

Abstract
Objective
Parkinson disease is characterized by motor and nonmotor symptoms, reduced striatal dopamine signaling, and loss of dopamine neurons in the substantia nigra. It is now known that the pathological process in Parkinson disease may begin decades before the clinical diagnosis and include a variety of neuronal alterations in addition to the dopamine system.
Methods
This study examined the density of all synapses with synaptic vesicle glycoprotein 2A (SV2A) in Parkinson disease subjects with mild bilateral disease (n = 12) and matched normal controls (n = 12) using in vivo high‐resolution positron emission tomographic imaging as well as postmortem autoradiography in an independent sample with Parkinson disease (n = 15) and normal controls (n = 13) in the substantia nigra and putamen.
Results
A group‐by‐brain region interaction effect (F 10, 22 = 3.52, p = 0.007) was observed in the primary brain areas with in vivo SV2A binding. Post hoc analyses revealed that the Parkinson disease group exhibited lower SV2A in the substantia nigra (−45%; p < 0.001), red nucleus (−31%; p = 0.03), and locus coeruleus (−17%; p = 0.03). Exploratory analyses also revealed lower SV2A binding in clinically relevant cortical areas. Using autoradiography, we confirmed lower SV2A in the substantia nigra (−17%; p < 0.005) and nonsignificant findings in the putamen (−4%; p = 0.06).
Interpretation
This work provides the first evidence of synaptic loss in brainstem nuclei involved in the pathogenesis of Parkinson disease in living patients. SV2A imaging holds promise for understanding synaptic changes central to the disease. Ann Neurol 2020;87:329–338
Parkinson disease (PD) is a neurodegenerative disease with prominent motor and nonmotor symptoms. Despite well‐documented dopamine neuron loss, the pathological process in PD is only partially understood. Alpha‐synuclein aggregates are thought to play a key role and are a main component of Lewy bodies and neurites. Braak and colleagues have proposed a Lewy pathology‐based staging of PD, with each stage characterized by the involvement of specific brain regions, starting from the olfactory system and lower brainstem and extending to subcortical and cortical regions in later disease stages.1 From early in the disease process, there is evidence of pathology involving multiple neurotransmitter systems and pathways, including the cholinergic, noradrenergic, and serotonergic systems, the cell bodies of which are located in the brainstem and project diffusely throughout the whole brain.2, 3, 4, 5 However, understanding the pathological changes in the brainstem and cortex during disease progression has been challenging due to the lack of suitable clinical research technologies.
Mounting evidence has indicated that damage to neurons may begin decades before symptoms appear. The majority of genes implicated in PD (eg, α‐synuclein, LRRK2, DJ‐1, PINK1, and PRKN) have a critical role in presynaptic function, and corresponding knockout mouse models have demonstrated disruption of synaptic plasticity and neurotransmitter function.6, 7, 8 A collection of presynaptic proteins important for vesicle transport has been found to interact with these gene products, including synaptic vesicle protein 2A (SV2A).6 Thus, it is hypothesized that these gene–protein interactions cause a decrease of vesicle transport that is a final common pathway leading to PD.9 As synaptic changes are centrally involved in PD and characteristic of the pathogenesis of the disease,10, 11 understanding the pathogenesis and progression of synaptic degeneration in PD could vastly facilitate characterization of disease pathology, monitoring of the disease process, and assessment of treatment efficacy.
We have recently developed a noninvasive in vivo imaging technique using [11C]UCB‐J positron emission tomography (PET) to measure the density of synapses in humans with SV2A, an essential vesicle membrane protein present in virtually all presynaptic terminals.12, 13 As SV2A has a relatively consistent copy number per vesicle,14 a reduction in [11C]UCB‐J binding is likely to reflect a reduction in synaptic vesicles and be attributable to synaptic density loss. In this study, we examined synaptic loss in individuals with mild bilateral PD in vivo with high‐resolution PET imaging of SV2A and postmortem in more advanced patients with autoradiography.
Subjects and Methods
Human Subjects
The study was performed under a protocol approved by the Yale University Human Investigation Committee and the Yale New Haven Hospital Radiation Safety Committee, and in accordance with the US federal policy for the protection of human research subjects contained in Title 45 Part 46 of the Code of Federal Regulations. Written informed consent was obtained from all participants after complete explanation of study procedures.
There was no restriction for gender, race, or ethnicity. To be eligible, all subjects had to be 40 to 80 years old and be in good general health as evidenced by comprehensive medical and psychiatric histories, physical examination, neurological examination (performed by a single movement disorders neurologist for PD subjects), routine laboratory studies, and electrocardiogram. Exclusions included any current or past clinically significant medical or neurological illness (other than PD) that could affect study outcome, a history of alcohol or substance abuse, current pregnancy (as documented by pregnancy testing at screening and on the day of the PET imaging study), breast feeding, and contraindications to magnetic resonance imaging (MRI). PD diagnosis was made according to the UK Parkinson's Disease Society Brain Bank Clinical Diagnosis Criteria.15 PD subjects had to be able to consent and tolerate PET scanning procedures. PD subjects were excluded if taking medications affecting SV2A binding (eg, levetiracetam) and were instructed to withhold their morning PD medication so that their last dose of medication was at least 12 hours prior to the scheduled PET radiotracer injection time. PD medications and total L‐dopa equivalent daily dose are listed in Table 1 for individual subjects.
