.0–23.2) | 13.5 (2.8–27.6) |
| Proportion ≥ 20% (%) | 37 (90.2) | 35 (94.6) | 2 (50) | 9 (50) | 4 (100) | 5 (35.7) | 4 (17.4) | 2 (33.3) | 2 (11.8) | 1 (0.8) | 25 (7.8) |
| Percentage of C:G>G:C | |||||||||||
| Median (range) | 0.3 (0.2–0.6) | 0.3 (0.2–0.6) | 0.3 (0.2–0.6) | 0.5 (0.2–9.5) | 0.7 (0.3–9.5) | 0.5 (0.2–2.0) | 1.0 (0.2–26.1) | 5.0 (0.3–26.1) | 0.9 (0.2–2.0) | 1.5 (0.0–8.8) | 8.9 (0.0–47.7) |
| Proportion < 0.6% (%) | 37 (90.2) | 34 (91.9) | 3 (75) | 11 (61.1) | 2 (50) | 9 (64.3) | 6 (26.1) | 1 (16.7) | 5 (29.4) | 5 (3.9) | 2 (0.6) |
| Percentage of C:G>T:A | |||||||||||
| Median (range) | 43.6 (26.2–77.4) | 40.7 (26.2–63.1) | 58.3 (50.8–77.4) | 52.1 (35.2–77.7) | 50.9 (35.2–55.3) | 52.1 (40.9–77.7) | 45.9 (20.5–77.7) | 47.2 (21.0–70.2) | 44.6 (20.5–77.7) | 46.0 (0.0–79.9) | 47.9 (4.7–85.5) |
| Percentage of T:A>A:T | |||||||||||
| Median (range) | 1.1 (0.5–1.7) | 1.1 (0.5–1.6) | 1.4 (0.8–1.7) | 1.5 (0.7–4.8) | 1.3 (1.1–4.8) | 1.5 (0.7–3.4) | 2.4 (1.1–6.8) | 4.3 (1.1–6.8) | 2.3 (1.1–5.9) | 1.9 (0.0–8.2) | 4.5 (0.0–12.4) |
| Percentage of T:A>C:G | |||||||||||
| Median (range) | 8.9 (5.2–29.5) | 8.4 (5.2–29.5) | 10.7 (7.4–11.8) | 8.9 (3.1–15.1) | 7.9 (4.6–12.1) | 9.5 (3.1–15.1) | 11.8 (9.2–52.2) | 10.7 (9.2–11.8) | 12.1 (9.5–52.2) | 11.7 (2.4–100.0) | 9.3 (1.3–30.3) |
| Percentage of T:A>G:C | |||||||||||
| Median (range) | 12.8 (2.9–21.7) | 13.0 (4.0–21.7) | 5.1 (2.9–7.0) | 2.3 (0.6–11.7) | 6.0 (3.2–11.7) | 1.6 (0.6–5.8) | 1.6 (0.6–20.5) | 1.8 (1.2–20.5) | 1.6 (0.6–4.5) | 1.4 (0.0–8.0) | 3.9 (0.0–13.1) |
| Proportion ≥ 4% (%) | 38 (92.7) | 36 (97.3) | 2 (50) | 6 (33.3) | 3 (75) | 3 (21.4) | 3 (13.0) | 2 (33.3) | 1 (5.9) | 6 (4.7) | 154 (48.0) |
| Percentage of small indels | |||||||||||
| Median (range) | 0.5 (0.2–6.0) | 0.5 (0.2–6.0) | 2.8 (1.8–4.0) | 5.2 (0.4–35.5) | 1.5 (0.4–3.2) | 6.7 (0.9–35.5) | 9.5 (0.4–35.1) | 8.9 (0.4–9.7) | 14.5 (1.9–35.1) | 24.8 (0.0–40.2) | 7.4 (0.0–80.9) |
| Proportion < 5% (%) | 39 (95.1) | 35 (94.6) | 4 (100) | 8 (80) | 4 (100) | 4 (28.6) | 4 (17.4) | 1 (16.7) | 3 (17.6) | 3 (2.4) | 76 (23.7) |
Of the 41 TCGA ECs with a somatic non‐hotspot POLE mutation, 18 were located within the exonuclease domain. Comparing these with the 23 tumours with non‐exonuclease domain mutations, non‐hotspot POLE exonuclease domain‐mutant ECs had a higher TMB (median 164.4 versus 42.8 mut/Mb) and C>A proportion (median 20.2% versus 10.8%), and a lower C>G proportion (median 0.5% versus 1.0%) and indel proportion (median 5.2% versus 9.5%) (Table 2).
