

Abstract
Free radical attack on C1′ of deoxyribose forms the oxidized abasic (AP) site 2-deoxyribonolactone (dL). In vitro, dL traps the major base excision DNA repair enzyme DNA polymerase beta (Polβ) in covalent DNA-protein crosslinks (DPC) via the enzyme’s N-terminal lyase activity acting on 5′-deoxyribose-5-phosphate residues. We previously demonstrated formation of Polβ-DPC in cells challenged with oxidants generating significant levels of dL. Proteasome inhibition under 1,10-copper-ortho-phenanthroline (CuOP) treatment significantly increased Polβ-DPC accumulation and trapped ubiquitin in the DPC, with Polβ accounting for 60-70% of the total ubiquitin signal. However, the identity of the remaining oxidative ubiquityl-DPC remained unknown. In this report, we surveyed whether additional AP lyases are trapped in oxidative DPC in mammalian cells in culture. Poly(ADP-ribose) polymerase 1 (PARP1), Ku proteins, DNA polymerase λ (Polλ), and the bifunctional 8-oxoguanine DNA glycosylase 1 (OGG1), were all trapped in oxidative DPC in mammalian cells. We also observed significant trapping of Polλ, PARP1, and OGG1 in cells treated with the alkylating agent methylmethane sulfonate (MMS), in addition to dL-inducing agents. Ku proteins, in contrast, followed a pattern of trapping similar to that for Polβ: MMS failed to produce Ku-DPC, while treatment with CuOP or (less effectively) H2O2 gave rise to significant Ku-DPC. Unexpectedly, NEIL1 and NEIL3 were trapped following H2O2 treatment, but not detectably in cells exposed to CuOP. The half-life of all the AP lyase-DPC ranged from 15-60 min, consistent with their active repair. Accordingly, CuOP treatment under proteasome inhibition significantly increased the observed levels of DPC in cultured mammalian cells containing PARP1, Ku protein, Polλ, and OGG1 proteins. As seen for Polβ, blocking the proteasome led to the accumulation of DPC containing ubiquitin. Thus, the ubiquitin-dependent proteolytic mechanisms that control Polβ-DPC removal may also apply to a broad array of oxidative AP lyase-DPC, preventing their toxic accumulation in cells.
Keywords: 1,10-copper-ortho-phenanthroline; methylmethane sulfonate; hydrogen peroxide; 2-deoxyribonolactone; AP lyase; DNA-protein crosslinks
1. Introduction
Abasic (AP) sites are a common type of DNA lesion, formed at an estimated rate of 10,000 or more lesions per day in mammalian cells (1–4). AP sites arise from a variety of chemical and biochemical processes including the loss of unstable modified nucleobases (5, 6), intermediates generated by DNA N-glycosylases during base excision DNA repair (BER) (5, 6), and through base elimination following free radical attack of deoxyribose (7, 8). If left unrepaired, AP sites can block key DNA transactions such as replication and transcription, which can contribute to mutagenesis and carcinogenesis (9). The selective pressure to repair AP sites is highlighted by the presence of AP endonuclease genes in the genomes of virtually all living organisms (10). The predominant such enzyme in mammalian cells is Ape1 protein (also called APEX1), which hydrolyzes the 5’ phosphodiester of the AP site itself (5, 6).
To date, >20 DNA repair enzymes, especially in BER, have been reported to have an associated lyase activity that incises AP sites or removes 5′-deoxyribose-5-phospate (5′-dRp) termini (11, 12). AP sites are generated by DNA glycosylases during BER by the hydrolysis or lyase-mediated cleavage of the N-glycosylic linkage between the nitrogenous bases and the deoxyribose moiety. DNA glycosylases exist as two general types: monofunctional glycosylases that only remove the damaged base by hydrolysis, and bifunctional enzymes that both eliminate a base lesion and can subsequently incise the phosphodiester bond immediately 3′ to the AP site. The latter reaction generates 3′ termini that must be further processed to enable gap-filling DNA repair synthesis (13, 14). Ape1 and other enzymes are thought to carry out this trimming step to generate a usable primer terminus; Ape1 cleavage of an AP site generates such active primers directly. For 5’-processing after Ape1 incision, the BER protein DNA polymerase beta (Polβ) and the “backup” BER enzyme DNA polymerase lambda (Polλ) harbor a 5′-dRp excision activity (5, 15, 16, 71). Excision of 5′-dRp residues by these enzymes occurs via β-elimination, which generates a normal 5’-nucleotide end (17). Bifunctional glycosylases and 5′-dRp lyases that catalyze β-, δ- or β-/δ-elimination reactions proceed via the formation of a covalent enzyme-DNA intermediate, a Schiff base, that is resolved by hydrolysis, often as the rate-limiting step (11, 12). These transient enzyme-DNA intermediates can be frozen mid-cycle in vitro upon addition of strong reducing agents such as sodium borohydride (NaBH4) to the reaction (12, 18).
With the reductive trapping approach, several DNA repair enzymes have been shown to form DPC in vitro via their respective 5′-dRp lyase or AP lyase activities. These include Ku antigen (19–22), poly(ADP-ribose) polymerases 1 and 2 (PARP1, PARP2) (23–25), and DNA polymerase lambda (Polλ) (15). In stark contrast to the transient intermediates formed by lyases acting on hydrolysis-generated AP sites, the oxidized abasic site 2-deoxyribonolactone (dL) poses a formidable obstacle to the lyase reaction (26). The major BER protein, Polβ, was the first mammalian enzyme shown to be trapped in stable DNA-protein complexes (DPC) by interaction with dL residues in vitro (27–30). Other AP lyases can also be trapped by dL or its β-elimination product butenolide in vitro, including the eukaryotic OGG1 and NEIL DNA glycosylases, and bacterial Nth1, which generated the highest yield of DPC compared to a panel of other lyases tested (31, 32) (see Fig. S1 for schematic of the NaBH4 trapping reaction and formation of AP lyase-dL-DPC).
Our preliminary experiments indicated that both Ku antigen and Polλ can form dL-DPC in vitro, and PARP1-DPC were recently reported in cells treated with MMS or with clinically used PARP inhibitors (24, 33, 34). However, the possible oxidative trapping of these AP lyases in DPC in mammalian cells had not been addressed. We have detailed the formation and processing of oxidative Polβ-DPC in mouse and human cells exposed to various dL-generating agents. That work (35) showed that Polβ-DPC are rapidly processed by the proteasome; when the proteasome is inhibited, ubiquitin is contained in the Polβ-DPC. Most of the ubiquitin in the oxidative DPC is lost in POLB−/− mouse embryonic fibroblasts (MEF) or in human cells with the lyase-inactive K72A form of Polβ, but 30-40% remains and may be associated with other lyases trapped in DPC and undergoing ubiquitylation prior to digestion by the proteasome. We have recently identified ubiquitylation sites on Polβ required for its proteolytic processing (Quinones et al., submitted). Biochemical analysis of whole-cell extracts (WCE) also indicates that a variety of other proteins can form DPC at dL sites, although Polβ appears to be the most reactive on a molar basis (27).
In the work presented here, we probed for the formation of DPC with specific AP lyase proteins in cells treated with the dL-inducing agent 1,10-copper-ortho-phenanthroline (CuOP). We found that, in addition to Polβ, Ku antigen, Polλ, PARP1, and OGG1 are also trapped in oxidative DPC, and rapidly processed by the proteasome in conjunction with ubiquitylation. These results suggest a general mechanism for cells to cope with the burden of oxidative DPC containing abasic lyases.
