

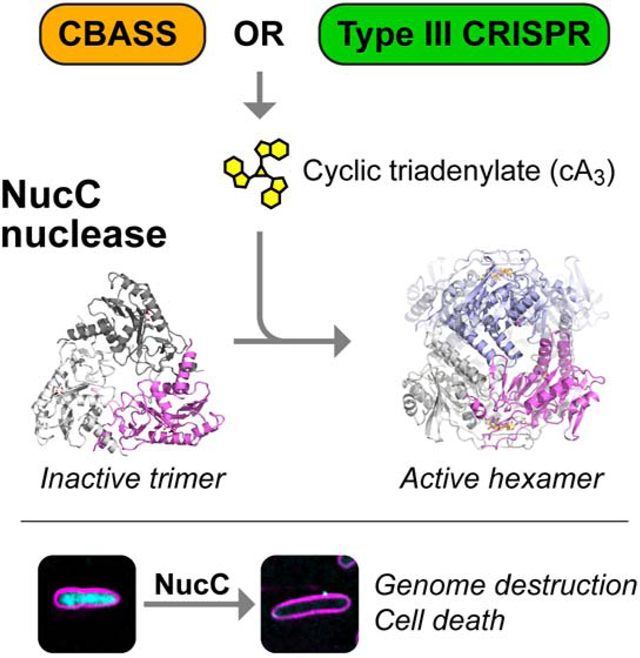
Summary
Bacteria possess an array of defenses against foreign invaders, including a broadly-distributed bacteriophage defense system termed CBASS (Cyclic oligonucleotide-Based Anti-phage Signaling System). In CBASS systems, a cGAS/DncV-like nucleotidyltransferase synthesizes cyclic di- or trinucleotide second messengers in response to infection, and these molecules activate diverse effectors to mediate bacteriophage immunity via abortive infection. Here, we show that the CBASS effector NucC is related to restriction enzymes but uniquely assembles into a homotrimer. Binding of NucC trimers to a cyclic tri-adenylate second messenger promotes assembly of a NucC homohexamer competent for non-specific double-strand DNA cleavage. In infected cells, NucC activation leads to complete destruction of the bacterial chromosome, causing cell death prior to completion of phage replication. In addition to CBASS systems, we identify NucC homologs in over 30 Type III CRISPR/Cas systems, where they likely function as accessory nucleases activated by cyclic oligoadenylate second messengers synthesized by these systems’ effector complexes.
Keywords: Second messenger signaling, bacteriophage immunity, abortive infection, CD-NTase, Endonuclease
Graphical Abstract
eTOC Blurb
Here, Lau et al. show that the CBASS-associated NucC nuclease causes death of phage-infected cells through genome destruction. They show how NucC is allosterically activated by a cyclic triadenylate second messenger synthesized by CBASS upon infection. NucC has also been integrated into Type III CRISPR/Cas systems as an accessory nuclease.
Introduction
Bacteria are locked in a continual struggle to protect themselves and their genomes from foreign mobile genetic elements and bacteriophages, and as such have evolved sophisticated defenses against these invaders. Two well-characterized bacterial defense systems are restriction-modification systems and CRISPR/Cas systems, both of which rely on targeted nuclease activity to destroy foreign nucleic acids. In a typical restriction-modification system, a bacterium encodes a sequence-specific DNA endonuclease and a corresponding DNA methylase that modifies the host genome to protect it against endonuclease cleavage (Arber and Linn, 1969; Wilson and Murray, 1991). While restriction-modification systems mount a standard response to all improperly-modified DNA and as such can be likened to an “innate” immune system, the more recently-characterized CRISPR/Cas systems are more akin to an “adaptive” immune system. In these systems, effector nucleases are loaded with guide sequences that specifically target foreign nucleic acids (Koonin et al., 2017; Sorek et al., 2013; Wright et al., 2016). In Type III CRISPR/Cas systems, the multi-protein effector complex not only recognizes and cleaves foreign RNA and DNA (Koonin et al., 2017; Pyenson and Marraffini, 2017), but also synthesizes 3′−5′ linked cyclic oligoadenylate second messengers upon target binding (Kazlauskiene et al., 2017; Niewoehner et al., 2017). These second messengers in turn activate nonspecific accessory nucleases encoded in the same operons, including the RNases Csm6 and Csx1, and the DNase Can1 (Han et al., 2018; Kazlauskiene et al., 2017; Mcmahon et al., 2019; Niewoehner et al., 2017).
The newly-described CBASS (Cyclic oligonucleotide-Based Anti-phage Signaling System) bacteriophage defense systems encode a cGAS/DncV-like nucleotidyltransferase (CD-NTase), one of several diverse effectors, and optionally a set of regulatory proteins putatively responsible for sensing an infection (Burroughs et al., 2015; Cohen et al., 2019; Whiteley et al., 2019). In related work, we determined the molecular mechanisms of a CBASS system encoding regulatory proteins related to eukaryotic HORMA domain proteins and the AAA+ ATPase Pch2/TRIP13 (Ye et al., 2019). In this system, detection of specific peptides by the CBASS-encoded HORMA domain protein stimulates synthesis of the cyclic trinucleotide second messenger cyclic AMP-AMP-AMP (cAAA), which activates the DNA endonuclease NucC (Nuclease, CD-NTase associated). Both cAAA production and NucC catalytic activity are required for bacteriophage immunity, which is mediated by an abortive infection mechanism in which infected cells die prior to completion of phage replication (Ye et al., 2019). Still unanswered, however, has been how NucC activation leads to cell death, and how this restriction endonuclease-related enzyme is controlled by the second messenger cAAA.
Here, we show that NucC is a homotrimeric DNA endonuclease that binds the second messenger cAAA in a three-fold symmetric allosteric pocket. We show that nuclease activation involves the assembly of two NucC trimers into a homohexamer, which juxtaposes pairs of endonuclease active sites and promotes coordinated double-strand DNA cleavage. NucC is largely sequence non-specific and is not inhibited by DNA methylation, and its activation in infected cells leads to complete destruction of cellular DNA. Finally, we find that NucC homologs have been incorporated into Type III CRISPR/Cas systems, defining a new family of Type III CRISPR accessory nucleases activated by cyclic oligoadenylate second messengers.
Results
NucC activation leads to genome destruction in infected cells
We have previously shown that the CBASS system from a patient-derived strain of E. coli (MS115–1; Figure 1A) provides immunity against bacteriophage λ infection by an abortive infection mechanism (Ye et al., 2019). To better understand the mechanism of CBASS-mediated cell death, and particularly the role of the effector nuclease NucC, we directly imaged infected cells carrying either the intact CBASS operon or a variant carrying a mutant of NucC (D73N) that lacks DNA cleavage activity in vitro (Figure 1B). We infected cells carrying these operons with bacteriophage λ at a high multiplicity of infection and visualized cell membranes and DNA in live cells. Cultures lacking CBASS show moderate cell lysis by 60 minutes post-infection, and large-scale lysis by 80 minutes post-infection (Figure 1C–D, Figure S1). In cultures carrying the intact CBASS system, however, by 40 minutes post-infection we observe a large fraction of cells in which cellular DNA appears to have been either partially or completely degraded (Figure 1C–D). These cultures show large-scale lysis by 80 minutes post-infection, but do not show the high levels of extracellular DNA characteristic of lysed cultures lacking CBASS (Figure 1C). This degradation depends on NucC’s endonuclease activity, as cultures carrying CBASS with the NucC D73N mutant show only cell lysis similar to controls (Figure 1 C–D). These data strongly indicate that NucC mediates cell death by destroying DNA within the infected cell upon activation.
Figure 1. NucC activation leads to chromosome destruction in infected cells.

(A) Schematic of the CBASS system from E. coli MS115–1 harboring NucC. Asterisk denotes the position of the D73N nuclease-dead mutant (Ye et al., 2019). (B) Digestion of a bacteriophage λ BstEII ladder by wild-type Ec NucC (10 nM) and nuclease-dead D73N mutant, in the presence of cAAA (0, 6.25, 25, 100, and 400 nM). (C) Fluorescence microscopy of E. coli JP313 cells transformed with empty pLAC22 vector or vectors encoding intact E. coli MS115–1 CBASS or a variant with the NucC D73N mutant. DAPI staining is shown in cyan, and FM4–64 staining (membranes) is shown in magenta. See Figure S1A for separate channels. Strong DAPI-staining dots represent bacteriophage particles. (D) Top: Representative images of cells in different classes: Intact nucleoid, Degraded nucleoid, Nucleoid remnant, or Empty Cell (no nucleoid). Bottom: Quantitation of the results shown in panel (C), showing that nucleoid destruction occurs in cells harboring the full CBASS operon (n=243–1385 cells per condition in ≥10 fields over two separate experiments). N/D: not determined (high levels of cell lysis precluded scoring at 80 minutes post-infection in Empty Vector and NucC D73N conditions).
NucC is related to restriction endonucleases but adopts a homotrimeric architecture
To understand the structure of NucC and its mechanism of activation, we overexpressed and purified E. coli MS115–1 (Ec) NucC, and a second NucC from a related operon found in Pseudomonas aeruginosa strain ATCC27853 (Pa). Ec NucC and Pa NucC share 77% overall sequence identity, and both proteins self-associate in yeast two-hybrid assays (Ye et al., 2019). Using size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS), we found that both proteins form homotrimers in solution (Figure S2A–B). Several families of DNA exonucleases are known to form homotrimers, including bacteriophage λ nuclease (Kovall and Matthews, 1997; Zhang et al., 2011) and the archaeal nuclease PhoExoI (Miyazono et al., 2015). In contrast, DNA endonucleases are more commonly homodimeric, with two active sites that cleave opposite DNA strands to effect double-strand cleavage (Pingoud et al., 2005).