MSI status was available for all TCGA ECs, of which 35/82 cases with somatic POLE mutations (42.7%) were MSI‐H. Comparison between ECs with hotspot mutations and non‐hotspot mutations within and outside the exonuclease domain revealed striking differences: only 4/41 (9.8%) of the TCGA ECs with one of the five hotspot mutations were MSI‐H, whereas 14/18 (78%) ECs with a non‐hotspot exonuclease domain mutation and 17/23 (74%) ECs with a non‐exonuclease domain mutation were MSI‐H (p < 0.0001). Analysis of the genomic architecture of these tumours revealed notable differences between groups. Tumours with hotspot POLE mutations and MSI had a high TMB (median TMB of 339.0 mut/Mb, > 100 mut/Mb in all four cases) and a high proportion of C>A and T>G substitutions (median 20.0% and 5.1%, respectively), with a low proportion of C>G substitutions (median 0.3%) and indels (median 2.8%) (Table 2). Tumours with non‐hotspot POLE EDM and MSI had a lower TMB (median 207.1 mut/Mb, > 100 mut/Mb in 9/14 cases) and C>A and T>G proportions (median 10.8% and 1.6%, respectively), a similar proportion of C>G substitutions (median 0.5%), and higher indel proportion (median 6.7%) (Table 2). These differences were greater in tumours with a POLE mutation outside the exonuclease domain and concomitant MSI, which had a median TMB of 48.5 mut/Mb (> 100 mut/Mb in 6/17 cases); C>A and T>G proportions of 9.9% and 1.6%, respectively; a C>G frequency of 0.9%; and a median indel proportion of 14.5%. For comparison, of 127 MSI‐H ECs without a POLE mutation (MSI‐H ECs), only one case had a TMB above 100 mut/MB (median 21.5) or a C>A proportion above 20% (median 9.1%); these cancers also had low T>G proportions (median 1.4%),a higher C>G proportion (median 1.5%,) and a high indel proportion (median 24.8%) (Table 2). Thus, the genomic characteristics of MSI‐H cancers with a POLE mutation outside the exonuclease domain are similar to those of MSI‐H tumours without a POLE mutation. Consequently, the frequency with which MSI co‐exists with POLE mutation varies by POLE mutation location and is reflected in differing genomic architecture – consistent with variable pathogenicity of POLE mutations.
These analyses confirm that ECs with one of the five somatic hotspot POLE EDMs carry characteristic genomic sequence alterations distinct from MSI‐H and MSS, POLE‐wild‐type EC. These genomic alterations are variably present in cases with non‐hotspot POLE EDM and are uncommon in ECs with POLE mutations outside the exonuclease domain. The variation in the genomic correlates of POLE mutations by their location is mirrored by variation in the prevalence of MSI in cancers carrying these mutations, and in differences in the genomic architecture of tumours harbouring both defects. Collectively, these data confirm that different POLE mutations vary in pathogenicity and underscore the need for its reliable estimation to ensure accurate patient classification.
Establishing a pathogenicity score for somatic POLE mutations
Motivated by our preliminary analyses, we next used the TCGA WES data to develop a scoring system to assess the pathogenicity of POLE mutations (defined as the likelihood that they are associated with the characteristic ultramutated phenotype), using the hotspot POLE mutations as a truth set. Taking TMB and C>A, T>G, C>G, and indel proportions as the most discriminating genomic alterations for these pathogenic mutations, and building on previous work 26, we developed a pragmatic scoring system in which tumours scored 1 point for each of the following: TMB > 100 mut/Mb; C>A ≥ 20%; T>G ≥ 4%; C>G ≤ 0.6%; and indels ≤ 5%. All 41 TCGA ECs with a hotspot POLE mutation scored 3–5 points, while 13/41 (31.7%) ECs with a non‐hotspot POLE mutation scored ≥ 3 points, including 8/18 with exonuclease domain mutations, while 19/23 tumours with POLE mutations outside the exonuclease domain had scores ≤ 2, the exceptions being three tumours with score 3 (each of which had likely pathogenic mutations in POLD1: D316G, S478N, and L606M) and one scoring 5 points with a POLE R705W mutation. We therefore chose to focus on mutations in the exonuclease domain, given the infrequent association of non‐exonuclease domain mutations with genomic alterations associated with the ultramutated phenotype.
To further refine this scoring system, we considered whether POLE mutations were recurrent in ECs within the COSMIC or TCGA databases, as recurrent mutations are more likely to be pathogenic (that is, causal of tumour ultramutation) 37. Forty‐eight out of 54 (88.9%) ECs scoring ≥ 3 points had a recurrent POLE mutation (including hotspot mutations), compared with 7/28 (25%) tumours scoring ≤ 2 points (p ≤ 0.001, χ2 statistics). Restricting the analysis to non‐hotspot POLE EDM, 7/8 (87.5%) tumours scoring ≥ 3 points had a recurrent mutation versus 5/10 (50%) scoring ≤ 2 points (p = 0.152, Fisher's exact test). Based on these results, ‘recurrence’ was incorporated into the final scoring system (Figure 2), which we termed the ‘POLE pathogenicity score’ (POLE‐score) (Table 1 and Figure 1).
Figure 2.
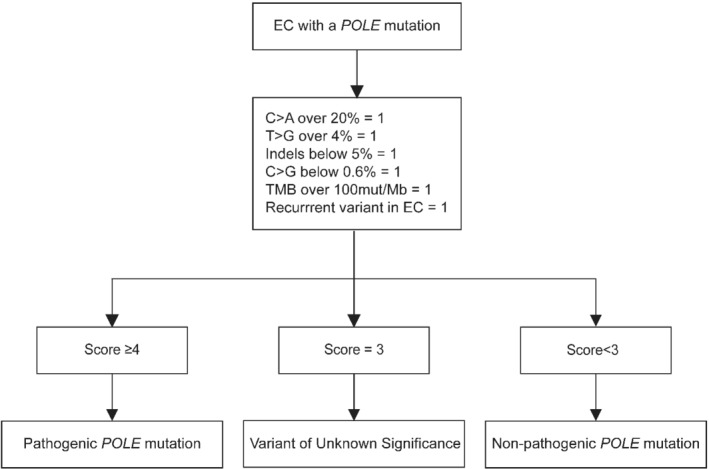
POLE genomic alteration score (POLE‐score). Diagnostic scoring system based on mutation type proportion and TMB of the five hotspot POLE mutations, as well as the variant recurrence.