Table 1.
PD Medications of Each Subject along with the Total LEDD
| Subject | PD Medications | LEDD, mg |
|---|---|---|
| 1 | CD/LD, amantadine, pramipexole, rasagiline | 675 |
| 2 | CD/LD ER, selegiline | 233 |
| 3 | Amantadine, pramipexole | 600 |
| 4 | CD/LD, selegiline, pramipexole ER | 1,000 |
| 5 | CD/LD, CD/LD CR, ropinirole | 460 |
| 6 | CD/LD, safinamide | 400 |
| 7 | CD/LD, ropinirole | 355 |
| 8 | CD/LD ER | 858 |
| 9 | CD/LD ER, ropinirole | 1,102 |
| 10 | CD/LD, rasagiline | 400 |
| 11 | CD/LD, rasagiline, amantadine ER | 970 |
| 12 | CD/LD, selegiline | 750 |
CD/LD = carbidopa/L‐dopa; CR = sustained release; ER = extended release; LEDD = L‐dopa equivalent daily dose.
PD subjects were assessed according to the Movement Disorders Society (MDS) Task Force Clinical Diagnostic Criteria for PD using the MDS‐Unified Parkinson's Disease Rating Scale.16 The Hoehn and Yahr scale was used for disease staging.17 The Montreal Cognitive Assessment test was administered to screen for dementia (cutoff <21 of 30 points).18 Neurocognitive assessments included the Trail Making Test (Parts A and B), Digit Span forward and backward, letter fluency tasks (F‐A‐S), and the 10‐point Clock Drawing. Assessments were used in the analysis if results were approximately normally distributed.
Demographic and clinical characteristics for the participants are shown in Table 2.
Table 2.
Demographic and Clinical Characteristics for Participants in the Positron Emission Tomography Study
| Subjects | Age, yr | Gender | BMI | Years since Dx [range] | Hoehn and Yahr | MDS‐UPDRS Part III |
|---|---|---|---|---|---|---|
| NC, n = 12 | 60 (10) | 8F/4M | 27 (4) | N/A | N/A | N/A |
| PD, n = 12 | 61 (9) | 8F/4M | 25 (4) | 4 yr (2 mo–10 yr) | 2 | 34 (8) |
Data are represented as mean (standard deviation).
BMI = body mass index; Dx = diagnosis; F = female; M = male; MDS‐UPDRS Part III = Movement Disorders Society Unified Parkinson's Disease Rating Scale motor examination scores; N/A = not applicable; NC = normal control; PD = Parkinson disease.
Imaging
All subjects received a structural MRI scan on a 3T Prisma system (Siemens Medical Systems, Erlangen, Germany) for purposes of excluding subjects with anatomical abnormalities, coregistering with the PET images and region of interest (ROI) definition. MRI scans were acquired using a T1‐weighted magnetization‐prepared rapid gradient echo sequence with 1mm3 isotropic resolution (flip angle = 7°, echo time = 3.34 milliseconds, inversion time = 1,100 milliseconds, repetition time = 2,500 milliseconds). Images were skull‐ and muscle‐stripped (Functional Magnetic Resonance Imaging of the Brain [FMRIB]‘s Brain Extraction Tool, http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/BET) before processing.
Each subject then underwent at least a 60‐minute PET measurement with [11C]UCB‐J on the High Resolution Research Tomograph (Siemens/CTI, Knoxville, TN) with 207 slices (1.2mm slice separation) and a reconstructed image resolution of ~3mm. Before tracer injection, a 6‐minute transmission measurement was performed for attenuation correction. [11C]UCB‐J was radiolabeled as previously described.19 Subjects either received an intravenous bolus or an intravenous bolus plus constant infusion to the end of the PET acquisition12; because kinetic modeling was performed, the different administration methods had no effect on the results. [11C]UCB‐J was injected at 565 ± 210MBq with a mass dose (0.03 ± 0.01μg/kg) for PD subjects and at 554 ± 190MBq with a mass dose (0.02 ± 0.02μg/kg) for normal control (NC) subjects.
Based on previous PD research,1, 20 primary ROIs were the substantia nigra (SN), locus coeruleus, raphe nucleus, red nucleus, putamen, caudate, globus pallidus, parahippocampal gyrus, primary and supplemental motor cortex, and cerebellum. Secondary (exploratory) regions included the ventral striatum, thalamus, amygdala, hippocampus, insula, anterior and posterior cingulum, and dorsolateral prefrontal, orbitofrontal, ventromedial prefrontal, parietal, temporal, and occipital cortices. The ROIs above were taken from the Anatomical Automatic Labeling for SPM2 (AAL) atlas with the exception of hand‐drawn templates in the following areas: the SN, which was defined with a dopamine tracer,21, 22, 23 the locus coeruleus, defined with a norepinephrine tracer,24 and the raphe nucleus, which was defined with a serotonin tracer,25 as these tracers show high uptake in the respective areas that are not defined in AAL. ROI delineation was described in more detail in previous work,23, 24 but briefly, the SN was drawn on 5 to 7 coronal slices for approximately a 1ml volume and the other non‐AAL ROIs were confirmed on the Talairach and Tournoux atlas.26, 27 All bilateral areas were defined with the right and left sides, and the average was used in analyses. The MRI‐based Hammer atlas28 was also used to independently confirm results in the SN. All cortical ROIs had gray matter segmentation that was conducted using SPM12 (Wellcome Trust Centre for Neuroimaging, London, UK) and then partial volume corrected using the Muller–Gartner algorithm to account for any possible gray matter differences.29 Regional volumes were calculated in cortical ROIs using the same gray matter segmentation with SPM12. The intracranial volume was used for normalizing the intersubject variance and estimated with T1‐weighted images analyzed with FreeSurfer 6.0. No volumetric decreases in examined cortical areas in PD subjects were found; increases are shown in Table 3
Table 3.
