

Abstract
The consistent observation of phosphorylated tau in the pathology of Alzheimer's disease has contributed to the emergence of a model where hyperphosphorylation triggers both tau disassociation from microtubules and its subsequent aggregation. Herein, we applied a total chemical synthetic approach to site‐specifically phosphorylate the microtubule binding repeat domain of tau (K18) at single (pS356) or multiple (pS356/pS262 and pS356/pS262/pS258) residues. We show that hyperphosphorylation of K18 inhibits 1) its aggregation in vitro, 2) its seeding activity in cells, 3) its binding to microtubules, and 4) its ability to promote microtubule polymerization. The inhibition increased with increasing the number of phosphorylated sites, with phosphorylation at S262 having the strongest effect. Our results argue against the hyperphosphorylation hypothesis and underscore the importance of revisiting the role of site‐specific hyperphosphorylation in regulating tau functions in health and disease.
Keywords: aggregation, hyperphosphorylation, native state stabilization, protein modifications, tau protein
A total chemical synthetic approach to site‐specifically phosphorylate multiple residues within the microtubule binding domain of tau is described. An increase in phosphorylation decreased the aggregation propensity and seeding efficiency, which argues against the pathogenic hyperphosphorylation hypothesis and underscores the critical importance of revisiting the role of site‐specific tau phosphorylation.
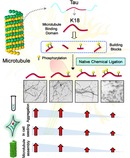
Introduction
The microtubule (MT) binding protein tau is the primary component of the neurofibrillary tangles (NFTs) found in the brain of Alzheimer's disease (AD) patients.1 Aggregated tau is also present in other pathological protein aggregates that characterize several other neurodegenerative diseases, including progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementias, and chronic traumatic encephalopathy (CTE), collectively known as tauopathies. Although initially thought to be a secondary and downstream effect of amyloid pathology in AD, increasing evidence from human clinical trials,2 animal models,3, 4 and longitudinal imaging studies5, 6 points towards a central and causative role for tau aggregation in the initiation and progression of AD.7 The exact mechanisms by which tau contributes to neurodegeneration in tauopathies, the role of phosphorylation and other posttranslational modifications (PTMs) on tau aggregation, pathology spreading, and toxicity, and the nature of the toxic species remain poorly understood.
Several aspects of tau function in health and disease are regulated by PTMs including phosphorylation, acetylation, ubiquitination, and truncation.5 However, deciphering the tau PTM code has proven to be challenging because of the large number of co‐occurring PTMs and the lack of methods that allow site‐specific introduction of these modifications. For example, there are at least 85 putative phosphorylation sites (80 serine/threonine and 5 tyrosine residues). Among those, 42 tau phosphorylation sites have been experimentally detected.5 Towards addressing these challenges, we recently developed a semisynthetic strategy that enables the site‐specific introduction of single or multiple PTMs within residues 246–441 of tau and used this approach to elucidate the effect of acetylation at K280 on tau aggregation and tau‐mediated tubulin polymerization.7, 8 In this work, we have leveraged these advances to optimize a synthetic method for producing the K18 fragment, consisting of the MTBD of tau. We applied this method to investigate the effects of phosphorylation at one, two, and three disease‐associated residues of the MTBD on MT binding, in vitro aggregation, and seeding in cell models.
We chose the K18 fragment to investigate the role of tau phosphorylation for the following reasons. 1) It contains all four microtubule binding repeats (R1, R2, R3, and R4) that are involved in the binding of tau to MTs and bears several of the disease‐causing mutations associated with tauopathies.9 2) It contains several PTM sites (Figure 1) that have been shown to strongly influence tau aggregation and pathology formation, tau tubulin binding, and MT assembly, stability, and dynamics, although other regions in the protein also influence the dynamics of these processes (Figure 1). 3) Cryo‐EM studies of AD10 and CTE11 derived tau filaments have revealed that the R3 and R4 repeats constitute the core of the PHFs in the AD brain; similarly, cryo‐EM structures derived from narrow pick filaments (NPFs) of Pick's disease brain revealed that the R1, R3, and R4 repeats constitute the core of the NPFs.12 4) The K18 fragments reproduce many of the key features of tau aggregation, exhibit rapid aggregation in vitro,13 and seed more efficiently in cells14 than the full‐length tau protein.15 Therefore, the development of a synthetic strategy that enables site‐specific modifications in the K18 fragment is highly relevant for elucidating the sequence and structural determinants of tau structure and aggregation.
Figure 1.