We crystallized and determined the structure of Ec NucC to a resolution of 1.75 Å (Table S1). In agreement with prior sequence analysis (Burroughs et al., 2015), the NucC monomer adopts a restriction endonuclease-like fold (Figure 2A). A database search using the DALI structure-comparison server (Holm and Rosenström, 2010) revealed structural similarity to type II restriction endonucleases including NgoMIV (Deibert et al., 2000), Kpn2I (PDB ID 6EKR; unpublished), and SgrAI (Dunten et al., 2008). Indeed, comparing NucC with a structure of SgrAI bound to DNA (Dunten et al., 2008) reveals an overall r.m.s.d. of 1.97 Å over 132 aligned Cα atoms (Figure 2A and Figure S2C–D). NucC also possesses a conserved active site motif of IDx30EAK (residues 72–106 in Ec NucC), and the conserved acidic residues Asp73 and Glu104 coordinate a Mg2+ ion in our structure (Figure 2B). Consistent with our SEC-MALS data, the structure of NucC reveals a tightly-packed homotrimer with an overall triangular architecture (Figure 2B). The three active sites are arrayed on the outer edge of the trimer. The trimer interface of NucC is composed largely of elements that are not part of the core nuclease fold, but are rather embellishments not shared with dimeric restriction endonucleases (Figure S2E).
Figure 2. NucC is a homotrimeric relative of restriction endonucleases.
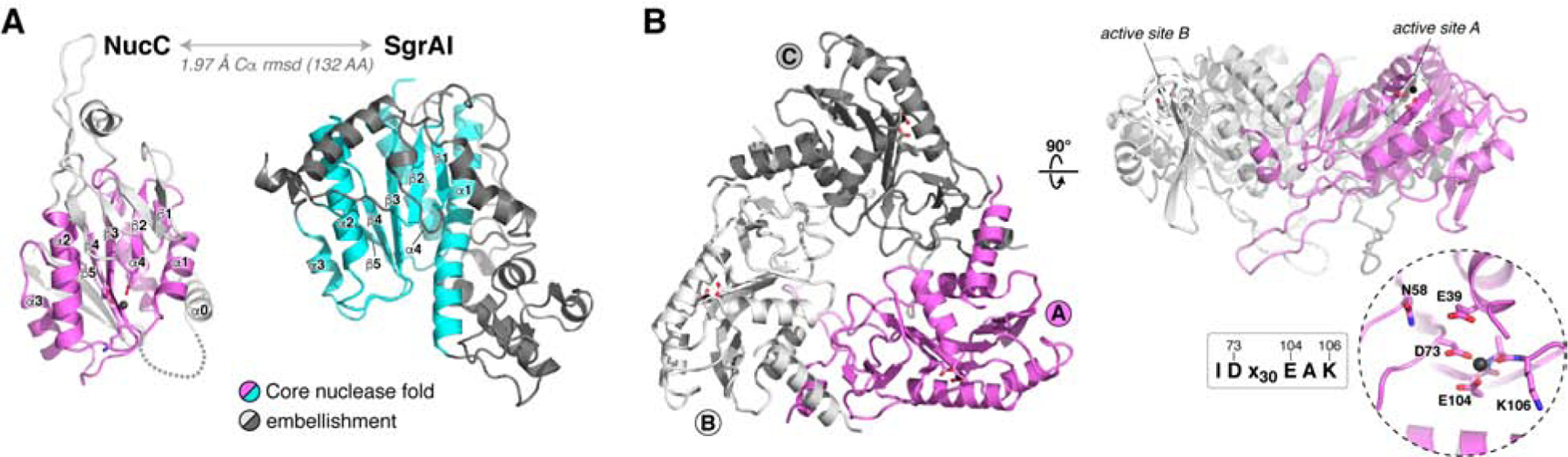
(A) Structure of an Ec NucC monomer and the related restriction endonuclease SgrAI (PDB ID 3DW9; (Dunten et al., 2008)). Shared secondary structural elements in the core nuclease fold are colored violet (NucC) or cyan (SgrAI), and family-specific embellishments are shown in gray. (B) Structure of an Ec NucC trimer with monomers colored violet (monomer A), white (B), and gray (C). The side view (right) shows the active sites of the A and B monomers. Inset: closeup view of the NucC active site, with ordered Mg2+ ion shown as a black sphere. NucC possesses a conserved IDx30EAK active-site consensus motif (See Figure S2C–D for comparison with the active site of SgrAI, and Figure S2E for NucC sequence alignment).
NucC binds cAAA in an allosteric pocket
In vitro, Ec NucC is strongly activated by cAAA, and is more weakly activated by cAAG and the cyclic dinucleotide 3′,3′ cyclic di-AMP (Ye et al., 2019). We used isothermal titration calorimetry (ITC) to directly test binding of Ec NucC to a range of dinucleotide and trinucleotide second messengers. We found that Ec NucC binds strongly to 3′,3′,3′ cAAA (Kd = 0.7 μM), and binds more weakly to both 3′,3′ cyclic di-AMP (Kd = 2.6 μM) and 3′−5′ linked linear di-AMP (5′-pApA; 4.4 μM). We did not detect binding to a mixed-linkage cyclic di-AMP (2′,3′ cyclic di-AMP) or AMP (Figure S3 and Figure S4). No second messenger bound with a stoichiometry higher than ~0.3 molecules per NucC monomer in our ITC assays, suggesting that the NucC trimer likely possesses a single second messenger binding site.
We next co-crystallized both Ec NucC and Pa NucC with 3′,3′,3′ cAAA and determined the structures of these complexes to a resolution of 1.66 and 1.45 Å, respectively (Table S1, Figure 3A–C, Figure S5A–C). We also determined a 1.66 Å-resolution structure of Ec NucC bound to 5′-pApA, which binds equivalently to cAAA in the crystals despite failing to stimulate nuclease activity in vitro (Table S1 and Figure S5D–F). The structures reveal that a single second messenger molecule binds in a conserved, three-fold symmetric allosteric pocket on the “bottom” of the NucC trimer, and that second messenger binding causes a series of conformational changes both within each NucC monomer and in the trimer as a whole (Figure 3A–B). Each adenine base is recognized through a hydrogen bond between its C6 amine group and the main-chain carbonyl of Val227, and through π-stacking with Tyr81 (Figure 3C–D). In addition, NucC residues Arg53 and Thr226 form a hydrogen-bond network with each phosphate group of the cyclic trinucleotide (Figure 3C). Binding of cAAA induces a dramatic conformational change in an extended hairpin loop in NucC, which we term the “gate loop”, resulting in the closure of all three loops over the conserved pocket to completely enclose the bound cAAA molecule (Figure 3E–F). Two aromatic residues, His136 and Tyr141, interact through π-stacking to stabilize the gate loop’s structure in both open and closed states. In the cAAA-bound structure, these two residues serve to “latch” the gate loops through tight association at the three-fold axis of the NucC trimer. Mutation of gate loop residues His136 or Tyr141, or cAAA-binding residues Arg53, Tyr81, or Thr226 completely disrupts Ec NucC’s cAAA-activated endonuclease activity (Figure 3G).
Figure 3. NucC binds cAAA in an allosteric pocket.