eTIV (ml) and eTIV‐Corrected Brain Regions with Significant Differences (Uncorrected) Shown for NC and PD Groups
| NC | PD | % Increase in PD | p | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| eTIV | 1,458.37 | 177.54 | 1,528.92 | 112.83 | 5% | 0.26 |
| Orbitofrontal cortex | 0.0144 | 0.0120 | 0.0175 | 0.0023 | 21% | <0.01 |
| Posterior cingulum | 0.0013 | 0.0003 | 0.0016 | 0.0004 | 24% | 0.04 |
| Ventromedial prefrontal cortex | 0.0148 | 0.0020 | 0.0168 | 0.0019 | 14% | 0.02 |
Mean and SD shown.
eTIV = estimated total intracranial volume; NC = normal control; PD = Parkinson disease; SD = standard deviation.
To apply ROIs to the PET data, 2 transformations were completed. First, a nonlinear transformation grid was estimated between the AAL template and each subject's magnetic resonance (MR) image, using BioImage Suite software (v2.5; http://www.bioimagesuite.com). This nonlinear coregistration algorithm is a method first described in 200130 and subsequently used in multiple studies.29, 31, 32, 33, 34, 35 Then, the subject's summed PET image (0–10 minutes postinjection) was coregistered to their MR image using a 6‐parameter rigid (linear) registration estimated with a mutual information algorithm (FMRIB's Linear Image Registration Tool, FMRIB Software Library). Please see Figure 1 for a diagram of image registration processing. Regional values were computed by applying the template ROIs to PET space. To create the group averaged PET images, the combined nonlinear and linear registrations were used to transform individual PET parametric images to AAL template space.
Figure 1.
A diagram of image registration processing used in the study. MR = magnetic resonance; PET = positron emission tomography. [Color figure can be viewed at http://www.annalsofneurology.org]
During PET scans, continuous head motion data were acquired using the Polaris Vicra optical tracking system (NDI Systems, Waterloo, Ontario, Canada).36 Data were reconstructed including corrections for attenuation, normalization, scatter, random data, dead time, and motion using the MOLAR algorithm.37 Arterial blood was collected for measurement of blood and plasma radioactivity in 9 NC and 8 PD subjects. The remaining subjects did not have arterial line measurements because of line failure or subject's preference. Thus, binding potential (BP ND) values were used as the primary outcome measure in this study with the simplified reference tissue model 2 (SRTM2). This was applied to compute BP ND parametric images using a fixed k 2′ value (clearance rate constant, k 2, of the reference region), which was computed as a k 2 population average of the centrum semiovale using the 1‐tissue compartment model (k 2 = 0.031 ± 0.005 [1/min], n = 17). The ROI for the centrum semiovale, a white matter region, was defined based on the white matter probability map (SPM12) of NC subjects’ MR images, and the centrum semiovale region was defined on the AAL template to minimize partial volume effects and used as the reference region. BP ND correlated well to values from the 1‐tissue compartment model, a model used in prior work,12 in subjects with arterial blood data (y = 1.0292x + 0.0199, R 2 = 0.9811, n = 17). In the voxel‐based approach, between‐group comparisons were performed using independent sample t tests at each voxel with SRTM2 parametric images using SPM12.
Postmortem Autoradiography
Human brain tissue samples were purchased from the Banner Sun Health Research Institute Brain and Body Donation Program. Donor demographics are summarized in Table 4. Tissues were provided as cryosections (10μm) of SN, pars compacta, and putamen and stored with desiccant at −80°C until use. [3H]UCB‐J was prepared with high radiochemical purity (98.9%) and specific activity (34.9Ci/mmol) and stored at −20°C in ethanol until use.
Table 4.
Demographic and Clinical Characteristics for Subjects in the Autoradiography Study
| Subjects | Age, yr | PMI, h | MMSE | LB Stage |
|---|---|---|---|---|
| NC, n = 15 | 84 (7) | 2.9 (0.9) | 28 (2) | 0, no LB |
| PD, n = 15 | 76 (17) | 3.5 (0.7) | 22 (5) | III–IV, neocortical |
Age of individuals, PMI, total MMSE score before death, and LB stage at autopsy are shown. Age, PMI, and MMSE are represented as mean (standard deviation).
LB = Lewy body; MMSE = Mini‐Mental Status Examination; PMI = postmortem interval.
