

Abstract
The paraherquamides are potent anthelmintic natural products with complex heptacyclic scaffolds. One key feature of these molecules is the spiro-oxindole moiety that lends a strained three-dimensional architecture to these structures. The flavin monooxygenase PhqK was found to catalyze spirocycle formation through two parallel pathways in the biosynthesis of paraherquamides A and G. Two new paraherquamides (K and L) were isolated from a ΔphqK strain of Penicillium simplicissimum, and subsequent enzymatic reactions with these compounds generated two additional metabolites paraherquamides M and N. Crystal structures of PhqK in complex with various substrates provided a foundation for mechanistic analyses and computational studies. While it is evident that PhqK can react with various substrates, reaction kinetics and molecular dynamics simulations indicated that the dioxepin-containing paraherquamide L was the favored substrate. Through this effort, we have elucidated a key step in the biosynthesis of the paraherquamides, and provided a rationale for the selective spirocyclization of these powerful anthelmintic agents.
Keywords: spirocycle, biocatalysis, monooxygenase, flavin, alkaloid
Graphical Abstract
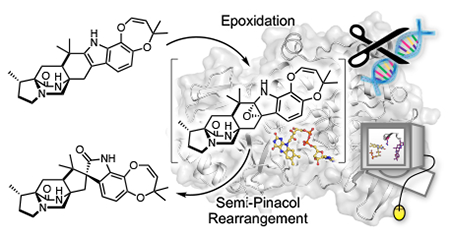
INTRODUCTION
The paraherquamide family of natural products contains metabolites isolated from various species of Penicillium including P. paraherquei,1 P. charlesii,2 P. cluniae,3 P. fellutanum,4 and P. IMI 332995.5 These molecules are of particular interest due to their anthelmintic therapeutic potential, and various analogs have been developed to improve their pharmacological properties.6–9 Derquantel, or 2-deoxyparaherquamide A, is currently used in combination with the established anthelmintic abamectin to combat gastrointestinal nematode infections in sheep.10 The significant biological activity of this family of molecules is complemented by the intriguing biosynthetic chemistry including intramolecular Diels-Alder cyclization, unusual oxidative substitutions, and spiro-oxindole formation.
Paraherquamide-type molecules belong to a family of fungal indole alkaloids containing a unique bicyclo[2.2.2]diazaoctane ring, which is formed through a [4+2] intramolecular Diels-Alder (IMDA) reaction.11 The spirocycle is a common functionality of many molecules within this family including the marcfortines,12, 13 brevianamides,14, 15 antiinsectan sclerotiamide,16 and cytotoxic notoamides.17 The respective enzymes involved in spirocycle formation are proposed to generate an initial C2=C3 indole epoxide with facial selectivity, and controlled collapse of the epoxide giving rise to the observed spiro-oxindoles. Very few enzymes responsible for this type of reaction within the bicyclo-ring containing family have been characterized.18 NotB catalyzes the 2,3-β-face epoxidation of notoamide E (1) to generate the non-spiro-cyclized terminal metabolites notoamides C (2) and D (3), which are not IMDA substrates. In this pathway, another flavin-dependent monooxygenase NotI performs a presumed 2,3-α-face epoxidation of stephacidin A (4) in the process of generating the bioactive natural product notoamide B (5) (Figure 1a). These flavin-dependent monooxygenases have evolved to perform facially selective epoxidation either before or after the IMDA cyclization, resulting in a divergence in the notoamide biosynthetic pathway.
Figure 1.
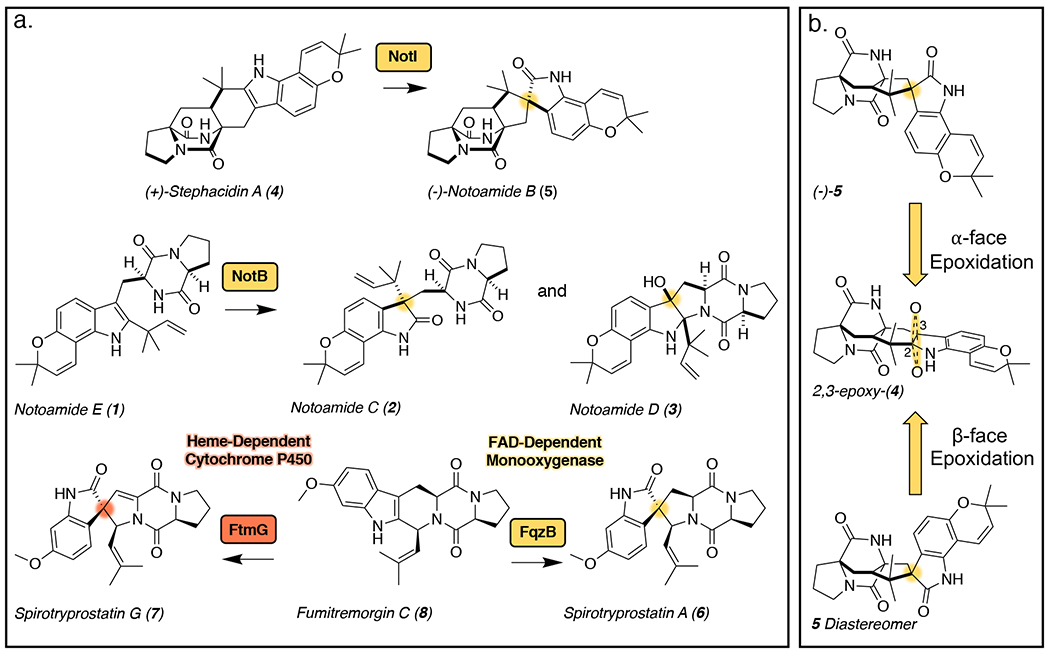
(a.) Indole alkaloid products resulting from the various enzyme selectivities for the indole epoxide collapse. (b.) Representation of the biocatalyst-controlled facial selectivity of the presumed epoxidation of stephacidin A (4) to generate (−)-notoamide B (5) through α-face epoxidation and the unnatural diastereomer of 5 through β-face epoxidation.
Biocatalytic epoxidation typically occurs through reactions with molecular oxygen which is activated either by a metal ion or flavin adenine dinucleotide (FAD) cofactor.19 The enzyme active site orients the substrate for facial selectivity of the epoxidation, and in some cases, subsequent collapse of the epoxide in a stereocontrolled manner to generate complex molecular structures (Figure 1b). Enantioselective oxidation is a common step in the syntheses of many natural products and bioactive molecules, including complex alkaloids.20, 21 In particular, indole-2,3-epoxidation serves as a synthetic intermediate to rearrangements that generate a variety of products. The collapse of this intermediate can be directed by the inherent reactivity of the substrate or through a catalyst-controlled mechanism. For example, synthetic methods have been developed to favor spirocyclization22–25 or other oxidized products20 in the total syntheses of select fungal indole alkaloids.
While synthetic methods for enantioselective epoxidation and spirocyclization are rapidly improving, the role of biocatalysis for solving synthetic challenges is also gaining greater visibility. The inherent selectivity of enzymes is unparalleled in synthetic chemistry and with each enzymatic discovery, the biocatalytic toolbox is broadening. In particular, there is evidence that oxygenases can be successfully employed in the generation of enantiomerically pure starting materials for total syntheses.27 To this end, a few enzymes that catalyze epoxidation and subsequent spirocyclization have been identified.