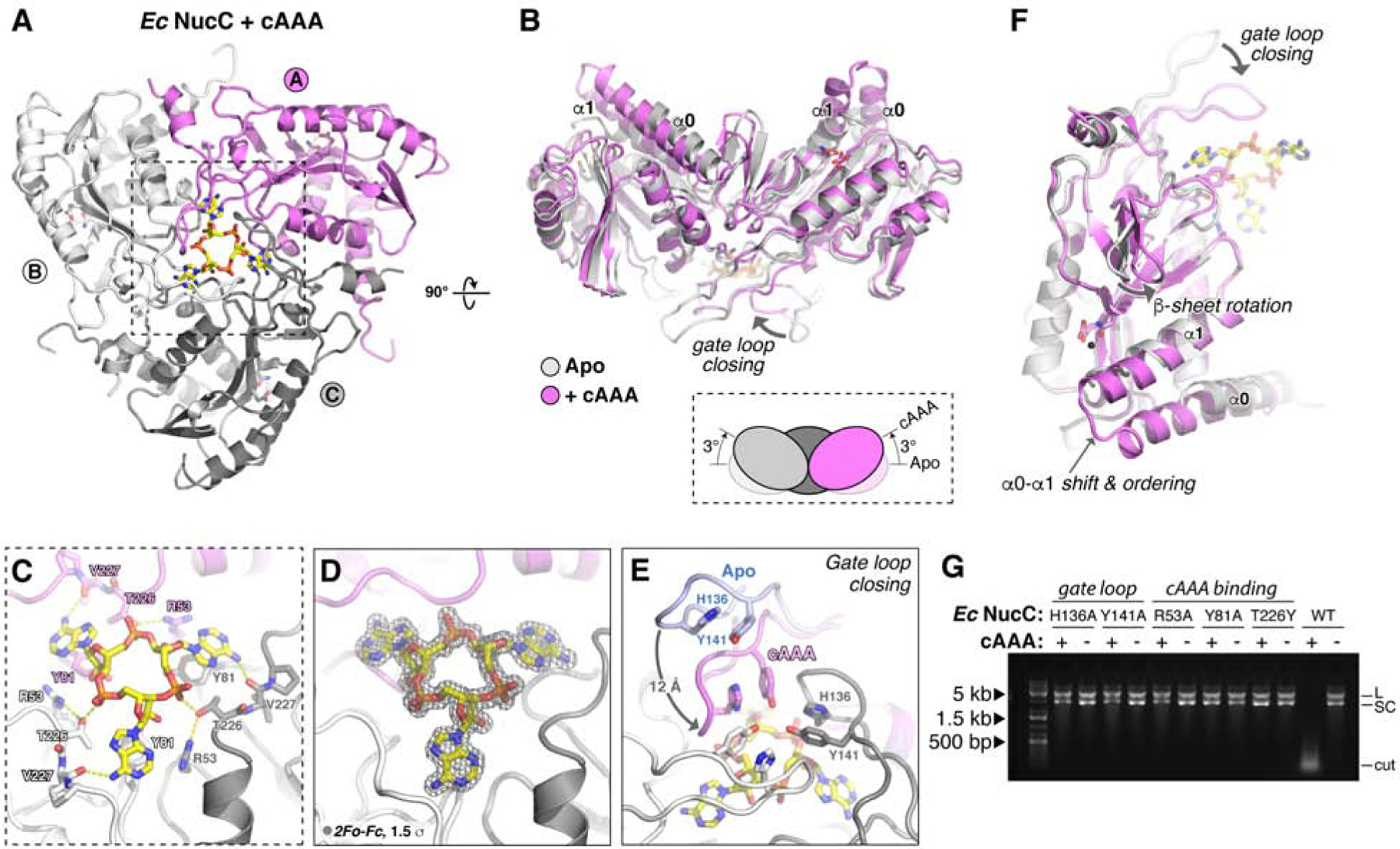
(A) Bottom view of an Ec NucC trimer bound to cAAA, with subunits colored violet/gray/white as in Figure 2B. See Figure S3 for synthesis, purification, and mass spectrometry analysis of cAAA, and Figure S4 for isothermal titration calorimetry measurements of Ec NucC binding second messengers. (B) Overlay of the NucC trimer in the Apo state (gray) and bound to cAAA (violet), showing the 3° upward rotation of each subunit induced by cAAA binding. (C) Overlay of the NucC monomer in the Apo state (gray) and bound to cAAA (violet), showing the major conformational differences within each subunit: gate loop closing, shifting and ordering of the α0-α1 region, and rotation of the small β-sheet. (D) Closeup view of cAAA binding NucC. (E) 2Fo-Fc electron density for cAAA at 1.66 Å resolution, contoured at 1.5 σ. (F) Closeup view of gate loop closure upon cAAA binding. For one monomer, the gate loop is shown in both open (Apo, blue) and closed (cAAA-bound, violet) conformations, showing the ~12 Å motion between the two states. Pi stacking of “latch” residues H136 and Y141 (labeled in gray monomer) stabilizes gate loop conformation in both states. See Figure S5 for structures of Pa NucC bound to cAAA, and of Ec NucC bound to 5′-pApA. (G) Plasmid digestion assay with wild-type and structure-based mutants of Ec NucC (10 nM) in the presence and absence of cAAA (100 nM).
cAAA-mediated NucC hexamerization activates double-strand DNA cleavage
cAAA binding causes several conformational changes within each NucC protomer, including closing of the gate loops, a shift in the position of α-helices 0 and 1, and rotation of a small β-sheet encompassing NucC residues 85–95 (Figure 3B, F). Within the NucC trimer, each subunit undergoes a rotation of ~3° “upward” away from the cAAA binding site compared to the unbound structure (Figure 3B). In the structures of both Ec NucC and Pa NucC bound to cAAA, we also observe that pairs of NucC trimers bind face-to-face to form symmetric homohexamers (Figure 4A–C and Figure S5A). The trimer-trimer interface involves a symmetric interaction of the α0 helices of facing NucC protomers, plus a hydrophobic cluster assembled from three different NucC protomers. In the α0 interface, Leu19 from two NucC protomers forms a symmetric hydrophobic interaction across the interface, and Glu15 of each NucC protomer forms a hydrogen bond with Lys26 of the opposite protomer (Figure 4B). In the hydrophobic cluster, Phe28 from one NucC protomer docks into a hydrophobic pocket on the opposite trimer, which involves Trp4 from one NucC protomer and Phe60, Phe68, and Phe86 of another (Figure 4C). The docking of Phe28 into this hydrophobic pocket causes residues 23–34 linking α0 and α1, which are disordered in our NucC Apo structure, to become well-ordered in the cAAA-bound structure. Our structure of Pa NucC bound to cAAA shows equivalent association of two NucC trimers into a hexamer (Figure S5A).
Figure 4. cAAA-stimulated NucC hexamerization is required for DNA cleavage activity and bacteriophage λ immunity.

(A) Cartoon view of two cAAA-bound NucC trimers (colored violet/gray/white and light blue, respectively) assembled into a homohexamer. Bound cAAA molecules are shown in yellow sticks. (B) Closeup view of the NucC α0-α0 interface mediating hexamer formation. (C) Closeup view of the NucC hydrophobic cluster interface mediating hexamer formation. (D) Size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) for Ec NucC in the absence of cAAA (gray line with molecular weight measurement in cyan) and in the presence of cAAA (black line with molecular weight measurement in violet). The molar mass of an Ec NucC homotrimer is 80.1 kDa, and a homohexamer is 160.3 kDa. See Figure S5G for SEC-MALS of Pa NucC in the presence and absence of cAAA. (E) SEC-MALS analysis of Ec NucC L19D mutant, which remains trimeric in the presence of cAAA. See Figure S6A–F for SEC-MALS analysis of other Ec NucC mutants. (F) Plasmid digestion assay with wild-type and F28A mutant Ec NucC (10 nM), in the presence of cAAA (for each set: 400, 100, 25, 6.25, and 0 nM cAAA). “L” denotes linear plasmid, “SC” denotes closed-circular supercoiled plasmid, and “cut” denotes fully-digested DNA. See Figure S6G for plasmid digestion data on other Ec NucC mutants. (G) Dilutions of bacteriophage λ on lawns of E. coli MS115–1 (top) and JP313 (wild-type laboratory strain) with the plasmid-encoded E. coli MS115–1 CBASS system encoding wild-type proteins or the indicated mutations. EV: empty vector. Six 10-fold bacteriophage dilutions are shown. (H) Summary of SEC-MALS, in vitro DNA cleavage activity, and bacteriophage λ immunity data for Ec NucC hexamer interface mutants. Shaded in gray are mutants that strongly disrupt both hexamer formation and DNA cleavage; NucC F28A further disrupts bacteriophage λ immunity. N/D: not determined.
We tested for NucC hexamer assembly in solution by SEC-MALS, and found that both Ec NucC and Pa NucC form hexamers when pre-incubated with cAAA (Figure 4D and Figure S5G). To test whether hexamer assembly and nuclease activation are functionally linked, we designed specific mutations in Ec NucC to disrupt hexamer assembly. We mutated Leu19 in the α0 helix to alanine or aspartate, and mutated Trp4 and Phe28 in the hydrophobic cluster to alanine. We also mutated Ala27, which borders the hydrophobic cluster and is tightly packed in the trimer-trimer interface, to either glutamate or lysine in order to disrupt this interface. We purified six mutant proteins and tested both their ability to undergo cAAA-mediated hexamerization (Figure 4E and Figure S6A–F) and their cAAA-stimulated DNA cleavage activity (Figure 4F and Figure S6G). Four of the six mutants (L19D, A27E, A27K, and F28A) strongly disrupt both hexamer formation and DNA cleavage activity (Figure 4H). The W4A mutant partially disrupts hexamer assembly but retains robust cAAA-activated DNA cleavage activity, while the L19A mutant does not affect either hexamer assembly or DNA cleavage (Figure 4G). Finally, we found by plaque assay that the NucC F28A mutation, which eliminates hexamer formation and DNA cleavage activity, also completely eliminates the protective effect of the CBASS system in cells (Figure 4G). Overall, these data strongly indicate that the active form of NucC is a hexamer whose assembly is promoted by cAAA binding.
To better understand why hexamer formation activates NucC, we modelled DNA binding to the NucC hexamer by overlaying each NucC monomer with a structure of DNA-bound SgrAI (Dunten et al., 2008). Hexamer formation juxtaposes pairs of NucC active sites from opposite trimers ~25 Å apart (Figure 5A), and we could model a continuous bent DNA duplex that contacts a pair of juxtaposed NucC active sites (Figure 5B). In this model, the putative scissile phosphate groups are positioned two base pairs apart on opposite DNA strands (Figure 5B), such that coordinated cleavage at these two sites would yield a double-strand break with a two-base 3′ overhang. To determine whether NucC generates single- or double-strand breaks, we used limiting Mg2+ conditions to enrich for singly-cut plasmid in a plasmid digestion assay (Figure 5C). While NucC did generate a nicked species in this assay, we also observed a significant fraction of linear DNA that could only arise through cleavage of both DNA strands at a single site. These data support the idea that hexamerization of NucC activates double-strand DNA cleavage through juxtaposition of pairs of active sites.
Figure 5. NucC generates double-strand cuts with two-base 3′ overhangs.