To define a cut‐off for pathogenicity, we applied the POLE‐score on hotspot POLE‐mutant, non‐hotspot POLE EDM and control POLE wild‐type ECs (MSS and MSI‐H) in the TCGA cohort. Thirty‐eight of 41 (92.7%) ECs with a hotspot POLE EDM had a POLE‐score of ≥ 5 points (Figure 1). The remaining three tumours, all of which harboured a V411L mutation, scored 4 points. In contrast, of the 18 tumours with a non‐hotspot POLE EDM, seven scored ≥ 4 points (all of which carried mutations recurrent in the TCGA or COSMIC EC databases: F367S, L424I, M295R, P436R, M444K, D368Y), five scored 3 points (four of which carried recurrent mutations: A465V, L424V, T278M, L424I; one with a non‐recurrent A428T substitution), and six scored ≤ 2 points (one of which had a recurrent mutation). For comparison, all 321 MSS, POLE wild‐type ECs scored ≤ 3 points and all 127 MSI‐H POLE wild‐type ECs scored ≤ 2.
Based on these data, we used a POLE‐score of ≥ 4 points to define pathogenicity of POLE mutations in EC. When applying this cut‐off, 48 ECs in the TCGA are classified as having pathogenic POLE EDM (all 41 cases with hotspot mutations and seven with non‐hotspot variants), comprising 11 unique mutations, all of which are recurrent in TCGA/COSMIC (Table 3). ECs with a POLE‐score ≤ 2 were classified as having non‐pathogenic POLE EDM, based on the absence of genomic alterations associated with ultramutated phenotype. Cancers with a score of 3 (A465V, L424V, T278M, and A428T) were classified as having a variant of uncertain significance.
Table 3.
Pathogenic POLE EDM based on POLE‐score
| Protein change | Nucleotide substitution |
|---|---|
| P286R | c.857C>G |
| V411L | c.1231G>T/C |
| S297F | c.890C>T |
| S459F | c.1376C>T |
| A456P | c.1366G>C |
| F367S | c.1100T>C |
| L424I | c.1270C>A |
| M295R | c.884T>G |
| P436R | c.1307C>G |
| M444K | c.1331T>A |
| D368Y | c.1102G>T |
To validate the POLE‐score, we noted the contribution of COSMIC signature 10 in ECs with a POLE EDM EC with a POLE‐score ≥ 4 points: signature 10 was present in 46 ECs (95.8%) and completely absent in two ECs (L424I and V411L) (mean 0.623, range 0.000–1.000). The contribution of signature 10 in ECs with one of the five hotspot POLE EDMs ranged from 0.978 to 0.123. Only in one (20.0%) EC with a POLE EDM classified as VUS (L424V) and one (16.7%) EC with a POLE EDM classified as non‐pathogenic (D368*) was activity of COSMIC signature 10 identified (mean contribution 0.106, range 0.00–0.529 and mean 0.002, range 0.000–0.011, respectively for each group). In comparison, COSMIC signature 10 was identified in 11 (8.7%) MSI‐POLEwt and 96 (29.9%) MSS‐POLEwt ECs (mean signature 10 contribution 0.002, range 0.000–0.048, and mean 0.017, range 0.000–0.218, respectively).
Relationship between pathogenicity of somatic POLE mutations, microsatellite instability, and clinical outcome
The co‐existence of POLE mutations and MMRd/MSI in EC 26, 38 and the variation in its prevalence by POLE mutation location raise important questions about which is the initial, presumably dominant factor determining tumour phenotype and clinical outcome. To further investigate this, we used the POLE‐score to stratify TCGA cases into predicted pathogenic and non‐pathogenic POLE mutations using a score of ≥ 4. Nine of 49 (18.4%) ECs with a predicted pathogenic POLE mutation (including four known hotspot mutations) were MSI‐H, compared with 26/33 (78.8%) tumours with a predicted non‐pathogenic mutation (p ≤ 0.0001, χ2 statistic). Restricting the analysis to tumours with POLE EDM, 9/48 (18.8%) cases with a predicted pathogenic EDM (including hotspot mutations) were MSI‐H, as opposed to 9/11 (81.8%) with a predicted non‐pathogenic EDM (p ≤ 0.0001, Fisher's exact test). Interestingly, further stratification suggested a similar variation between likely pathogenic POLE mutations, as only 2/34 ECs with a P286R or V411L mutation were MSI‐H, compared with 7/14 ECs with one of the other nine predicted pathogenic mutations (p = 0.0012). Thus, POLE mutations co‐existent with MSI in EC are more likely to be non‐exonuclease, non‐pathogenic mutations, though this is not universally the case.