Of the currently characterized natural products that include spiro-centers, most structures fall outside of the bicyclo[2.2.2]diazaoctane ring-containing family, and the predicted biosynthetic enzymes utilize either metal-dependent radical-based mechanisms28,29 or flavin-dependent redox chemistry (Figure 1).30 In some cases, the spirocyclic scaffold is known to be critical for biological activity.29 Studies detailing spirocycle formation in the spirotryprostatins revealed intricate enzymatic details for this conversion.31 Two distinct routes for the formation of this spirocyclic moiety involve either a flavin monooxygenase (with homology to NotB18), which employs an indole epoxidation route to generate spirotryprostatin A (6), or a cytochrome P450 that involves a radical route to generate spirotryprostatin G (7), both starting from fumitremorgin C (8). Cross talk between the flavin-dependent monooxygenase from the fumiquinazoline biosynthetic pathway and enzymes from the fumitremorgin biosynthetic pathway have led to the generation of unique diketopiperazine-containing spirocyclic metabolites in Aspergillus fumigatus. While these functional studies provided important insights regarding the various mechanisms of formation for the spirocyclized natural products, stmctural data for the key biocatalysts has remained elusive.
The paraherquamide class of natural products contains a spiro-oxindole center within a complex heptacyclic ring system. The biosynthesis of these molecules starts with the condensation of l-tryptophan and l-β-methyl-proline,32–34 followed by spontaneous oxidation to generate zwitterion 9 (Scheme 1). 35, 36 Reverse prenylation at the indole C2 position provides the prenylated zwitterion substrate 10, which is reduced to azadiene 11 and cyclized by the Diels-Alderase11 to produce preparaherquamide (12). While precursor incorporation studies demonstrated that 12 is incorporated into the final product (−)-paraherquamide A (13),36 it was unclear whether the spirocyclization occurred before or after the formation of the pyran and dioxepin rings. Through disruption of phqK in Penicillium simplicissimum using a CRISPR-Cas9 system, we identified that 12 undergoes a series of oxidation and prenylation reactions to generate paraherquamides K and L (14 and 15) prior to spiro-cyclization by PhqK.4, 35, 36 This enzyme was characterized in vitro as a flavin-dependent monooxygenase, which acts on native substrates 14 and 15 to generate the corresponding spirocyclized products. Crystal structures of PhqK in complex with its native substrates revealed precise substrate orientation to promote selective epoxidation thereby enabling stereospecific formation of the spiro-oxindole moiety.
Scheme 1.
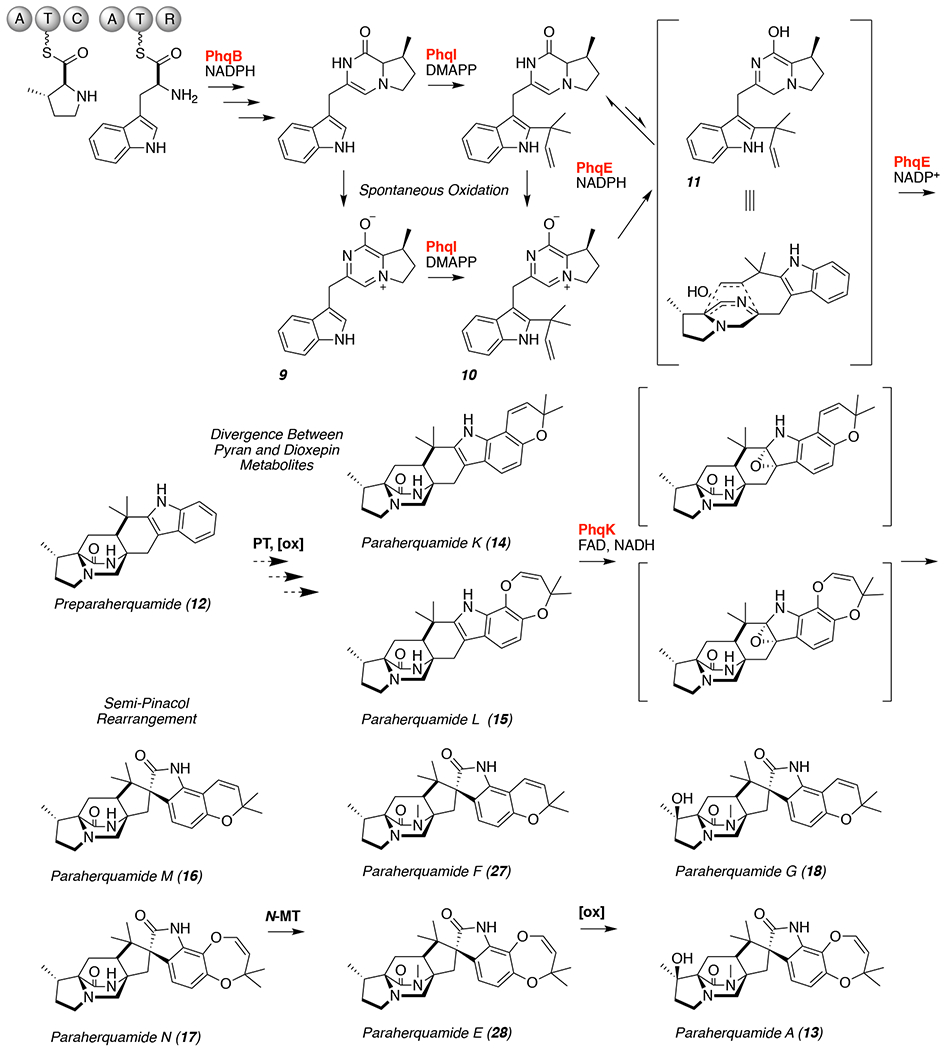
Proposed paraherquamide A (13) and G (18) biosynthetic pathways. The functions of enzymes highlighted in red have been confirmed.11
Consistent with previous hypotheses, we have proposed a mechanism based on indole α-epoxidation and collapse of the putative epoxide at C3 to generate a C2 hydroxyl carbocation.26 Subsequent migration of the reverse prenyl group from C2 to C3 would generate the 2-oxindole product. The work described here provides the first structural data for this unique class of spirocycle forming flavin monooxygenases, and sheds light on biocatalytic control of epoxide formation and mode of Pinacol rearrangement.
RESULTS
Parallel efforts were pursued in vitro and in vivo to identify the enzyme responsible for the spirocyclization reaction in paraherquamide biosynthesis. Paraherquamide K (14) and paraherquamide L (15) accumulated from the ΔphqK strain of Penicillium simplicissimum (Supplemental Figures 32 and 33), indicating that both molecules are natural substrates of the enzyme. The in vitro reactions of 14 and 15 with PhqK confirmed their conversion into the respective spirocyclized products paraherquamide M (16) and paraherquamide N (17). This information provided evidence to support a branched biosynthetic scheme where the pyran and dioxepin rings are both formed prior to the spirocyclization (Scheme 1). The identification of two natural substrates for PhqK indicates that functionalization of 12 is the divergent step between two parallel pathways leading to paraherquamides G (18) and A (13). In the first case, a putative prenyltransferase and oxidative enzyme build the pyran ring, whereas the latter involves generation of the dioxepin ring by a poorly understood process.35 PhqK accepts both substrates, thus the downstream N-methyltransferase and β-methyl proline hydroxylase must also display the same level of substrate flexibility.