2. Materials and Methods
2.1. Mammalian Cell Culture Conditions
Human MDA-MB-231 mammary tumor cells expressing flag-tagged Polβ were maintained in a humidified incubator at 37°C under 5% CO2 in RPMI 1640 (Lonza) supplemented with 1% penicillin-streptomycin (Life Technologies), 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies), (0.5 μg/ml) puromycin (Sigma-Aldrich), (700 μg/ml) G418 (Sigma-Aldrich) and (10 μg/ml) gentamycin (Sigma-Aldrich) as previously described . The human glioma LN-428 cell lines expressing WT and mutant versions of Polβ or shRNA against Trip 12 were maintained and grown in α-MEM (Life Technologies) supplemented with 10% heat-inactivated FBS (Life Technologies), (5 μg/ml) gentamycin (Sigma-Aldrich), 80 u penicillin/80 μg Streptomycin/0.32 μg amphotericin/ml (Life Technologies), and (2 mM) L-glutamine (Sigma-Aldrich) as previously described (37). MEF cells WT or carrying a homozygous deletion in the POLL gene were cultured in a humidified incubator at 37°C under 5% CO2 in DMEM containing GlutaMAX (Thermo Fisher Scientific), 10% FBS (Life Technologies) and 1% penicillin-streptomycin (Life Technologies) as previously described (38). The Ape1-deleted murine B-cell line, CH12F3, was cultured in RPMI 1640 medium (Lonza), at 37°C under 5% CO2 supplemented with 10% fetal bovine serum and 50 μM β-mercaptoethanol (Sigma-Aldrich) as previously described (39).
2.2. Detection of DPC in Mammalian Cells by the RADAR Assay
DPC were generated, isolated, and analyzed as previously described (35). Typically, 5.0 x 104 cells were treated with CuOP (CuSO4 and 1,10 phenanthroline mixed in a 1:2 molar ratio immediately before use), MMS, or H2O2 at the concentrations indicated in the figures. Where indicated, these treatments were combined with treatment with the proteasome inhibitor MG132. Following treatment, the cells were immediately lysed in DPC lysis buffer containing [4 M guanidinium thiocyanate (Sigma-Aldrich), 1% Sarkosyl, 2% Triton X-100 (Sigma-Aldrich), 1% dithiothreitol (DTT); (Roche), 0.1 M sodium acetate, pH 5.0 (G-Biosciences), 20 mM Tris-HCI, pH 8.0 (G-Biosciences), 20 mM EDTA, pH 8.0 (G-Biosciences), adjusted to pH 6.5 using 2 M NaOH], After lysis, genomic DNA was precipitated with ice-cold 100% ethanol, pelleted, then washed three times with 70% ethanol. The washed DNA pellets were resuspended in 8 mM NaOH by passing the DNA through a 21G syringe ten times. The resulting DPC-containing solution was quantified by PicoGreen (Life Technologies) fluorescence, then equivalent amounts of DNA were blotted onto nitrocellulose membranes using a slot blot vacuum manifold (Bio-Rad).
Membranes were probed for 1-2 h with primary antibodies specific for the protein target of interest using the following dilutions: rabbit polyclonal anti-Polβ (Novus Biologicals; cat.#NB100-91734), 1:1,000; rabbit polyclonal anti-Ape1 (Novus Biologicals’ cat.#NB100-101), 1:10,000; mouse monoclonal anti-ubiquitin (BioLegend; cat/#838704), 1:1,000; rabbit polyclonal anti-Polλ (S.H. Wilson laboratory), 1:2,000; rabbit polyclonal anti-Ku antigen (D.A. Ramsden laboratory), 1:5,000; rabbit polyclonal anti-PARP1 (Proteintech Group, Inc; cat.#229999-1-AP), 1:1,000; rabbit polyclonal anti-OGG1, (Abcam; cat.# ab135940) 1:1,000; rabbit polyclonal anti-NEIL1, (Proteintech Group, Inc; cat.#12145-1-AP) 1:1,000; rabbit polyclonal anti-NEIL3, (Proteintech Group, Inc; cat.#11621-1-AP) 1:1,000. Blots were then incubated with secondary IR dye-conjugated goat anti-mouse IR680 (LI-COR Biosciences; cat.# 925-68070), or with goat anti-rabbit IR800 dye (LI-COR Biosciences; cat.#926-32211) at a dilution of 1:10,000 for 1 h at room temperature. Slot blots were imaged using an Odyssey laser scanner in grey-scale mode, and the bands were quantified using NIH ImageJ software.
2.3. Immunoblot analysis
Whole cell extracts were prepared by lysing cells in 10 mM Tris-HCI (Roche), pH 7.4, 150 mM NaCl, 20 mM EDTA, pH 8.0, 10% Triton X-100 supplemented with protease cocktail inhibitor (Roche) as recommended. Total protein was quantified with the Bradford assay, using bovine serum albumin (BSA; Sigma-Aldrich) as the standard. For immunoblot analysis of Polβ, Polλ, Ku antigen, PARP1, OGG1, NEIL1, and NEIL3 in MDA-MB-231 cells and for analysis of Polλ in MEF cells, the samples were electrophoresed on on either 12% or 4-12% pre-cast NuPAGE protein gels (Life Technologies) in 1X 3-(N-morpholino) propanesulfonic acid) (MOPS) (Life Technologies) running buffer. Proteins were electroblotted onto polyvinylidene difluoride membranes using 1X NuPAGE transfer buffer (Life Technologies) supplemented with 20% methanol and 0.1% sodium dodecyl sulfate. Protein binding was confirmed by staining with Ponceau S, and the membranes were washed three times with Tris-buffered saline (TBS) containing 0.1% Tween-20 for 10 min each cycle, then blocked in 3% BSA dissolved in phosphate-buffered saline (PBS) containing 0.1% Tween 20 for 1 h. Next, the membranes were probed for 1 h with the primary antisera diluted in blocking buffer at the dilutions indicated above. For testing olaparib activity, the membranes were probed with monoclonal rabbit anti-PAR antibody (Santa Cruz Biotechnology, SCBT) at a 1:500 dilution for 1 h at room temperature. As loading controls, the membranes were probed with rabbit polyclonal anti-GAPDH (Rockland Immunochemicals, Inc.) at a 1:1,000 dilution, or with mouse monoclonal anti-α tubulin, (LI-COR Biosciences) at a 1:2,000 dilution. Finally, secondary goat anti-mouse conjugated to IR dye 680 (LI-COR Biosciences), or goat anti-rabbit conjugated to IR dye 800 (LI-COR Biosciences) were incubated with the membranes at a dilution of 1:10,000 for 1 h at room temperature in blocking buffer.
3. Results
3.1. Oxidative AP Lyase-DPC Formation in Mammalian Cells
To test whether AP lyase enzymes beyond Polβ form oxidative DPC in cultured mammalian cells, the human breast cancer cell line MDA-MB-231 (35) was exposed to the dL-inducing oxidant CuOP. Immunoblot analysis of WCE prepared from oxidant-treated MDA-MB-231 cells was used to confirm the specific detection of each type of DPC using antisera directed against each of the proteins under study (specificities verified in Fig. S2). Using the DNA isolation and blotting “RADAR” assay as in our studies of Polβ (35), we observed a CuOP dose-dependent increase in DPC containing Ku antigen (Fig. 1A), PARP1 (Fig. 1B), and Polλ (Fig. 1C) . The formation of oxidative Polλ-DPC was also observed in wild-type (WT) mouse embryonic fibroblasts (MEF) treated with CuOP (Fig. 1D), which was reduced to background level in genomic DNA isolated from POLL−/− MEF and not increased by CuOP treatment (Fig. 1D).
Figure 1. Oxidative Trapping of AP Lyases in DPC in vivo.