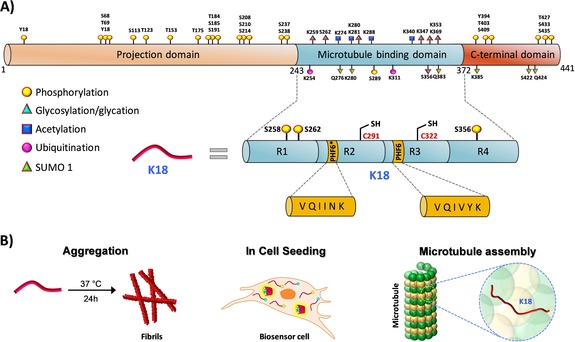
A) Schematic representation of the longest tau isoform (2N4R), which contains an N‐terminal projection domain, MTBD, and a C‐terminal domain (PTMs highlighted). The MTBD is composed of four microtubule binding repeats, R1, R2, R3, and R4. Two hexapeptides, PHF6* and PHF6 in the R2 and R3 repeats, play critical roles in tau fibrillization. Among the 85 putative phosphorylation sites (80 serine/threonine and 5 tyrosine residues) on tau, 28 sites were identified to be exclusively phosphorylated in AD brains. In the case of MTBD, the sites S258, S262, S289, and S356, which occur in the MTBD, are heavily phosphorylated in AD pathology. B) Schematic depiction illustrating the different experimental approaches used in this study to elucidate the effect of single and multiple phosphorylation states on the functional (tubulin assembly) and pathological (aggregation and seeding) aspects of K18.
Phosphorylation is one of the most actively investigated tau PTMs5 because pathological tau aggregates observed in the AD brain are heavily hyperphosphorylated.16 Furthermore, phosphorylation at several specific sites17 has been shown to be correlated with the progression of tau aggregation and is thus used as a marker of pathology. These observations have led to the hypothesis that phosphorylation disrupts tau–MT interactions and enhances its misfolding and aggregation. Previous attempts to test this hypothesis have relied on the use of phosphomimetic mutations,18, 19 which do not fully recapitulate all aspects of phosphorylation or kinases.16 The latter approach gives rise to mixtures of tau species with different phosphorylation patterns, rendering it impossible to elucidate the role of specific phosphorylation sites or patterns on the regulation of tau aggregation and MT binding.
Herein, we describe a novel, total chemical synthetic strategy that allows the preparation of site‐specific and homogeneously modified forms of K18 (consisting of 130 amino acids (AAs)). We used this method to determine the effect of K18 phosphorylation at both single (pS356) and multiple residues (pS262, pS356 or pS262, pS258, pS356) on aggregation in vitro, tubulin assembly, and seeding activity in a cellular model of tauopathy. Our studies demonstrate that phosphorylation within the MTBD inhibits rather than promotes tau fibrillization. We also show that the phosphorylation at S262 is a major contributor to disrupting tau–MT binding. On the basis of these findings and the modest effect of phosphorylation at S262 on K18 aggregation, we propose that inhibitors of kinases that regulate phosphorylation‐dependent disassociation of tau from microtubules could provide a viable strategy to stabilize the native state of tau and inhibit its aggregation.
Results
Design and Total Synthesis of Site‐Specifically Phosphorylated K18 Proteins
Our strategy for the chemical synthesis of K18 is based on a three‐fragment approach utilizing native chemical ligation (NCL;20 Scheme 1). To maintain the native sequence of K18, we designed our strategy by taking advantage of the two native cysteine residues (Cys291 and Cys322) to serve as ligation sites. The three fragments were synthesized by Fmoc solid phase peptide synthesis (Fmoc‐SPPS). As SPPS is limited to peptides that contain ≤40 AAs,20 we first tested the feasibility of synthesizing the longest fragment tau 1 (322–372), which is composed of 51 AAs. During our initial efforts to prepare fragment 1 using rink amide resin (loading 0.55 mmol g−1) and HCTU as the coupling reagent, we observed deletions of several amino acids, especially glycine (Gly) residues in regions 333–335 and 367–369, resulting in poor yields. To overcome these challenges, we used a low‐loading rink amide resin (0.27 mmol g−1), double coupling of all AAs, and incorporated pseudoproline dipeptides at Ile360‐Thr361 and Lys340‐Ser341. Under these conditions, we succeeded in obtaining fragment 1 in good yield (25 %) and high purity (see the Supporting Information, Figure S1). Next, we synthesized fragments 2 (291–321) and 3 (244–290). For both fragments, C‐terminal thioesterification was performed using the 3‐(Fmoc‐amino)‐4‐(methylamino) benzoic acid (Fmoc‐MeDbz) linker as a precursor, as described previously by Dawson and co‐workers.21 After introducing the last amino acid coupled with N‐terminal Boc protection, the C‐terminus of the peptide was activated by acylation with p‐nitrophenyl chloroformate and cyclized using a solution of 0.5 m DIEA in DMF (Figures S3 and S4). As fragment 2 has both an N‐terminal and a C‐terminal Cys thioester, the N‐terminal Cys residue was introduced as thiazolidine (Thz) to avoid undesired inter‐ and/or intramolecular ligations.
Scheme 1.