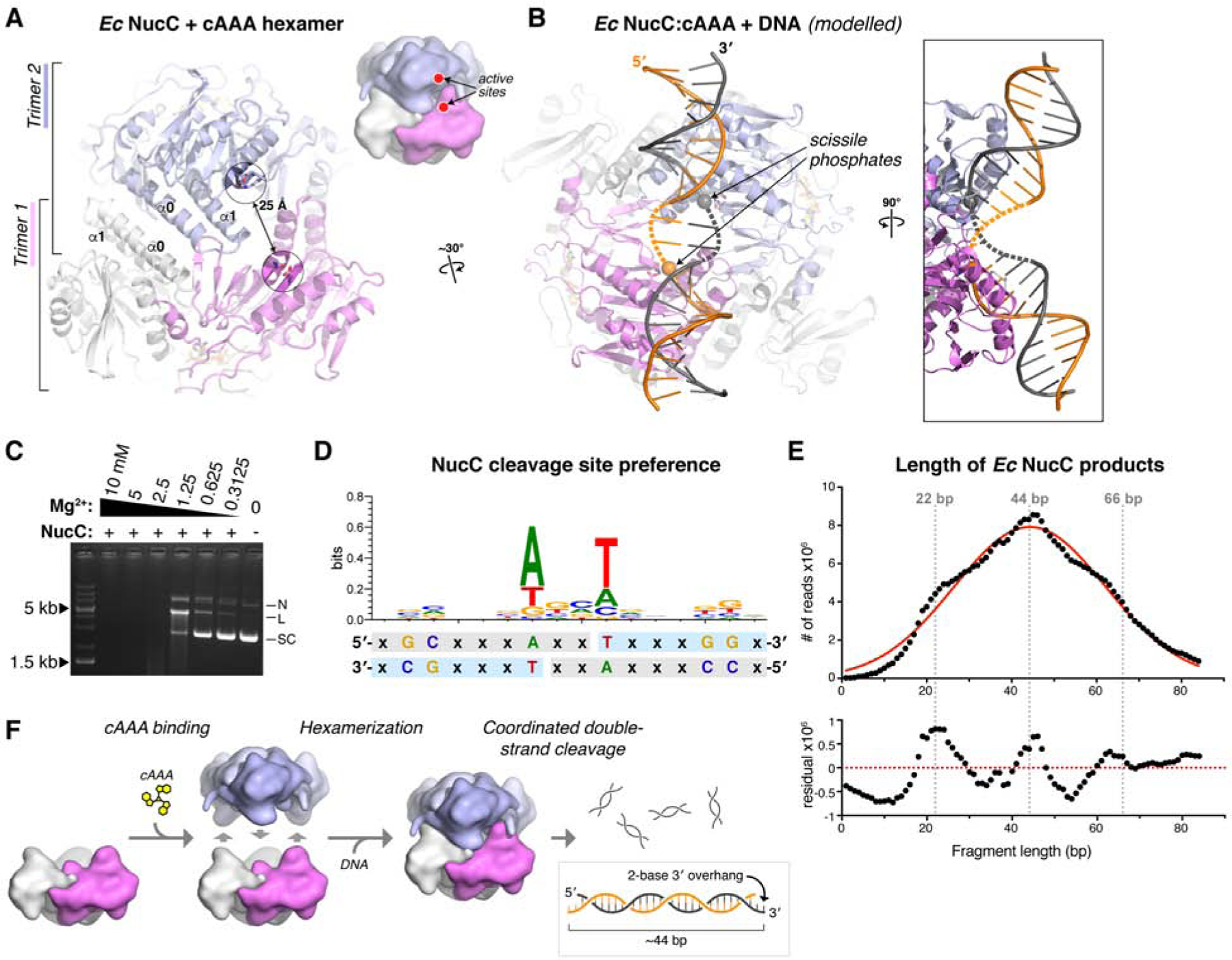
(A) View of the cAAA-bound Ec NucC hexamer, showing the juxtaposition of two active sites ~25 Å apart. (B) Model of a NucC hexamer bound to DNA, assembled from separately overlaying the NucC monomers colored violet and blue with a structure of SgrAI bound to DNA (PDB ID 3DW9; (Dunten et al., 2008)). The two modelled DNAs can be joined by a bent-two base-pair linker (dotted lines) to generate a contiguous dsDNA (two strands colored gray and orange). Cleavage of the scissile phosphates (spheres) at each active site would generate a double-strand break with a two-base 3′ overhang. (C) Plasmid cleavage by Ec NucC in limiting Mg2+. Supercoiled DNA (SC) is converted to both nicked (N) and linear (L) forms, demonstrating significant double-strand cleavage. (D) Analysis of NucC DNA cleavage site preference, from mapping ~600,000 cleavage sites from fully-digested plasmid DNA. Light-blue indicates the sequenced strand; the two-fold symmetry of the cleavage site supports a model of double-strand cleavage resulting in two-base 3′ overhangs. (E) Length distribution of Ec NucC products after complete digestion, using high-throughput sequencing read lengths from a 343 million-read dataset. Red line shows a Gaussian fit, and bottom panel shows residual values from the Gaussian fit. (F) Model for hexamerization and coordinated double-strand cleavage by NucC. Inactive NucC trimers associate upon binding cAAA to form active hexamers. Coordinated cleavage at pairs of NucC active sites juxtaposed by hexamerization results in double-strand breaks with two-base 3′ overhangs.
To gain further information about NucC sequence-specificity and cleavage products, we next completely digested a plasmid with NucC and deep-sequenced the resulting short fragments. To avoid losing information about DNA ends by “blunting” during sequencing library preparation, we denatured the fragments and sequenced individual DNA strands (see STAR Methods). We thereby mapped ~1 million cleavage events, enabling us to assemble a picture of any preferred sequence motifs at NucC cleavage sites. We observed a striking two-fold symmetry in the preferred cleavage sites, with a strong preference for “T” at the 5′ end (+1 base) of the cleaved fragment, and an equally strong preference for “A” at the −3 posiUon (Figure 5D). Similarly, we observed a weak preference for “G” at the +5 and +6 bases, and for “C” at the −7 and −8 bases. The two-fold symmetry of preferred NucC cleavage sites strongly suggests that two NucC active sites cooperate to cleave both strands of DNA. Moreover, the location of the breaks on each strand (as judged by the first sequenced base of the fragments) suggests that double-stranded cleavage by NucC results in two-base 3′ overhangs, consistent with our structural modeling (Figure 5D and Figure 5F).
We used the same sequencing dataset to precisely measure the length distribution of NucC products. NucC product sizes followed a roughly Gaussian distribution with a mean of ~44 bases, in agreement with our electrophoretic results (Figure 5E). Curiously, the size distribution showed a systematic variation from a Gaussian distribution, with enrichment of product lengths around 22, 44, and 66 base pairs in length. These data suggest that in addition to coordinated DNA cleavage by pairs of NucC active sites, cleavage by the three pairs of active sites in a NucC hexamer may be weakly coordinated, perhaps through wrapping of a single DNA around the complex to contact multiple sets of active sites.
Type III CRISPR systems encode NucC homologs
While exploring the distribution of NucC homologs in bacteria, we identified a group of NucC homologs with ~50% sequence identity to Ec NucC and Pa NucC, that are encoded not within CBASS systems but rather within Type III CRISPR/Cas systems (Figure 6A–C). Prompted by this finding, we comprehensively searched the gene neighborhoods of bacterial NucC homologs and identified a total of 31 distinct NucC proteins encoded within Type III CRISPR/Cas systems (Figure 6A, Table S4). These proteins are found predominantly in Gammaproteobacteria and Firmicutes, but are also found in Alpha-, Beta-, and Deltaproteobacteria (Table S4). Their distribution within a phylogenetic tree of more than 500 predominantly CBASS-associated NucC proteins suggests that NucC was independently incorporated into Type III CRISPR/Cas systems at least ten times (Figure 6A, Table S5). Supporting this finding, a recent bioinformatics analysis of protein families associated with Type III CRISPR/Cas systems identified a group of putative nucleases (denoted PD-DExK after the conserved active-site motif) that includes members of NucC and a second, likely unrelated nuclease family (Table S4) (Shah et al., 2019).
Figure 6. NucC is an accessory nuclease in Type III CRISPR systems.

(A) Phylogenetic tree of 531 bacterial NucC proteins, with 31 NucC homologs encoded within Type III CRISPR/Cas systems highlighted in orange. Numbers denote clusters of Type III CRISPR-associated NucC homologs, whose distribution within the larger NucC tree suggests that each cluster represents an independent integration of NucC into a Type III CRISPR system. See Table S4 for a complete list of Type III CRISPR-associated NucC homologs, and Table S5 for the complete list of proteins used in tree construction. (B) Schematic of Type III CRISPR effector complexes (adapted from (Makarova et al., 2017)). The large subunit (Cmr2/Cas10; green) produces cyclic oligoadenylates that in some type III systems activate accessory RNases (Csm6 or Csx1) or DNases (Can1). (C) Schematics of representative Type III CRISPR operons from each cluster shown in panel (A). Core Type III genes are colored as in panel (B), with NucC homologs magenta. CRISPR arrays are denoted with red lines, predicted adaptation module genes are shown in cyan, non-core or unassigned genes are shown in gray, and two predicted SAVED domain-containing proteins (Burroughs et al., 2015) are shown in white. RT: predicted reverse transcriptase. The operon for Acetivibrio cellulolyticus CD2 was poorly annotated, and the gene names shown are based on the closest match from BLAST searches.