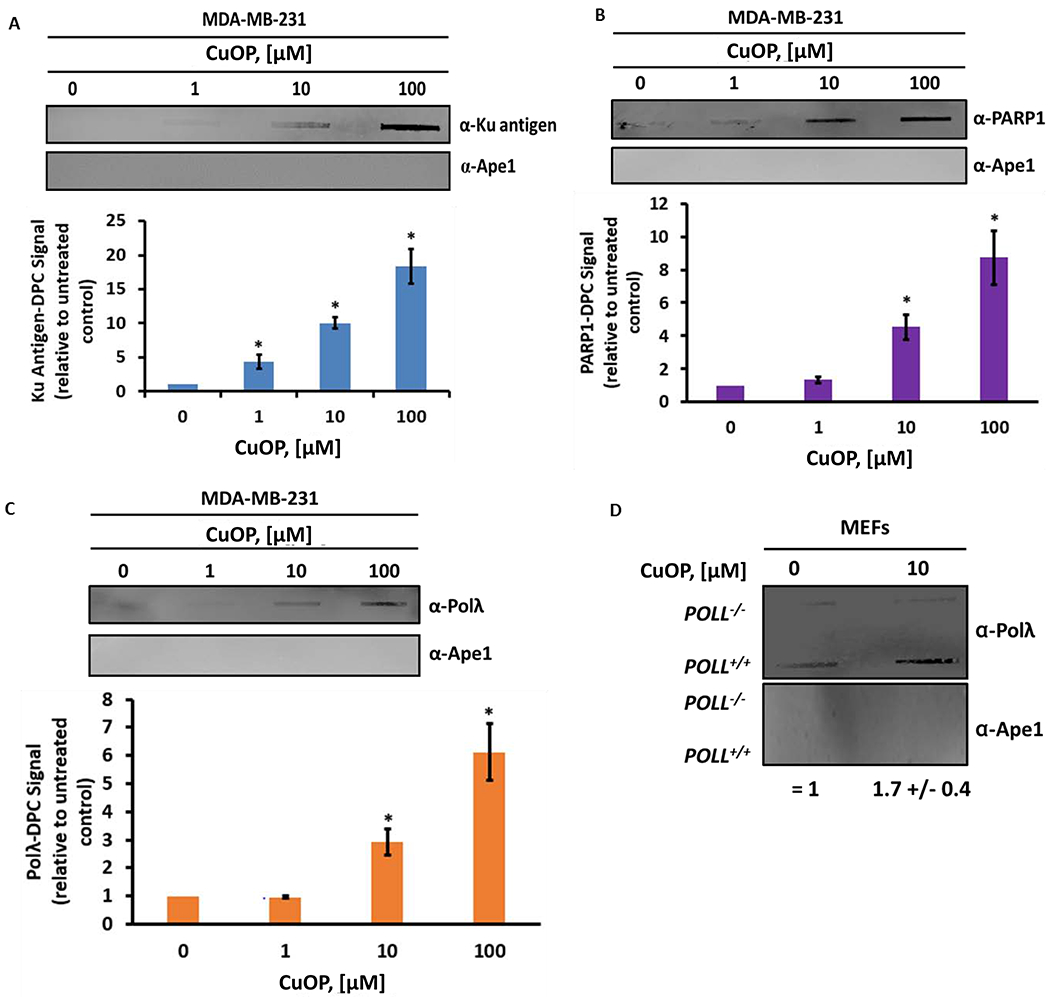
MDA-MB-231 cells were treated with increasing concentrations of CuOP for 1 h (under conditions described in section 2.1), genomic DNA (gDNA) immediately isolated, and 1-μg aliquots assayed for DPC using the appropriate antisera. A, Ku antigen; B, PARP1; C, Polλ. For panel D, WT or POLL−/− MEF were treated and assayed for Ku-DPC. For each set of experiments, a representative blot is shown, with quantification for n= 3 independent experiments, normalized to untreated cells. Error bars show +/− S.D. * indicates p<0.05 (two-tailed Student’s t-test) compared to untreated cells.
Previous data showed that Polβ-DPC formation requires Ape1 in CH12F3 cells (35). We hypothesized that, in the absence of Ape1, more dL would be available to trap bifunctional DNA glycosylases such as OGG1. The formation of OGG1-DPC formation in CuOP-treated CH12F3 cells did not require the presence of Ape1, in contrast to the Ape1 requirement for Polβ-DPC formation (35), and the observed OGG1-DPC level was ~2-fold higher in Ape1-deficient than in Ape1-containing cells (Fig. 2). This result suggests that more DPC-forming substrate was available in the absence of Ape1, allowing other AP lyases such as OGG1 to become trapped in oxidative DPC. Because Ape1 does not form DPC with dL in vitro (28), nor were Ape1-DPC detectable in CuOP-treated cells (35), the blots were also probed with anti-Ape1 antiserum to control for adventitious trapping by the various agents. Ape1 protein was not detected in these samples under the experimental conditions that detected other DPC in the same samples (Fig. 1A–C, 2). Collectively, these data indicate that Ku antigen, Polλ, PARP1, and OGG1 can be trapped in DPC by oxidative lesions in cultured mammalian cells.
Figure 2. APE1-Dependence of oxidative OGG1-DPC formation.

CH12F3 cells were treated where indicated with 5 μM CuOP for 1 h (under conditions described in section 2.1), and gDNA was immediately isolated. Samples (1 μg) were assayed for OGG1-DPC. For each set of experiments, a representative slot blot is shown, with quantification for n= 3 independent experiments, comparing DPC signals between CH12F3 Ape1-expressing (APE1+/−/− and Ape1-knockout (APE1−/−/−) cells. Error bars show +/− S.D. * indicates p<0.05 (two-tailed Student’s t-test) compared to CH12F3 APE1−/−/− cells.
3.2. Chemical Specificity for Trapping AP Lyases in DPC in Mammalian Cells
Recent data showing PARP1-DPC formation in cells treated with MMS suggest that agents that generate AP sites or BER intermediates could trap other enzymes with AP lyase activity (41–43). To determine whether different agents that generate BER substrates can trap AP lyases in mammalian cells in culture, we assayed for DPC formation in cells treated for 1 h with MMS, H2O2, or CuOP. The results (Fig. 3A) revealed that significant Ku-DPC were detected only in the oxidant-treated cells, similar to Polβ-DPC in this study and our published work (35). In contrast, DPC were detected for PARP1 (Fig. 3B), Polλ (Fig. 3C), and OGG1 (Fig. 3D) in cells exposed to any of the three agents tested.
Figure 3. Chemical specificity of AP Lyase-DPC formation.
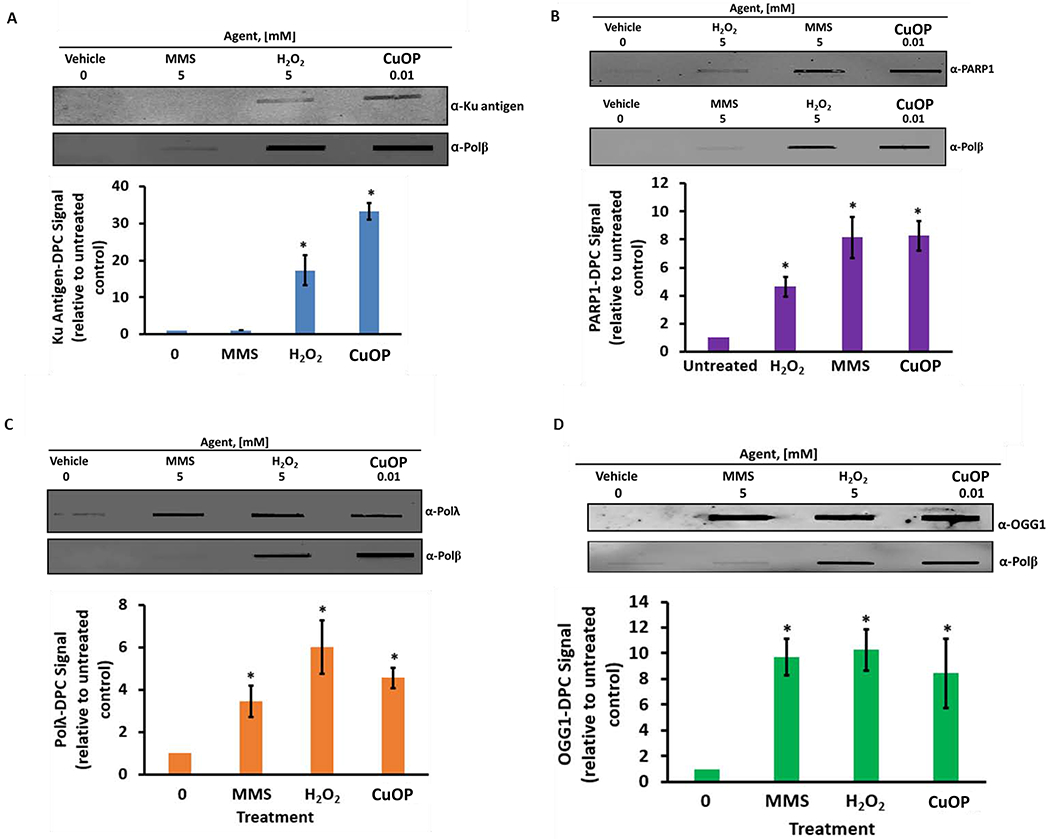
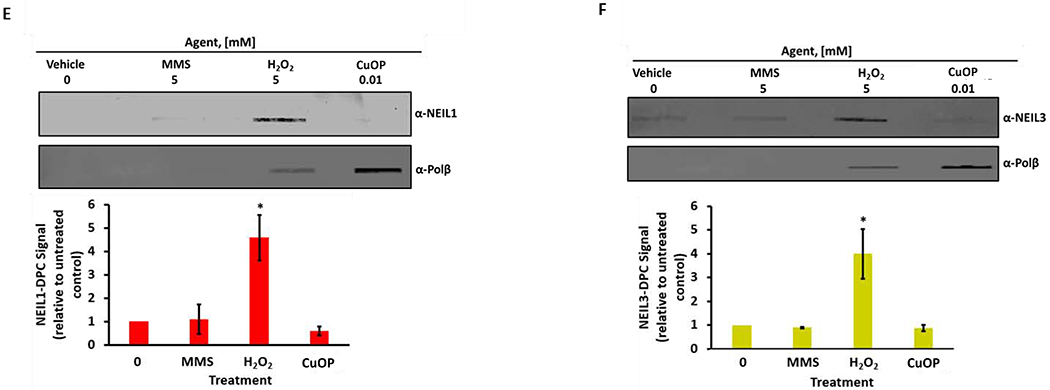
MDA-MB-231 cells were exposed for 30 min to 5 mM MMS or 5 mM H2O2, or to 10 μM CuOP for 1 h (under conditions described in section 2.1), and gDNA was immediately isolated. Samples (1 μg) were assayed for DPC containing: A, Ku antigen; B, PARP1; C, Polλ; D, OGG1; E, NEIL1; F, NEIL3. For each set of experiments, a representative blot is shown, with quantification for n= 3 independent experiments, normalized to untreated cells. Error bars show +/−S.D. * indicates p<0.05 (two-tailed Student’s t-test) compared to untreated control.