To obtain further information about the selectivity of PhqK, we employed Michaelis-Menten kinetic analysis (Supplemental Figures 34 and 35). Although the pyran-containing 14 displayed a higher turnover number (kcat), the Km for 15 (19.4 ± 5.2 μM) was significantly lower compared to 14 (92.1 ± 29.4 μM). This resulted in nearly equivalent catalytic efficiencies (kcat/Km) for both substrates (0.05 ± 0.01 μM−1min−1 for 15 and 0.04 ± 0.01 μM−1min−1 for 14), however based on the low probability that these secondary metabolites are present at saturating concentrations in vivo, we reasoned that 15 is likely the favored substrate.
The high yields and activity of PhqK indicated its potential as a biocatalyst, thus the substrate scope was analyzed on a series of structurally related malbrancheamide analogs. The halogenated malbrancheamides are produced by two fungal species, including the terrestrial strain Malbranchea aurantiaca37 and the marine-derived Malbranchea graminicola.38 The corresponding biosynthetic gene clusters for malbrancheamide (19) and spiromalbramide (20) from these two fungi possess 99% amino acid sequence identity.39 Intriguingly, the spirocyclized malbrancheamide analog spiromalbramide has only been isolated from the marine fungus.38 Therefore, we tested the reactivity of PhqK on the halogenated malbrancheamide and analogs malbrancheamides B (21), C (22), E (23), and isomalbrancheamide D (24).39 The reactions generated a variety of spirocyclized malbrancheamides (20, 25-28) including the natural product spiromalbramide (20), indicating that a PhqK homolog in M. graminicola may be involved in spiromalbramide biosynthesis (Figure 2, Supplemental Figures 11–31, and Supplemental Tables 7–16).38 PhqK catalyzes formation of products with a relative stereochemical outcome matching that identified in the natural metabolites (5aR, 6aS, 12aS, 13aS) and demonstrated its ability to accept non-native substrates such as the malbrancheamides. While efficient conversion of the malbrancheamides was achieved, the conversion of smaller, less structurally complex substrates was not observed (Figures S47–S49). This indicates an evolutionary distinction between early- and late-stage Pinacolase enzymes, as previously demonstrated in the notoamide biosynthetic pathway.18
Figure 2.
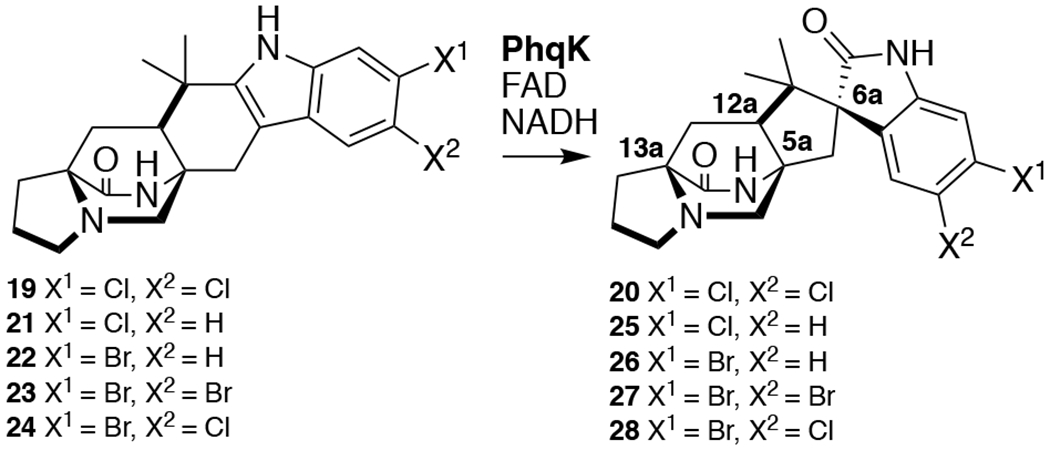
Spirocyclization of malbrancheamides.
In an effort to improve the efficiency of PhqK, we analyzed reactions in the presence of a cofactor regeneration system (glucose-6-phosphate dehydrogenase) and catalase to eliminate the detrimental effects of hydrogen peroxide resulting from the flavin redox chemistry. In the presence of stoichiometric FAD concentrations, the use of the regeneration system (Figure S45) and catalase (Figure S44) significantly increased the conversion of non-native substrates, while conversions of 14 and 15 remained unaffected (Figures S40–S43). This system may mimic the enzyme’s natural environment in the producing organism, and we expect that with the aid of other enzymes commonly found in fungi, PhqK may be more efficient.
With this foundational knowledge, we proceeded to elucidate the molecular basis for stereospecificity by analyzing the structure of the enzyme. PhqK was cocrystallized as four distinct substrate complexes, bearing the natural pyran-containing 14 (1.89 Å) and dioxepin-containing 15 (2.09 Å), and non-native substrates monochlorinated malbrancheamide B (21) (1.69 Å), and monobrominated malbrancheamide C (22) (1.25 Å). A ligand-free structure was also obtained (1.71 Å), aiding the visualization of how the active site changes to accommodate the hexa- and heptacyclic alkaloids (Figure 3). The FAD binding pocket and substrate binding pocket are connected by a long tunnel that appears to put significant distance between the cofactor and the substrate. This can be explained by the well-characterized flavin dynamics described in previous work.40,41 Flavin monooxygenases commonly display a dynamic cofactor with an orientation that is dependent on discrete parameters, including substrate binding or cofactor oxidation state. In both substrate bound and unbound PhqK structures, the flavin was observed in the “out” conformation. It is hypothesized that the “in” conformation may be induced by reduction of the FAD cofactor, but our attempts to obtain a structure bound to reduced flavin using sodium dithionite and NADH were unsuccessful. As an alternative, we modelled the “in” conformation of the cofactor from HpxO42 into the PhqK structure (Figures 4 and 5). HpxO is a FAD-dependent urate oxidase from Klebsiella pneumoniae and its secondary structure overlays well with PhqK (26% sequence identity, RMSD 1.54 Å), with the major differences attributed to FAD conformation. Divergence in the FAD monooxygenase family is based on evolutionary domain fusion events leading to differences in the substrate binding pocket and reactivity,43 thus this structural similarity in the cofactor binding region is expected.
Figure 3.
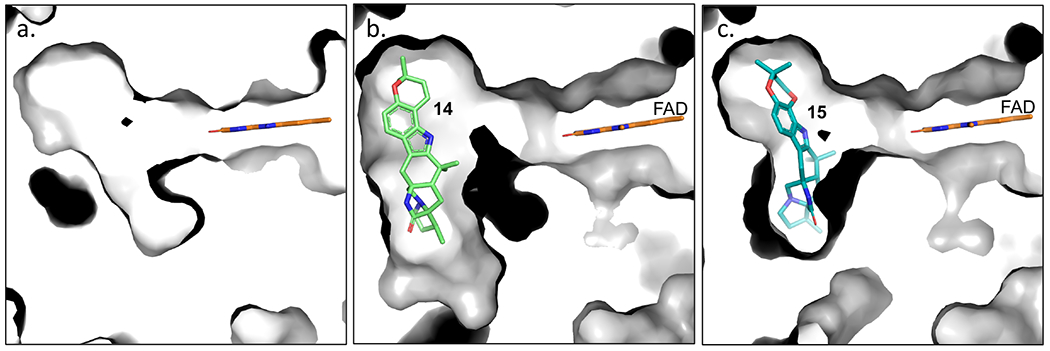
Changes in the active site pocket from no ligand (a.), to accommodate substrates paraherquamide K (14) (b.), and L (15) (c.).