Tissue autoradiography was performed in 50mM Tris HCl pH 7.4, 138mM NaCl, 2.7mM KCl and solutions of 2.5nM [3H]UCB‐J. Incubations were 60 minutes at room temperature. Washes (2 × 10 minutes) used cold assay buffer followed by 5 dips in cold distilled water. Slides were allowed to dry under a fan, then stored overnight in a vacuum desiccator. Tritium‐sensitive storage phosphor screens were exposed for 1 week and imaged at 600 dots per inch using a PerkinElmer (Waltham, MA) Cyclone phosphor imager. Autoradiography data were analyzed in ImageJ by quantifying the mean pixel intensity within manually defined anatomical regions of interest that sampled consistent gray matter substructures in SN, pars compacta, and putamen tissue sections. Tritium standards (American Radiolabeled Chemicals, St Louis, MO) were used to convert data from mean pixel intensity to nCi/mg. Data visualization and statistical analyses were performed using Prism (GraphPad Software, San Diego, CA).
Statistical Analysis
All outcomes were summarized descriptively and assessed for normality prior to analysis using normal probability plots and Shapiro–Wilk test statistics. All outcomes were approximately normally distributed. Linear mixed models were used to examine the independent and joint effects of group (between‐subjects factor) and ROI (within‐subject factor) on BP ND values. Within‐subject correlations were accounted for by fitting 3 variance–covariance structures to the data (unstructured, compound symmetry, and heterogeneous compound symmetry) with an unstructured form fitting the data best according to the Bayesian information criterion. Potential associations between clinical assessments and measures with ROIs were estimated using Pearson correlations. Multiple comparison corrections were not performed on post hoc and secondary analyses given their exploratory nature. All analyses were considered significant at the 2‐tailed α < 0.05 threshold and were conducted using SAS version 19 (IBM, Armonk, NY).
Results
In Vivo PET Imaging with [11C]UCB‐J Shows Lower Synaptic Density in Primary PD‐Related Brain Regions
We found evidence of group differences in the primary brain regions of PD (n = 12) compared with NC subjects (n = 12; group‐by‐brain region interaction effect, F 10, 22 = 3.52, p = 0.007). Figure 2 shows post hoc analyses, with PD subjects exhibiting significantly lower SV2A‐specific binding in the SN (−45%; p < 0.001), red nucleus (−31%; p = 0.03), locus coeruleus (−17%; p = 0.03), and parahippocampal gyrus (−12%; p < 0.01). Lateralized effects were seen in the caudate on the contralateral side to symptom onset (11% lower; p = 0.04), but not in the putamen or SN.
Figure 2.
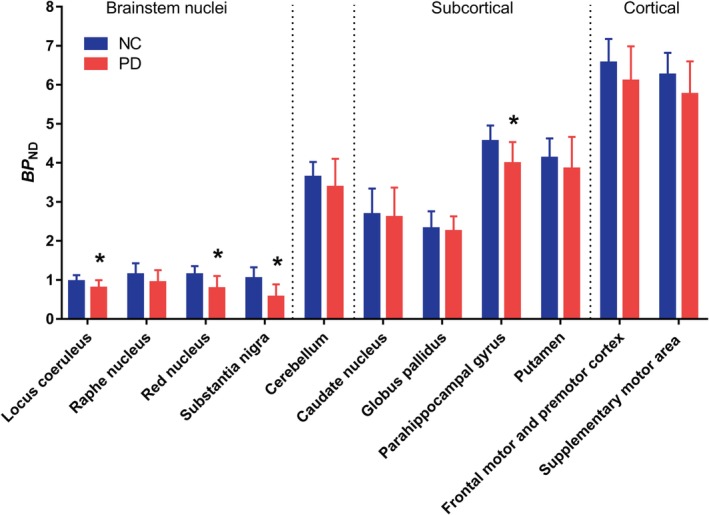
Region of interest analysis of binding potential (BP ND) values for normal control (NC) and Parkinson disease (PD) subjects. BP ND values were lower across all primary brain regions between the NC and PD subjects. *Statistical significance at p = 0.05 or better on post hoc tests. Error bars denote standard deviation. [Color figure can be viewed at http://www.annalsofneurology.org]
Averaged group parametric images of BP ND values denoting the location of the SN region and displaying differences between the 2 groups are shown in Figure 3A. Figure 3B provides a scatterplot showing demographically matched NC and PD subjects (indicated by lines), with lower BP ND on the individual subject level in all cases with 1 exception. Analyses based upon an independently defined atlas15 similarly found lower SV2A binding (−24%, p = 0.02).
Figure 3.
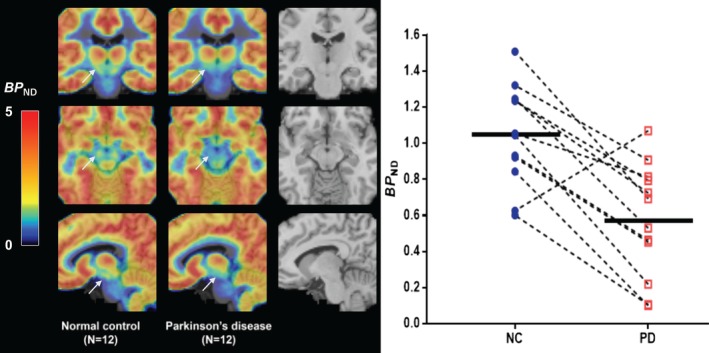
(A) The midbrain area with arrows denoting the substantia nigra (SN) is shown on the averaged group parametric images of [11C]UCB‐J binding potential (BP ND) values between normal control (NC; left) and Parkinson disease (PD; center) subjects. The color scale denotes BP ND. The magnetic resonance imaging template is also shown for comparison purpose (right). (B) Scatterplot results of SN between NC (blue circles) and PD (red squares) subjects. Connected lines indicate demographically matched subjects. Horizontal bars indicate group means.