We also assayed whether two of the NEIL-family glycosylases could form DPC in mammalian cells. Significant amounts of both NEIL1-DPC and NEIL3-DPC were detected in cells exposed to H2O2, but not for CuOP or MMS treatment (Fig. 3E, F). That result indicates that these enzymes do not interact significantly with dL or AP sites in chromosomal DNA, or that the linkages they may form with these lesions are somehow readily reversed. The effectiveness of H2O2 in forming the NEIL-DPC is, however, consistent with a role for these enzymes in processing oxidized base lesions, as the enzyme intermediates are relatively long-lived in vitro(44–46).
3.3. PARP Inhibition and the Formation of PARP1-DPC in CuOP-Treated Cells
Combining alkylating agent treatment with clinical PARP inhibitors enhances the trapping of PARP1 in DPC (34, 42). We hypothesized that PARP inhibition would likewise enhance the trapping of PARP1 at oxidative DNA damage. We confirmed the activity of olaparib by performing immunoblot analysis for poly(ADP-ribosyl)ated proteins in WCE from MDA-MB-231 cells treated with olaparib (Fig. S3). In cells treated with CuOP, a strong poly(ADP-ribosyl)ation signal was detected, which was significantly diminished in the presence of olaparib (Fig. S3). In analysis of PARP1-DPC formation in MDA-MB-231 cells, exposure to olaparib or CuOP alone generated easily detectable PARP1-DPC. When cells were treated simultaneously with both CuOP and olaparib, trapping of PARP1 in DPC was significantly enhanced compared to CuOP or olaparib treatment alone (Fig. 4), demonstrating an additive effect. This finding is consistent with the ability for both agents to produce significant PARP1-DPC in mammalian cells in culture, though likely through different mechanisms.
Figure 4. Effect of PARP inhibitor on oxidative PARP1-DPC formation.
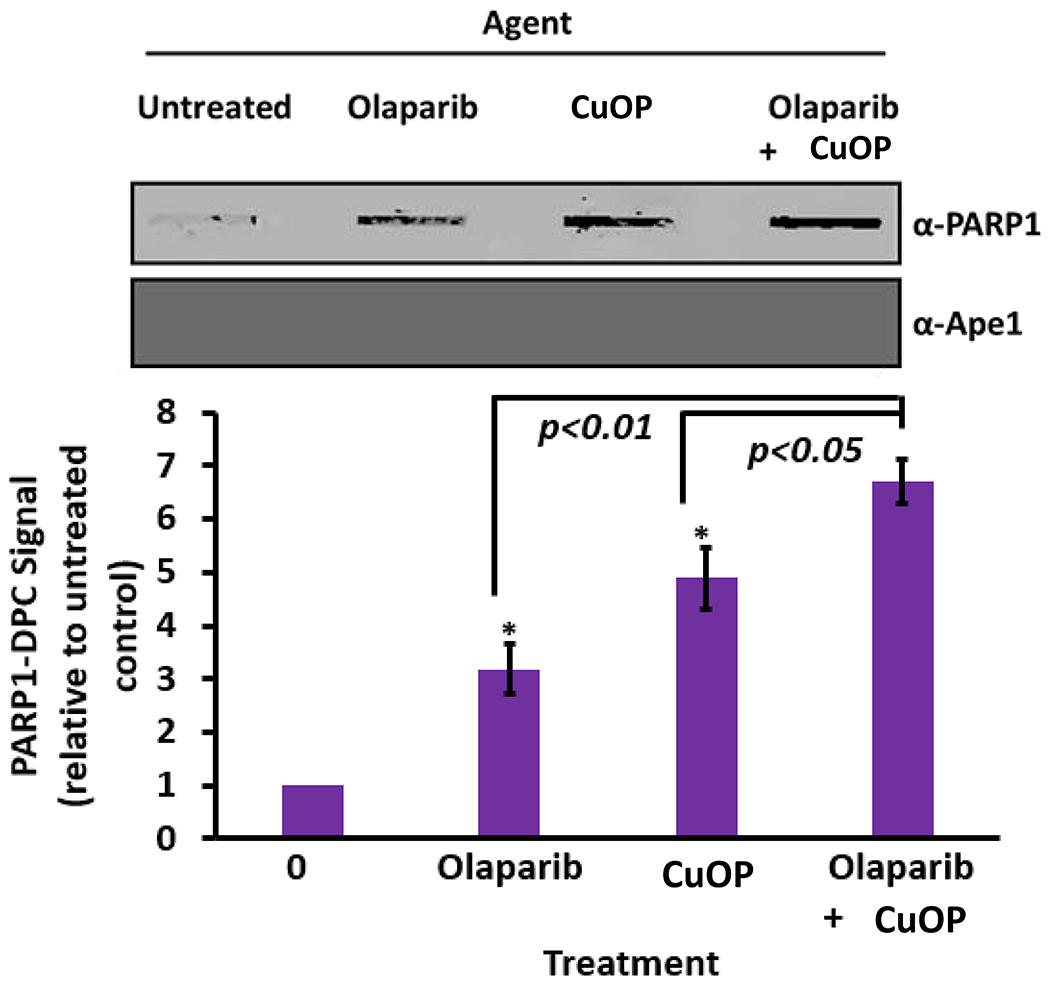
MDA-MB-231 cells were exposed for 1.5 h to olaparib or CuOP, or to both sequentially (under conditions described in section 2.1). For the combination treatment, the cells were first exposed to 10 μM CuOP for 1 h, followed by addition of 10 μM olaparib for 30 min. Immediately after the treatments, gDNA was isolated. Aliquots of gDNA (1 μg) were assayed for PARP1-DPC; blots were also probed for Ape1 as a negative control. Representative blots are shown, with quantification for n= 3 independent experiments, normalized to untreated cells. Error bars show +/−S.D. for n= 3 independent experiments. * indicates p<0.05 or p<0.01 (two-tailed Student’s t-test) compared to the indicated treated controls.
3.4. Clearance of Oxidative AP Lyase-DPC in Mammalian Cells Dependent on the Proteasome
We previously showed that the Polβ in oxidative DPC is rapidly processed by proteasome-dependent digestion in mammalian cells [with a half-life 15-30 min in both human and mouse cells (35)]. We therefore tested whether the oxidative DPC containing Ku antigen, PARP1, Polλ, and OGG1 are also processed rapidly. Indeed, the signals for Ku-, Polλ-, or OGG1-DPC all displayed half-lives of 15-20 min following a 1-h CuOP treatment (Fig. 5A,C,D). The processing of PARP1-DPC was significantly slower, with a half-life ≥60 min (Fig. 5B). Thus, the various AP lyase-DPC are actively removed in cells, with some variation in the rate of removal for different DPC.