Figure 4.
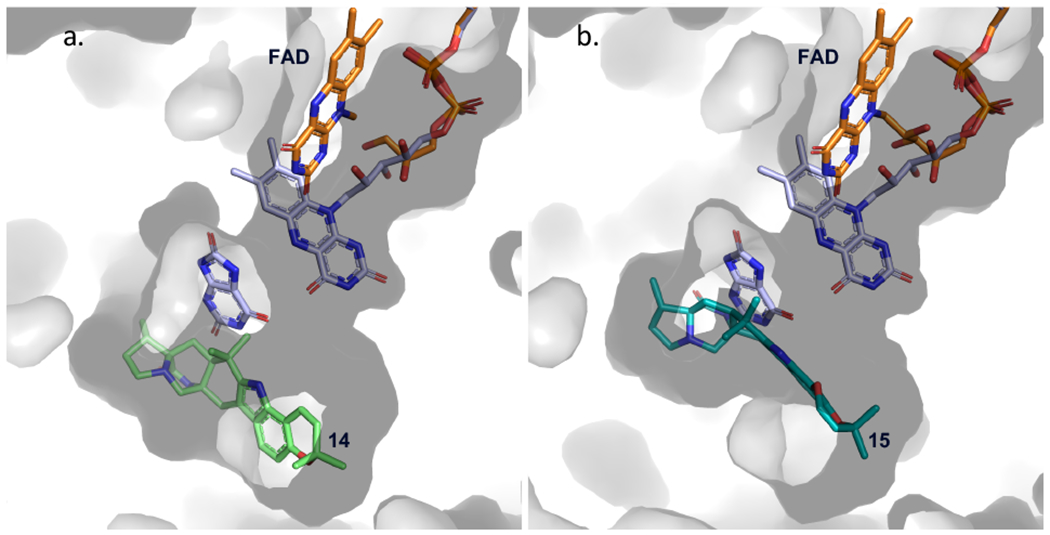
Modeled cofactor dynamics in PhqK. Flavin dynamics were modeled from HpxO crystal structure (FAD and substrate in lavender) (PDB: 3rp7) to depict the “in” conformation of the flavin relative to the substrate position. The surface representation of PhqK is shown along with the substrate and FAD from the PhqK structure (orange). Substrates 14 (a.) and 15 (b.) are shown in green and teal.
Figure 5.
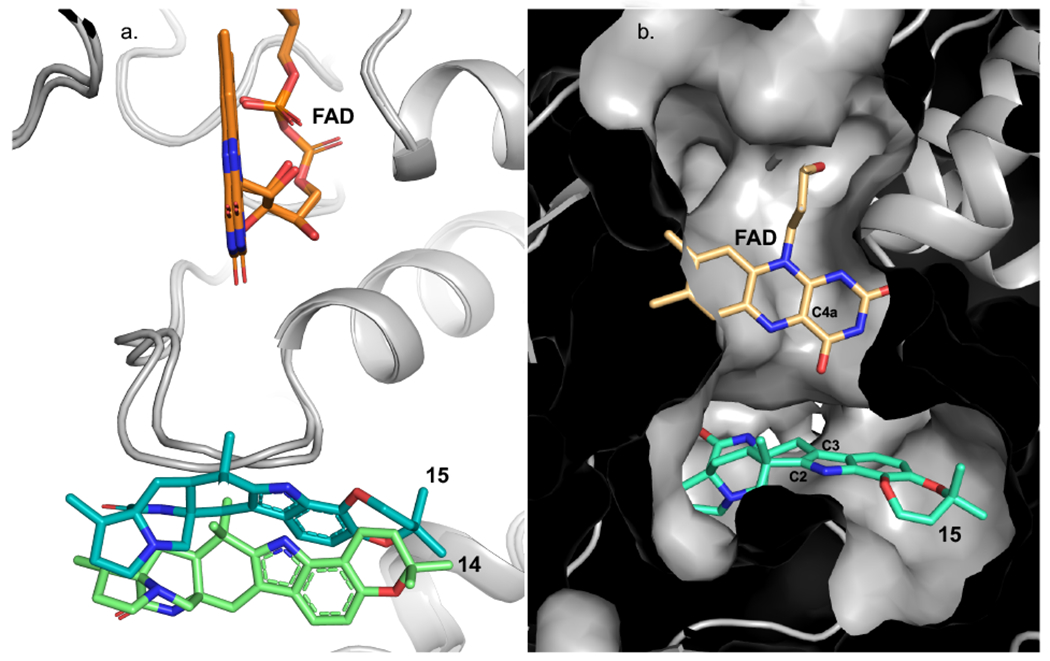
(a.) Overlay of PhqK cocrystal structures. The structures are complexed with 14 (green) and 15 (teal). (b.) The “in” FAD cofactor was modeled from the HpxO structure (PDB: 3rp7) into the PhqK cocrystal structure with 15, where the FAD-C4a is perfectly aligned to perform the epoxidation of the indole C2=C3.
When inspecting the overlay of PhqK with HpxO, we observed that the main changes are localized to the substratebinding region where two loops, one near the cofactor and one near the substrate, are altered in HpxO. Whereas the FAD cofactor in the PhqK structure reveals the “out” position, the FAD in HpxO is found as the “in” position, making it a suitable model for generating a visual representation of this phenomenon in PhqK. The “in” flavin from HpxO was modelled into the PhqK structure and fit well in the active site tunnel. In this orientation, the C4a-OOH-FAD would reside sufficiently close to the substrate (6.5 Å between C4a and indole C2/C3) to catalyze epoxidation at the indole C2=C3 position (Figures 4 and 5). Inspection of the substrates in PhqK structural complexes revealed that each one binds in the same manner except for 15, which is oriented for direct α-epoxidation. It is possible that the individual substrates sample both conformations in solution. Since 15 appears to be the favored substrate, it may preferentially bind in the active conformation, explaining performed 1,500 ns MD simulations which demonstrated that 15 maintains a more rigid binding pose with better facial selectivity for epoxidation. By contrast, 14 is more flexible within the binding pocket and adopts less productive conformations for epoxidation (Supplemental Figure 54). Site-directed mutagenesis experiments revealed the importance of specific amino acid residues in the active site. Most of the mutants led to significant losses in flavin incorporation within the protein (indicated as %FAD in Supplemental Table 17) and a decrease in enzyme stability (indicated as [PhqK] in Supplemental Table 17). Mutation of certain active site residues abolished enzyme activity, indicating that they may perform essential catalytic roles. For example, Gln232, which interacts with the indole N-H, is necessary for conversion of both 14 and 15 to the corresponding spiro-oxindole forms. Substitutions of alanine and glutamate at this site abrogated catalytic activity toward 15, while 14 exhibited about 2% conversion. Additionally, Arg192 and Asp47 are two essential amino acid residues within the tunnel between the substrate and cofactor. When Arg192 was substituted with alanine and lysine, 15 was not converted to product, while 14 showed 5-10% conversion.