Nonsignificant lower BP ND values were found in every examined brain region, ranging from 3 to 10%.
Correlations between Synaptic Density and Neurocognitive Function
Neurocognitive assessment scores and disease duration were not correlated with BP ND values in the significant brain regions reported.
Autoradiography Studies
Using [3H]UCB‐J autoradiography in a separate sample (n = 15 in PD, n = 13 in NC), we also detected lower SV2A binding in the SN (−17%; p < 0.005). As with the in vivo data, nonsignificant findings were found in the putamen (−4%; p = 0.06; see Fig 4).
Figure 4.
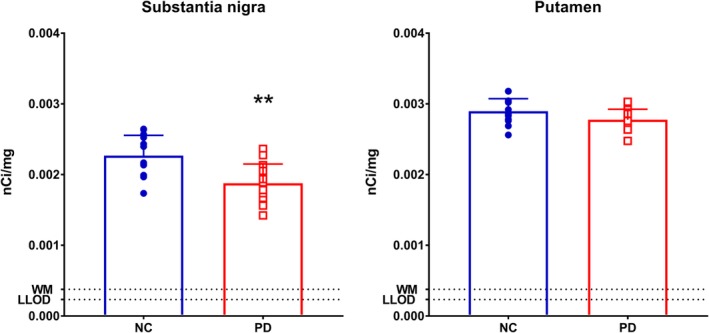
Autoradiography results of the substantia nigra and putamen with [3H]UCB‐J between normal control (NC) and Parkinson disease (PD) subjects. **Statistically significant at p < 0.005. Error bars denote standard deviation. LLOD = lower limit of detection; WM = white matter. Units are in nanocuries/milligram. [Color figure can be viewed at http://www.annalsofneurology.org]
PET Imaging with [11C]UCB‐J in Secondary PD‐Related Brain Regions
Secondary post hoc analyses were performed in additional brain regions for exploratory purposes. Significantly lower BP ND values (all p values are uncorrected) in PD subjects were found in the posterior cingulate cortex (−15%; p = 0.01), orbitofrontal cortex (−11%; p = 0.01), and ventromedial prefrontal cortex (−11%; p = 0.02). An averaged group image of the whole brain is shown for visual comparison in Figure 5.
Figure 5.
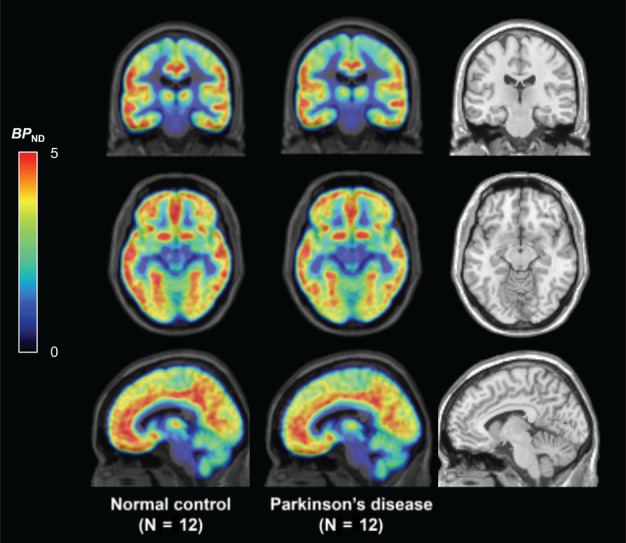
The whole brain is shown in averaged group binding potential (BP ND) [11C]UCB‐J parametric images for normal control (left) and Parkinson disease (middle) subjects along with the magnetic resonance imaging template (right). Color scale denotes BP ND.