To investigate the clinical outcome of POLE exonuclease domain‐mutant EC with concomitant MMRd, we identified 30 such patients from a pooled analysis of 3236 ECs (Table 4). Five‐year recurrence‐free survival (RFS) for this subgroup was 83.2%, with a 5‐year overall survival (OS) of 80.9% (Figure 3) (corresponding figures for 24 patients with stage I disease were 84.2% and 85.4%, respectively) (supplementary material, Figure S1), seemingly contrasting with the 5‐year RFS and OS of 92–100% previously reported for POLE exonuclease domain‐mutant EC 4, 5, 7. To clarify this, we stratified patients according to predicted pathogenic versus non‐pathogenic EDM using the POLE‐score and analysed their clinical outcome. For cases that lacked WES data and for which POLE EDM had not been previously described in the TCGA, we considered all mutations different to the ones present in Table 3 (mutations deemed pathogenic using the POLE‐score) as VUS. This revealed that the 13 cases with one of the 11 mutations classified as likely pathogenic by POLE‐score (Table 3) had a 5‐year RFS of 92.3%, while the corresponding value for the 14 patients with EDM classified as likely non‐pathogenic/VUS was 76.2% (p = 0.40, log‐rank test) (Figure 3). While the clinical behaviour of tumours with combined MMRd/MSI and POLE EDM may vary based on the pathogenicity of the latter, this difference was not statistically significant, possibly owing to insufficient power/small numbers of cases, and it is not possible to determine the prognosis of this subgroup with certainty at present.
Table 4.
Clinicopathological features of MMRd–POLEmut ECs
| MMRd–POLEmut ECs | |
|---|---|
| n = 30 (100%) | |
| Age, years | |
| Mean [range] | 66.5 [27–87] |
| < 60 | 9 (30) |
| 60–70 | 8 (26.7) |
| > 70 | 13 (43.3) |
| Stage | |
| IA | 14 (46.6) |
| IB | 10 (33.3) |
| II | 3 (10) |
| III | 4 (10) |
| IV | 0 (0) |
| Histology | |
| Endometrioid | 25 (83.3) |
| Serous | 1 (3.3) |
| Mixed | 2 (6.7) |
| Clear cell | 3 (6.7) |
| Grade | |
| 1–2 | 19 (63.3) |
| 3 | 11 (36.7) |
| Myometrium invasion | |
| < 50% | 13 (43.3) |
| > 50% | 15 (56.7) |
| LVSI | |
| Absent | 21 (70) |
| Present | 4 (13.3) |
| Missing | 5 (16.7) |
| Treatment | |
| None | 7 (23.3) |
| Radiotherapy | 10 (33.3) |
| Chemotherapy | 1 (3.3) |
| Radiochemotherapy | 5 (16.7) |
| Unknown | 7 (23.3) |
| POLE mutations | |
| Pathogenic mutation | 14 (46.7) |
| Non‐pathogenic mutation/variant of unknown significance | 16 (53.3) |
Figure 3.
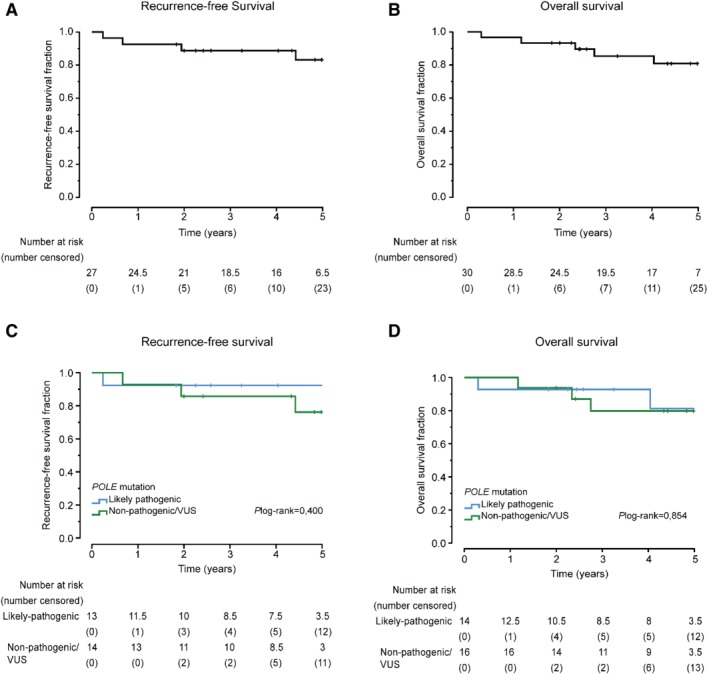
Clinical outcome of MMRd–POLEmut ECs. Kaplan–Meier survival curves for RFS (A) and OS (B) of MMRd–POLEmut ECs. RFS and OS of MMRd–POLEmut ECs with a pathogenic POLE EDM (mutation present in Table 3) versus all other tumours MMRd–POLEmut (C and D).