Figure 5. Processing of oxidative AP Lyase-DPC.
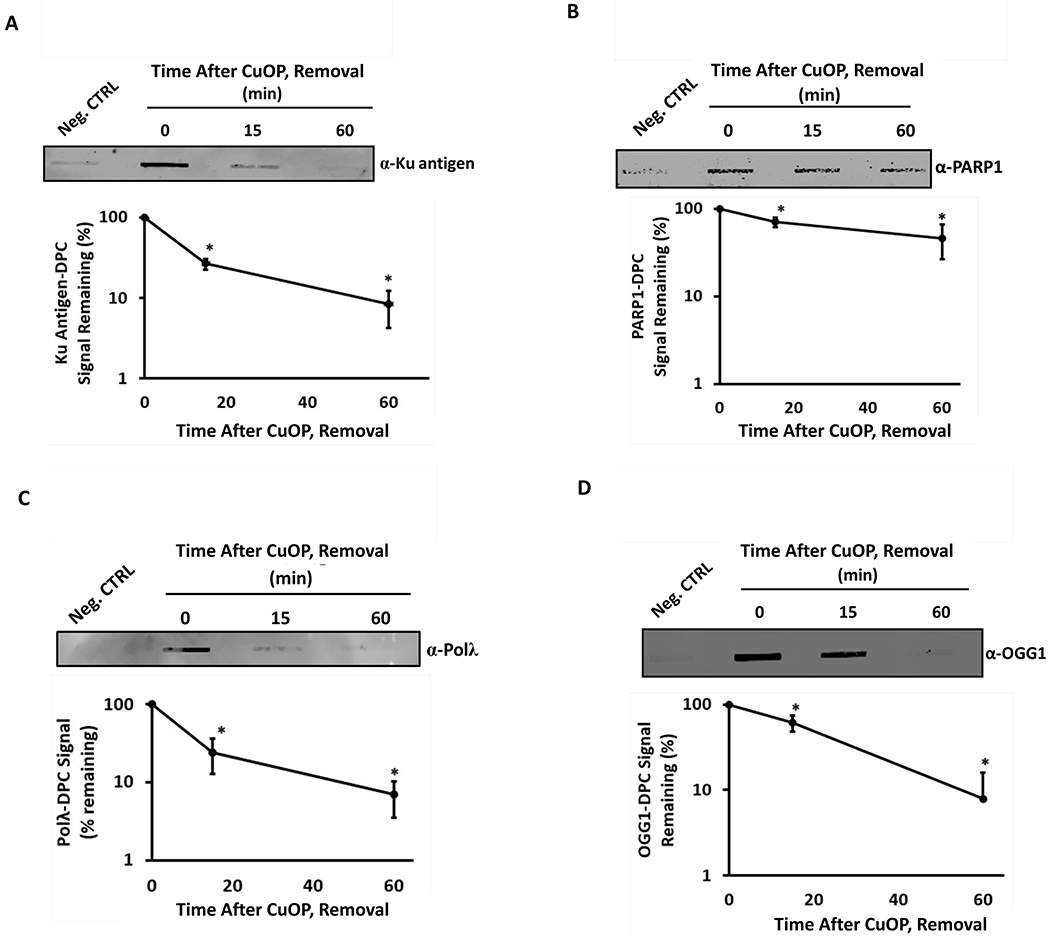
MDA-MB-231 cells were treated with 10 μM CuOP for 1 h (under conditions described in section 2.1), then transferred to fresh medium. gDNA was isolated either immediately after the treatment, or at the indicated times of the subsequent incubation. Samples (1μg) were assayed for: A, Ku antigen; B, PARP1; C, Polλ; and D, OGG. Quantification for each set of experiments is shown for each blot. Representative blots are shown, with quantification for n= 3 independent experiments, normalized to untreated cells. Error bars show +/−S.D. for n= 3 independent experiments. * indicates p<0.05 (two-tailed Student’s t-test) compared to the ‘0 min’ time point (defined as 100%).
In light of the active removal of Ku-, PARP1-, Polλ-, and OGG1-DPC, we tested whether their processing depended on the activity of the proteasome, as do Polβ-DPC (35). Compared to CuOP treatment alone, a combined treatment with CuOP and MG 132 resulted in ~2-fold increase in the levels of all the AP lyase-DPC assayed (Fig. 6A–D). Importantly, treatment with MG132 also generated a significant ubiquitin immunosignal in each case (Fig. 6A–D). These data are also consistent with the finding that additional oxidative ubiquityl-DPC are formed along with Polβ-DPC under CuOP treatment (35), and that, once the DPC are ubiquitylated, the proteasome acts quickly to digest them.
Figure 6. Oxidative AP Lyase-DPC increased by proteasome inhibition.
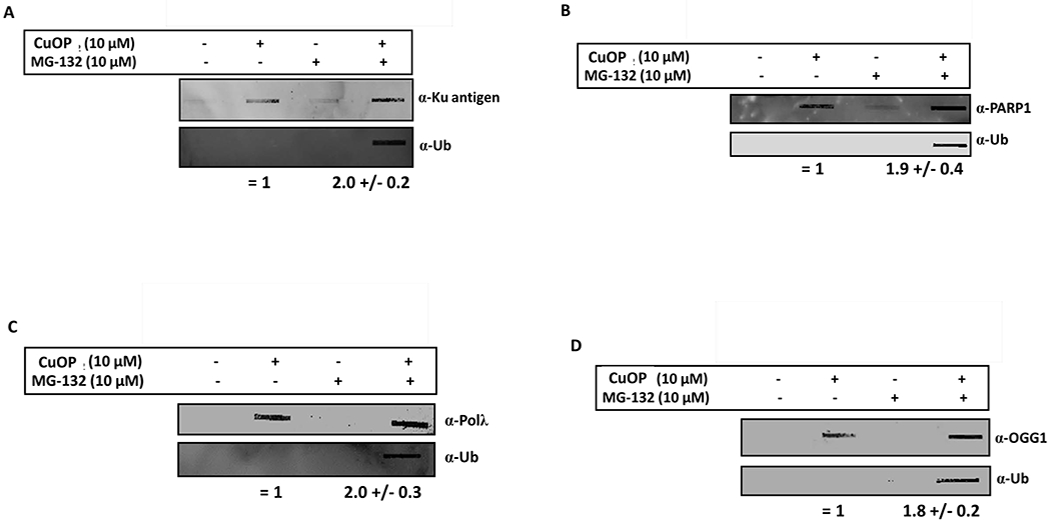
MDA-MB-231 cells were treated with 10 μM CuOP for 1.5 h, with or without MG132 present (under conditions described in section 2.1), and gDNA isolated immediately. Aliquots of gDNA (1μg) were assayed for: A, Ku antigen; B, PARP1; C, Polλ; or D, OGG1. Separate blots of the same samples were also probed for ubiquitin in each experiment. Representative blots are shown, with quantification for n= 3 independent experiments. Error bars show +/−S.D. for n= 3 independent experiments. * indicates p<0.05 (two-tailed Student’s t-test) comparison for CuOP versus CuOP + MG132.
4. Discussion
Oxidative DNA damage is a significant by-product of aerobic metabolism and has been implicated in the etiology of aging and neurodegenerative disorders (47, 48). Additionally, oxidative base and sugar lesions are commonly generated by a variety of antitumor agents that are of clinical importance (47, 49). Although many of the lesions generated by ROS can be efficiently processed by BER, certain lesions can pose an obstacle to some of the core components of the BER pathway (50).
A significant oxidative lesion that cannot be processed via single-nucleotide BER is the oxidized AP site, dL. This lesion can irreversibly trap DNA repair lyases in covalent DPC in vitro (26, 51). We recently showed that the core BER enzyme, Polβ, forms DPC via its 5′-dRp lyase activity, specifically in cells exposed to dL-inducing oxidants (35) [(recently reviewed here (52–55)]. Although Polβ-DPC account for the bulk of ubiquityl-DPC in cells treated with CuOP, the nature of the remaining 30-40% of ubiquitin-DPC remained unknown (35).