Discussion
In this study, we detected significantly lower synaptic density for the first time in living PD subjects using in vivo SV2A PET imaging. The largest reductions were found in the SN, red nucleus, and locus coeruleus. Lower SV2A binding in the SN in PD was corroborated with postmortem autoradiography using brain tissue slices from a separate cohort of patients with PD. These findings provide in vivo evidence of synaptopathy in brainstem nuclei in earlier disease manifestation. This, along with previous work in animal models of the disease, further indicates the likelihood that clinical symptomology of PD may begin in the synapses of brainstem nuclei and remain present throughout the course of the disease.10, 11, 38
The largest relative difference in SV2A‐specific binding was found in the SN, a region central in PD pathology. Postmortem tissue studies estimate that 30 to 80% of dopamine neurons in the SN are lost when motor symptoms are present.39 Previous PET studies have used radiotracers more specific to the monoamine systems, and found differences of around 20 to 40% in mild to advancing PD for vesicular monoamine transporter type 240 and 30% for dopamine transporter in early PD patients in the SN.41
According to Braak staging, dopamine deficits are present from Braak stage 3 on. In the current study, we evaluated all synapses and found lower SV2A binding in the locus coeruleus, a region that is densely populated with norepinephrine neurons and affected in Braak stage 2,1 confirming the presence of neurodegeneration in nondopaminergic brainstem nuclei. Similarly, lower SV2A binding was found in the red nucleus, a region adjacent to the dorsal raphe nucleus known to contain high serotonin transporter density,42 although the SV2A binding in the raphe nuclei was not significantly different between the 2 groups. Taken as a whole, synaptic differences in brainstem nuclei are present in symptomatic patients, and this may extend into the preclinical stage, as was recently shown for the serotonin transporter in A53T SNCA carriers.43
SV2A BP ND values in the striatum showed smaller differences that were not significantly different between the PD and NC groups in both the in vivo and postmortem samples. There could be different reasons for this. For example, the SV2A radiotracer signal is nonspecific to neuronal type, and the nondopaminergic neuron population in the striatum might be differentially affected. The neurons expressing tyrosine hydroxylase are usually identified as being dopaminergic in the striatum, but recently this has been contested, as they also have γ‐aminobutyric acidergic (GABAergic) markers and function suggesting that dopaminergic terminals might constitute a smaller fraction of striatal synapses.44, 45 Thus, the lack of significant differences in the striatum may reflect synapses other than nigrostriatal dopamine terminals or alternatively reflect a depletion of SV2A in the presynaptic dopamine terminals (ie, those arriving from the striatum and cortex) and not the cell bodies of dopamine neurons themselves. In addition, during the early stages of PD, increases have been shown in synapses in animal models, and human PET studies have shown upregulation of D2 and D3 receptor sites in the striatum.20, 46 It is thus possible that a similar compensatory mechanism from dopaminergic denervation is present in the striatum in this cohort.47 It is also important to note that [11C]UCB‐J has previously been found to be specific for SV2A and not for SV2B or SV2C.19 There is preclinical evidence of SV2C disruption in PD pathogenesis.48 It is possible that SV2C is selectively expressed in dopamine neurons and therefore a more sensitive marker of synaptic loss in the striatum, which would make an SV2C‐specific PET tracer potentially valuable in this regard.
An exploratory analysis revealed numerically lower SV2A binding in cortices including the posterior cingulate and key frontal regions that have previously been implicated with changes in dopamine, serotonin, GABA, and other neurotransmitter systems throughout the course of the disease.1 This is an advantage of SV2A PET, as all synapses in the brain can be examined with one measurement. This is particularly relevant as 90% of alpha‐synuclein in PD is located presynaptically in much smaller aggregates than Lewy bodies and can be found throughout the brain.38 Pathologically, alpha‐synuclein, as well as other PD genes and proteins, mainly involve synaptic machinery and play a critical role at the presynaptic site, causing alterations in neurotransmitter reuptake, vesicular storage, and release.49, 50, 51, 52 Models based on the most prevalent genetic lesions that underlie familial forms of PD, LRRK2 lesions, also have severe neurotransmission defects due to interactions of various presynaptic proteins, including SV2A.6, 7 These findings are also consistent with Braak classification, suggesting that the pathological progression follows a subcortical–limbic–neocortical gradient. Because our PD cohort had bilateral disease and an average of 4 years since disease diagnosis, it is conceivable that the PD pathology involved the limbic and neocortical structures as well. It is important to note that volumetric measurements in brain regions showed either no significant differences or an increase in the PD group (see Table 3), supporting the uniqueness and specificity of SV2A methodology in measuring synapses devoid of confounding factors such as glia that may influence other imaging modalities. Current work is focusing on the possibility of using SV2A binding as a biomarker to track synaptic changes in vivo in PD across the whole brain at different disease stages as well as comparing results vis‐à‐vis imaging biomarkers measuring the dopamine system.
There are several limitations that warrant mentioning. First, although our sample size is modest for a PET investigation, it is well within the range for a PET study evaluating a new radiotracer in PD.41, 46 The limited sample size, combined with the lack of range in the clinical Hoehn and Yahr scale across PD subjects, may have obscured possible correlations with clinical measures or duration of disease. These homogeneous scores were a design of the study, however, and a relative strength for a preliminary study in PD. Future studies should use larger PD cohorts that are stratified according to disease stage. This might clarify possible SV2A differences in secondary regions and demonstrate clinical relationships. Second, the primary outcome measure BP ND was calculated using the centrum semiovale as a reference region. Ideally, reference regions are completely devoid of specific binding, but in previous studies we have shown a 10 to 20% contribution of SV2A‐specific binding to [11C]UCB‐J centrum semiovale total binding (volume of distribution [V T]).12 Importantly however, the centrum semiovale V T was very similar for both subject groups (V T = 3.90 in NC; 3.85 in PD; −1%; p = 0.84) and was significantly lower than the V T of the primary regions studied; it was therefore suitable to be used as a reference region. Third, medication effects cannot be ruled out as contributing to the current results, although brain penetrative medications were not administered for an average of 13 hours before PET measurements, and medications affecting SV2A were exclusionary criteria. Fourth, although we interpret SV2A differences as evidence of synaptic decreases, as noted recently, other interpretations are possible (eg, synaptic vesicle dysfunction and/or cycling35). Preclinical work has shown, however, that synaptic vesicle pools are stable within synapses.53 Last, it should be noted that other studies have used similar methodology with SV2A imaging at our center and also found disease‐specific differences in SV2A in epilepsy, Alzheimer disease, and major depressive disorder.12, 29, 35 Thus, emerging regional patterns in these diseases foster interest in the utility of SV2A as a wide‐ranging neuropsychiatric biomarker for diagnosis, disease progression, and possibly treatment monitoring.