Estimation of pathogenicity of somatic POLE mutations in the absence of exome or genome sequencing
Somatic mutation profiling in clinical practice is typically performed by targeted panel sequencing, rather than WES/WGS approaches at present. To develop a classification tool for ECs with somatic non‐hotspot POLE mutations that can be implemented using such data, we used mutation location, prior data, and in silico tools which estimate the probability that a mutation is damaging. We first noted that nearly all (> 95%) POLE mutations outside the exonuclease domain are classified as non‐pathogenic by POLE‐score. We next noted that in the case of exonuclease domain mutations reported in the TCGA, the POLE‐score can be used to estimate pathogenicity (Table 3). We finally noted that for POLE EDM not present in the TCGA, in silico prediction tools could be used to estimate pathogenicity. Further exploration of this revealed that 10/11 POLE EDMs classified as pathogenic by POLE‐score in the TCGA cases were universally predicted to be disruptive by six in silico tools, the exception being an L424I substitution predicted to be deleterious by five tools but benign by one (Table 1 and supplementary material, Table S2). However, of five POLE EDMs present in the TCGA but classified as non‐pathogenic by POLE‐score, one (S461L, POLE‐score 2) was predicted to be damaging by all six tools, while another variant (E396G, POLE‐score 1) was predicted to be damaging by four tools. Furthermore, of four mutations classified as uncertain pathogenicity with a POLE‐score of 3, three (A465V, L424V, T278M) were considered damaging by all in silico prediction tools, while the other (A428T) was considered benign by five out of six prediction tools. Thus, in silico tools appear sensitive but not specific for prediction of pathogenic POLE EDMs, in the sense of their being likely causal for tumour ultramutation.
To define the extent of the problem of ascribing pathogenicity to POLE mutations in clinical practice, we identified 296/3840 (7.7%) tumours with a somatic POLE mutation from EC cohorts other than TCGA 6, 7, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20, 21, 22, 24, 25 (Table 5 and supplementary material, Table S3). Of 296 non‐TCGA POLE‐mutant ECs reported in the literature, 15 had mutations outside the exonuclease domain, and 254 carried mutations in the exonuclease domain previously detected in the TCGA and classified by POLE‐scores as pathogenic (249 cases), of uncertain pathogenicity (four cases), or non‐pathogenic (one case). The remaining 27 cancers with POLE EDM could not be classified by POLE‐score because their genomic correlates are yet to be determined by WES. This represents 9.1% of all reported POLE mutations, or 0.7% of non‐TCGA molecularly subtyped ECs to date. Of these 27 unique POLE EDMs, one was predicted to be benign by most in silico tools, while the others were predicted to be damaging by four tools or more (Table 5). The greater negative predictive value than positive predictive value of these tools, noted above, suggests that benign predictions should carry more weight and that the former are non‐pathogenic mutations, while the latter should be regarded as of uncertain pathogenicity. Cases such as these could be prioritised for more comprehensive sequencing, such as WES to provide sufficient data to determine their POLE‐score.
Table 5.
POLE EDMs in ECs not described previously in the TCGA
| Protein change | No. of cases | Nucleotide substitution | Exon | MSI cases (%) | Mutation recurrent in EC | Mutation recurrent pan‐cancer | No. of benign results by in silico tools |
|---|---|---|---|---|---|---|---|
| A426V | 1 (2.4) | c.1277C>T | 13 | Unknown | Recurrent | Recurrent | 1 |
| A456G | 1 (2.4) | c.1367C>G | 14 | Unknown | Novel | Novel | 1 |
| A456V | 1 (2.4) | c.1367C>T | 14 | 0 (0) | Novel | Recurrent | 0 |
| D275V | 1 (2.4) | c.824A>T | 9 | Unknown | Novel | Novel | 0 |
| D287E | 2 (4.9) | c.861T>A/G | 9 | 1 (50) | Novel | Novel | 1 |
| D462E | 1 (2.4) | c.1386T>A/G | 14 | 0 (0) | Novel | Novel | 1 |
| F367C | 1 (2.4) | c.1100T>G | 11 | 0 (0) | Novel | Novel | 0 |
| F367L | 1 (2.4) | c.1101T>A/G | 11 | 1 (100) | Novel | Novel | 0 |
| F367V | 1 (2.4) | c.1099T>G | 11 | 0 (0) | Novel | Novel | 0 |
| G364V | 1 (2.4) | c.1091G>T | 11 | 1 (100) | Novel | Novel | 0 |
| G388S | 1 (2.4) | c.1162G>A | 12 | 0 (0) | Novel | Novel | 0 |
| H342R | 1 (2.4) | c.1025A>G | 11 | Unknown | Novel | Novel | 5 |
| L283F | 1 (2.4) | c.847C>T | 9 | 1 (100) | Novel | Novel | 1 |
| L424P | 1 (2.4) | c.1271T>C | 13 | 0 (0) | Novel | Novel | 0 |
| M299I | 1 (2.4) | c.897G>A/C/T | 9 | 0 (0) | Novel | Novel | 0 |
| M405I | 1 (2.4) | c.1215G>A/C/T | 12 | 0 (0) | Novel | Novel | 2 |
| P286L | 1 (2.4) | c.857C>T | 9 | 1 (100) | Novel | Recurrent | 0 |
| P286S | 1 (2.4) | c.856C>T | 9 | 0 (0) | Novel | Recurrent | 0 |
| P436S | 2 (4.9) | c.1306C>T | 13 | 1 (50) | Novel | Novel | 0 |
| P441L | 1 (2.4) | c.1322C>T | 13 | 0 (0) | Novel | Recurrent | 1 |
| R375Q | 1 (2.4) | c.1124G>A | 12 | 0 (0) | Novel | Novel | 1 |
| S297Y | 1 (2.4) | c.889T>G | 9 | 0 (0) | Novel | Recurrent | 0 |
| T323A | 1 (2.4) | c.967A>G | 10 | 1 (100) | Novel | Novel | 1 |
| T457M | 1 (2.4) | c.1370C>T | 14 | 0 (0) | Novel | Recurrent | 2 |
| T483I | 1 (2.4) | c.1448C>T | 14 | 0 (0) | Novel | Novel | 0 |
Recommendations for classification of somatic POLE mutations in clinical practice
Based on the analyses above, we developed a pragmatic tool to classify EC with somatic POLE mutations in clinical practice, shown in Table 6 39, 40. For cases with WES/WGS, POLE‐score and the presence or absence of MSI/MMRd can be used to stratify cases into POLEmut, MMRd, or one of the other two TCGA subgroups depending on p53 status (Singh et al 41). It is important to note that the presence of a POLE mutation alone is insufficient to classify tumours as ‘POLEmut’, and that classification of tumours with combined POLE mutation and MMRd/MSI depends on the POLE‐score (i.e. genomic correlates) of the POLE mutation. For cases without WES/WGS, POLE‐score can be used if the POLE mutation has previously been reported in the TCGA. Where this is not the case, in silico tools can be used to triage tumours for more comprehensive WES/WGS to permit calculation of POLE‐score and subsequent classification.