Type III CRISPR/Cas systems encode a multi-subunit effector complex, that when activated by target binding synthesizes a range of cyclic oligoadenylate second messengers with 2–6 bases (cA2-cA6), often with a high proportion of cAAA/cA3 (Grüschow et al., 2019; Kazlauskiene et al., 2017; Niewoehner et al., 2017). These cyclic oligoadenylate second messengers have been shown to activate the accessory RNases Csm6 and Csx1 in the CRISPR operons (Han et al., 2018; Kazlauskiene et al., 2017; Niewoehner et al., 2017). More recently, a cA4-activated DNA endonuclease was identified in Type III CRISPR/Cas systems, which generates single-stranded nicks thought to slow viral replication by causing replication fork collapse (Mcmahon et al., 2019). The presence of NucC homologs in Type III CRISPR/Cas systems suggests that these enzymes may function similarly to their relatives in CBASS systems, with activation dependent on cAAA synthesized by the CRISPR effector complexes. We cloned several Type III CRISPR-associated NucC proteins, and successfully purified one such enzyme from Vibrio metoecus, a close relative of V. cholerae (Kirchberger et al., 2014). We found that V. metoecus (Vm) NucC is a DNA endonuclease that is strongly activated by cAAA, and weakly activated by both cAA and cAAG, mirroring our findings with Ec NucC (Figure 7A–B) (Ye et al., 2019). By SEC-MALS, we found that Vm NucC forms a mix of trimers and hexamers on its own, and forms solely hexamers after pre-incubation with cAAA (Figure 7C).
Figure 7. Type III CRISPR-associated NucC from V. metoecus is a cAAA-activated endonuclease.

(A) Schematic of the Type III CRISPR operon from V. metoecus sp. RC341. This operon’s NucC shares 53% overall sequence identity with the CBASS-associated NucC from E. coli MS115–1 (B) Plasmid digestion assay with V. metoecus NucC in the presence of nucleotide-based second messengers. (C) Two views of of the V. metoecus NucC apo-state trimer. The Vm NucC trimer shows an overall r.m.s.d. of 1.48 Å (over 607 equivalent Cα atoms) with the Ec NucC apo-state trimer. See Figure S7A for a structural comparison with Ec NucC, and Figure S7B for a close-up view of the Vm NucC active site. (D) SEC-MALS of V. metoecus NucC in either the Apo state (gray/cyan) or after pre-incubation with cAAA (black/magenta). Calculated MW of the NucC homotrimer = 84.1 kDa; homohexamer = 168.1 kDa. (E) Structure of the Vm NucC hexamer with trimer 1 colored as in panel (A), and trimer 2 in blue. One pair of active sites that become juxtaposed after hexamer formation are highlighted with dotted circles.
We next determined crystal structures of Vm NucC in both the trimer and hexamer states. In the absence of cAAA, Vm NucC crystallizes as a homotrimer with an overall structure equivalent to the Ec NucC apo-state trimer (Figure 7D). Vm NucC shares a common active-site structure with Ec NucC (Figure S7A–B), and the Vm NucC trimer overlays with the Ec NucC trimer with an overall Cα r.m.s.d. of 1.48 Å (over 607 equivalent atoms). In the presence of cAAA, Vm NucC crystallizes as a homohexamer, with an overall structure equivalent to the hexamer structures of both Ec NucC and Pa NucC (Figure 7E). Vm NucC shares both the symmetric α0-α0 interface and hydrophobic cluster interactions that mediate trimer-trimer association in Ec NucC. Curiously, while Vm NucC required cAAA to crystallize in the hexamer form, we observed no electron density for cAAA in the allosteric pocket despite 100% conservation of cAAA-binding residues with Ec NucC and Pa NucC. Inspection of crystal packing interactions in these crystals revealed a symmetric interaction between the gate loops of neighboring hexamers that pulls the gate loops into an open conformation (Figure S7C–D). We propose that this open gate-loop conformation allows bound cAAA to diffuse out of the allosteric pocket after crystallization, resulting in an empty binding site. Nonetheless, we could model cAAA binding to Vm NucC without clashes, indicating that the Vm NucC trimer can accommodate cAAA.
Together, our biochemical and structural data on Vm NucC indicate that CRISPR-associated NucC homologs share a common activation and DNA cleavage mechanism with their relatives in CBASS systems. Thus, NucC homologs have been incorporated into Type III CRISPR/Cas systems, where they are likely activated upon target recognition and cyclic oligoadenylate synthesis by these systems’ effector complexes.
Discussion
Bacteria possess an extraordinary variety of pathways to defend themselves against foreign threats including bacteriophages, invasive DNA elements, and other bacteria. A large class of defense pathways, including restriction-modification systems and CRISPR-Cas systems, specifically defend the bacterial genome through the targeted action of DNA and RNA nucleases. Here, we describe the structure and activation mechanism of a restriction endonuclease-like protein, NucC, found primarily in the CBASS bacteriophage defense system. The intact NucC-containing CBASS system from the patient-derived E. coli strain MS115–1 confers immunity to bacteriophage λ infection, and this immunity requires both cAAA synthesis by this operon’s CD-NTase and the DNA cleavage activity of NucC (Ye et al., 2019). Moreover, the E. coli MS115–1 CBASS system functions via abortive infection, in which infected cells die prior to completion of the phage replication cycle (Ye et al., 2019). Here, we show that death of infected cells is caused by NucC-mediated destruction of the bacterial chromosome. Thus, NucC is part of a widespread bacterial defense pathway that mediates cell death in response to bacteriophage infection.
While evolutionarily related to restriction endonucleases, NucC adopts a unique homotrimeric structure with an allosteric binding pocket for its cyclic trinucleotide activator. This pocket is assembled from embellishments to the core nuclease fold, which include a “gate loop” that closes over the bound second messenger. cAAA binding mediates a subtle conformational change within the NucC trimer, enabling the symmetric association of two NucC trimers into a hexamer and juxtaposing pairs of active sites to activate double-strand DNA cleavage. This exquisitely specific activation mechanism likely ensures that NucC is kept inactive in cells prior to an infection. Once activated by cAAA binding, the nuclease possesses only very weak sequence specificity and in vitro, cleaves DNA to an average length of ~50 base pairs.
Bacterial CBASS systems are widespread among environmental and pathogenic bacteria, but are sparsely distributed and often associated with transposase or integrase genes and/or conjugation systems, suggesting that they are mobile DNA elements that confer some selective advantage on their bacterial host (Burroughs et al., 2015; Whiteley et al., 2019). These operons encode diverse putative effector proteins including proteases, exonucleases, and two families of endonucleases (Burroughs et al., 2015). Given the structure of these operons, it is likely that each effector is specifically activated by the second messenger synthesized by its associated CD-NTase (Whiteley et al., 2019). Bacterial CD-NTases have been classified into eight major clades, and while members of several clades have been shown to synthesize specific cyclic di- and trinucleotides (Whiteley et al., 2019), the full spectrum of products synthesized by these enzymes remains an important open question. Given our data on NucC structure and activation, we propose that all bacterial CD-NTases associated with NucC effectors – which includes members of clades C01 (including E. coli MS115–1 CdnC), C03, and D05 (including P. aeruginosa ATCC27853 CdnD) (Whiteley et al., 2019) – likely synthesize cAAA. While CD-NTases in clade C03 are primarily associated with NucC effectors, clade C01 and D05 CD-NTases are associated with a range of effectors (Whiteley et al., 2019). One key question is whether all of these effectors are activated by cAAA, or whether their associated CD-NTases synthesize distinct second messengers. More broadly, a full accounting of bacterial CD-NTase products, and of the effector proteins they activate, remains an important area for future study.
Type III CRISPR/Cas systems uniquely encode nonspecific accessory RNases (Csm6 and Csx1) and DNases (Can1) that are activated by cyclic tetra- and hexa-adenylates synthesized by their effector complexes (Han et al., 2018; Kazlauskiene et al., 2017; Mcmahon et al., 2019; Niewoehner et al., 2017). The accessory RNases aid immunity by non-specifically degrading both host and invader transcripts, causing growth arrest in an infected cell (Rostøl and Marraffini, 2019), while Can1 is thought to slow viral DNA replication (Mcmahon et al., 2019). We identified a set of Type III CRISPR/Cas systems that encode clear homologs of NucC, with their distribution suggesting that NucC has been independently incorporated into these systems at least ten times. We show that one such NucC, from Vibrio metoecus, is a cAAA-stimulated double-stranded DNA endonuclease, demonstrating a common mechanism of action with CBASS-associated NucC. Notably, NucC is the first reported Type III CRISPR effector nuclease to be activated by cAAA, a major second messenger product of many Type III CRISPR effector complexes. The biological role of NucC in Type III CRISPR systems remains unclear. It is possible that incorporation of NucC into a CRISPR system converts this system from a specific defensive pathway into an abortive infection system; indeed, recent reports have shown that certain type IF and type VI CRISPR systems can act via abortive infection (Meeske et al., 2019; Watson et al., 2019). Alternatively, NucC’s cleavage activity may be spatially or temporally restrained in these systems to limit its potential to destroy the host genome. Discriminating between these models will require direct tests with NucC-encoding Type III CRISPR/Cas systems.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kevin D. Corbett (kcorbett@ucsd.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
All proteins were produced in E. coli strain Rosetta2 pLysS (EMD Millipore). Cells were grown in standard media (2XYT broth) with appropriate antibiotics, and standard temperatures (37°C for growth, 20°C for protein expression induction). For cellular assays with E. coli MS115–1 (BEI Resources #HM-344) and JP313 (Economou et al., 1995), cells were grown in LB broth at 30°C with appropriate antibiotics.