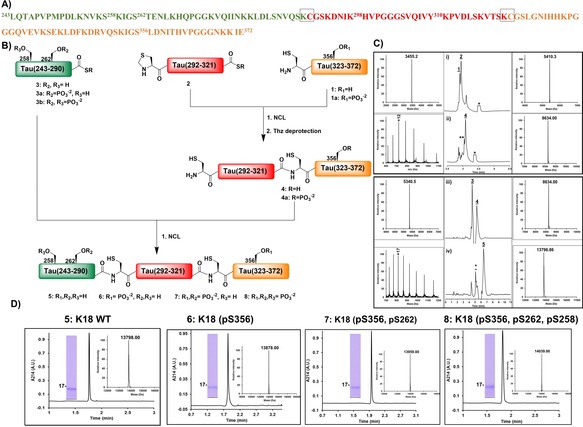
Strategy for the total chemical synthesis of the WT and mono‐, di‐, and triphosphorylated K18. A) K18 amino acid sequence with the three fragments shown in green, red, and orange and the ligation sites highlighted by gray boxes. B) Schematic representation of the synthesis of hyperphosphorylated K18 from the three synthetic fragments. C) Analytical RP‐HPLC traces and mass spectrometry (ESI‐MS) analysis of the ligation reactions between the different fragments of K18. Ligation of fragments 1 and 2 i) at 0 h and ii) at 4.5 h followed by in situ thiazolidine deprotection. Ligation of fragments 3 and 4 iii) at 0 h and iv) 2 h. D) Characterization of the purified K18 proteins by analytical RP‐HPLC, ESI‐MS, and SDS‐PAGE.
With the three tau peptide fragments in hand, we then started the assembly of K18 by NCL of fragments 1 and 2. The ligation reaction was complete after 4.5 h, and the resulting crude product bearing an N‐terminal Thz was then treated with 0.2 m methoxyamine hydrochloride for 12 h at pH 4 to free the N‐terminal cysteine required for the subsequent ligation. After completion of the Thz deprotection, the desired product was purified by RP‐HPLC to provide fragment 4 (291–372) in approximately 17 % overall yield (Figure S7). Next, the second ligation was carried out by dissolving thioester peptide 3 (243–290)‐SR and fragment 4 (291–372) in 8 m urea followed by the addition of 50 mm tris(2‐carboxyethyl)phosphine (TCEP) as a reducing agent and 50 mm 4‐mercaptophenylacetic acid (MPAA) as a thiol additive. The purification of the final product proved to be difficult because of its hydrophilicity, which led to its coelution with MPAA. To circumvent this problem, we used the volatile alkyl thiol trifluoroethanethiol (TFET),22 which can be easily removed after completion of the ligation reaction by evaporation with nitrogen gas for 30 min. The final product, WT K18 5, was purified by RP‐HPLC and obtained in high purity and about 20 % yield (Figure S8).
To confirm that synthetic K18 adopts the correct conformation and is functional, we assessed and compared its secondary structure and tubulin polymerization activity to that of the recombinant K18 produced in E. coli as described previously.23, 24 Synthetic and recombinant K18 exhibited virtually identical CD spectra (Figure 2 B) and tubulin polymerization activity (Figure 2 C).25 Next, we achieved the synthesis of K18 phosphorylated at single (pS356), double (pS356, pS262), or multiple sites (pS356, pS262, pS258), following the same synthetic strategy (Figures S9–S12) as was used for the WT K18, using one or a combination of the following phosphorylated peptide fragments: peptide 1 a (322–372, pS356), 3 a (243–290, pS262)‐SR, and 3 b (243–290, pS262, pS258)‐SR (Figures S2, S5, and S6).
Figure 2.
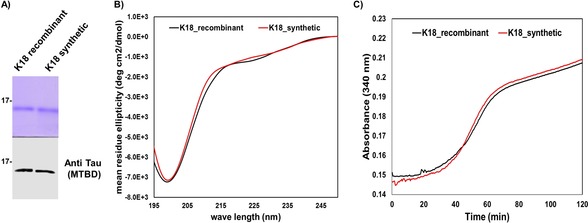
Characterization of synthetic and recombinant WT K18. A) SDS‐PAGE analysis (top) of WT recombinant and synthetic K18 as well as a western blot using the tau specific antibody (polyclonal rabbit anti‐total tau antibody, generated in house; bottom). B) Circular dichroism spectra of the WT recombinant (10 μm, black) and synthetic (10 μm, red) K18. C) Tubulin polymerization assay in the presence of synthetic and recombinant WT K18.
An Increase in Phosphorylation Decreases the Aggregation of K18
To study the effect of site‐specific phosphorylation on the aggregation of K18, synthetic K18 proteins (10 μm) bearing one (pS356), two (pS356, pS262), or three (pS356, pS262, pS258) phosphoserine residues were incubated at 37 °C in the presence of the polyanion heparin, which promotes fibril formation.24 The kinetics of fibril formation were assessed by monitoring the changes in thioflavin S (ThS) fluorescence over time. As shown in Figure 3 A, the WT K18 protein reaches the aggregation plateau within 4–5 h. Phosphorylation of serine 356 (K18_pS356) did not significantly affect the aggregation kinetics of K18 but resulted in significantly lower ThS values compared to the unmodified K18. In contrast, phosphorylation of both S262 and S356 significantly delayed but did not inhibit the aggregation of K18, as observed by the longer nucleation phase and slower growth slope. Interestingly, phosphorylation at three residues (pS356, pS262, and pS258) completely abolished fibril formation, as no increase in the ThS fluorescence was observed over time, underscoring the inhibitory impact of phosphorylation on K18 aggregation.