Based on their roles in DNA repair, we analyzed Ku antigen, Polλ, PARP1, OGG1, NEIL1 and NEIL3 as possible targets to form oxidative DPC in mammalian cells. In this report, we show that all the AP lyase proteins tested can form oxidative DPC in mammalian cells in culture, albeit to varying extents and with different selectivity for the type of agent. CuOP was an effective trapping agent for Ku protein, PARP1, Polλ, and OGG1 in DPC. However, in contrast to Polβ (35), the increased trapping of OGG1 in APE1-knockout cells compared to Ape1-proficient cells indicates that the glycosylase can be trapped at the uncleaved dL residues that are expected to accumulate in the absence of Ape1. Alternatively, the increased OGG1-DPC could reflect the accumulation of the dL breakdown product butenolide at DNA 3’ ends (31), which would also be considerably diminished when Ape1 acts first to cleave dL efficiently (31, 56).
Similar to what has been shown for PARP1 (24), MMS induced significant DPC with both Polλ and OGG1. In contrast, Ku antigen followed the same pattern as Polβ in forming DPC with the dL-inducing oxidants H2O2 and (more effectively) CuOP, but not with MMS-induced lesions. The similar pattern for Polβ and Ku protein forming DPC was somewhat unexpected, in that Polβ shows the strongest affinity for lesions at nicks or small gaps in double-stranded DNA (57, 58), while Ku has a strong preference for 5′-dRp residues located at or near the 5′ ends of double-strand breaks (19, 22). This observation suggests that the lesion chemistry is a key factor in the DPC formation we have observed, with both Polβ and Ku being trapped at dL sites generated by H2O2 or CuOP, but not at ordinary AP sites, unlike PARP1 and PARP2 (23, 24). At the same time, it may be that the Polβ-DPC and Ku-DPC generated in CuOP- or H2O2-treated cells are in different structural contexts (single- vs. double-strand breaks, respectively).
DPC formation by PARP1, OGG1 and Polλ in MMS-treated cells indicates that these proteins can be trapped at normal AP sites. This result is consistent with the slow turnover of the lyase activity of PARP1 for both incised and intact AP sites in vitro and in MMS-treated cells (24). In contrast, Polλ does not possess a detectable AP lyase activity, but the enzyme has a 5′-dRp lyase activity that is ~5-fold lower than that of Polβ (15). Thus, the detected Polλ-DPC may simply reflect the slow catalytic turnover of the 5’-dRp lyase, such that a detectable amount of the Schiff base intermediate has accumulated in MMS-treated cells at the time of their lysis.
The finding that OGG1 forms DPC in MMS-treated cells is also somewhat unexpected. OGG1 is a bifunctional glycosylase acting on oxidative base lesions such as 8-oxoguanine and the ring-opened formamidopyrimidine (59). However, overexpression of OGG1 provides some resistance both to MMS and to more complex alkylating agents (60, 61). It may be that the reported weak 5’-dRp lyase activity of OGG1 or its AP incision activity (11, 62) account for this role via the accumulation of the Schiff base intermediate during the treatment time, as we proposed for Polλ.
The bifunctional glycosylases NEIL1 and NEIL3 gave still another trapping pattern: DPC were found only for H2O2-treated cells, but not upon treatment with MMS or CuOP. Both of these enzymes have roles in processing oxidative base lesions (45, 63), notably the formamidopyrimidine derivatives of adenine and guanine (47). Thus, the DPC observed for NEIL1 and NEIL3 could represent slowly-resolving intermediates of the excision reactions, as we have argued for PARP1 and Polλ. Free radical attack also generates the thymine-derived lesion 5-hydroxy-5-methylhydantoin, which traps bifunctional DNA glycosylases such as Fpg and NEIL1 in vitro (64). Therefore, the observed trapping could also arise from suicidal crosslinking of these enzymes to 5-hydroxy-5-methylhydantoin sites formed in H2O2-treated cells. Because NEIL1 and NEIL3 were not significantly trapped by the more selective dL-inducing oxidant CuOP, the data suggest that dL is not a significant lesion for trapping these AP lyases in mammalian cells.
The disappearance of oxidative AP lyase-DPC was rapid, with half-lives ranging from 15-60 min. These rates are similar to that observed for Polβ-DPC (35), consistent with an active repair process. Two aspects of this processing point to an essential role for the proteasome: the increased DPC levels observed when cells are co-treated with both CuOP and MG132 compared to CuOP alone; and the accumulation of ubiquitylated DPC when the inhibitor is present. These aspects are the same as for the initial processing steps found for Polβ-DPC.
The main nucleophilic residues responsible for 5′-dRp/AP lyase activity in Ku antigen (20, 22), PARP1 (24), Polλ (15), and OGG1 (65, 66) have been predicted through mutational and biochemical analyses. While beyond the scope of this paper, we anticipate that future work using cell lines expressing mutated lyase proteins will shed light on the mechanistic basis for AP lyase-DPC formation and their effects on cell survival. Along these lines, we realize that one limitation of this study is the specificity of the antibody-based approach, in that the antisera used could have limited sensitivity for detecting some DPC, and possible cross-reaction with other proteins must be considered. For the latter possibility, such cross-reactivity was not detected by immunoblotting (Fig. S2). To address these issues more deeply, future studies investigating the mechanisms of oxidative AP lyase-DPC formation should also include, if possible, cell lines harboring homozygous deletions of the AP lyase under study, or cells expressing epitope-tagged versions of the proteins under study, and where possible, mutant proteins lacking the lyase activity.
With respect to disease, mutations in various AP lyases involved in BER have been associated with cancer in humans (9, 67). For example, polymorphic variants of OGG1 that are associated with a higher risk of disease have been identified in prostate, lung, and renal carcinomas (67), suggesting these polymorphisms could be exploited (assuming they significantly affect the lyase activities) by challenging them with oxidative agents. The development of small molecule inhibitors that target the lyase domain of DNA glycosylases is currently underway (68, 69), and may be useful in combating neoplasms that aberrantly express these proteins.
Lastly, our data also suggest a possible combination therapy using clinical PARP inhibitors and oxidative agents that trap PARP1 in order to target certain human cancers (e.g., BRCA-negative breast cancer). Cells that express low levels of Ku antigen are hypersensitive to chemotherapy and radiation therapy (70), suggesting that oxidative agents that trap this enzyme may further potentiate the cell killing effect of these antitumor strategies. Collectively, these studies imply that the development of novel copper-based complexes could be used to enhance treatment of human neoplasms that express mutant or imbalanced levels of these 5′-dRp/AP lyases.
Supplementary Material
Highlights.
In vivo DPC formation with oxidative lesions is a common feature of 5′-dRp lyases involved in DNA repair
The efficiency of DPC formation varies according to the oxidant treatment
Some lyases can be trapped at non-oxidative lesions formed by MMS
These differences appear to reflect the in vivo functions of the enzymes
The lyase-DPC are rapidly ubiquitylated and targeted for digestion by the proteasome
Acknowledgements
Support for this work was provided by the National Institutes of Health (NIH) [grant number F31GM109747 to J.L.Q., and grants R01GM040000 and R21CA198752 to B.D.]. Additional funding for the research presented here was granted by the National Aeronautics and Space Administration [RIS4E, P.I. Dr. Glotch]. This work was also funded by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences project number Z01 ES050159 to S.H.W.
Author statement
J.L.Q. and B.D. designed the experimental approach;
J.L.Q. performed the experiments except those with CH12F3 cells, which was accomplished with the help of U.T.
The manuscript was written by J.L.Q. and edited by B.D., S.H.W., D.A.R., and U.T. Funding for this research was obtained by J.L.Q. (fellowship) and B.D. (research grant).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflict of interest.