METHOD DETAILS
Bacterial Growth and Microscopy
pLAC22 vectors were constructed encoding either the full CBASS operon from E. coli strain MS115–1 or a variant encoding the NucC D73N mutant under the control of an IPTG-inducible promoter, as described (Ye et al., 2019). E. coli strain JP313 (a derivative of E. coli MC4100) (Economou et al., 1995) was transformed with these vectors and incubated in 5 mL LB broth with 0.1 mg/ml ampicillin in culture tubes at 30°C with rolling. OD at 600nm was measured after 3 hours and cells were diluted to an OD600 of 0.1 in a reservoir containing LB with 100ug/ml ampicillin and 0.2mM IPTG. Bacteriophage λcI- diluted in phage buffer with 1mM CaCl2 was added for a final multiplicity of infection of 2.5, then incubated at 30°C with rolling.
For fluorescence microscopy, samples of each culture were taken at 0, 60, 90, and 120 minutes post-infection for imaging by fluorescence microscopy. Harvested samples were stained with 1 μg/mL FM4–64 (Pogliano et al., 1999) and 2 μg/mL DAPI. Cultures were centrifuged at 3,300 × g for 30 s in a microcentrifuge and resuspended in 1/10 volume of the original cultures. Three microliters of concentrated cells were transferred onto an agarose pad containing 1.2% agarose and 20% LB medium for microscopy. Microscopy was performed using a Deltavision Elite System (GE Healthcare) and images were deconvolved with SoftWoRx software. For each condition, at least five fields from each of two independent experiments were manually scored (n=243–1385 cells for each condition)
Protein Expression, Purification, and Characterization
Full length NucC genes from E. coli MS115–1 (Genbank ID EFJ98159; synthesized by Invitrogen/Geneart), P. aeruginosa ATCC 27853 (Genbank ID WP_003050273; amplified from genomic DNA), Vibrio metoecus sp. RC341 (IMG ID 647193063; synthesized by Invitrogen/Geneart), and Gynuella sunshinyii YC6258 (IMG ID 2632356247; synthesized by Invitrogen/Geneart) were cloned into UC Berkeley Macrolab vector 1B (Addgene #29653) to generate N-terminal fusions to a TEV protease-cleavable His6-tag. Mutants of Ec NucC were generated by PCR mutagenesis.
Proteins were expressed in E. coli strain Rosetta2 pLysS (EMD Millipore) by induction with 0.25 mM IPTG at 20°C for 16 hours. For selenomethionine derivatization of Ec NucC, cells were grown in M9 minimal medium, then supplemented with amino acids (Leu, Ile and Val (50 mg/L), Phe, Lys, Thr (100 mg/L) and Selenomethionine (60 mg/L)) upon induction with IPTG.
For protein purification, cells were harvested by centrifugation, suspended in resuspension buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 10 mM imidazole, 1 mM dithiothreitol (DTT) and 10% glycerol) and lysed by sonication. Lysates were clarified by centrifugation (16,000 rpm 30 min), then supernatant was loaded onto a Ni2+ affinity column (HisTrap HP, GE Life Sciences) pre-equilibrated with resuspension buffer. The column was washed with buffer containing 20 mM imidazole and 100 mM NaCl, and eluted with a buffer containing 250 mM imidazole and 100 mM NaCl. The elution was loaded onto an anion-exchange column (Hitrap Q HP, GE Life Sciences) and eluted using a 100–600 mM NaCl gradient. Fractions containing the protein were pooled and mixed with TEV protease (1:20 protease:NucC by weight) plus an additional 100 mM NaCl, then incubated 16 hours at 4°C for tag cleavage. Cleavage reactions were passed over a Ni2+ affinity column, and the flow-through containing cleaved protein was collected and concentrated to 2 mL by ultrafiltration (Amicon Ultra-15, EMD Millipore), then passed over a size exclusion column (HiLoad Superdex 200 PG, GE Life Sciences) in a buffer containing 20 mM Tris-HCl pH 8.0, 300 mM NaCl, and 1 mM DTT. Purified proteins were concentrated by ultrafiltration and stored at 4°C for crystallization, or aliquoted and frozen at −80°C for biochemical assays. All mutant proteins were purified as wild-type.
For characterization of oligomeric state by size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS), 100 μL of purified protein at 2–5 mg/mL was injected onto a Superdex 200 Increase 10/300 GL column (GE Life Sciences) in a buffer containing 20 mM HEPES-NaOH pH 7.5, 300 mM NaCl, 5% glycerol, and 1 mM DTT. For samples with second messengers, protein was pre-incubated with 0.1 mM second messenger for 16 hours at 4°C prior to analysis. Light scattering and refractive index profiles were collected by miniDAWN TREOS and Optilab T-rEX detectors (Wyatt Technology), respectively, and molecular weight was calculated using ASTRA v. 6 software (Wyatt Technology). SEC-MALS analysis of all Ec NucC mutants (Figure 3 and Figure S5) was performed with protein also containing the D73N active-site mutation.
Second messenger molecules
Cyclic and linear dinucleotides, including 5′-pApA, 3′,3′-cGAMP, and 3′,3′-cyclic di-AMP, were purchased from Invivogen. Cyclic trinucleotides were generated enzymatically by Ec-CdnD02 from Enterobacter cloacae, which was cloned and purified as described (Whiteley et al., 2019). Briefly, a 40 mL synthesis reaction contained 500 nM Ec-CdnD02 and 0.25 mM each ATP and GTP (ATP alone for large-scale synthesis of cAAA) in reaction buffer with 12.5 mM NaCl, 20 mM MgCl2, 1 mM DTT, and 10 mM Tris-HCl pH 9.0. The reaction was incubated at 37°C for 16 hours, then 2.5 units/mL reaction volume (100 units for 40 mL reaction) calf intestinal phosphatase was added and the reaction incubated a further 4 hours at 37°C. The reaction was heated to 65°C for 30 minutes, and centrifuged 20 minutes at 4,000 RPM to remove precipitated protein. Reaction products were separated by ion-exchange chromatography (Mono-Q, GE Life Sciences) using a gradient from 0 to 2 M ammonium acetate. Three major product peaks (Figure S2A) were pooled, evaporated using a speedvac, then resuspended in water and analyzed by LC MS/MS. cAAA synthesized by the E. coli MS115–1 CdnC:HORMA complex was similarly purified and verified to activate NucC equivalently to that synthesized by Ec-CdnD02 (Ye et al., 2019).
Mass spectrometry
Whiteley et al. previously identified the major product of Ec-CdnD02 as cAAG by NMR (Whiteley et al., 2019). To further characterize the products of Ec-CdnD02, we performed liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure S2B–D). LC-MS/MS analysis was performed using a Thermo Scientific Vanquish UHPLC coupled to a Thermo Scientific Q Exactive™ HF Hybrid Quadrupole-Orbitrap™ Mass Spectrometer, utilizing a ZIC-pHILIC polymeric column (100 mm × 2.1 mm, 5 μm) (EMD Millipore) maintained at 45 °C and flowing at 0.4 mL/min. Separation of cyclic trinucleotide isolates was achieved by injecting 2 μL of prepared sample onto the column and eluting using the following linear gradient: (A) 20 mM ammonium bicarbonate in water, pH 9.6, and (B) acetonitrile; 90% B for 0.25 minutes, followed by a linear gradient to 55% B at 4 minutes, sustained until 6 minutes. The column was re-equilibrated for 2.50 minutes at 90% B.
Detection was performed in positive ionization mode using a heated electrospray ionization source (HESI) under the following parameters: spray voltage of 3.5 kV; sheath and auxiliary gas flow rate of 40 and 20 arbitrary units, respectively; sweep gas flow rate of 2 arbitrary units; capillary temperature of 275 °C; auxiliary gas heater temperature of 350°C. Profile MS1 spectra were acquired with the following settings; mass resolution of 35,000, AGC volume of 1×106, maximum IT of 75 ms, with a scan range of 475 to 1050 m/z to include z=1 and z=2 ions of cyclic trinucleotides. Data dependent MS/MS spectra acquisition was performed using collision-induced dissociation (CID) with the following settings: mass resolution of 17,500; AGC volume of 1×105; maximum IT of 50 ms; a loop count of 5; isolation window of 1.0 m/z; normalized collision energy of 25 eV; dynamic exclusion was not used. Data reported is for the z=1 acquisition for each indicated cyclic trinucleotide.
Isothermal Titration Calorimetry
Isothermal titration calorimetry was performed the Sanford Burnham Prebys Medical Discovery Institute Protein Analysis Core. Measurements were performed at 20°C on a Microcal ITC 200 (Malvern Panalytical) in a buffer containing 20 mM Tris-HCl (pH 8.5), 200 mM NaCl, 5 mM MgCl2, 1 mM EDTA, and 1 mM tris(2-carboxyethyl)phosphine (TCEP). Nucleotides at 1 mM were injected into an analysis cell containing 100 μM Ec NucC (Figure S3A–G, Table S2). A second round of ITC was performed with 5′pApA and 3′,3′ c-di-AMP in a buffer containing 10 mM Tris-HCl (pH 7.5), 25 mM NaCl, 10 mM MgCl2, and 1 mM TCEP (Figure S3H–K, Table S3).