The substitutions of alanine and asparagine for Asp47 also led to a complete loss of activity. All of these residues are located on the indole N-H side of the substrate and thus could play a role in directing the collapse of the epoxide and why we captured that pose in the crystal structure. We semi-Pinacol rearrangement to this side of the molecule. Arg192 clashes with the modeled “in” flavin, indicating that it must move in concert with the swing of the cofactor. We hypothesize that Arg192 moves closer to the substrate upon epoxidation and can serve as a general acid to catalyze epoxide opening (Figure 6). The distance between Arg192 and the indole C2 is around 7 Å. With a full extension of Arg192, it would be close enough to the substrate to facilitate general acid catalysis (Figure 6 and Scheme 2). An analysis of Arg192 using PROPKA on the PDB2PQR server44 indicated that the pKa of this general acid decreased as the conformation of the enzyme approached the catalytically competent pose (Table S20). The well-established conformational flexibility of arginine and dynamics of these flavin-dependent enzymes are indicative of a drastic change in the active site during catalysis. As we observed the pKa value decreasing upon binding 15 in a more catalytically competent pose, we hypothesize that once the FAD cofactor moves to the “in” position, the environment of this hydrophobic pocket would be adequate for lowering the pKa of Arg192 for the protonation event.
Figure 6.
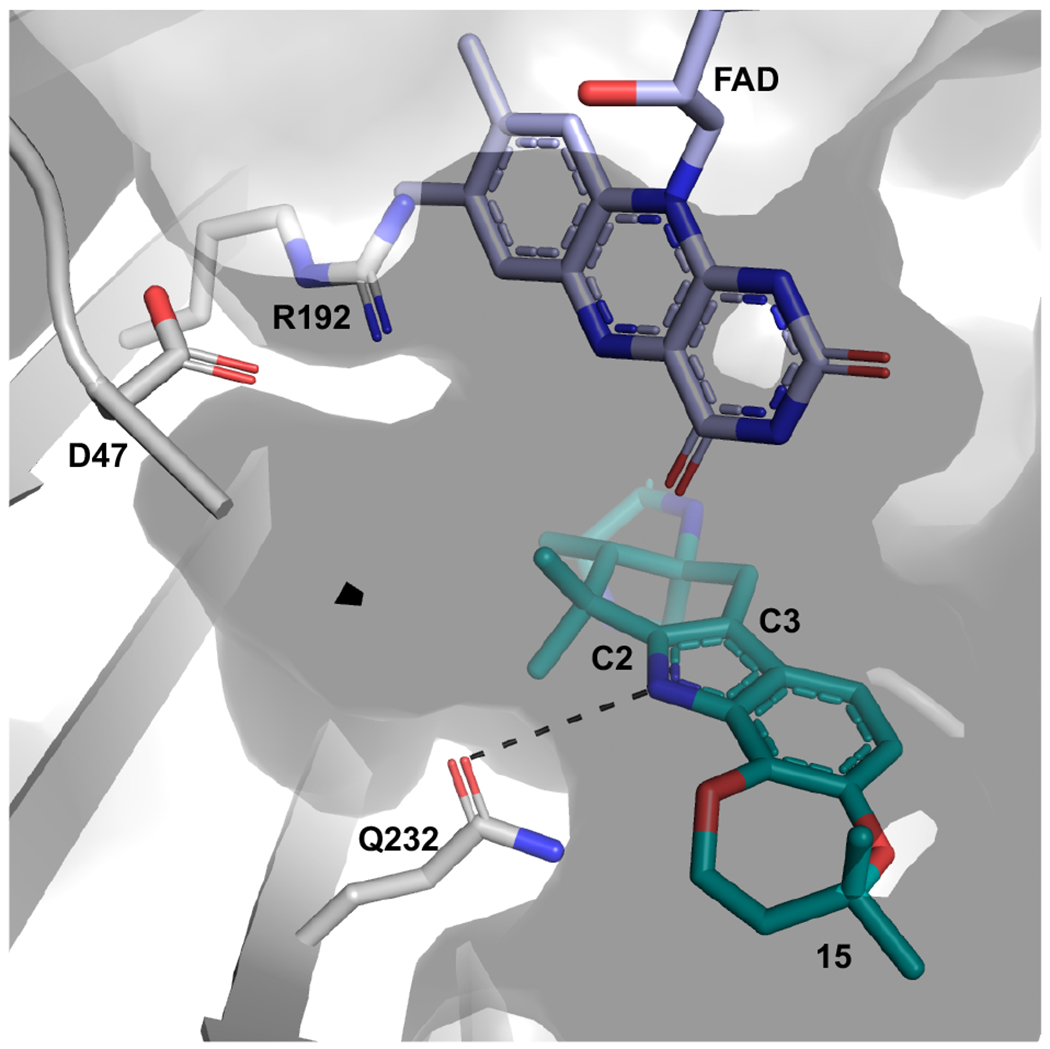
Essential amino acids in PhqK active site. Cocrystal structure of PhqK with paraherquamide L (15) displaying the active site residues that are pertinent to the activity. The FAD is modeled in the “in” orientation.
Scheme 2.
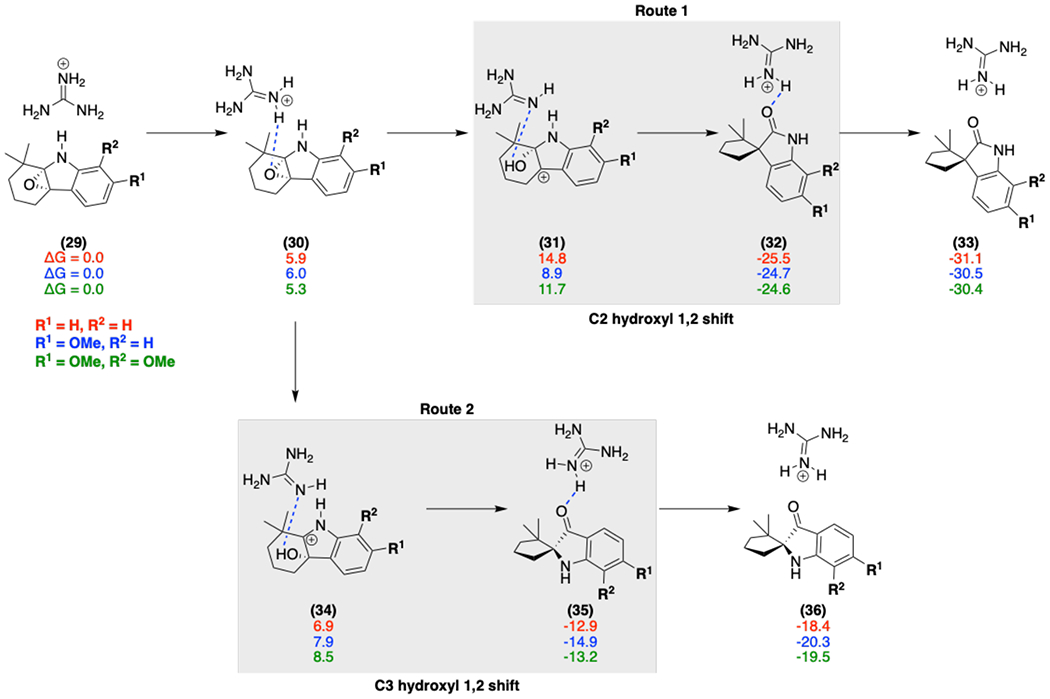
Free energies (calculated at the M06-2X/6-311++G(d,p)-ECFPCM(water)//M06-2X/6-31G(d)-IEFPCM (water) level) for various models (red, blue, green) with the Arg192 general acid (modeled by a guanidinium moiety) catalyzed epoxide ring-opening mechanism; initial protonation of the epoxide leads to ring opening to form a hydroxy cation (31 and 34), and deprotonation accompanies the 1,2 shift to generate the final spirocycle product.