Author Contributions
D.M., I.S., R.A.C., Y.H., S.J.F., and R.E.C. contributed to the conception and design of the study; D.M., S.T., K.C.W., M.N., T.T., M.D., S.H., B.P., J.R., N.N., and S.J.F. contributed to the acquisition and analysis of data; D.M., S.T., K.C.W., M.N., T.T., S.J.F., and R.E.C. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
K.C.W., I.S., R.A.C., and S.J.F. were full‐time employees of AbbVie during the conception, acquisition and reporting of this study.
Acknowledgment
This article was made possible by Clinical and Transitional Science Award grant number UL1 TR000142 from the NIH National Center for Advancing Translational Science. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH. We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of postmortem brain tissues. This work was supported by AbbVie and internal funding of the Yale Positron Emission Tomography Research Center.
We thank the staff of the Yale Positron Emission Tomography Research Center and Yale Magnetic Resonance Research Center; and Dr T. Montavon at AbbVie for preparing [3H]UCB‐J.
References
- 1. Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 2. Todorova A, Jenner P, Ray Chaudhuri K. Non‐motor Parkinson's: integral to motor Parkinson's, yet often neglected. Pract Neurol 2014;14:310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sauerbier A, Jenner P, Todorova A, Chaudhuri KR. Non motor subtypes and Parkinson's disease. Parkinsonism Relat Disord 2016;22(suppl 1):S41–S46. [DOI] [PubMed] [Google Scholar]
- 4. Jellinger KA. Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord 2012;27:8–30. [DOI] [PubMed] [Google Scholar]
- 5. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- 6. Belluzzi E, Greggio E, Piccoli G. Presynaptic dysfunction in Parkinson's disease: a focus on LRRK2. Biochem Soc Trans 2012;40:1111–1116. [DOI] [PubMed] [Google Scholar]
- 7. Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature 2016;539:207–216. [DOI] [PubMed] [Google Scholar]
- 8. Plowey ED, Chu CT. Synaptic dysfunction in genetic models of Parkinson's disease: a role for autophagy? Neurobiol Dis 2011;43:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Esposito G, Ana Clara F, Verstreken P. Synaptic vesicle trafficking and Parkinson's disease. Dev Neurobiol 2012;72:134–144. [DOI] [PubMed] [Google Scholar]
- 10. Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson's disease. Adv Exp Med Biol 2012;970:553–572. [DOI] [PubMed] [Google Scholar]
- 11. Bellucci A, Mercuri NB, Venneri A, et al. Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 2016;42:77–94. [DOI] [PubMed] [Google Scholar]
- 12. Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med 2016;8:348ra96. [DOI] [PubMed] [Google Scholar]
- 13. Finnema SJ, Nabulsi NB, Mercier J, et al. Kinetic evaluation and test‐retest reproducibility of [(11)C]UCB‐J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab 2018;38:2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bajjalieh SM, Peterson K, Linial M, Scheller RH. Brain contains two forms of synaptic vesicle protein 2. Proc Natl Acad Sci U S A 1993;90:2150–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahsan RL, Allom R, Gousias IS, Habib H, Turkheimer FE, Free S, Lemieux L, Myers R, Duncan JS, Brooks DJ, Koepp MJ, Hammers A. Volumes, spatial extents and a probabilistic atlas of the human basal ganglia and thalamus. Neuroimage 2007;38:261‐270. [DOI] [PubMed] [Google Scholar]
- 16. Movement Disorder Society Task Force on Rating Scales for Parkinson's Disease . The Unified Parkinson's Disease Rating Scale (UPDRS): status and recommendations. Mov Disord 2003;18:738–750. [DOI] [PubMed] [Google Scholar]
- 17. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 18. Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 19. Nabulsi NB, Mercier J, Holden D, et al. Synthesis and preclinical evaluation of 11C‐UCB‐J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med 2016;57:777–784. [DOI] [PubMed] [Google Scholar]
- 20. Nikolaus S, Antke C, Muller HW. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav Brain Res 2009;204:1–31. [DOI] [PubMed] [Google Scholar]
- 21. Matuskey D, Gaiser EC, Gallezot JD, et al. A preliminary study of dopamine D2/3 receptor availability and social status in healthy and cocaine dependent humans imaged with [(11)C](+)PHNO. Drug Alcohol Depend 2015;154:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaiser EC, Gallezot JD, Worhunsky PD, et al. Elevated dopamine D2/3 receptor availability in obese individuals: a PET imaging study with [11C](+)PHNO. Neuropsychopharmacology 2016;41:3042–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallezot JD, Zheng MQ, Lim K, et al. Parametric imaging and test‐retest variability of (1)(1)C‐(+)‐PHNO binding to D(2)/D(3) dopamine receptors in humans on the high‐resolution research tomograph PET scanner. J Nucl Med 2014;55:960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding YS, Singhal T, Planeta‐Wilson B, et al. PET imaging of the effects of age and cocaine on the norepinephrine transporter in the human brain using (S,S)‐[(11)C]O‐methylreboxetine and HRRT. Synapse 2010;64:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naganawa M, Nabulsi N, Planeta B, et al. Tracer kinetic modeling of [(11)C]AFM, a new PET imaging agent for the serotonin transporter. J Cereb Blood Flow Metab 2013;33:1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lancaster JL, Rainey LH, Summerlin JL, et al. Automated labeling of the human brain: a preliminary report on the development and evaluation of a forward‐transform method. Hum Brain Mapp. 1997;5:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lancaster JL, Woldorff MG, Parsons LM, et al. Automated Talairach atlas labels for functional brain mapping. Hum Brain Mapp 2000;10:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahsan RL, Allom R, Gousias IS, et al. Volumes, spatial extents and a probabilistic atlas of the human basal ganglia and thalamus. Neuroimage 2007;38:261–270. [DOI] [PubMed] [Google Scholar]