References
- 1.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11( 19):3610–8. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura J, Walker VE, Upton PB, Chiang S-Y, Kow YW, Swenberg JA. Highly Sensitive Apurinic/Apyrimidinic Site Assay Can Detect Spontaneous and Chemically Induced Depurination under Physiological Conditions. Cancer Research. 1998;58(2):222–5. [PubMed] [Google Scholar]
- 3.Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB. Endogenous versus Exogenous DNA Adducts: Their Role in Carcinogenesis, Epidemiology, and Risk Assessment. Toxicological Sciences. 2011;120(Suppl 1):S130–S45. doi: 10.1093/toxsci/kfq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura J, Swenberg JA. Endogenous Apurinic/Apyrimidinic Sites in Genomic DNA of Mammalian Tissues. Cancer Research. 1999;59(ll):2522–6. [PubMed] [Google Scholar]
- 5.Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Research. 2015. doi: 10.1093/nar/gkv1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schermerhorn KM, Delaney S. A Chemical and Kinetic Perspective on Base Excision Repair of DNA. Accounts of Chemical Research. 2014;47(4):1238–46. doi: 10.1021/ar400275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dedon PC. The Chemical Toxicology of 2-Deoxyribose Oxidation in DNA. Chemical Research in Toxicology. 2007;21(1):206–19. doi: 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg MM. Reactivity of Nucleic Acid Radicals Advances in Physical Organic Chemistry: Academic Press; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markkanen E, Fischer R, Ledentcova M, Kessler BM, Dianov GL. Cells deficient in base-excision repair reveal cancer hallmarks originating from adjustments to genetic instability. Nucleic Acids Research. 2015. doi: 10.1093/nar/gkv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grasso S, Tell G. Base excision repair in Archaea: Back to the future in DNA repair. DNA Repair. 2014;21:148–57. doi: 10.1016/j.dnarep.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Khodyreva S, Lavrik O. New Players in Recognition of Intact and Cleaved AP Sites: Implication in DNA Repair in Mammalian Cells In: (Ed.) PCC, editor. Selected Topics in DNA Repair: InTech; 2011. [Google Scholar]
- 12.Piersen CE, McCullough AK, Lloyd RS. AP lyases and dRPases: commonality of mechanism. Mutation Research/DNA Repair. 2000;459(1):43–53. doi: 10.1016/S0921-8777(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 13.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Recent advances in the structural mechanisms of DNA glycosylases. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2013;1834(1):247–71. doi: http://dx.doi.Org/10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs A, Schär P. DNA glycosylases: in DNA repair and beyond. Chromosoma. 2012;121(1):1–20. doi: 10.1007/s00412-011-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Díaz M, Bebenek K, Kunkel TA, Blanco L. Identification of an Intrinsic 5′-Deoxyribose-5-phosphate Lyase Activity in Human DNA Polymerase λ. Journal of Biological Chemistry. 2001;276(37):34659–63. doi: 10.1074/jbc.M106336200. [DOI] [PubMed] [Google Scholar]
- 16.Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA Polymerase β Deoxyribose Phosphate Lyase: SUBSTRATE SPECIFICITY AND CATALYTIC MECHANISM. Journal of Biological Chemistry. 1998;273(24):15263–70. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 17.Demple B, Sung J-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair. 2005;4(12):1442–9. doi: http://dx.doi.Org/10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Verdine Gregory L., Norman DPG. Covalent Trapping of Protein-DNA Complexes. Annual Review of Biochemistry. 2003;72(1):337–366. doi: doi: 10.1146/annurev.biochem.72.121801.161447. [DOI] [PubMed] [Google Scholar]
- 19.Kosova AA, Khodyreva SN, Lavrik OI. Ku antigen displays the AP lyase activity on a certain type of duplex DNA. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2016;1864(9):1244–52. doi: 10.1016/j.bbapap.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 20.Strande NT, Carvajal-Garcia J, Hallett RA, Waters CA, Roberts SA, Strom C, Kuhlman B, Ramsden DA. Requirements for 5′dRP/AP lyase activity in Ku. Nucleic Acids Research. 2014. doi: 10.1093/nar/gku796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strande N, Roberts SA, Oh S, Hendrickson EA, Ramsden DA. Specificity of the dRP/AP Lyase of Ku Promotes Nonhomologous End Joining (NHEJ) Fidelity at Damaged Ends. Journal of Biological Chemistry. 2012;287(17):13686–93. doi: 10.1074/jbc.M111.329730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464(7292):1214–7. doi: http://www.nature.com/nature/journal/v464/n7292/suppinfo/nature08926_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutuzov MM, Khodyreva SN, Ilina ES, Sukhanova MV, Amé J-C, Lavrik OI. Interaction of PARP-2 with AP site containing DNA. Biochimie. 2015;112:10–9. doi: 10.1016/j.biochi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Prasad R, Horton JK, Chastain PD, Gassman NR, Freudenthal BD, Hou EW, Wilson SH. Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Research. 2014. doi: 10.1093/nar/gku288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khodyreva SN, Prasad R, Ilina ES, Sukhanova MV, Kutuzov MM, Liu Y, Hou EW, Wilson SH, Lavrik OI. Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc Natl Acad Sci USA. 2010;107(51):22090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quiñones JL, Demple B. When DNA Repair Goes Wrong: BER-Generated DNA-protein Crosslinks to Oxidative Lesions. DNA Repair. 2016. doi: 10.1016/j.dnarep.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Son M-Y, Jun H-I, Lee K-G, Demple B, Sung J-S. Biochemical Evaluation of Genotoxic Biomarkers for 2-Deoxyribonolactone-Mediated Cross-Link Formation with Histones. Journal of Toxicology and Environmental Health, Part A. 2009;72(21-22):1311–7. doi: 10.1080/15287390903212402. [DOI] [PubMed] [Google Scholar]
- 28.DeMott MS, Beyret E, Wong D, Bales BC, Hwang JT, Greenberg MM, Demple B. Covalent trapping of human DNA polymerase beta by the oxidative DNA lesion 2-deoxyribonolactone. J Biol Chem. 2002;277(10):7637–40. [DOI] [PubMed] [Google Scholar]
- 29.Sung JS, Demple B. Analysis of Base Excision DNA Repair of the Oxidative Lesion 2-Deoxyribonolactone and the Formation of DNA-Protein Cross-Links. In: Campbell J, Modrich P, editors. Methods in Enzymology: Academic Press; 2006. p. 48–64. [DOI] [PubMed] [Google Scholar]
- 30.Sung JS, Park IK. Formation of DNA-protein cross-links mediated by C1′-oxidized abasic lesion in mouse embryonic fibroblast cell-free extracts. Integrative Biosciences. 2005;9(2):79–85. doi: 10.1080/17386357.2005.9647255. [DOI] [Google Scholar]
- 31.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Cross-Linking of 2-Deoxyribonolactone and Its β-Elimination Product by Base Excision Repair Enzymest†. Biochemistry. 2003;42(8):2449–55. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 32.Faure V, Saparbaev M, Dumy P, Constant J-F. Action of multiple base excision repair enzymes on the 2′-deoxyribonolactone. Biochemical and Biophysical Research Communications. 2005;328(4):1188–95. doi: 10.1016/j.bbrc.2005.01.082. [DOI] [PubMed] [Google Scholar]
- 33.Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, Digiammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, Stolarik DF, Osterling DJ, Johnson EF, Maag D. Mechanistic Dissection of PARP1 Trapping and the Impact on in vivo Tolerability and Efficacy of PARP Inhibitors. Molecular Cancer Research. 2015. doi: 10.1158/1541-7786.mcr-15-0191-t. [DOI] [PubMed] [Google Scholar]