Next, we performed density functional theory (DFT) calculations to explore different mechanisms and indole substituent effects on epoxide ring opening for the generation of the spiro-oxindole products. Our calculations involved the truncated indole fragment 29, as a model substrate with varying methoxy substitutions to model the electronic effects of substrates 14 and 15 (Scheme 2). Under general acid catalysis by Arg192 (modelled by a guanidinium moiety), the epoxide opening step in Route 1 is preferred over Route 2 when there is a C6 methoxy substituent due to the additional electronic stabilization of C2 hydroxyl carbocation 31, whereas in the alternative pathway C3 hydroxyl carbocation 34 does not benefit from this stabilization.
While this substitution plays a role, the subsequent carbon bond migration is the rate-limiting step. We evaluated the effect of the general acid and the indole substitutions on the Gibbs energy of the transition state (Figure 7). The general acid, Arg192, significantly lowers the barrier by 6.5 kcal/mol for Route 1 and 3.9 kcal/mol for Route 2. While both Routes 1 and 2 have lower barriers, Route 1 always has a lower transition state Gibbs energy regardless of the substitution pattern due to the ground state stabilization of C3 hydroxyl carbocation 34 in Route 2. This indicates that the formation of the experimentally observed spiro-oxindoles is intrinsically preferred and PhqK lowers the transition state energy for this 1,2 shift through a general acid catalyzed mechanism.
Figure 7.
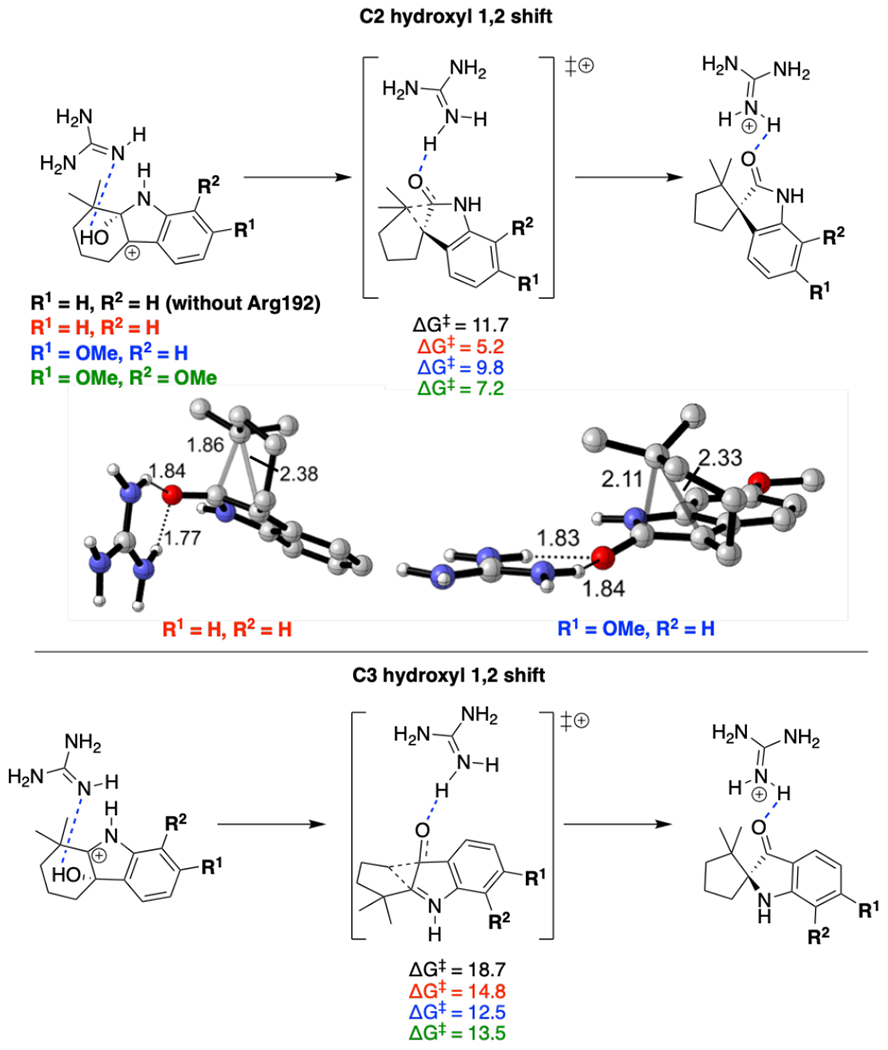
General acid catalyzed transition states for 1,2 shift on C2 hydroxyl carbocation and C3 hydroxyl carbocation on a paraherquamide model system. Various indole substituents are modeled (red, blue, green) as compared to a model without general acid (black). Energies are given in kcal/mol and distances in Å.
DISCUSSION
The spiro-oxindole moiety occurs in a variety of natural products and its biosynthesis is of significant interest due to the therapeutic potential of these metabolites.29 The spirocyclic scaffold has increasingly been utilized in the drug development process due to its unique structural attributes.45 Additionally , the chemistry of the reaction is intriguing from a biochemical standpoint due to the numerous factors contributing to the selectivity. A variety of enzymes have been found to catalyze spirocycle forming reactions including copper-dependent oxidases, cytochromes P450, and FAD-dependent monooxygenases. In the metal-dependent enzymes, the reaction is proposed to proceed through a radical-based mechanism, while the flavin-dependent enzymes proceed through FAD redox chemistry.
We have characterized the FAD-dependent monooxygenase PhqK as the spirocycle generating enzyme within the paraherquamide biosynthetic pathway. Through elucidation of this key step, we have developed a revised biosynthetic scheme for the production of paraherquamides A and G in which spirocyclization occurs after formation of the pyran and dioxepin moieties. We demonstrated that PhqK accepts both 14 and 15, confirming the existence of two parallel pathways (branching from 12) within the paraherquamide-producing Penicillium strains. While both substrates are accepted by the enzyme, the kinetic data and molecular dynamics simulations indicate that 15, bearing the dioxepin, is likely the favored substrate in vivo. This provides evidence that the difference in the orientation of 15 observed in the PhqK cocrystal structure is due to its persistence in the catalytically competent pose, whereas the less favored substrates might occupy a resting state until the flavin redox chemistry is initiated. The orientation of the molecule within the active site sets selectivity for α-epoxidation as demonstrated by the 15-bound structure.
Moreover, directed collapse of the initial 2,3-indole epoxide is likely mediated through general acid catalysis via Arg192 and has an energetic preference for migration of the prenyl group to C3 with oxidation at C2 of the indole. This is in excellent agreement with results from a similar system in the brevianamide family where the authors observed exclusive formation of the indoxyl product 36 resulting from the C3 hydroxyl carbocation.45 From DFT calculations, they concluded that the enzyme should favor a general base mechanism via a glutamate residue. This makes the transition state barrier lower for the 1,2 shift involving the C3 hydroxyl intermediate 34, over the general acid mechanism which favors the product from C2 hydroxyl intermediate 31. These results indicate that stereocontrol of the spiro-oxindole product results from the mechanistic mode dictated by the particular enzyme rather than a substituent effect on the indole ring.