Crystallization and structure determination
E. coli NucC
We obtained crystals of E. coli NucC in the Apo state in hanging drop format by mixing protein (8–10 mg/mL) in crystallization buffer (25 mM Tris-HCl pH 7.5, 200 mM NaCl, 5 mM MgCl2, 1 mM TCEP) 1:1 with well solution containing 27% PEG 400, 400 mM MgCl2, and 100 mM HEPES pH 7.5 (plus 1 mM TCEP for selenomethionine-derivatized protein). For cryoprotection, we supplemented PEG 400 to 30%, then flash-froze crystals in liquid nitrogen and collected diffraction data on beamline 14–1 at the Stanford Synchrotron Radiation Lightsource (support statement below). We processed all datasets with the SSRL autoxds script, which uses XDS (Kabsch, 2010) for data indexing and reduction, AIMLESS (Evans and Murshudov, 2013) for scaling, and TRUNCATE (Evans, 2006) for conversion to structure factors. We determined the structure by single-wavelength anomalous diffraction methods with a 1.82 Å resolution dataset from selenomethione-derivatized protein. We identified heavy-atom sites using hkl2map (Pape et al., 2004) (implementing SHELXC and SHELXD (Sheldrick, 2010)), then provided those sites to the PHENIX Autosol wizard (Terwilliger et al., 2009), which uses PHASER (McCoy et al., 2007) for phase calculation and RESOLVE for density modification (Terwilliger, 2003a) and automated model building (Terwilliger, 2003b). We manually rebuilt the model in COOT (Emsley et al., 2010), followed by refinement in phenix.refine (Afonine et al., 2012) using positional, individual B-factor, and TLS refinement (statistics in Table S1).
We obtained crystals of E. coli NucC bound to 5′-pApA or cAAA in hanging drop format by mixing protein (8–10 mg/mL) in crystallization buffer plus 0.1 mM 5′-pApA (Invivogen) or cAAA 1:1 with well solution containing 17–24% PEG 3350, 0.1 M Na/K tartrate, and 25 mM Tris-HCl pH 8.0. For cryoprotection, we added 10% glycerol, then flash-froze crystals in liquid nitrogen and collected diffraction data on beamline 24ID-E at the Advanced Photon Source at Argonne National Lab (support statement below). We processed datasets with the RAPD data-processing pipeline, which uses XDS, AIMLESS, and TRUNCATE as above. We determined the structures by molecular replacement using the structure of Apo NucC, manually rebuilt the model in COOT, and refined in phenix.refine using positional, individual B-factor, and TLS refinement (statistics in Table S1). For both 5′-pApA and cAAA, ligands were fully built and assigned an occupancy of 0.33 to account for their location on a three-fold crystallographic rotation axis. The ligands were assigned a “custom-nonbonded-symmetry-exclusion” in phenix.refine to allow symmetry overlaps during refinement.
For analysis of Ec NucC trimer and hexamer interfaces (Figure S2E), we used the PISA structure-analysis server (Krissinel and Henrick, 2007).
P. aeruginosa NucC
We obtained crystals of P. aeruginosa ATCC27853 NucC D73N mutant bound to cAAA by mixing protein (12 mg/mL) in crystallization buffer plus 0.1 mM cAAA 1:1 with well solution containing 0.1 M HEPES pH 7.5, 0.1 M L-proline, and 10% PEG 3350. Crystals were cryoprotected with 20% glycerol, flash-frozen in liquid nitrogen, and a 1.45 Å diffraction dataset was collected at beamline 8.3.1 at the Advanced Light Source at Lawrence Berkeley National Lab. We used XDS (Kabsch, 2010) for data indexing and reduction, scaling, and conversion to structure factors, and determined the structure by molecular replacement in PHASER using the structure of cAAA-bound Ec NucC as a search model. We manually rebuilt the model in COOT, and refined in phenix.refine using positional, individual B-factor, and TLS refinement (statistics in Table S1).
V. metoecus NucC
For Vm NucC hexamer crystals, Vm NucC (20 mg/mL) in crystallization buffer plus 0.1 mM cAAA was mixed with well solution containing 0.1 M HEPES pH 7.5, 0.1 M sodium formate, and 14% PEG 3350 in hanging-drop format. Crystals were cryoprotected with the addition of 20% glycerol and flash-frozen in liquid nitrogen. Diffraction data were collected to 1.65 Å resolution at the Advanced Light Source (Berkeley, CA) beamline 8.3.1, and processed in space group P6322 by XDS. Molecular replacement was performed with PHASER (McCoy et al., 2007) using the structure of E. coli MS115–1 NucC. The model was manually rebuilt in COOT (Emsley et al., 2010), followed by refinement in phenix.refine (Afonine et al., 2012) using positional, individual B-factor, and TLS refinement (statistics in Table S1). For Vm NucC Apo trimer crystals, Vm NucC (20 mg/mL) in crystallization buffer (25 mM Tris-HCl, 0.2 M NaCl, 5 mM MgCl2, 1 mM Tris-carboxyethylphosphine (TCEP)) was mixed with well solution containing 0.1 M sodium acetate pH 4.5 and 1.8 M ammonium sulfate in hanging-drop format. Crystals were cryoprotected with the addition of 30% glycerol and flash-frozen in liquid nitrogen.
Diffraction data were collected to 2.15 Å resolution at the Advanced Light Source (Berkeley, CA) beamline 8.3.1, and processed in space group P41212 by XDS. Molecular replacement was performed with PHASER using a single chain from the Vm NucC Apo hexamer structure, then the model was rebuilt in COOT and refined in phenix.refine.
Synchrotron Support Statements
SSRL Support Statement
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
APS NE-CAT Support Statement
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
ALS Beamline 8.3.1 Support Statement
Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15–328599 the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc. and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02–05CH11231, Office of Basic Energy Sciences.
Nuclease assays
For all nuclease assays, a λ-BstEII ladder (New England Biolabs) or UC Berkeley Macrolab plasmid 2AT (Addgene #29665; 4,731 bp; sequence in Supplemental Information) was used. Ec NucC (10 nM unless otherwise indicated) and second messenger molecules (10 nM unless otherwise indicated) were mixed with 1 μg DNA in a buffer containing 10 mM Tris-HCl (pH 7.5), 25 mM NaCl, 10 mM MgCl2, and 1 mM DTT (50 μL reaction volume), incubated 10 min at 37°C, then separated on a 1.2% agarose gel. Gels were stained with ethidium bromide and imaged by UV illumination.
High-throughput sequencing and data analysis
For high-throughput analysis of NucC products, 1 μg of vector 2AT (see Methods S1 for sequence) was digested to completion by Ec NucC, separated by agarose gel, then the prominent ~50 bp band was removed from the gel and gel-purified (Qiagen Minelute). 80 ng of purified DNA was prepared for sequencing using the Swift Biosciences Accel-NGS 1S Plus DNA Library kit, then sequenced on an Illumina HiSeq 4000 (paired-end, 100 base reads), yielding ~402 million read pairs. The Accel-NGS 1S kit: (1) separates DNA fragments into single strands; (2) ligates the Adapter 1 oligo to the 3′ end of the single-stranded fragment using an “adaptase” step that adds ~8 bp of low-complexity sequence to the fragment before adapter ligation; (3) ligates the Adapter 2 oligo to the 5′ end of the original fragment. After sequencing, read 1 (R1) represents the 5′ end of the original fragment, and read 2 (r2) represents the 3′ end (preceded by ~ 8 bp of low-complexity sequence resulting from the adaptase step).
For NucC fragment length analysis, we took advantage of the fact that most NucC products are less than 100 base pairs in length. We identified the (reverse complement of the) 3′ end of Adapter 1 (full sequence: 5′ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT 3′) in R1, searching for the sequence “AGATCGGA”. We calculated the length of each NucC fragment as: (length of R1 prior to AGATCGGA) minus 8 (average length of adaptase-added low-complexity sequence). The graph in Figure 5E represents the lengths of 343 million NucC fragments.
For NucC cleavage site identification (Figure 5D), we truncated a 1-million read subset of R1 to 15 bases, then mapped to the 2AT plasmid sequence (with ends lengthened by 100 bases to account for circularity of the plasmid) using bowtie (Langmead et al., 2009) with the command: “bowtie -a -n 0 -m 3 -best -t”. Of the successfully-mapped reads (594,183 of 1 million, 59.4%), 288,609 reads mapped to the forward strand and 305,574 mapped to the reverse strand. We extracted 20 bp of sequence surrounding each mapped 5′ end (first base of R1), and input the resulting alignment of 594,183 sequences into WebLogo 3 (http://weblogo.threeplusone.com) (Crooks et al., 2004).
Identification of CRISPR-associated NucC homologs
To identify NucC homologs associated with Type III CRISPR/Cas systems, we first used the Integrated Microbial Genomes (IMG) database at the DOE Joint Genome Institute (https://img.jgi.doe.gov) to identify NucC homologs throughout bacteria. We used four diverged NucC proteins as search queries to assemble a group of 744 likely NucC proteins, each of which contained the conserved active-site motif IDx30EAK and the characteristic gate loop. We downloaded the genomic sequence +/−10 kb for each NucC homolog, then used NCBI Genome Workbench (https://www.ncbi.nlm.nih.gov/tools/gbench/) to perform a custom TBLASTN searches for proteins related to the Type III CRISPR protein Cmr2. We verified each hit by examining its gene neighborhood for a complete Type III CRISPR/Cas system. Verified hits were included in a set of 531 bacterial NucC proteins, aligned using MAFFT, then an average-distance tree (using the BLOSUM62 scoring matrix) was generated in Jalview (Waterhouse et al., 2009) and visualized in Dendroscope (Huson and Scornavacca, 2012). See Table S4 for the complete list of Type III CRISPR associated NucC proteins, and Table S5 for the complete list of bacterial NucC proteins used for the phylogenetic tree in Figure 6A.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of primary diffraction datasets was performed with standard measures including Rsym, Rmeas, and CC1/2 to determine data quality and resolution cutoffs. Statistical analysis of refined crystal structures used standard measures including R and Rfree factors, and geometry analysis by the MolProbity server (Chen et al., 2010).