Table 6.
Recommendations for the interpretation of somatic POLE mutations in ECs. Recommendations to classify ECs with POLE mutations with (A) POLE‐score available or (B) POLE‐score absent
| A | |||
|---|---|---|---|
| POLE mutation | Predicted pathogenicity | MSI/MMR status | Treatment recommendation |
| Exonuclease domain mutation | Pathogenic | MSS/MMRp | POLEmut EC |
| POLE‐score ≥ 4 | Pathogenic | MSI/MMRd | POLEmut EC* |
| Exonuclease domain mutation | Non‐pathogenic | MSS/MMRp | POLEwt EC |
| POLE‐score < 4 | Non‐pathogenic | MSI/MMRd | MMRd EC |
| Non‐exonuclease domain mutation | – | MSS/MMRp | NSMP/p53abn EC† |
| – | MSI/MMRd | MMRd EC | |
| If tumours‐only sequencing is performed, detection of L424V variant should prompt consideration of germline testing 39, 40. | |||
| B | |||
|---|---|---|---|
| POLE mutation | Predicted pathogenicity | MSI/MMR status | Treatment recommendation |
| Exonuclease domain mutation predicted to be pathogenic by ≥ 4 in silico prediction tools | VUS | MSS/MMRp | WES or NSMP/p53abn EC†, ‡ |
| VUS | MSI/MMRd | WES or MMRd EC‡ | |
| Exonuclease domain mutation predicted to be non‐pathogenic by > 1 in silico prediction tool | Non‐pathogenic | MSS/MMRp | NSMP/p53abn EC† |
| Non‐pathogenic | MSI/MMRd | MMRd EC | |
| Non‐exonuclease domain mutation | – | MSS/MMRp | NSMP/p53abn EC† |
| – | MSI/MMRd | MMRd EC | |
If tumours‐only sequencing is performed, detection of L424V variant should prompt consideration of germline testing 39, 40.
Treat as POLEmut EC (based on genomic alteration) independently of MMR status (insufficient data to suggest otherwise).
p53 IHC should be performed to exclude a p53abn EC.
Treat conservatively, i.e. as MMRd/NSMP or send for WES.
NSMP, no specific molecular profile; VUS, variant of unknown significance.
Discussion
The development of pragmatic surrogate markers has accelerated the clinical implementation of molecular EC classification. The presence of a pathogenic POLE EDM is causal for ultramutated EC, a subtype associated with enhanced immune response 2, 42 and excellent clinical outcome 6, 7, 13. De‐escalating adjuvant treatment in these patients is currently under investigation in the randomised PORTEC4a trial. However, interpretation of POLE sequence variants is challenging due to lack of standardized criteria, other than for the most common ‘hotspot’ mutations for which pathogenicity is reliably established. We aimed to generate tools to estimate the pathogenicity of POLE mutations using WES data, and to guide the management of cases where comprehensive genomic profiling is not available.
Using cases with recurrent ‘hotspot’ POLE EDMs as a truth set, we identified their characteristic genomic correlates to generate a ‘POLE‐score’. In addition to correctly classifying all cases with POLE hotspot mutations in the TCGA cohort, it classified a further six POLE EDMs as likely pathogenic. Four exonuclease domain mutations had a POLE‐score of 3 and were classified as being of uncertain pathogenicity, while three cases with POLE mutations outside the exonuclease domain had a POLE‐score of 3 – all of which carried a plausibly pathogenic POLD1 mutation that could explain the mutational spectrum 8. Intriguingly, a single case with a POLE mutation outside the exonuclease domain (R705W) was classified as pathogenic by POLE‐score. The location of the mutation within the catalytic domain, close to the polymerase active sites, may explain this mutational spectrum; however, the clinical significance of this is unclear at present.