Figure 3.
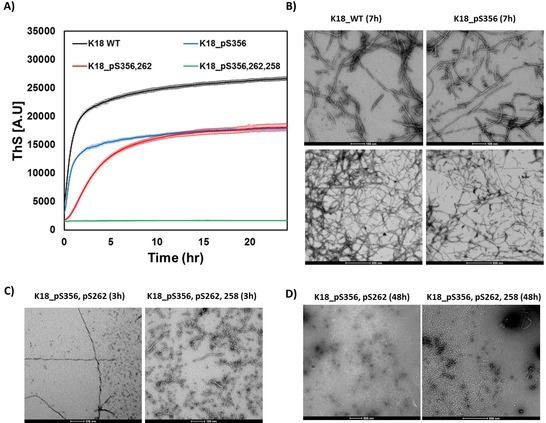
In vitro aggregation of WT and phosphorylated K18 proteins. A) Aggregation kinetics measured by ThS fluorescence at 490 nm (mean±SEM, n=3). B) TEM images of WT and K18_pS356 after 7 h (scale bars are 100 nm (upper panels) and 500 nm (lower panels)). C) TEM images of K18_pS356, pS262 and K18_pS356, pS262, pS258 after 3 h (scale bar is 100 nm) and D) 48 h of incubation (scale bar is 500 nm).
To further corroborate the ThS data and assess the effect of mono‐, di‐, and triphosphorylation on K18 fibril morphology, the aggregates were examined by transmission electron microscopy (TEM). As shown in Figure 3 B–D, both mono‐ and diphosphorylated K18 rapidly form fibrillar aggregates similar to those previously reported for K18. In the case of the diphosphorylated K18, both fibrils and oligomers are observed at 3 h (Figure 3 C, left panel), while only fibrils are detected at longer incubation times (48 h, Figure 3 D, left panel). The lower ThS values for the phosphorylated K18 proteins suggest that the phosphorylated fibrils might exhibit lower binding to ThS. Interestingly, triphosphorylated K18 also self‐assembles into oligomeric structures within the first 3 h (Figure 3 C, right panel), but these aggregates persist over time and do not go on to form fibrils; however, we observed a very small amount of short fibrils at long incubation times (Figure 3 D, right panel). The negligible number of fibrils observed by EM likely explains the absence of a ThS signal in the kinetic aggregation assay (Figure S14). These findings, derived from the homogeneously phosphorylated synthetic proteins, counter the conventional thinking that associates hyperphosphorylation with the induction or promotion of tau aggregation.6
An Increase in Phosphorylation Decreases the Seeding Efficiency of K18
To investigate the effect of phosphorylation at these sites on the seeding activity of K18, we used the previously described HEK293T biosensor cell reporter lines.26 As depicted in Figure 4 A, these cells stably express the K18 4R tau repeat domain (RD, K18) containing the disease‐associated mutation P301S fused to either cyan or yellow fluorescent protein (RD‐CFP/RD‐YFP, respectively). RD‐CFP and RD‐YFP exist exclusively as monomers unless the cells are exposed to exogenous tau aggregates, which are taken up by micropinocytosis.27 Here, we introduced the aggregates directly into the cells using a cationic lipid transfection reagent (Lipofectamine), which increases the assay sensitivity by approximately 100‐fold.26 The uptake of the exogenous aggregates induces intracellular aggregation of RD‐CFP/YFP, which creates a fluorescence resonance energy transfer (FRET) signal. Within a population of cells, the percentage that contain intracellular aggregates can be accurately measured by FRET‐flow cytometry. To determine the titer of tau seeding activity, a FRET value was derived for a cell population using the integrated FRET density (IFD), which is the product of the percentage of positive cells and the mean fluorescence intensity of the FRET‐positive cells.26
Figure 4.
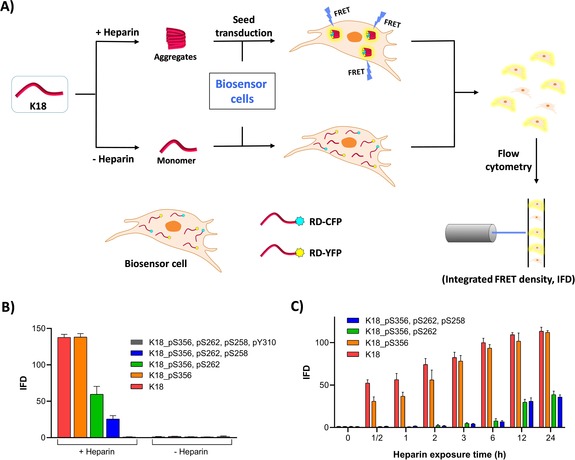
A) Schematic representation of the FRET‐based seeding assay using HEK293T biosensor cell reporter lines. Heparin‐induced aggregates are transfected into the biosensor cells using Lipofectamine. Aggregation seeded by the K18 proteins is detected as FRET emission and quantified using IFD. B) IFD measured following transduction of WT, mono‐, di‐, tri‐, and tetraphosphorylated K18 previously incubated with or without heparin for 24 h at 37 °C (N=3 repeats). C) Effect of different heparin exposure times with K18 proteins prior to transfection of the aggregates (N=3 repeats).