The current study has provided new insights into the structure and mechanism of flavin-dependent spirocycle-generating enzymes, and the evidence supports a selective epoxidation of the indole C2=C3 followed by a stereoselective collapse of the epoxide. While PhqK catalyzes spirocyclization at a late-stage in paraherquamide biosynthesis, homologous enzymes within the brevianamide, notoamide, and other indole alkaloid pathways can generate Pinacol-rearrangement products with varying stereochemistry, or bearing non-spirocyclic functionality. Some of these transformations even require multiple enzymes, while PhqK operates as a dual function oxidase/Pinacolase to generate the spirocyclized paraherquamides.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Institutes of Health for financial support, including R01 CA070375 to R.M.W. and D.H.S., R35 GM118101 and the Hans W. Vahlteich Professorship to D.H.S., R01 GM124480 to K.N.H., R01 DK042303 and the Margaret J. Hunter Professorship to J.L.S., The Rackham Predoctoral Fellowship to A.E.F., and NIH Predoctoral Training Grant GM067555 to K.C.H. GM/CA@APS is supported by the National Institutes of Health, National Institute of General Medical Sciences (AGM-12006) and National Cancer Institute (ACB-12002). The Advanced Photon Source is a U.S. Department of Energy (DOE) Office of Science User Facility operated by Argonne National Laboratory under Contract DE-AC02-06CH11357. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1 053575). The authors also thank Rajani Arora, UM LSI Multimedia and Social Media Specialist, for contributions in preparation of the Supplemental cover art.
Footnotes
Full experimental details and computational methods, NMR spectra, tables, and figures.
PDB IDs: 6pvf, 6pvg, 6pvh, 6pvi, 6pvj
REFERENCES
- 1.Yamazaki M; Okuyama E; Kobayashi M; Inoue H, The structure of paraherquamide, a toxic metabolite from Penicillium paraherquei. Tetrahedron Lett 1981, 22 (2), 135–136. [Google Scholar]
- 2.Ondeyka JG; Goegelman RT; Schaeffer JM; Kelemen L; Zitano L, Novel antinematodal and antiparasitic agents from Penicillium charlesii 1. Fermentation, isolation and biological activity. J Antibiot 1990, 43 (11), 1375–1379. [DOI] [PubMed] [Google Scholar]
- 3.López-Gresa MP; González MC; Ciavatta L; Ayala I; Moya P; Primo J, Insecticidal activity of paraherquamides, including paraherquamide H and paraherquamide I, two new alkaloids isolated from Penicillium cluniae. J Agr Food Chem 2006, 54 (8), 2921–2925. [DOI] [PubMed] [Google Scholar]
- 4.Sommer K; Williams RM, Studies on paraherquamide biosynthesis: synthesis of deuterium-labeled 7-hydroxy-preparaherquamide, a putative precursor of paraherquamides A, E, and F. Tetrahedron 2009, 65 (16), 3246–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchflower SE; Banks RM; Everett JR; Reading C, Further novel metabolites of the paraherquamide family. J Antibiot 1993, 46 (9), 1355–1363. [DOI] [PubMed] [Google Scholar]
- 6.Blizzard TA; Margiatto G; Mrozik H; Schaeffer JM; Fisher MH, Chemical modification of paraherquamide 3. Vinyl ether modified analogs. Tetrahedron Lett 1991, 32 (22), 2437–2440. [Google Scholar]
- 7.Blizzard TA; Marino G; Mrozik H; Fisher MH; Hoogsteen K; Springer JP, Chemical modification of paraherquamide 1. Unusual reactions and absolute stereochemistry. J Org Chem 1989, 54 (11), 2657–2663. [Google Scholar]
- 8.Blizzard TA; Mrozik H; Fisher MH; Schaeffer JM, Chemical modification of paraherquamide 2. Replacement of the C-14 methyl group. J Org Chem 1990, 55 (7), 2256–2259. [Google Scholar]
- 9.Blizzard TA; Margiatto G; Mrozik H; Schaeffer JM; Fisher MH, Chemical modification of paraherquamide 4. 1-N-Substituted analogs. Tetrahedron Lett 1991, 32 (22), 2441–2444. [Google Scholar]
- 10.Little PR; Hodge A; Watson TG; Seed JA; Maeder SJ, Field efficacy and safety of an oral formulation of the novel combination anthelmintic, derquantel-abamectin, in sheep in New Zealand. New Zeal Vet J 2010, 58 (3), 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dan Q; Newmister SA; Klas KR; Fraley AE; A. MT; Somoza AD; Sunderhaus JD; Ye Y; Shende VV; Yu F; Sander JN; Brown WC; Zhao L; Paton RS; Houk KN; Smith JL; Sherman DH; Williams RM, Fungal indole alkaloid biogenesis through evolution of a bifunctional reductase/Diels-Alderase. Nat Chem 2019, 11, 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polonsky J; Merrien MA; Prange T; Pascard C; Moreau S, Isolation and structure (X-ray analysis) of marcfortine A, a new alkaloid from Penicillium roqueforti. J.C.S. Chem. Comm 1980, 601–602. [Google Scholar]
- 13.Prangé T; Billion MA; Vuilhorgne M; Pascard C; Polonsky J; Moreau S, Structures of marcfortine B and marcfortine C (X-ray analysis), alkaloids from Penicillium roqueforti. Tetrahedron Lett 1981, 22 (21), 1977–1980. [Google Scholar]
- 14.Birch AJ; Wright JJ, Brevianamides - a new class of fungal alkaloid. Chem Commun 1969, (12), 644–645. [Google Scholar]
- 15.Birch AJ; Wright JJ, Studies in relation to biosynthesis. XLII. Structural elucidation and some aspects of biosynthesis of brevianamides A and brevianamides E. Tetrahedron 1970, 26 (10), 2329–3244. [DOI] [PubMed] [Google Scholar]
- 16.Whyte AC; Gloer JB, Sclerotiamide: a new member of the paraherquamide class with potent antiinsectan activity from the sclerotia of Aspergillus sclerotiorum. J Nat Prod 1996, 59 (11), 1093–1095. [DOI] [PubMed] [Google Scholar]
- 17.Kato H; Yoshida T; Tokue T; Nojiri Y; Hirota H; Ohta T; Williams RM; Tsukamoto S, Notoamides A-D: Prenylated indole alkaloids isolated from a marine-derived fungus, Aspergillus sp. Angew Chem Int Edit 2007, 46 (13), 2254–2256. [DOI] [PubMed] [Google Scholar]
- 18.Li SY; Finefield JM; Sunderhaus JD; McAfoos TJ; Williams RM; Sherman DH, Biochemical characterization of NotB as an FAD-dependent oxidase in the biosynthesis of notoamide indole alkaloids. J Am Chem Soc 2012, 134 (2), 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grüschow S; Sherman DH, The biosynthesis of epoxides In Aziridines and epoxides in organic synthesis, Yudin AK, Ed. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006; pp 349–398. [Google Scholar]
- 20.Kolundzic F; Noshi MN; Tjandra M; Movaddaghi M; Miller SJ, Chemoselective and enantioselective oxidation of indoles employing aspartyl peptide catalysts. J. Am. Chem. Soc 2011, 133 (23), 9104–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han L; Zhang W; Shi XX; You SL, Dearomatization of indoles via a phenol-directed vanadium-catalyzed asymmetric epoxidation and ring-opening cascade. Adv. Synth. Catal 2015, 357, 3064–3068. [Google Scholar]