Because POLE‐score relies on WES or WGS to estimate TMB and mutation proportions, it is unable to assign pathogenicity in the case of novel POLE mutations detected by targeted sequencing, where breadth is typically inadequate to estimate these parameters. Although this represents a potential challenge in clinical practice where targeted approaches are common, our pooled analysis suggests that this situation is uncommon – only 0.7% of ECs at the time of writing, a figure that will drop over the coming years as more WES/WGS data are accrued. We found that pathogenicity of such variants is not reliably predicted by in silico tools, which have low specificity. We suggest an approach to these tumours (outlined in Table 6), which may guide the use of additional sequencing (e.g. WES) to permit calculation of POLE‐score in these cases. Although WES remains relatively costly compared with targeted approaches, such outlay is modest against that of local or systemic therapy, and thus remains a possible approach for cases where a significant treatment decision hangs in the balance.
Our study confirms the complex relationship between POLE mutations and DNA mismatch repair deficiency/microsatellite instability in EC. Perhaps most straightforward are those with POLE mutations outside the exonuclease domain: these appear to be passengers secondary to the hypermutator phenotype and should be classified as MMRd ECs. Co‐existence of POLE EDM with MSI/MMRd is relatively uncommon, occurring in 3.4% cases in TCGA and 0.9% cases of molecularly subtyped tumours in our pooled series (this variation probably reflects a combination of targeted sequencing with enrichment for pathogenic POLE mutations in the latter cases). This group of tumours is heterogeneous. Those with POLE mutations predicted as pathogenic by POLE‐score and MSI had genomic architecture similar to POLE hotspot‐mutant/MSS tumours, supporting their classification as POLEmut EC. Those with POLE mutations predicted as non‐pathogenic by POLE‐score and MSI more closely resembled POLE‐wild‐type MSI cases, supporting their classification as MMRd EC. POLE EDM in combination with MMR loss causes a distinct mutational signature in EC (COSMIC signature 14) 1, 38 – the observation that this is not universal in cases with both defects supports the notion that these tumours are a heterogeneous group, where MSI/MMRd could be acquired after POLE EDM and vice versa, with differing impacts on prognosis. Interestingly, while data were limited, patients with combined pathogenic POLE EDM and MSI appeared to have a good clinical outcome in our pooled cohort (5‐year RFS 92.3%), though additional cases are required before this can be concluded.
In conclusion, our work provides guidance in the diagnostic interpretation of POLE mutations in EC in the presence and absence of WES data. Tumours with any of the 11 POLE EDMs identified in the TCGA and classified as pathogenic by POLE‐score should be classified as ‘POLE ultramutated’ EC, independently of MMRd/MSI status. For cases where a POLE EDM not present in the TCGA is identified and WES data are available, POLE‐score can be used for classification. In the absence of WES data, classification should be informed by the results of POLE‐score on mutations reported in the TCGA and classified in Table 3. In silico prediction tools have limited value but may be able to identify benign changes and triage cases for WES/WGS. The guidelines that we provide will evolve over time but will allow for almost all tumours encountered to be classified into a molecular subtype based on currently available information.
Author contributions statement
AL and HB carried out experiments and analysed data. TB, CBG, DNC, and MM conceived experiments and analysed data. JNM, RN, SK, SYB, JWC, EE, and TTR analysed data. All the authors were involved in writing the paper and had final approval of the submitted and published versions.
Supporting information
Figure S1. Clinical outcome of MMRd–POLEmut ECs
Table S1. POLE mutations reported in ECs in COSMIC or TCGA
Table S2. In silico tools results for POLE mutations found in ECs in TCGA
Table S3. In silico tools results for non‐hotspot POLE mutations found in EC cohorts published not present in TCGA
Acknowledgements
This work was supported by the Dutch Cancer Society (KWF‐YIG 8232‐31648; AL and TB), the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) (DNC), and the BC Cancer Foundation and Michael Smith Foundation for Health Research (JM). DNC is funded by an Advanced Clinician Scientist Fellowship from Cancer Research UK (C26642/A27963) and was previously funded by a Clinician Scientist Fellowship from the Academy of Medical Sciences/Health Foundation. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. We would like to thank Tissue Bank Bern for providing tissues for study purposes.
No conflicts of interest were declared.