When the WT and phosphorylated K18 variants were incubated in the absence of heparin for 24 or 72 hours at 37 °C or 25 °C and then added to the biosensor cell reporter line, we did not observe seeding activity (Figure 4 B). This is consistent with the fact that tau does not spontaneously form fibrils in the absence of polyanionic inducers. However, when WT and K18_pS356 were incubated in the presence of heparin for 24 or 72 hours at 37 °C, significant seeding activity was observed, while constructs bearing double (pS356, pS262) or triple (pS356, pS262, pS258) phosphorylated sites showed significantly lower seeding activity (Figures 4 B and S15). Interestingly, the tetraphosphorylated K18 protein bearing one additional phosphate group at Y310, which occurs within one of the key aggregation motifs (PHF6; Figure 1), did not exhibit any seeding capacity (Figure 4 B).
When the fibrils were incubated in the presence of heparin for shorter periods of time before the addition to the biosensor cells, we observed that WT K18 and K18_pS356 have different seeding activity when added to the biosensor cells at shorter incubation times (0–12 h, Figure 4 C), whereas WT K18 developed seeding activity more rapidly than K18_pS356 (Figure S16). Together, these findings suggest a direct correlation between the in vitro aggregation propensity of the K18 proteins and their seeding activity in the cell‐based biosensor assay.
Phosphorylation at S262 Disrupts Tau Binding to MTs
One of the key physiological functions of tau is its role in promoting tubulin polymerization and the stabilization of MTs.17 Several studies have shown that phosphorylation at single or multiple residues in the MTBD could regulate or negatively influence tubulin polymerization. However, all of these studies were based on experimental approaches that did not allow for efficient and site‐specific phosphorylation of tau or relied on the use of phosphomimetic mutations, which do not reproduce the effects of bona fide phosphorylation. To assess the effect of authentic phosphorylation at single or multiple residues within the MTBD on the modulation of MT assembly, we performed a tubulin polymerization assay.28 The tubulin protein (40 μm) was incubated with K18 proteins (30 μm) bearing one (pS356), two (pS356, pS262), or three (pS356, pS262, pS258) phosphoserine residues at 37 °C in the presence of GTP, as previously described.29, 30
The kinetics of the MT assembly were assessed by monitoring the changes in absorbance at 350 nm over time. As expected, the native K18 protein accelerated the rate of MT polymerization (Figure 5 A).28 In contrast, the phosphorylated K18 proteins exhibited significantly perturbed tubulin assembly. Phosphorylation at S356 slightly impaired the K18‐induced MT polymerization, whereas the di‐ and triphosphorylated forms of K18 bearing phosphorylation at S262 and S356 or S258, S262, and S356 strongly reduced MT polymerization. Similarly, microtubule binding analyzed using the sedimentation assay showed that K18_pS356, pS262 and K18_pS356, pS262, pS258 have significantly lower binding towards MTs compared to K18 or K18_pS356, which is consistent with the results from the MT polymerization assay (Figure S17). These findings suggest that phosphorylation at S262 has a dominant effect on the disruption of tau binding to MTs.
Figure 5.
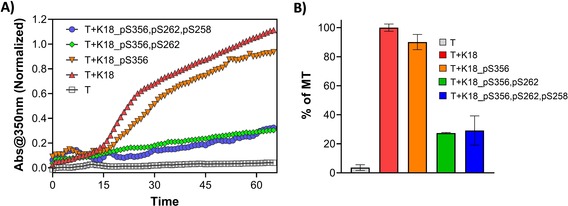
K18 protein binding to MT. A) MT assembly in the presence of K18 proteins (K18, K18_pS356, K18_pS356, pS262, and K18_pS356, pS262, pS258) was evaluated by measuring light scattering at 350 nm over time. B) The percentage of MT formed after 60 min of incubation with K18 proteins determined by measuring the absorbance at 350 nm (3 repeats, represented as the mean±SD).
Discussion
Despite the consistent observation that pathological tau species in the brains of patients with AD and other tauopathies are hyperphosphorylated, the role of tau phosphorylation in tau pathology formation and the pathogenesis of AD remains unclear. For decades, the prevailing hypothesis has been that hyperphosphorylation of tau induces its disassociation from MTs, disrupts axonal transport, and increases aggregation into PHFs. Testing this hypothesis has proven to be challenging for several reasons: 1) The exact residues and patterns of phosphorylation that define each hyperphosphorylated state remain poorly defined, especially as the state of phosphorylation is defined primarily using a limited set of antibodies, and the impact of the presence of multiple PTMs on the detection by these antibodies has not been systematically investigated. 2) There is a lack of synthetic strategies or in vitro phosphorylation conditions that enable the site‐specific introduction of PTMs and the preparation of homogeneously modified forms of tau.