- 22.Mercado-Marin EV; Garcia-Reynaga P; Romminger S; Pimenta EF; Romney DK; Lodewyk MW; Williams DE; Anderson RJ; Miller SJ; Tantillo DJ; Berlinck RGS; Sarpong R, Total synthesis and isolation of citrinalin and cyclopiamine congeners. Nature 2014, 509 (7500), 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Artman GD; Grubbs AW; Williams RM, Concise, asymmetric, stereocontrolled total synthesis of stephacidins A, B and notoamide B. J Am Chem Soc 2007, 129 (19), 6336–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greshock TJ; Grubbs AW; Jiao P; Wicklow DT; Gloer JB; Williams RM, Isolation, structure elucidation, and biomimetic total synthesis of versicolamide B, and the isolation of antipodal (−)-stephacidin A and (+)-notoamide B from Aspergillus versicolor NRRL 35600. Angew Chem Int Edit 2008, 47 (19), 3573–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greshock TJ; Grubbs AW; Tsukamoto S; Williams RM, A concise, biomimetic total synthesis of stephacidin A and notoamide B. Angew Chem Int Edit 2007, 46 (13), 2262–2265. [DOI] [PubMed] [Google Scholar]
- 26.Grubbs AW; Artman GD; Tsukamoto S; Williams RM, A concise total synthesis of the notoamides C and D. Angew Chem Int Edit 2007, 46 (13), 2257–2261. [DOI] [PubMed] [Google Scholar]
- 27.Faber K; Orru RVA, In Enzyme catalysis in organic chemistry, 2nd ed.; Drauz K; Waldmann H, Eds. Wiley-VCH: Weinheim, 2002; Vol. II, pp 579–608. [Google Scholar]
- 28.Huang KX; Fujii I; Ebizuka Y; Gomi K; Sankawa U, Molecular cloning and heterologous expression of the gene encoding dihydrogeodin oxidase, a multicopper blue enzyme from Aspergillus terreus. J Biol Chem 1995, 270 (37), 21495–21502. [DOI] [PubMed] [Google Scholar]
- 29.Cacho RA; Chooi YH; Zhou H; Tang Y, Complexity generation in fungal polyketide biosynthesis: A spirocycleforming P450 in the concise pathway to the antifungal drug griseofulvin. Acs Chem Biol 2013, 8 (10), 2322–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ames BD; Haynes SW; Gao X; Evans BS; Kelleher NL; Tang Y; Walsh CT, Complexity generation in fungal peptidyl alkaloid biosynthesis: Oxidation of fumiquinazoline A to the heptacyclic hemiaminal fumiquinazoline C by the flavoenzyme Af12070 from Aspergillus fumigatus. Biochemistry 2011, 50 (40), 8756–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsunematsu Y; Ishikawa N; Wakana D; Goda Y; Noguchi H; Moriya H; Hotta K; Watanabe K, Distinct mechanisms for spiro-carbon formation reveal biosynthetic pathway crosstalk. Nat Chem Biol 2013, 9 (12), 818–825. [DOI] [PubMed] [Google Scholar]
- 32.Stocking EM; Sanz-Cervera JF; Williams RM; Unkefer CJ, Studies on the biosynthesis of paraherquamide A. Origin of the beta-methylproline ring. J Am Chem Soc 1996, 118 (29), 7008–7009. [DOI] [PubMed] [Google Scholar]
- 33.Stocking EM; Martinez RA; Silks LA; Sanz-Cervera JF; Williams RM, Studies on the biosynthesis of paraherquamide: Concerning the mechanism of the oxidative cyclization of L-isoleucine to beta-methylproline. J Am Chem Soc 2001, 123 (14), 3391–3392. [DOI] [PubMed] [Google Scholar]
- 34.Stocking EM; Sanz-Cervera JF; Unkefer CJ; Williams RM, Studies on the biosynthesis of paraherquamide. Construction of the amino acid framework. Tetrahedron 2001, 57 (25), 5303–5320. [Google Scholar]
- 35.Stocking EM; Williams RM; Sanz-Cervera JF, Reverse prenyl transferases exhibit poor facial discrimination in the biosynthesis of paraherquamide A, brevianamide A, and austamide. J Am Chem Soc 2000, 122 (38), 9089–9098. [Google Scholar]
- 36.Stocking EM; Sanz-Cervera JF; Williams RM, Studies on the biosynthesis of paraherquamide: Synthesis and incorporation of a hexacyclic indole derivative as an advanced metabolite. Angew Chem Int Edit 2001, 40 (7), 1296–1298. [DOI] [PubMed] [Google Scholar]
- 37.Martínez-Luis S; Rodríguez R; Acevedo L; González MC; Lira-Rocha A; Mata R, Malbrancheamide, a new calmodulin inhibitor from the fungus Malbranchea aurantiaca. Tetrahedron 2006, 62 (8), 1817–1822. [Google Scholar]
- 38.Watts KR; Loveridge ST; Tenney K; Media J; Valeriote FA; Crews P, Utilizing DART mass spectrometry to pinpoint halogenated metabolites from a marine invertebratederived fungus. J Org Chem 2011, 76 (15), 6201–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraley AE; Garcia-Borrás M; Tripathi A; Khare D; Mercado-Marin EV; Tran H; Dan Q; Webb GP; Watts KR; Crews P; Sarpong R; Williams RM; Smith JL; Houk KN; Sherman DH, Function and structure of MalA/MalA’, iterative halogenases for late-stage C-H functionalization of indole alkaloids. J Am Chem Soc 2017, 139 (34), 12060–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ballou DP; Entsch B; Cole LJ, Dynamics involved in catalysis by single-component and two-component flavin-dependent aromatic hydroxylases. Biochem Bioph Res Co 2005, 338 (1), 590–598. [DOI] [PubMed] [Google Scholar]
- 41.Entsch B; Cole LJ; Ballou DP, Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Archives of Biochemistry and Biophysics 2005, 433, 297–311. [DOI] [PubMed] [Google Scholar]
- 42.Hicks KA; O’Leary SE; Begley TP; Ealick SE, Structural and mechanistic studies of HpxO, a novel flavin adenine dinucleotide-dependent urate oxidase from Klebsiella pneumoniae. Biochemistry 2013, 52 (3), 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mascotti ML; Ayub MJ; Furnham N; Thornton JM; Laskowski RA, Chopping and changing: the evolution of the flavin-dependent monooxygenases. J Mol Biol 2016, 428 (15), 3131–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolinksy TJ; Nielsen JE; McCammon JA; Baker NA PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004. 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng YJ; Tice CM; Singh SB, The use of spirocyclic scaffolds in drug discovery. Bioorganic & Medicinal Chemistry Letters 2014, 24 (16), 3673–3682. [DOI] [PubMed] [Google Scholar]
- 46.Ye Y; Du L; Zhang X; Newmister SA; Zhang W; Mu S; Minami A; McCauley M; Alegre-Requena JV; Fraley AE; Androver-Castellano ML; Shende VV; Oikawa H; Kato H; Tsukamoto S; Paton R; Williams RM; Sherman DH; Li S, Cofactor-independent Pinacolase directs non-Diels-Alderase biogenesis of the brevianamides. ChemRxiv 2019. 10.2634/chemrxiv.9122009.v1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.