- 34.Murai J, Huang S-yN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Research. 2012;72(21):5588–99. doi: 10.1158/0008-5472.can-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B. Enzyme mechanism-based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proceedings of the National Academy of Sciences. 2015;112(28):8602–7. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trivedi RN, Wang X-h, Jelezcova E, Goellner EM, Tang J, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase β expression collectively predict sensitivity to temozolomide 2008. doi: 10.1124/mol.108.045112%.J Molecular Pharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang Q, Inane B, Schamus S, Wang XH, Wei L, Brown AR, Svilar D, Sugrue KF, Goellner EM, Zeng X, Yates NA, Lan L, Vens C, Sobol RW. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nature communications. 2014;5:5513 Epub 2014/11/27. doi: 10.1038/ncomms6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase lambda mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J Biol Chem. 2005;280( 18): 18469–75. Epub 2005/03/08. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- 39.Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. 2013;33(7):1468–73. Epub 2013/02/04. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y-j, DeMott MS, Hwang JT, Greenberg MM, Demple B Action of human apurinic endonuclease (Ape1) on C1′-oxidized deoxyribose damage in DNA. DNA Repair. 2003;2(2):175–85. doi: 10.1016/sl568-7864(02)00194-5. [DOI] [PubMed] [Google Scholar]
- 41.Prasad R, Horton JK, Chastain IIPD, Gassman NR, Freudenthal BD, Hou EW, Wilson SH. Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Research. 2014;42(10):6337–51. doi: 10.1093/nar/gku288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murai J, Pommier Y. Classification of PARP Inhibitors Based on PARP Trapping and Catalytic Inhibition, and Rationale for Combinations with Topoisomerase I Inhibitors and Alkylating Agents In: Curtin NJ, Sharma RA, editors. PARP Inhibitors for Cancer Therapy. Cham: Springer International Publishing; 2015. p. 261–74. [Google Scholar]
- 43.Prasad R, Horton JK, Dai D-P, Wilson SH. Repair pathway for PARP-1 DNA-protein crosslinks. DNA Repair. 2019;73:71–7. doi: 10.1016/j.dnarep.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takao M, Oohata Y, Kitadokoro K, Kobayashi K, Iwai S, Yasui A, Yonei S, Zhang Q-M. Human Nei-like protein NEIL3 has AP lyase activity specific for single-stranded DNA and confers oxidative stress resistance in Escherichia coli mutant. Genes to Cells. 2009;14(2):261–70. doi: 10.1111/j.l365-2443.2008.01271.x. [DOI] [PubMed] [Google Scholar]
- 45.Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of Formamidopyrimidines in DNA Involves Different Glycosylases: ROLE OF THE OGG1, NTH1, AND NEIL1 ENZYMES. Journal of Biological Chemistry. 2005;280(49):40544–51. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 46.Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair. 2005;4(1):41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Dizdaroglu M Oxidatively induced DNA damage and its repair in cancer. Mutation Research/Reviews in Mutation Research. 2015;763:212–45. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mechanisms of Ageing and Development. 2012;133(4):157–68. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20–33. doi: 10.1038/nrc.2015.210.1038/nrc.2015.2http://www.nature.com/nrc/journal/v16/n1/abs/nrc.2015.2.html#supplementary-informationhttp://www.nature.com/nrc/journal/v16/n1/abs/nrc.2015.2.html#supplementary-information . [DOI] [PubMed] [Google Scholar]
- 50.Demple B, DeMott MS. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene. 2002;21(58):8926–34. doi: 10.1038/sj.onc.1206178. [DOI] [PubMed] [Google Scholar]
- 51.Ide H, Nakano T, Salem AMH, Shoulkamy MI. DNA–protein cross-links: Formidable challenges to maintaining genome integrity. DNA Repair. 2018;71:190–7. doi: 10.1016/j.dnarep.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 52.Klages-Mundt NL, Li L. Formation and repair of DNA-protein crosslink damage. Science China Life Sciences. 2017;60(10):1065–76. doi: 10.1007/s11427-017-9183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mentegari E, Kissova M, Bavagnoli L, Maga G, Crespan E. DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair. Genes. 2016;7(9):57. doi: 10.3390/genes7090057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stingele J, Bellelli R, Boulton SJ. Mechanisms of DNA–protein crosslink repair. Nature Reviews Molecular Cell Biology. 2017;18:563. doi: 10.1038/nrm.2017.56. [DOI] [PubMed] [Google Scholar]
- 55.Ide H, Nakano T, Shoulkamy MI, Salem AM. Formation, repair, and biological effects of DNA-protein cross-link damage Advances in DNA Repair: InTech; 2015. [Google Scholar]
- 56.Xu YJ, DeMott MS, Hwang JT, Greenberg MM, Demple B. Action of human apurinic endonuclease (Ape1) on C1′-oxidized deoxyribose damage in DNA. DNA Repair (Amst). 2003;2(2):175–85. Epub 2003/01/18. [DOI] [PubMed] [Google Scholar]
- 57.Singhal RK, Wilson SH. Short gap-filling synthesis by DNA polymerase beta is processive. Journal of Biological Chemistry. 1993;268(21):15906–11. [PubMed] [Google Scholar]
- 58.Chagovetz AM, Sweasy JB, Preston BD. Increased Activity and Fidelity of DNA Polymerase β on Single-nucleotide Gapped DNA. Journal of Biological Chemistry. 1997;272(44):27501–4. doi: 10.1074/jbc.272.44.27501. [DOI] [PubMed] [Google Scholar]
- 59.Ba X, Boldogh I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biology. 2018;14:669–78. doi: 10.1016/j.redox.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee M-R, Kim S-H, Cho H-J, Lee K-Y, Moon AR, Jeong HG, Lee J-S, Hyun J-W, Chung M-H, You HJ. Transcription Factors NF-YA Regulate the Induction of Human OGG1 Following DNA-alkylating Agent Methylmethane Sulfonate (MMS) Treatment. Journal of Biological Chemistry. 2004;279(11):9857–66. doi: 10.1074/jbc.M311132200. [DOI] [PubMed] [Google Scholar]
- 61.Xu Y, Hansen WK, Rosenquist TA, Williams DA, Limp-Foster M, Kelley MR. Protection of Mammalian Cells against Chemotherapeutic Agents Thiotepa, 1,3-N,N′-Bis(2-chloroethyl)-N-nitrosourea, and Mafosfamide Using the DNA Base Excision Repair Genes Fpg and α-hOgg1: Implications for Protective Gene Therapy Applications. Journal of Pharmacology and Experimental Therapeutics. 2001;296(3):825–31. [PubMed] [Google Scholar]
- 62.Sandigursky M, Yacoub A, Kelley MR, Xu Y, Franklin WA, Deutsch WA. The yeast 8-oxoguanine DNA glycosylase (Ogg1) contains a DNA deoxyribophosphodiesterase (dRpase) activity. Nucleic Acids Research. 1997;25(22):4557–61. doi: 10.1093/nar/25.22.4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ide H, Kotera M. Human DNA Glycosylases Involved in the Repair of Oxidatively Damaged DNA. Biological and Pharmaceutical Bulletin. 2004;27(4):480–5. doi: 10.1248/bpb.27.480. [DOI] [PubMed] [Google Scholar]
- 64.Le Bihan Y-V, Angeles Izquierdo M, Coste F, Aller P, Culard F, Gehrke TH, Essalhi K, Carell T, Castaing B. 5-Hydroxy-5-methylhydantoin DNA lesion, a molecular trap for DNA glycosylases. Nucleic Acids Research. 2011. doi: 10.1093/nar/gkr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Girard P-M, Guibourt N, Boiteux S. The Ogg1 protein of Saccharomyces cerevisiae: a 7,8-dihydro-8-oxoguanine DNA glycosylase/AP lyase whose lysine 241 is a critical residue for catalytic activity. Nucleic Acids Research. 1997;25(16):3204–11. doi: 10.1093/nar/25.16.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nash HM, Lu R, Lane WS, Verdinel GL. The critical active-site amine of the human 8-oxoguanine DNA glycosylase, hOgg1: direct identification, ablation and chemical reconstitution. Chemistry & Biology. 1997;4(9):693–702. doi: 10.1016/S1074-5521(97)90225-8. [DOI] [PubMed] [Google Scholar]
- 67.Sidorenko VS, Zharkov DO. Role of base excision repair DNA glycosylases in hereditary and infectious human diseases. Mol Biol. 2008;42(5):794–805. doi: 10.1134/S0026893308050166. [DOI] [PubMed] [Google Scholar]
- 68.Donley N, Jaruga P, Coskun E, Dizdaroglu M, McCullough AK, Lloyd RS. Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (OGG1). ACS Chemical Biology. 2015;10(10):2334–43. doi: 10.1021/acschembio.5b00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacobs AC, Calkins MJ, Jadhav A, Dorjsuren D, Maloney D, Simeonov A, Jaruga P, Dizdaroglu M, McCullough AK, Lloyd RS. Inhibition of DNA Glycosylases via Small Molecule Purine Analogs. PLoS ONE. 2013;8(12):e81667. doi: 10.1371/journal.pone.0081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fell VL, Schild-Poulter C. The Ku heterodimer: Function in DNA repair and beyond. Mutation Research/Reviews in Mutation Research. 2015;763:15–29. doi: 10.1016/j.mrrev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 71.Thapar U, Demple B, Deployment of DNA polymerases beta and lambda in single- nucleotide and multinucleotide pathways of mammalian base excision DNA repair, DNA Repair 76 (2019) 11–19, 10.1016/j.dnarep.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.