To address these limitations and pave the way for deciphering the tau PTM code, we developed and optimized a total chemical synthesis method for the K18 fragment and used it to generate K18 fragments that were site‐specifically phosphorylated at single or multiple physiologically and pathologically relevant sites. The flexibility of this method allowed us for the first time to begin to answer with great precision the long‐standing question of whether phosphorylation of tau within the MTBD inhibits or accelerates its aggregation. In addition, the availability of these homogeneously phosphorylated proteins enabled us to identify which phosphorylation sites dominantly regulate tau–MT binding and aggregation, thus providing critical insight for developing novel therapeutic strategies on the basis of site‐specific regulation of tau phosphorylation states.
Our results demonstrate that the effect of phosphorylation on K18 aggregation and K18 fibril seeding activity is sequence‐context‐dependent and is dramatically reduced with an increasing number of phosphorylation sites. We showed that phosphorylation of K18 within the MTBD strongly inhibits fibril formation in vitro and leads to the accumulation of nonfibrillar oligomers. The reduced seeding activity for the triphosphorylated K18 suggests that the phosphorylated oligomers exhibited very low seeding activity. Our findings are consistent with previous studies by Mandelkow and co‐workers demonstrating that in vitro hyperphosphorylation of tau by MARK and PKA kinases, which phosphorylate tau at multiple residues including S262 and S356, inhibits its aggregation.31 However, these kinases phosphorylate tau at several additional sites, thus making it difficult to correlate the inhibitory effects observed with specific phosphorylation patterns.16 In our work, we addressed this major limitation using protein total chemical synthesis, which enabled the generation of site‐specifically and homogeneously hyperphosphorylated forms of K18.
In an attempt to gain insight into the structural basis underlying the inhibitory effects of phosphorylation and the relative contribution of each phosphorylation site, we reviewed the recent high‐resolution atomic cryo‐EM structures of the tau filaments derived from the brains of patients with AD and Pick's disease.11, 12 Close examination of the structures suggests that phosphorylation of S258 and S262 are likely to influence the Tau assembly (see Figure S18 A).
Previous models that sought to establish a link between the disruption of tau normal function (stabilization of MTs) and its propensity to aggregate speculated that the phosphorylation events responsible for inducing tau disassociation from MTs are likely to also increase aggregation and pathology formation. Our work here shows that this is not the case. For example, phosphorylation at S356, which does not influence K18 aggregation and seeding, results in small but significant inhibition of tau‐mediated MT assembly. On the other hand, phosphorylation of the S262 residue, which results in a dramatic reduction of MT polymerization, resulted in significant inhibition of tau fibril formation and cellular seeding activity. Interestingly, phosphorylation at both S258 and S262 resulted in the near abolishment of K18 fibrillization in vitro, seeding activity in cells, and tau‐mediated MT polymerization. These results show that phosphorylation events within the MTBD that lead to the disruption of tau–MT binding also inhibit its aggregation and seeding activity. These findings are consistent with previous studies showing that the nonspecific phosphorylation of tau using kinases that phosphorylated at S262 or the introduction of point mutations that mimic phosphorylation at this residue disrupt tau binding to MTs and inhibit tau‐mediated tubulin assembly.32 Together, these observations suggest that S262 plays a key role in the tubulin–tau interaction and the stabilization of this native conformation of the protein. This is consistent with a recent report on the atomic structure of tubulin and tau interactions, which shows that both residues S262 and S258 of tau are directly involved in hydrogen bonding interactions with tubulin residue E434 (Figure S18 B), which further highlights the role of S262 in MT binding.33 Introducing phosphorylation at S262 and S258 would disrupt the hydrogen bonding and further introduce electrostatic repulsion between pS262 or pS258 of tau and tubulin E434, which could potentially destabilize the tau–MT interaction. These attributes clearly support our experimental results that K18 phosphorylated at S262 and S258 significantly disrupts MT assembly and MT binding (Figure S17).
Implications for Tau Therapeutic Strategies
Disassociation of tau from MTs has been proposed as one of the key early pathological events that lead to increasing the pool of free tau, which then aggregates and forms PHFs.6 Therefore, stabilizing tau interactions with MTs or interfering with events that lead to its disassociation from MTs (i.e., native‐state stabilization) could constitute an effective strategy to prevent tau aggregation and pathology formation. On the basis of our results and previous findings discussed above, we propose that inhibiting the kinases responsible for the phosphorylation events that lead to the disruption of the tau interaction with MTs represents an attractive strategy for preventing tau accumulation and aggregation by stabilizing its native MT‐bound conformation. Given the dominant role of S262 phosphorylation in disrupting tau interactions with MTs, we propose that inhibiting the natural kinases responsible for phosphorylating tau at S262 or S258 and S262 constitutes a viable therapeutic strategy for the treatment of AD and potentially other tauopathies. This would not only stabilize the native state of the tau–tubulin complex (Figure 6) but would also decrease the levels of unbound tau available for the formation of potentially toxic species (oligomers and fibrils). As phosphorylation at S262 alone had only a modest effect on tau aggregation compared to the di‐ and triphosphorylated K18 protein, we do not anticipate that selective inhibition of phosphorylation at this residue would promote tau aggregation. However, further studies are required to validate these results in the context of full‐length tau (in vitro and in vivo) and to assess whether modulation or phosphorylation at S262 or other sites that directly or indirectly disrupt tau–MT binding is sufficient to inhibit tau aggregation and toxicity.