- 29. Chen MK, Mecca AP, Naganawa M, et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol 2018;75:1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Papademetris X, Jackowski AP, Schultz RT, et al. Integrated intensity and point‐feature nonrigid registration. Med Image Comput Comput Assist Interv 2001;3216:763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radhakrishnan R, Nabulsi N, Gaiser E, et al. Age‐related change in 5‐HT6 receptor availability in healthy male volunteers measured with 11C‐GSK215083 PET. J Nucl Med 2018;59:1445–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matuskey D, Worhunksy P, Correa E, et al. Age‐related changes in binding of the D2/3 receptor radioligand [(11)C](+)PHNO in healthy volunteers. Neuroimage 2016;130:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matuskey D, Pittman B, Planeta‐Wilson B, et al. Age effects on serotonin receptor 1B as assessed by PET. J Nucl Med 2012;53:1411–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matuskey D, Dias M, Naganawa M, et al. Social status and demographic effects of the kappa opioid receptor: a PET imaging study with a novel agonist radiotracer in healthy volunteers. Neuropsychopharmacology 2019;44:1714–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holmes SE, Scheinost D, Finnema SJ, et al. Lower synaptic density is associated with depression severity and network alterations. Nat Commun 2019;10:1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin X, Mulnix T, Gallezot JD, Carson RE. Evaluation of motion correction methods in human brain PET imaging—a simulation study based on human motion data. Med Phys 2013;40:102503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carson RE, Barker WC, Liow J‐S, Johnson CA. Design of a motion‐compensation OSEM list‐mode algorithm for resolution‐recovery reconstruction of the HRRT. In: 2003 IEEE Nuclear Science Symposium: conference record: nuclear science symposium: medical imaging conference: 19–25 October, 2003, Portland, Oregon, USA. Piscataway, NJ: IEEE, 2004:3281–3285.
- 38. Schulz‐Schaeffer WJ. The synaptic pathology of alpha‐synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol 2010;120:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsiao IT, Weng YH, Hsieh CJ, et al. Correlation of Parkinson disease severity and 18F‐DTBZ positron emission tomography. JAMA Neurol 2014;71:758–766. [DOI] [PubMed] [Google Scholar]
- 41. Fazio P, Svenningsson P, Cselenyi Z, et al. Nigrostriatal dopamine transporter availability in early Parkinson's disease. Mov Disord 2018;33:592–599. [DOI] [PubMed] [Google Scholar]
- 42. Son YD, Cho ZH, Choi EJ, et al. Individually differentiated serotonergic raphe nuclei measured with brain PET/MR imaging. Radiology 2014;272:541–548. [DOI] [PubMed] [Google Scholar]
- 43. Wilson H, Dervenoulas G, Pagano G, et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α‐synuclein parkinsonism: a cross‐sectional study. Lancet Neurol 2019;18:748–759. [DOI] [PubMed] [Google Scholar]
- 44. Xenias HS, Ibanez‐Sandoval O, Koos T, Tepper JM. Are striatal tyrosine hydroxylase interneurons dopaminergic? J Neurosci 2015;35:6584–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huot P, Parent A. Dopaminergic neurons intrinsic to the striatum. J Neurochem 2007;101:1441–1447. [DOI] [PubMed] [Google Scholar]
- 46. Boileau I, Guttman M, Rusjan P, et al. Decreased binding of the D3 dopamine receptor‐preferring ligand [11C]‐(+)‐PHNO in drug‐naive Parkinson's disease. Brain 2009;132(pt 5):1366–1375. [DOI] [PubMed] [Google Scholar]
- 47. Paille V, Picconi B, Bagetta V, et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci 2010;30:14182–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dunn AR, Stout KA, Ozawa M, et al. Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci U S A 2017;114:E2253–E2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scherfler C, Khan NL, Pavese N, et al. Striatal and cortical pre‐ and postsynaptic dopaminergic dysfunction in sporadic parkin‐linked parkinsonism. Brain 2004;127(pt 6):1332–1342. [DOI] [PubMed] [Google Scholar]
- 50. Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ‐1/PINK1 triple knockout mice. J Neurochem 2009;111:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Madeo G, Schirinzi T, Martella G, et al. PINK1 heterozygous mutations induce subtle alterations in dopamine‐dependent synaptic plasticity. Mov Disord 2014;29:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beccano‐Kelly DA, Volta M, Munsie LN, et al. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum Mol Genet 2015;24:1336–1349. [DOI] [PubMed] [Google Scholar]
- 53. Südhof TC. Composition of synaptic vesicles In: Siegel GJ, Agranoff BW, Albers RW, et al, eds. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th ed. Philidelphia, PA: Lippincott‐Raven, 1999:175‐ 190. [Google Scholar]