References
- 1. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Gool IC, Eggink FA, Freeman‐Mills L, et al POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res 2015; 21: 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howitt BE, Shukla SA, Sholl LM, et al Association of polymerase e‐mutated and microsatellite‐instable endometrial cancers with neoantigen load, number of tumor‐infiltrating lymphocytes, and expression of PD‐1 and PD‐L1. JAMA Oncol 2015; 1: 1319–1323. [DOI] [PubMed] [Google Scholar]
- 4. Stelloo E, Nout RA, Osse EM, et al Improved risk assessment by integrating molecular and clinicopathological factors in early‐stage endometrial cancer – combined analysis of the PORTEC cohorts. Clin Cancer Res 2016; 22: 4215–4224. [DOI] [PubMed] [Google Scholar]
- 5. Talhouk A, McConechy MK, Leung S, et al A clinically applicable molecular‐based classification for endometrial cancers. Br J Cancer 2015; 113: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McConechy MK, Talhouk A, Leung S, et al Endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Clin Cancer Res 2016; 22: 2865–2873. [DOI] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network , Kandoth C, Schultz N, et al Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rayner E, van Gool IC, Palles C, et al A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer 2016; 16: 71–81. [DOI] [PubMed] [Google Scholar]
- 9. Bellone S, Bignotti E, Lonardi S, et al Polymerase ϵ (POLE) ultra‐mutation in uterine tumors correlates with T lymphocyte infiltration and increased resistance to platinum‐based chemotherapy in vitro . Gynecol Oncol 2017;144:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bellone S, Centritto F, Black J, et al Polymerase ϵ (POLE) ultra‐mutated tumors induce robust tumor‐specific CD4+ T cell responses in endometrial cancer patients. Gynecol Oncol 2015;138:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Billingsley CC, Cohn DE, Mutch DG, et al Polymerase ϵ (POLE) mutations in endometrial cancer: clinical outcomes and implications for Lynch syndrome testing. Cancer 2015;121:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Church DN, Briggs SE, Palles C, et al DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet 2013;22:2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Church DN, Stelloo E, Nout RA, et al Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst 2015; 107: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eggink FA, Van Gool IC, Leary A, et al Immunological profiling of molecularly classified high‐risk endometrial cancers identifies POLE‐mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology 2017; 6: e1264565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Espinosa I, Lee CH, D'Angelo E, et al Undifferentiated and dedifferentiated endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Am J Surg Pathol 2017; 41: 1121–1128. [DOI] [PubMed] [Google Scholar]
- 16. Hussein YR, Weigelt B, Levine DA, et al Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Mod Pathol 2015; 28: 505–514. [DOI] [PubMed] [Google Scholar]
- 17. Jansen AM, van Wezel T, van den Akker BE, et al Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet 2016; 24: 1089–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Gallo M, O'Hara AJ, Rudd ML, et al Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin‐remodeling and ubiquitin ligase complex genes. Nat Genet 2012; 44: 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meng B, Hoang LN, McIntyre JB, et al POLE exonuclease domain mutation predicts long progression‐free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol 2014; 134: 15–19. [DOI] [PubMed] [Google Scholar]
- 20. Wong A, Kuick CH, Wong WL, et al Mutation spectrum of POLE and POLD1 mutations in South East Asian women presenting with grade 3 endometrioid endometrial carcinomas. Gynecol Oncol 2016; 141: 113–120. [DOI] [PubMed] [Google Scholar]
- 21. Zhao S, Choi M, Overton JD, et al Landscape of somatic single‐nucleotide and copy‐number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A 2013; 110: 2916–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soumerai TE, Donoghue MTA, Bandlamudi C, et al Clinical utility of prospective molecular characterization in advanced endometrial cancer. Clin Cancer Res 2018; 24: 5939–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stelloo E, Bosse T, Nout RA, et al Refining prognosis and identifying targetable pathways for high‐risk endometrial cancer; a TransPORTEC initiative. Mod Pathol 2015; 28: 836–844. [DOI] [PubMed] [Google Scholar]
- 24. Talhouk A, McConechy MK, Leung S, et al Confirmation of ProMisE: a simple, genomics‐based clinical classifier for endometrial cancer. Cancer 2017; 123: 802–813. [DOI] [PubMed] [Google Scholar]
- 25. Kommoss S, McConechy MK, Kommoss F, et al Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population‐based case series. Ann Oncol 2018; 29: 1180–1188. [DOI] [PubMed] [Google Scholar]
- 26. Shinbrot E, Henninger EE, Weinhold N, et al Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res 2014; 24: 1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Umar A, Boland CR, Terdiman JP, et al Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petljak M, Alexandrov LB. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 2016; 37: 531–540. [DOI] [PubMed] [Google Scholar]
- 29. Kim J, Mouw KW, Polak P, et al Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016; 48: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 31. Choi Y, Sims GE, Murphy S, et al Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012; 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adzhubei IA, Schmidt S, Peshkin L, et al A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mi H, Poudel S, Muruganujan A, et al PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 2016; 44: D336–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics 2015; 16((suppl 8)): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ioannidis NM, Rothstein JH, Pejaver V, et al REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016; 99: 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. León‐Castillo A, Gilvazquez E, Nout R, et al Clinicopathological and molecular characterisation of ‘multiple classifier’ endometrial carcinomas. J Pathol 2020; 250: 312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell BB, Light N, Fabrizio D, et al Comprehensive analysis of hypermutation in human cancer. Cell 2017; 171: 1042–1056.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haradhvala NJ, Kim J, Maruvka YE, et al Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun 2018; 9: 1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Valle L, Hernandez‐Illan E, Bellido F, et al New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet 2014; 23: 3506–3512. [DOI] [PubMed] [Google Scholar]
- 40. Palles C, Cazier JB, Howarth KM, et al Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh N, Piskorz A, Bosse T, et al p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol 2020; 250: 336–345. [DOI] [PubMed] [Google Scholar]
- 42. Talhouk A, Derocher H, Schmidt P, et al Molecular subtype not immune response drives outcomes in endometrial carcinoma. Clin Cancer Res 2019; 25: 2537–2548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinical outcome of MMRd–POLEmut ECs
Table S1. POLE mutations reported in ECs in COSMIC or TCGA
Table S2. In silico tools results for POLE mutations found in ECs in TCGA
Table S3. In silico tools results for non‐hotspot POLE mutations found in EC cohorts published not present in TCGA