Figure 6.
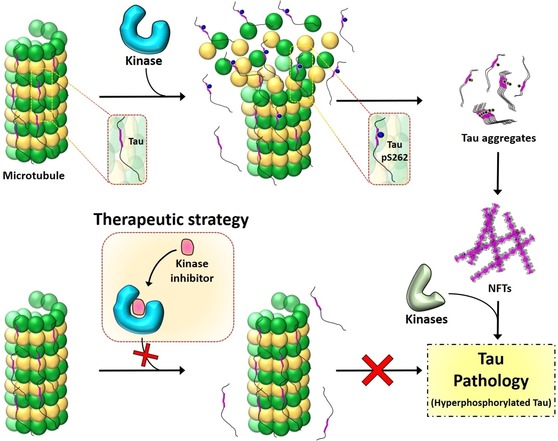
The residue S262 plays a vital role in stabilizing the binding of tau to tubulin of MTs. Phosphorylation at S262 disrupts the tau–MT interaction (top), leading to tau accumulation and pathology formation. Inhibiting the phosphorylation at S262 or other sites that directly or indirectly disrupt the tau–MT interaction by targeting the kinases involved could be an effective therapeutic strategy (bottom) to prevent tau dissociation from MTs by stabilizing the native state of the tau–tubulin complex.
Conclusion
Taken together, our results argue against the prevailing hypothesis that phosphorylation promotes tau aggregation and pathology formation. Instead, our data show that phosphorylation within the MTBD inhibits tau aggregation and seeding activity in cells, with the inhibitory effects increasing with an increasing number of phosphorylated sites, with phosphorylation at S258 and S262 having a dominant effect on K18 aggregation. The fact that the level of phosphorylation at these sites has been found to be elevated in the pathology of AD and other tauopathies may reflect post‐aggregation phosphorylation events rather than the primary role of phosphorylation at these residues in driving pathology formation. Although our results indicate that introducing disease‐relevant site‐specific phosphorylation in K18 inhibits its aggregation and fibril formation, further studies are required to understand the effect of phosphorylation at these sites in the context of the full‐length tau protein both in vitro and in vivo.
Together, our findings highlight the potential of targeting tau phosphorylation for the treatment of AD and tauopathies and underscore the critical importance of revisiting the role of site‐specific phosphorylation and hyperphosphorylation in regulating tau interactions with microtubules and binding partners with tau and transcellular pathology propagation. Furthermore, our results demonstrate that the oligomerization, fibrillization, and tubulin binding properties of K18 are highly sensitive to its phosphorylation state and pattern. Thus, our results underscore the importance of reassessing the phosphorylation patterns associated with the different physiological and pathogenic hyperphosphorylated states of tau with an emphasis on determining which phosphorylation sites co‐occur on the same molecule. This, combined with the advances made by our group7 and others34, 35 to enable site‐specific modifications of tau, should enable the generation and characterization of the different, modified tau species in homogeneous forms, thus paving the way for more systematic investigations of the tau phosphorylation code in health and disease. Our data also provide strong evidence in support of pursuing native‐state stabilization strategies for the treatment of AD and tauopathies. While tau does not have a stable folded monomeric conformation in solution or ligand‐binding pockets that could be targeted by small molecules, it does engage in highly specific and stable molecular interactions where it becomes highly ordered.36 Developing and identifying strategies for stabilizing these interactions through the modulation of phosphorylation or other modifications could provide viable small‐molecule‐based strategies for targeting tau aggregation and toxicity.
Conflict of interest
Hilal A. Lashuel is the founder and CSO of ND BioSciences.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the École Polytechnique Fédérale de Lausanne. We thank Dr. Nadine Ait Bouziad for her feedback and review of the manuscript.
M. Haj-Yahya, P. Gopinath, K. Rajasekhar, H. Mirbaha, M. I. Diamond, H. A. Lashuel, Angew. Chem. Int. Ed. 2020, 59, 4059.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.1101/261362).