DATA AND CODE AVAILABILITY
All primary diffraction datasets are available at the SBGrid Data Bank (https://data.sbgrid.org) under the accession numbers noted in Table S1 and the Key Resources Table. All reduced diffraction datasets and refined molecular coordinates are available at the RCSB Protein Data Bank (https://www.rcsb.org) under the accession numbers noted in Table S1 and the Key Resources Table. The original imaging data for Figure 1 and Figure S1 is available at Mendeley Data (https://data.mendeley.com) under DOI 10.17632/dr8mjtwh2d.1. All high-throughput sequencing data used for Figure 5 is available from the Lead Contact without restriction.
Supplementary Material
Table S1. (Related to Figure 2, Figure 3, Figure 4, and Figure 7) Crystallographic data collection and refinement
See attached Microsoft Excel spreadsheet
Table S4. (Related to Figure 6) Type III CRISPR/Cas-associated NucC homologs
See attached Microsoft Excel spreadsheet
Table S5. (Related to Figure 6) NucC proteins used for sequence alignment
See attached Microsoft Excel spreadsheet
Highlights.
The CBASS-associated nuclease NucC kills phage-infected cells by genome destruction
Binding of a cyclic triadenylate second messenger causes NucC homohexamer assembly
NucC hexamer assembly juxtaposes pairs of active sites to activate DNA cleavage
NucC is found in both CBASS systems and Type III CRISPR/Cas systems
Acknowledgements
The authors thank the staffs of the Stanford Synchrotron Radiation Lightsource, the Advanced Photon Source NE-CAT beamlines, and the Advanced Light Source beamline 8.3.1 for assistance with crystallographic data collection; A. Bobkov (Sanford Burnham Prebys Medical Discovery Institute, Protein Analysis Core) for assistance with isothermal titration calorimetry; and A. Desai and M. Daugherty for critical reading and helpful suggestions. RKL was supported by the UC San Diego Quantitative and Integrative Biology training grant (NIH T32 GM127235). ITM was supported by NIH T32 HL007444, T32 GM007752 and F31 CA236405. JDW was supported by NIH K01 DK116917 and P30 DK063491. MJ was supported by NIH S10 OD020025 and R01 ES027595. ATW was supported as a fellow of The Jane Coffin Childs Memorial Fund for Medical Research. BL was supported as a Herchel Smith Graduate Research Fellow. JJM was supported by NIH/NIAID R01AI018045 and R01AI026289. PJK was supported from the Richard and Susan Smith Foundation and the Parker Institute for Cancer Immunotherapy. JP was supported by NIH R01 GM129245. KDC was supported by the Ludwig Institute for Cancer Research and the University of California, San Diego.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, et al. (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D, Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber W, and Linn S (1969). DNA Modification and Restriction. Annu. Rev. Biochem 38, 467–500. [DOI] [PubMed] [Google Scholar]
- Burroughs AM, Zhang D, Schäffer DE, Iyer LM, and Aravind L (2015). Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res. 43, gkv1267--10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, Doron S, Amitai G, and Sorek R (2019). Cyclic GMP-AMP signaling protects bacterial against viral infection. Nature 574, 691–695. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deibert M, Grazulis S, Sasnauskas G, Siksnys V, and Huber R (2000). Structure of the tetrameric restriction endonuclease NgoMIV in complex with cleaved DNA. Nat. Struct. Biol 7, 792–799. [DOI] [PubMed] [Google Scholar]
- Dunten PW, Little EJ, Gregory MT, Manohar VM, Dalton M, Hough D, Bitinaite J, and Horton NC (2008). The structure of SgrAI bound to DNA; recognition of an 8 base pair target. Nucleic Acids Res. 36, 5405–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou A, Pogliano JA, Beckwith J, Oliver DB, and Wickner W (1995). SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83, 1171–1181. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P (2006). Scaling and assessment of data quality. Acta Crystallogr. Sect. D, Biol. Crystallogr 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. Sect. D, Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüschow S, Athukoralage JS, Graham S, Hoogeboom T, and White MF (2019). Cyclic oligoadenylate signalling mediates Mycobacterium tuberculosis CRISPR defence. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Stella S, Zhang Y, Guo T, Sulek K, Peng-Lundgren L, Montoya G, and She Q (2018). A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 46, 10319–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Rosenström P (2010). Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545--9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, and Scornavacca C (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol 61, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G, and Siksnys V (2017). A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Sci. (New York, NY) 357, 605–609. [DOI] [PubMed] [Google Scholar]
- Kirchberger PC, Turnsek M, Hunt DE, Haley BJ, Colwell RR, Polz MF, Tarr CL, and Boucher Y (2014). Vibrio metoecus sp. nov., a close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int. J. Syst. Evol. Microbiol 64, 3208–3214. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovall R, and Matthews BW (1997). Toroidal structure of lambda-exonuclease. Science 277, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Zhang F, and Koonin EV (2017). SnapShot: Class 1 CRISPR-Cas Systems. Cell 168, 946–946.e1. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcmahon SA, Zhu W, Graham S, Rambo R, White MF, and Gloster TM (2019). Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. BioRxiv 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ, Nakandakari-Higa S, and Marraffini LA (2019). Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature 570, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Ishino S, Tsutsumi K, Ito T, Ishino Y, and Tanokura M (2015). Structural basis for substrate recognition and processive cleavage mechanisms of the trimeric exonuclease PhoExo I. Nucleic Acids Res. 43, 7122–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O, Garcia-Doval C, Rostøl JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, and Jinek M (2017). Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548. [DOI] [PubMed] [Google Scholar]
- Pape T, Schneider TR, and IUCr (2004). HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr 37, 843–844. [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud V, and Wende W (2005). Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci 62, 685–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogliano J, Osborne N, Sharp MD, Abanes-De Mello A, Perez A, Sun Y-L, and Pogliano K (1999). A vital stain for studying membrane dynamics in bacteria: a novel mechanism controlling septation during Bacillus subtilis sporulation. Mol. Microbiol 31, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenson NC, and Marraffini LA (2017). Type III CRISPR-Cas systems: when DNA cleavage just isn’t enough. Curr. Opin. Microbiol 37, 150–154. [DOI] [PubMed] [Google Scholar]
- Rostøl JT, and Marraffini LA (2019). Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR–Cas immunity. Nat. Microbiol 4, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SA, Alkhnbashi OS, Behler J, Han W, She Q, Hess WR, Garrett RA, and Backofen R (2019). Comprehensive search for accessory proteins encoded with archaeal and bacterial type III CRISPR- cas gene cassettes reveals 39 new cas gene families. RNA Biol. 16, 530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM (2010). Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Lawrence CM, and Wiedenheft B (2013). CRISPR-Mediated Adaptive Immune Systems in Bacteria and Archaea. Annu. Rev. Biochem 82, 237–266. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2003a). SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 374, 22–37. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2003b). Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. Sect. D, Biol. Crystallogr 59, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, and Hung L-W (2009). Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. Sect. D, Biol. Crystallogr 65, 582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, and Barton GJ (2009). Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson BNJ, Vercoe RB, Salmond GPC, Westra ER, Staals RH., and Fineran PC (2019). Type I-F CRISPR-Cas resistance against virulent phage infection triggers abortive infection and provides population-level immunity. BioRxiv 679308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ, et al. (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GG, and Murray NE (1991). Restriction and Modification Systems. Annu. Rev. Genet 25, 585–627. [DOI] [PubMed] [Google Scholar]
- Wright AV, Nuñez JK, and Doudna JA (2016). Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164, 29–44. [DOI] [PubMed] [Google Scholar]
- Ye Q, Lau RK, Mathews IT, Watrous JD, Azimi CS, Jain M, and Corbett KD (2019). HORMA domain proteins and a Trip13-like ATPase regulate bacterial cGAS-like enzymes to mediate bacteriophage immunity. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, McCabe KA, and Bell CE (2011). Crystal structures of lambda exonuclease in complex with DNA suggest an electrostatic ratchet mechanism for processivity. Proc. Natl. Acad. Sci. U. S. A 108, 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. (Related to Figure 2, Figure 3, Figure 4, and Figure 7) Crystallographic data collection and refinement
See attached Microsoft Excel spreadsheet
Table S4. (Related to Figure 6) Type III CRISPR/Cas-associated NucC homologs
See attached Microsoft Excel spreadsheet
Table S5. (Related to Figure 6) NucC proteins used for sequence alignment
See attached Microsoft Excel spreadsheet
Data Availability Statement
All primary diffraction datasets are available at the SBGrid Data Bank (https://data.sbgrid.org) under the accession numbers noted in Table S1 and the Key Resources Table. All reduced diffraction datasets and refined molecular coordinates are available at the RCSB Protein Data Bank (https://www.rcsb.org) under the accession numbers noted in Table S1 and the Key Resources Table. The original imaging data for Figure 1 and Figure S1 is available at Mendeley Data (https://data.mendeley.com) under DOI 10.17632/dr8mjtwh2d.1. All high-throughput sequencing data used for Figure 5 is available from the Lead Contact without restriction.