References
- 1. Seubert P. et al., J. Biol. Chem. 1995, 270, 18917–18922. [DOI] [PubMed] [Google Scholar]
- 2. Cummings J., Clin. Transl. Sci. 2018, 11, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He Z. et al., Nat. Med. 2018, 24, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. LaFerla F. M., Green K. N., Cold Spring Harbor Perspect. Med. 2012, 2, a006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iqbal K., Liu F., Gong C.-X., Nat. Rev. Neurosci. 2016, 12, 15. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y., Mandelkow E., Nat. Rev. Neurosci. 2016, 17, 22. [DOI] [PubMed] [Google Scholar]
- 7. Haj-Yahya M., Lashuel H. A., J. Am. Chem. Soc. 2018, 140, 6611–6621. [DOI] [PubMed] [Google Scholar]
- 8. Ellmer D., Brehs M., Haj-Yahya M., Lashuel H. A., Becker C. F. W., Angew. Chem. Int. Ed. 2019, 58, 1616–1620; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1630–1634. [Google Scholar]
- 9. Goedert M., Ghetti B., Spillantini M. G., Cold Spring Harbor Perspect. Med. 2012, 2, a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Falcon B. et al., Nature 2019, 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., Crowther R. A., Ghetti B., Goedert M., Scheres S. H. W., Nature 2017, 547, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falcon B., Zhang W., Murzin A. G., Murshudov G., Garringer H. J., Vidal R., Crowther R. A., Ghetti B., Scheres S. H. W., Goedert M., Nature 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barghorn S., Mandelkow E., Biochemistry 2002, 41, 14885–14896. [DOI] [PubMed] [Google Scholar]
- 14. Lim S., Haque M. M., Kim D., Kim D. J., Kim Y. K., Comput. Struct. Biotechnol. J. 2014, 12, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ambadipudi S., Biernat J., Riedel D., Mandelkow E., Zweckstetter M., Nat. Commun. 2017, 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Augustinack J. C., Schneider A., Mandelkow E.-M., Hyman B. T., Acta Neuropathol. 2002, 103, 26–35. [DOI] [PubMed] [Google Scholar]
- 17. Liu F., Li B., Tung E.-J., Grundke-Iqbal I., Iqbal K., Gong C.-X., Eur. J. Neurosci. 2007, 26, 3429–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fischer D., Mukrasch M. D., Biernat J., Bibow S., Blackledge M., Griesinger C., Mandelkow E., Zweckstetter M., Biochemistry 2009, 48, 10047–10055. [DOI] [PubMed] [Google Scholar]
- 19. Schwalbe M., Kadavath H., Biernat J., Ozenne V., Blackledge M., Mandelkow E., Zweckstetter M., Structure 2015, 23, 1448–1458. [DOI] [PubMed] [Google Scholar]
- 20. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 21. Blanco-Canosa J. B., Nardone B., Albericio F., Dawson P. E., J. Am. Chem. Soc. 2015, 137, 7197–7209. [DOI] [PubMed] [Google Scholar]
- 22. Thompson R. E., Liu X., Alonso-García N., Pereira P. J. B., Jolliffe K. A., Payne R. J., J. Am. Chem. Soc. 2014, 136, 8161–8164. [DOI] [PubMed] [Google Scholar]
- 23. Ait-Bouziad N., Lv G., Mahul-Mellier A.-L., Xiao S., Zorludemir G., Eliezer D., Walz T., Lashuel H. A., Nat. Commun. 2017, 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.“Purification of Recombinant Tau Protein and Preparation of Alzheimer Paired Helical Filaments In Vitro”: Barghorn S., Biernat J., Mandelkow E. in Amyloid Proteins: Methods and Protocols (Ed.: E. M. Sigurdsson), Humana Press, Totowa, NJ, 2005, pp. 35–51. [DOI] [PubMed] [Google Scholar]
- 25. Goode B. L., Feinstein S. C., J. Cell Biol. 1994, 124, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holmes B. B., Furman J. L., Mahan T. E., Yamasaki T. R., Mirbaha H., Eades W. C., Belaygorod L., Cairns N. J., Holtzman D. M., Diamond M. I., Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holmes B. B. et al., Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar]
- 28. Cohen T. J., Guo J. L., Hurtado D. E., Kwong L. K., Mills I. P., Trojanowski J. Q., Lee V. M. Y., Nat. Commun. 2011, 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J. C., Timasheff S. N., Biochemistry 1975, 14, 5183–5187. [DOI] [PubMed] [Google Scholar]
- 30. Shelanski M. L., Gaskin F., Cantor C. R., Proc. Natl. Acad. Sci. USA 1973, 70, 765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schneider A., Biernat J., von Bergen M., Mandelkow E., Mandelkow E.-M., Biochemistry 1999, 38, 3549–3558. [DOI] [PubMed] [Google Scholar]
- 32. Biernat J., Gustke N., Drewes G., Mandelkow E.-., Mandelkow E., Neuron 1993, 11, 153–163. [DOI] [PubMed] [Google Scholar]
- 33. Kellogg E. H., Hejab N. M. A., Poepsel S., Downing K. H., DiMaio F., Nogales E., Science 2018, 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reimann O., Smet-Nocca C., Hackenberger C. P. R., Angew. Chem. Int. Ed. 2015, 54, 306–310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 311–315. [Google Scholar]
- 35. Broncel M., Krause E., Schwarzer D., Hackenberger C. P. R., Chem. Eur. J. 2012, 18, 2488–2492. [DOI] [PubMed] [Google Scholar]
- 36. Kadavath H., Hofele R. V., Biernat J., Kumar S., Tepper K., Urlaub H., Mandelkow E., Zweckstetter M., Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary