

Abstract
Members of the mammalian inflammatory caspase family, including caspase-1, caspase-4, caspase-5, caspase-11, and caspase-12 are key regulators of the innate immune response. Most studies to date have focused on the role of caspase-1 in the maturation of the pro-inflammatory cytokine interleukin-1β and its upstream regulation by the inflammasome signaling complexes. However, an emerging body of research has supported a role for caspase-4, caspase-5, and caspase-11 in both regulating caspase-1 activation and inducing the inflammatory form of cell death called pyroptosis. This non-canonical inflammatory caspase pathway appears essential for the regulation of cytokine processing and, in turn, may provide important and, to date, understudied targets for the treatment of auto-inflammatory disorders where the inflammasome pathway is dysregulated. Here, we will discuss the mechanisms of inflammasome and inflammatory caspase activation and how these pathways intersect to promote pathogen clearance.
Keywords: Inflammasome, caspase-1, Interleukin-1β, NLRP3, ASC
Introduction
The innate immune system serves to sense microbial, host-derived, and foreign danger signals. It employs pattern recognition receptors that respond to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) released by cells in conditions of cellular damage or stress. Pattern recognition receptors are expressed by cells that make contact with invading microbes, including macrophages, monocytes, dendritic cells, neutrophils, endothelial cells, and cells of the adaptive immune system. Three types of pattern recognition receptors exist: Toll like receptors (TLR) that are expressed at the cell surface, intracellular NOD (nucleotide binding oligomerization domain) receptors, and receptors that provide the scaffolds for inflammasome assembly. Engagement of receptors in the first two groups primarily results in the activation of the transcription factor NFκB in response to extracellular or intracellular proinflammatory signals, respectively. The third group initiates the assembly of inflammasomes, cytosolic multi-protein signaling complexes that are essential for the maturation and release of certain cytokines during the innate immune response. A number of distinct inflammasomes exist but they all converge on the activation of caspase-1, a protease required to ensure correct regulation of inflammatory signaling.
Caspase-1 and its role in cytokine maturation
Caspase-1 was the first caspase identified and belongs to the subgroup of caspases termed inflammatory caspases that also includes caspase-4, caspase-5, caspase-11, and caspase-12 [1]. It was originally named interleukin (IL)-1β-converting enzyme (ICE), and was discovered as a protease primarily synthesized in the monocytic cell lineage [2]. Caspase-1 is an endopeptidase with a catalytically active cysteine residue responsible for the nucleophilic attack and cleavage of target proteins after an aspartic acid residue.
Caspase-1 is synthesized as an inactive zymogen (pro-caspase-1) that requires activation [3]. The prodomain of caspase-1 contains a conserved protein-protein interaction motif known as a CARD (caspase recruitment domain) that is essential for the recruitment of caspase-1 to inflammasomes [4]. Recruitment to inflammasomes induces oligomerization, which is sufficient to activate caspase-1 [5]. Three dimensional structures have revealed that the active enzyme is a homodimer of the catalytic domains [6]. Each catalytic domain is comprised of a p20 and a p10 subunit, generated as a result of auto-processing of the enzyme dimer (Figure 1). The p20 subunit contains the catalytic cysteine residue that is conserved among all caspase family members. Based on the structural similarity between caspase-1 and the apoptotic initiator caspases, caspase-2, −8, and −9, dimerization is considered to be essential for the activation of caspase-1, while auto-processing likely serves to stabilize the active enzyme [7, 8].
Figure 1:
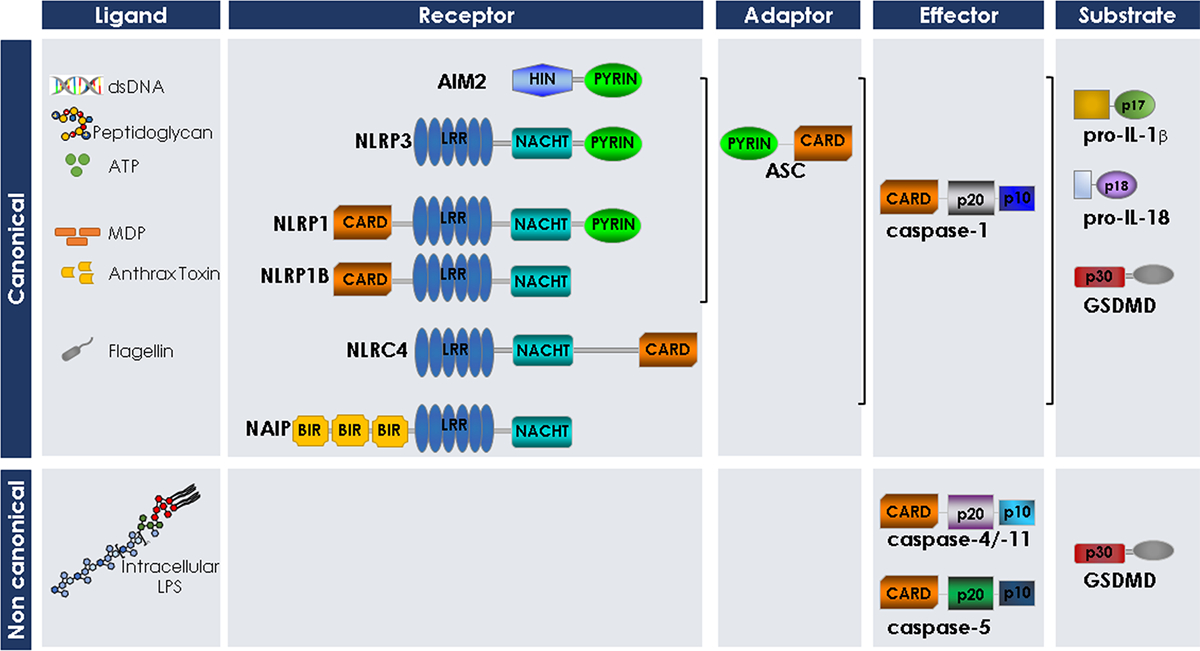
Domain organization of the inflammasome components, the inflammatory caspases and the inflammatory caspase substrates described in this review.
The main substrates for caspase-1 are the proinflammatory cytokines, IL-1β and IL-18 [2, 9]. These two cytokines share structural similarities, but IL-18 and IL-1β display different biological activities that are mediated through specific receptors [10]. IL-1β is an endogenous pyrogen that triggers fever, leukocyte tissue migration, and expression of other cytokines and chemokines [11]. It is produced as an inactive cytoplasmic precursor (proIL-1β, p35), which must be cleaved at Asp116 to generate the mature active form (p17) that is subsequently released from the cell [10]. IL-18 was first described in 1989 as an endotoxin-induced factor that stimulates the production of interferon(IFN)-γ in splenocytes [12]. It was first cloned in 1995 and was then shown to have an IL-1 signature sequence [13]. Its functions include the induction of other pro-inflammatory cytokines, upregulation of adhesion molecules, and activation of natural killer cell activity [14]. Like IL-1β, it is synthesized as an inactive precursor protein (proIL-18, p24) that is processed by caspase-1 at Asp36 to produce the mature 18 kDa peptide that is readily released from the cell [9, 15] (Figure 1, 2).
Figure 2:
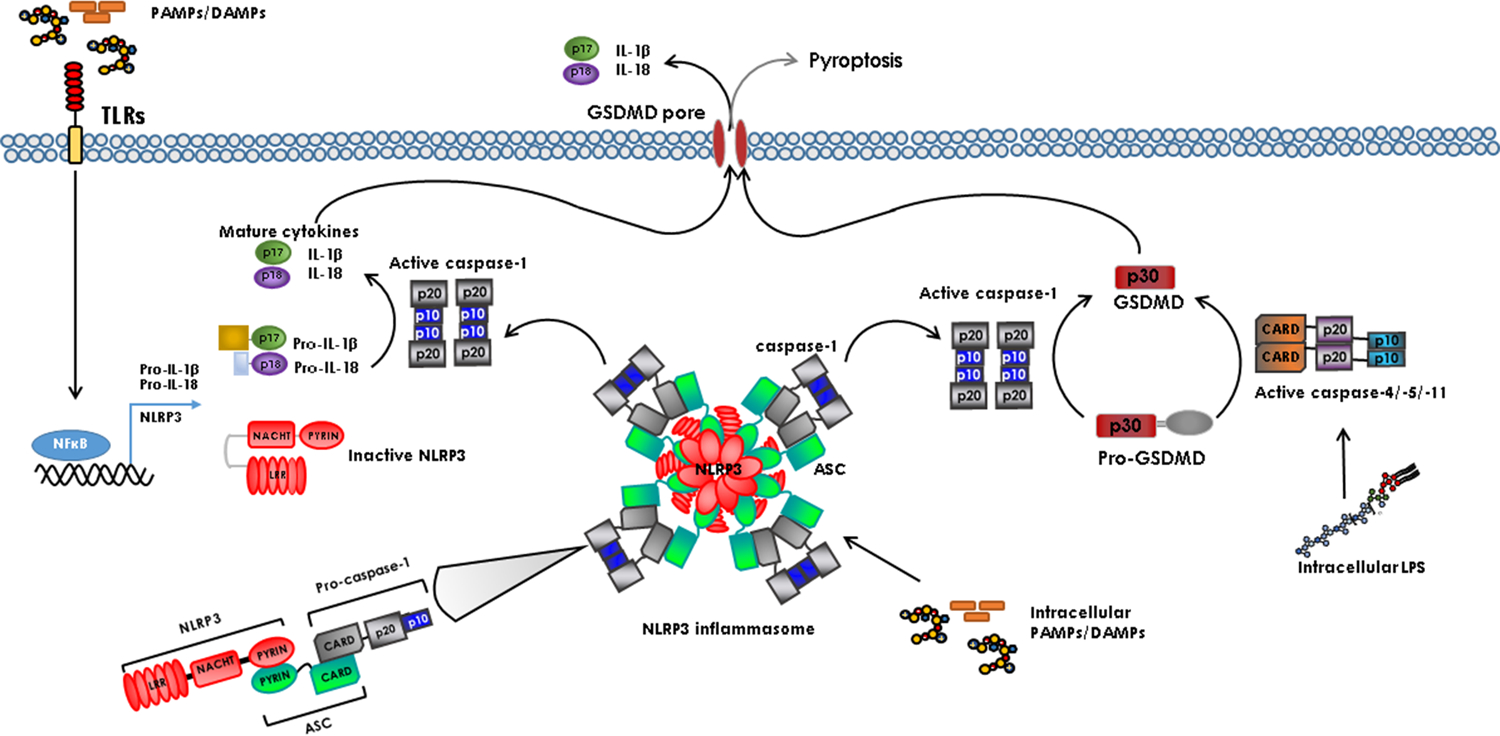
Model for inflammatory caspase activation. PAMPs and DAMPs activate TLRs on the plasma membrane, which induces expression of pro-IL-18, pro-IL-1β, and certain upstream inflammasome components including NLRP3. PAMPs and DAMPs trigger inflammasome assembly by inducing a conformational change in the NLR protein (NLRP3 is shown as an example) followed by oligomerization. NLRP3 recruits ASC bringing several caspase-1 molecules into proximity facilitating dimerization and activation. Once activated, caspase-1 cleaves IL-1β and IL-18 that are released from the cell through the GSDMD pore. Caspase-4/−5/−11 are activated directly by intracellular LPS to cleave GSDMD.
It is generally established that caspase-1 does not play a central role in apoptosis because the Casp1 knockout mouse does not display a phenotype consistent with disruption of a cell death pathway, unlike the gross abnormalities resulting from loss of Casp3, 8, or 9 (increased brain size, embryonic lethality, etc. [16]). In contrast, mice with targeted disruption of Casp1 have no apparent anatomical or developmental abnormalities, with normal counts of erythrocytes, leukocytes, and platelets in the peripheral blood. The mice showed no apparent defect in apoptosis and Casp1-deficient macrophages and thymocytes preserved their capacity to undergo apoptosis in response to multiple stimuli [17]. However, these mice have an acute defect in their capability to convert proIL-1β to its mature form and are resistant to lethal doses of endotoxin/lipopolysaccharide (LPS) [17]. These features of the knockout mouse indicate a more prominent role for caspase-1 in inflammation and associated processes than in apoptotic cell death.
There are some circumstances where caspase-1 may induce cell death. Caspase-1 has been shown to cleave the pro-apoptotic executioner caspase, caspase-7, which appears to be required for LPS-induced lymphocyte apoptosis [18]. In addition, caspase-1 has been shown to induce a form of cell death known as pyroptosis [19]. This type of cell death is morphologically distinct from apoptosis, which manifests as cell shrinkage, membrane blebbing and DNA fragmentation, resulting in cells being packaged into apoptotic bodies and phagocytized without causing inflammation. Pyroptosis is initiated in response to microbial pathogens and is characterized by cell swelling, membrane rupture, and release of proinflammatory cellular contents [20]. It is characterized by the formation of plasma membrane pores between 1.1 and 2.4 nm in diameter, large enough to allow the influx of extracellular calcium ions required for lysosome exocytosis [21]. These pores dissipate cellular ionic gradients, inducing a net increased osmotic pressure, water influx, and cell swelling that eventually results in osmotic lysis and release of inflammatory intracellular contents [21]. Thus, cells undergoing pyroptosis display a measurable increase in size [21, 22].
The mechanism by which mature IL-1β is released from the cell is unclear. This was originally thought to be an active secretion event, but IL-1β release does not depend on the conventional ER-Golgi secretion machinery [23]. More recently, it has been proposed that IL-1β release is a byproduct of pyroptotic cell death [24, 25]. However, IL-1β release from living cells has been reported [26, 27] and, therefore, it is possible to uncouple IL-1β release from lytic cell death. Pyroptosis is often thought to be a caspase-1-dependent type of cell death [20, 28], but caspase-1-independent pyroptosis has been observed (see next section). Emerging evidence suggests that this type of cell death is equally dependent on additional members of the inflammatory caspase family.
Caspases-4, −5, −11 and their role in inducing pyroptosis
The inflammatory caspases, including caspase-1, caspase-4, caspase-5, caspase-11, and caspase-12, are encoded by four genes in humans: CASP1, CASP4, CASP5, and CASP12; and three genes in mouse: Casp1, Casp11 and Casp12. Caspase-1, −4, −5, and −12 are clustered on human chromosome 11q22 and arose by tandem gene duplication. Like caspase-1, each of these proteins encodes a CARD in their prodomain (Figure 1). Sequence analysis of the human inflammatory caspases shows that caspase-1 and caspase-4 share 55% identity, while pro-caspase-4 and −5 share an amino acid sequence identity of 77% [1]. Even though human and mouse caspase-1 are likely orthologs, it is still unclear if human caspase-4, caspase-5, or both, are duplicated counterparts of murine caspase-11. Comparison of the amino acid sequences of human and murine caspases reveals that procaspase-4 and −5 are 59 and 54% identical to procaspase-11, respectively, and 48 and 45% identical to procaspase-12, respectively [1]. Caspase-4 and caspase-11 mRNA have similar tissue distribution patterns [29], but caspase-5 and caspase-11 are both LPS- or IFN-γ-inducible [30]. Despite the similarities between caspase-11 and caspase-5, most publications consider caspase-4 to be the functional homolog of caspase-11. Because caspase-4 and −5 are not expressed in mice, many of the conclusions regarding their functions have been based on research on their closest murine homolog, caspase-11.
Caspase-11 is a murine caspase that has much in common with caspase-1. The phenotype of the Casp-11 knockout mouse is similar to that of Casp-1 displaying resistance to endotoxic shock and defects in IL-1β processing [31]. However, caspase-11 did not directly cleave pro IL-1β. Consequently, caspase-11 was initially described as an upstream regulator of caspase-1 activation. It was not until a number of years later, when it was discovered that the published Casp1 mouse model was also deficient in caspase-11 [32], that it became clear that these two caspases have distinct as well as overlapping functions. The Casp1−/− mouse was generated using strain 129 embryonic stem cells, which has an incidental deletion in the Casp11 locus. Because Casp11 is only 1500 base pairs away from Casp1, multiple generations of backcrossing to C57BL/6 mice could not segregate the two mutant genes. Casp11−/− and Casp1−/−/11−/− mice were equally resistant to LPS-induced septic shock. However, when caspase-11 expression was reconstituted in Casp1−/−/Casp11−/− mice using a BAC-derived transgene, essentially creating a true caspase-1 knockout, the mice were no longer resistant to LPS challenge [32]. Therefore, sensitivity to LPS is conferred by caspase-11 rather than by caspase-1. Mice lacking both IL-1β and IL-18 do not phenocopy the LPS resistance of the Casp11 knockout mouse [33]. Thus, the LPS-induced lethality is not considered to be a result of elevated levels of these pro-inflammatory cytokines in the blood. Rather, it is thought that this form of septic shock is actually a direct result of caspase-11-induced tissue damage. While this idea has not been directly tested, it is supported by the fact that caspase-11 appears to be a major driver of pyroptosis in response to certain inflammatory stimuli.
Both caspase-1 and caspase-11 can induce pyroptosis, but the stimuli that engage each of these caspases to do so differ. Caspase-1-dependent cell death was induced by stimuli, such as ATP, that engage known inflammasomes and this was accompanied by IL-1β release. These effects were not dependent on caspase-11. In contrast, IL-1β release in LPS-primed macrophages in response to cholera toxin B (CTB), required both caspase-1 and caspase-11. However, CTBinduced cell death was blocked in the absence of caspase-11. These results suggest that caspase-11 regulates caspase-1 activation and the resulting processing of IL-1β/IL-18, but the role of caspase-11 in death signaling appears to be independent of known inflammasome components [32]. This pathway has been termed the non-canonical inflammasome pathway.
While CTB was originally considered a specific inducer of caspase-11, a follow up study showed that CTB was functioning to deliver LPS into the macrophages. Dixit and colleagues observed that CTB only induced caspase-11 activation when macrophages were primed with a specific E.coli serotype of LPS, O111:B4 [34]. They went on to show that CTB was dispensable for caspase-11 activation, but rather bound to this specific type of LPS and promoted endocytosis of the complex into the cell. Because the activation of caspase-11 was shown to be TLR4-independent, this appears to be a response to intracellular LPS, specifically the lipid A moiety of LPS. Intracellular LPS-induced pyroptosis in human monocytes is similarly dependent on caspase-4, the human homolog of caspase-11 [35]. It has been proposed that LPS directly activates caspase-4, caspase-5, and caspase-11 by binding to the CARD of the caspase and inducing oligomerization [35] (Figure 2). Hence, caspase-4, caspase-5, and caspase-11 appear to be intracellular receptors for LPS, triggering pyroptosis and caspase-1 activation in response to invasive bacteria. Indeed, studies have shown that in the absence of caspase-1, caspase-11 promotes more severe disease in mice infected with Salmonella typhimurium. Infected Casp11−/− mice had a higher bacterial load than Casp1−/−/Casp11−/− mice [36]. This is thought to result from excess pyroptosis, releasing bacteria that are not effectively cleared by neutrophils due to the absence of caspase-1.
Inflammatory caspase-induced pyroptosis is dependent on a substrate called gasdermin D (GSDMD) that is cleaved by caspase-1, caspase-4, caspase-5, and caspase-11 [37, 38] (Figure 2). GSDMD belongs to the family of gasdermins (GSDMs), which are pore-forming proteins expressed in a variety of cell types and tissues [39]. After cleavage, the N-terminal fragment of GSDMD travels to the plasma membrane to bind to acidic lipids on the inner leaflet, which results in a conformational change that allows GSDMD to form ring shaped structures inserted in the plasma membrane [40, 41]. Based on the solved structure of the GSDMA3 pore (a related member of the family), the pore is estimated to have an inner diameter of 180Å [42]. It has been shown that GSDMD is required for IL-1β release even if cell lysis is prevented [26], which suggests that the GSDMD pore allows IL-1β efflux in addition to the ion influx that causes cell lysis. IL-1β release from living cells has been reported in response to induced caspase-1 activation and in hyperactivated macrophages [26, 27]. It is likely that the extent of GSDMD pore formation determines if pyroptosis occurs but the exact stimuli and physiological conditions that result in IL-1β release without pyroptosis as well as the impact of such an event on the immune response have not been fully elucidated.
Caspase-12 and its role in sepsis
Initial studies in caspase-12-deficient mice suggested that caspase-12 was an ER stress-sensing protease that mediated an ER-specific apoptosis pathway [43]. These studies were later challenged and indicated that caspase-12 played no role in the mitochondrial pathway of cell death [44, 45]. Later studies showed that Casp12−/− mice display marked resistance to septic shock suggesting that caspase-12 may function to inhibit caspase-1 [46]. In humans, caspase-12 is expressed predominantly as a truncated version containing only the N-terminal CARD domain (caspase-12S). Only approximately 20% of people of African descent express the full-length molecule (caspase-12L), which is linked to hypo-responsiveness to LPS-induced production of cytokines such as IL-1β and increased severity of sepsis [47]. Thus, the evolutionary loss of the catalytic domains of caspase-12 may have conferred a selective advantage by increasing resistance to sepsis in human populations.
Inflammasomes: the caspase-1 signaling platforms
The inflammasome is a large intracellular signaling platform composed of a NOD-like receptor (NLR), an adaptor protein (ASC [apoptosis-associated speck-like protein containing a CARD]), and the protease (caspase-1) [4]. The central scaffold NLR proteins of inflammasomes are innate cytosolic receptors that recognize diverse PAMPs and DAMPs. These NLR monomers remain in an inactive conformation until an external or internal signal triggers their conformational change and assembly. NLRs characteristically possess three main domains: the leucine-rich repeat (LRR) domain, which is a region involved in ligand sensing; a central nucleotide-binding domain (NACHT), which drives oligomerization, and a domain involved in recruiting the caspase. The latter domain can be a pyrin domain (PYD), which forms a complex with ASC that, in turn, recruits caspase-1, or a CARD that directly recruits caspase-1 (Figure 1). The CARD and PYD are specific conserved protein interaction domains that mediate these protein-protein interactions. They are characterized by 6 antiparallel α-helices with a hydrophobic core and an outer surface composed of charged residues. There are variations in the length and orientation of these α-helices, and the specificity of protein-protein interactions largely depends on the charged and hydrophobic pockets on the surface [48]. The NLR molecules are thus named NLR with the suffix P or C in reference to the N terminal moiety, PYD or CARD respectively, followed by a number denoting the order in which the NLRs were discovered [49].
A number of distinct inflammasomes exist, the most-well studied being the NLRP1, NLRP3, NLRC4, and AIM2 (absent in melanoma 2) inflammasomes. They each respond to specific stimuli. For example, ATP engages the NLRP3 inflammasome; flagellin triggers the NLRC4-dependent inflammasome; and poly(dA:dT) or double-stranded DNA (dsDNA) activates the AIM2-dependent inflammasome. Common to most of the characterized inflammasomes is the adaptor protein ASC. ASC is required for NLRP3, and AIM2 activation of caspase-1. Like Casp1−/− cells, Asc-deficient cells display inefficient cytokine processing, and therefore lower levels of mature IL-1β and IL-18 release in response to a range of stimuli [50]. In response to most inflammasome activating stimuli, endogenous ASC assembles in the cytoplasm to form a single speck with a diameter of over 2 μm [51–53]. This ASC speck is often interpreted as representing the platform for recruitment and activation of caspase-1 [54, 55]. In vitro reconstitution of the ternary inflammasome suggests caspase-1 is over-stoichiometric to ASC, by approximately 3.5-fold [52].
Cryo-EM structures of the AIM2 and NLRP3 inflammasomes show that AIM2 and NLRP3 are able to induce full length ASC to convert to a self-perpetuating prion form [52, 56]. This prion-like polymerization of ASC provides a mechanism of response to a range of noxious agents. NLRP3 or AIM2 activation induces a conformational change that promotes the oligomerization of their individual PYDs. This is followed by the recruitment of multiple ASC proteins through PYD-PYD interactions resulting in prion nucleation, a process otherwise prevented from occurring spontaneously due to a high-energy barrier. These ASC prions guide other ASC molecules resulting in the formation of large polymers or filaments. Finally, the ASC filaments recruit multiple caspase-1 molecules through CARD-CARD interactions, bringing them into close proximity to induce their dimerization and activation. Further amplifying the signal, the ASC-CARD and caspase-1-CARD can each self-assemble into filaments [57, 58]. Nucleation of ASC prions is tightly regulated by heterotypic interactions with upstream protein sensors. The tight regulation of nucleation followed by efficient polymerization produces a highly sensitive and robust response to harmful signals.
The NLRP3 Inflammasome
Although it was not the first inflammasome discovered, the NLRP3 (NLR family pyrin-domain containing 3) inflammasome (also known as cryopyrin, NALP3, PYPAF1, or CIAS1) is the one that is best characterized. This is because a large number of mechanisms have been shown to play a role in the activation of the NLRP3 inflammasome, providing it a central role in anti-microbial immunity and sterile inflammation. Upon activation, NLRP3 nucleates ASC helical clusters through PYD-PYD interactions. The oligomerized ASC CARDs then form the platform for caspase-1 CARD to nucleate into filaments, which in turn activates caspase-1 (Figure 2) [59]. Pore formation is a common step shared by a number of NLRP3 activating stimuli. Consequently, NLRP3 inflammasome activation has been observed in response to many bacterial pore-forming toxins including: the marine toxin, maitotoxin, listeriolysin O from Listeria monocytogenes, aerolysin from Aeromonas hydrophila and Staphylococcus aureus hemolysins [60, 61]. NLRP3 is also activated by a range of DAMPs including uric acid crystals, extracellular ATP, silica, aluminium adjuvants, and asbestos [62–64].
One event that is generally required, but not sufficient, for NLRP3 inflammasome activation is the efflux of intracellular potassium [65]. Thus, the bacterial potassium ionophore, nigericin, is a potent inducer of the NLRP3 inflammasome [65, 66]. ATP also activates caspase-1 in an NLRP3-dependent fashion that requires K+ efflux [61, 67]. It has been proposed that ATP binds to the P2X7 receptor to recruit the channel pannexin-1, leading to the formation of a membrane channel mediating K+ efflux [66, 68]. However, a more recent study has identified TWIK2 as an ATP-responsive K+ efflux channel. Using gene expression analysis and targeted gene depletion, it was established that TWIK2 is required for the regulation of the ATP-induced NLRP3 inflammasome activation and sepsis-induced inflammatory lung injury in mice [69]. How K+ efflux induces NLRP3 activation is unclear. It has been shown that K+ efflux promotes the binding of the kinase NEK7 to NLRP3 [70, 71]. This interaction appears to be essential for NLRP3 inflammasome assembly and it is possible that binding of NEK7 to the LRRs of NLRP3 induces a conformational change allowing oligomerization of NLRP3.
The requirement of generation of reactive oxygen species (ROS) for NLRP3 activation is subject to some debate. Most stimuli that activate the NLRP3 inflammasome increase mitochondrial ROS (mtROS) and the use of ROS scavengers or inhibitors is known to block NLRP3-dependent caspase-1 activation [62]. One study showed that mtROS generation induced by treatment with the complex I inhibitor rotenone was sufficient to activate NLRP3 [72]. However, studies using cells from patients with mutations in subunits of NADPH-oxidase (NOX), which are essential for the generation of mtROS, have no defects in NLRP3 signaling [73–75]. Unique among the inflammasome receptors, NLRP3 requires a priming step for activation [76, 77]. In experimental practice, this is often in the form of LPS treatment, which is needed to induce NLRP3 expression. Inhibition of ROS inhibits this priming step [78]. Therefore, while mitochondrial ROS generation may be required to induce sufficient NLRP3 expression to allow the inflammasome to assemble, it does not appear to be a direct activator of NLRP3 and a second signal is required. This second signal may be oxidized mitochondrial DNA (mtDNA) that has been shown to bind and activate the NLRP3 inflammasome [79, 80]. The production of oxidized mtDNA results from TLR-dependent CMPK2 expression which is required for de novo mtDNA synthesis. mtDNA is highly susceptible to oxidization and fragmentation and the release of the mtDNA fragments from mitochondria lead to NPRP3 activation [79]. Thus, it appears that mtROS production and mitochondrial damage play an important, if indirect role in the assembly of the NLRP3 inflammasome providing the priming signal for NLRP3 expression and for production of the NLRP3 intracellular ligand.
The NLRC4 inflammasome
NLRC4 (also known as IPAF, CARD12, CLR2.1, or CLAN) is also capable of initiating caspase-1 activation and IL-1β processing. Flagellin is the canonical stimulus for the NLRC4-dependent inflammasome, but the NLRC4 inflammasome can also be activated independently of flagellin [81]. For example, the non-flagellated bacterium S. flexneri has been shown to activate caspase-1 in an NLRC4-dependent manner [82]. NLRC4 lacks a PYD but contains a CARD and is thus able to directly interact with and activate caspase-1, independently of ASC [83] (Figure 1). Therefore, the NLRC4 inflammasome can induce caspase-1 activity in cells that are deficient in Asc [55]. Nevertheless, it has been shown that ASC greatly enhances the efficiency of NLRC4-mediated pro-IL-1β and pro-IL-18 maturation [55, 84]. Activation of the NLRC4 inflammasome by flagellated bacteria such as S. typhimurium induces pyroptosis. This type of NLRC4-dependent cell death is induced by S.typhimurium in logarithmic phase and requires the type III secretion system (T3SS) encoded by Salmonella pathogenicity island 1 (SPI-1), which causes the bacteria to secrete flagellin [84]. Cell death through this mechanism was independent of ASC and caspase-1 [55]. This suggests that while NLRC4 can recruit ASC and caspase-1 to induce cytokine processing, it can also form as separate complex independent of caspase-1 that induces cell death. We previously demonstrated that caspase-4 or capase-5 dimerization is induced upon NLRC4 overexpression [53]. Thus, it is possible that pyroptosis induced by this logarithmic phase S.typhimurium assembles an NLRC4 inflammasome that recruits and activates caspase-4 or caspase-5 to induce pyroptosis. Evidence also suggests that triggers for NLRC4 can independently induce pyroptosis. When cells are infected with S. typhimurium in stationary phase, such that the bacteria can invade the cell, it induces a slower form of lytic cell death. Intracellular S. typhimurium engaged both the NLRC4 and NLRP3 inflammasomes to induce caspase-1 activation and cytokine processing [54], but cell death induced in this context was only partially dependent on NLRC4 and independently required caspase-11 [36].
NLRC4 does not sense bacterial ligands directly, but instead does so via NLR apoptosis-inhibitory proteins (NAIPs). NAIPs are a family of NLR proteins found in the cytosol that directly recognize pathogenic ligands and trigger assembly of the NLRC4 inflammasome in a ligand specific fashion. The NAIPs are homologues of NLRC4, containing a BIR (baculovirus inverted repeat) domain in place of the CARD (Figure 1). Mice express up to 7 NAIPs, all of them found within the same genomic locus, with Naip1, Naip2, Naip5 and Naip6 expressed in the commonly used C57BL/6 strain. Humans express a single NAIP gene. In mice, NAIP5 was shown to be required for NLRC4 activation by the C-terminus of flagellin [85], while NAIP2 was required for the NLRC4 response to the inner rod protein TTSS, such as PrgJ in Salmonella enterica or BsaK in B. thailandensis [86, 87]. The LRRs of NAIP5 or NAIP2 protein bind to flagellin or the TTSS rod protein respectively, allowing it to assume an open confirmation, which sequentially recruits NLRC4 monomers, similarly forcing each one in turn to overcome its auto-inhibited confirmation [88, 89]. This active conformation propagates to 8–10 more NLRC4 molecules creating a wheel like structure that provides the platform for caspase-1 CARD recruitment.
The NLRC4 inflammasome can also recruit the pro-apoptotic initiator, caspase-8, in an ASC-dependent fashion, but independent of caspase-1 and caspase-11 [90]. This appears to result in activation of caspase-8, which contributes to the regulation of pro-IL-1β expression but not IL-1β processing or cell death. It seems, therefore, that caspase-8 serves as a checkpoint to modulate the amount of pro-IL-1β available for caspase-1-dependent maturation. This NLRC4-ASC-caspase-8 complex was shown to be important in the inflammatory response of intestinal epithelial cells (IECs) to Salmonella [91]. Caspase-8 was required for the non-lytic expulsion of IECs and release of IL-18, which could compensate for the loss of caspase-1. In macrophages, inhibition of caspase-1 or GSDMD, favors the formation of the NLRC4-ASC-caspase-8 complex [92] and Casp8 deficiency in Casp1−/−Casp11−/− macrophages conferred resistance to NLRC4-mediated cell death [93]. In cells that either lacked caspase-1 expression or displayed low caspase-1 activity, the NLRC4/ASC/caspase-8 complex led to induction of apoptosis, an event that could be inhibited by TLR-mediated upregulation of the caspase-8 inhibitor cFLIP [93, 94]. Similar observations have been made for the AIM2, NLRP1B, and NLRP3 inflammasomes, suggesting that caspase-8 recruitment is a more general cell death response of inflammasomes when the caspase-1 pathway is blocked [94–96]. However, the significance of this for controlling inflammation or clearance of pathogens is unclear.
NLRP1 inflammasome
NLRP1 (NLR family Pyrin domain containing 1 [also known as NALP1, DEFCAP, NAC, CARD7 and CLR17.1]) was initially characterized as a member of the CED-4 family of mammalian apoptotic proteins, based on its structural similarity with APAF1 (apoptotic peptidase activating factor 1). Human NLRP1 contains an N-terminal PYD, followed by five tandem LRRs, a centrally located NACHT domain, and a C-terminal CARD (Figure 1) [97, 98].
The NLRP1 inflammasome was the first inflammasome identified and was originally reported to assemble in response to LPS [4]. Later, it was found that the activating component was actually a common contaminant of LPS preparations, the peptidoglycan component muramyl dipeptide (MDP) [99]. Removal of the LRR domain activates the NLRP1 inflammasome indicating that this domain has an auto-inhibitory function [4]. Therefore, a two-step activation mechanism has been proposed where first, MDP binds to the LRR inducing a conformational change in NLRP1, and second, NTPs bind to the NACHT domain inducing oligomerization to create a platform for caspase activation [99]. ASC is not required for NLRP1-mediated caspase-1 activation, but it does increase caspase-1 activity in a concentration-dependent manner. This improved efficiency is apparently dependent on ASC interacting with the PYD, allowing each molecule of NALP1 to recruit two procaspase-1 molecules [99]. In addition to its recruitment of caspase-1, NLRP1 has also been shown to bind caspase-5, mediated by the C-terminal CARD [4]. However, the functional significance of this interaction remains unclear.
In mice, the dominant form of NLRP1 expressed is Nalp1b which lacks the N-terminal PYD and therefore activates caspase-1 by a direct binding mechanism via the CARD (Figure 1). This isoform of NLRP1 is activated by the lethal factor (LF) component of B. anthracis lethal toxin (LT) [100]. LF cleaves a less than 10 kDa fragment off the N-terminal end of NLRP1B, a cleavage event that appears essential for NLRP1B inflammasome assembly [101]. Activation of NALP1B protein by anthrax LT in the absence of ASC induces caspase-1 activation, IL-1β release, and cell death, but not caspase-1 processing [102]. Thus, autocleavage of caspase-1 is not essential for activation by the NLRP1B inflammasome.
AIM2 inflammasome
AIM2 (also known as PYHIN4) is an interferon-inducible gene that was identified during a screening for tumor suppressor genes associated with melanoma [103]. AIM2 is a member of the HIN-200 family of IFN-inducible proteins and is the only one capable of activating caspase-1. It is a cytoplasmic DNA sensor with a high affinity for dsDNA. DNA sensing is mediated by the C-terminal HIN-200 domain for oligonucleotide/oligosaccharide-binding [104]. AIM2 recruits ASC and caspase-1 via its N-terminal PYD to facilitate DNA-mediated IL-1β processing and pyroptosis [105]. Intramolecular interaction between the HIN-200 domain and the PYD maintains the receptor in an auto-inhibited state in the absence of ligand binding [106]. Crystal structures of the PYD and HIN-200 domain demonstrate a strong preference for the two domains to interact through their respective charged surfaces. This auto-inhibition serves to ensure that AIM2 interacts with the downstream adapter ASC only upon activation by the dsDNA ligand [107].
Initial studies showed that silencing of AIM2 in human or mouse macrophages led to decreased inflammasome activation, caspase-1 activation, and cell death induced by vaccinia virus, cytoplasmic DNA, or poly (dA:dT) [105, 108]. This was confirmed in the Aim2−/− mice as the mice had impaired IL-1β release in response to synthetic DNA but not to activators of other inflammasomes [109–111]. Thus, the AIM2 inflammasome is essential for the inflammatory response to DNA viruses [109, 110]. The Aim2−/− mice also have impaired inflammation and survival in response to certain bacteria such as Francisella tularensis, the virulence of which is dependent on its ability to replicate in the host cell [109–111]. AIM2 is activated by other bacteria such as L. monocytogenes and F. novicida that release their DNA in the host cytosol after lysis of the bacteria within the host cell [112–117]. AIM2 is also activated by DNA released from necrotic cells. For example, in a murine model of chronic kidney injury, Aim2 deficiency diminished renal injury, fibrosis, and inflammation due to impaired macrophage uptake of DNA from necrotic cells [118].
In addition to activating the AIM2 inflammasome, cytosolic DNA can engage the cGAS/STING pathway to produce an interferon response. Cleavage of GSDMD downstream of the AIM2 inflammasome has been shown to induce K+ efflux that inhibits the cGAS/STING pathway [119]. This may be a means to mitigate damage resulting from sustained IFN signaling. Similarly, the cGAS/STING pathway itself can induce K+ efflux downstream of lysosomal damage that activates the NLRP3 inflammasome pathway [120]. Therefore, there is considerable crosstalk and cross-regulation between different inflammasomes and different inflammatory pathways.
Additional Inflammasomes
Other inflammasomes exist, including IFI-6, NLRP6, NLRP7, NLRP12, and NLRC5, but are less well characterized. While these atypical inflammasomes all have been documented to include ASC and caspase-1, the NLR components of each have more restrictive or additional, more dominant, roles. IFI-6 (interferon-inducible protein 16) is similar to AIM2 but is predominantly expressed in the nucleus to respond to DNA from viruses that enter the nucleus [121]. NLRP6 and NLRP12 have both been shown to dampen the immune response by suppressing NFκB signaling [122–124]. However, in the context of inflammasome activation these proteins have both been reported to have proinflammatory roles. NLRP6 is a key regulator of intestinal homeostasis [125] and has been shown to respond to lipoteichoic acid to recruit ASC and both caspase-1 and capsase-11 [126]. NLRP12 activates caspase-1 and IL-18 in response to Yersinia Pestis, the causative agent of plague, but the precise ligand for NLRP12 is unknown [127]. NLRP7 is expressed in oocytes and is important for embryonic development [128]. Finally, NLRC5 can interact with NLRP3, potentially enhancing the function of the NLRP3 inflammasome [129].
Genetic alteration of inflammasome pathways in auto-inflammatory diseases
Correct regulation of the inflammasome pathways has been implicated in the prevention of multiple disease states including sepsis, cardiovascular disease, and neurodegeneration [130–132]. The essential nature of inflammasomes is particularly demonstrated in a rare collection of monogenic human auto-inflammatory disorders that have been associated with mutations in the scaffold proteins of inflammasomes [55] (Table 1). The most well-known of these disorders, the cryopyrin-associated periodic syndromes (CAPS), are the result of mutations in NLRP3, which result in a constitutively active protein and lead to increased activation of the NLRP3 inflammasome and resultant increased secretion of IL-1β [133]. The clinical phenotypes of patients with CAPS vary in organ involvement and severity but can span from recurrent fevers and inflammatory skin rashes to hearing loss and devastating neuro-inflammation [134–136]. Fortunately, therapeutic blockade of the exacerbated IL-1β production has proven to be highly successful in abrogating disease activity in CAPS [137, 138]. Human disease is also caused by gain-of-function mutations in NLRC4, which seem to similarly release the protein from its baseline state of auto-inhibition and lead to high serum levels of both IL-1β and IL-18 [139]. As in CAPS, patients with NLRC4 inflammasomopathies present with a varied spectrum of inflammatory findings from recurrent skin rashes across the clinical continuum to life-threatening neonatal entercolitis [140]. Some patients with NLRC4 mutations appear to partially respond to IL-1 blockade [140], but elevated free IL-18 levels seem to uniquely distinguish this disorder from CAPS [141], and targeted therapeutics are being pursued (clinicaltrials.gov NCT03113760). Variants in and around NLRP1 have also been described in association with human disease [142–144] and in some cases enhanced NLRP1 inflammasome activation from these mutations has been confirmed and results in elevated IL-1β and IL-18 [142, 144]. The clinical phenotype associated with NLRP1 mutations appears to be predominantly cutaneous disease, spanning from vitiligo to dyskeratosis [142–144]. NLRP1 mutations only appear to be additionally associated with auto-inflammatory features, such as fever, in a subset of patients, one of whom was successfully treated with IL-1 blockade [144]. Human mutations in AIM2 as a cause of monogenic auto-inflammation have not been reported, perhaps due to the dual role of AIM2 as a tumor suppressor [103]. The finding that gain-of-function mutations in critical inflammasome proteins leads to human auto-inflammation underscores the importance of regulating inflammatory caspase activation. Further understanding of the mechanisms behind inflammasome activation will expand the therapeutic targets available for controlling human inflammatory disease [145, 146].
Table 1.
Conditions involving inflammatory caspases and inflammasome components
| Condition/Disease | Inflammasome involved | Characteristics | Citations |
|---|---|---|---|
| Vitiligo | NLRP1 | Loss of pigment-forming melanocytes; patches of white skin | [143, 147] |
| Addison disease | NLRP1 | Adrenocortical insufficiency. Requires lifelong glucocorticoid and mineralocorticoid replacement therapy | [148, 149] |
| Systemic lupus erythematosus | NLRP1 | Immune response against double-stranded DNA; cell death; organ failure. | [150] |
| NLRP1-associated auto-inflammation with arthritis and dyskeratosis (NAIAD) | NLRP1 | Dermatological anomalies; recurrent fever; chronic synovial inflammation of multiple joints | [144] |
| Kawasaki disease | NLRP1 | Acute febrile illness of childhood. Systemic vasculitis with infiltration of lymphocytes, macrophages and neutrophils in the vascular walls | [151] |
| Celiac disease | NLRP1 and NLRP3 | Abnormal immune response to gluten; gastrointestinal problems; failure to grow normally | [152] |
| Muckle-Wells syndrome (MWS) | NLRP3 | Frequent nonspecific limb pain, acute febrile inflammatory episodes; abdominal pain, joint inflammation, myalgia, urticaria, and conjunctivitis. | [134, 136] |
| Familial cold urticaria (FCU/FACS) | NLRP3 | Hypersensitivity to cold air, skin lesions, urticarial lesions, petechiae, joint pain, conjunctivitis, chills, nausea, fever. | [136] |
| Chronic infantile neurological cutaneous and articular syndrome (CINCA, NOMID) | NLRP3 | Onset very early in life; severe dermatologic, rheumatologic, and neurologic manifestations; multisystem inflammation. | [135] |
| NLRC4 macrophage activation syndrome (NLRC4-MAS) | NLRC4 | Human NLRC4 gain-of-function mutations (H443P, T337A and V341A) | [139] |
| Syndrome of enterocolitis and auto-Inflammation associated with mutation in NLRC4 (SCAN4) | NLRC4 | Gain-of-function mutations (H443P, T337A and V341A) of the human NLRC4 | [153] |
| Inflammation in Hepatitis B | AIM2 | Increased inflammation in chronic hepatitis B patients | [154] |
Closing Remarks
IL-1β targeted therapy has shown great success as a therapeutic intervention for a number of auto-inflammatory conditions. However, it is not effective for every patient and for every disease. Over the last two decades, research into the role of inflammasomes in the regulation of capsase-1 activation and the consequent inflammatory cytokine signaling has provided insight into the crucial role of these complexes both in protecting from infection and injury, and in preventing auto-inflammation. Recent discoveries regarding the crucial inflammatory roles of caspase-4, −5, −11 and GSDMD has renewed the focus on these important pathways. The non-canonical inflammasome pathway that leads to caspase-4, −5, and −11 activation and the inflammasome-dependent caspase-1 pathway appear to be both mechanistically divergent and interdependent. Thus, it is critical to continue to clarify the mechanisms of these two pathways and the cross-talk between them. Further understanding of these complex pathways will illuminate the crucial role of inflammatory caspases in regulation of inflammation, and will no doubt provide novel druggable targets so the therapeutic potential of these pathways can soon be fully realized.
Acknowledgements
We thank Ashley Boice for careful reading of the manuscript. Research in the Bouchier-Hayes laboratory is funded by grants from the Children’s Leukemia Research Association, the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM121389 and support from Texas Children’s Cancer and Hematology Centers.
Abbreviations
- AIM2
absent in melanoma 2
- APAF1
apoptotic peptidase activating factor 1
- ASC
apoptosis-associated speck-like protein containing a CARD
- BIR
baculovirus inverted repeat
- CAPS
cryopyrin-associated periodic syndromes
- CARD
caspase recruitment domain
- CTB
cholera toxin B
- DAMP
damage-associated molecular patterns
- dsDNA
double-stranded DNA
- GSDMD
gasdermin D
- ICE
interleukin (IL)-1β-converting enzyme
- IEC
intestinal epithelial cell
- IFI-6
interferon-inducible protein 16
- IFN
interferon
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- MDP
muramyl dipeptide
- mtROS
mitochondrial ROS
- NAIP
NLR apoptosis-inhibitory protein
- NLR
NOD-like receptor
- NLRC
NLR family CARD containing
- NLRP
NLR family Pyrin domain containing
- NOD
nucleotide binding oligomerization domain
- NOX
NADPH-oxidase
- PAMP
pathogen-associated molecular pattern
- PYD
pyrin domain
- ROS
reactive oxygen species
- TLR
toll like receptor
References
- 1.Lamkanfi M, Declercq W, Kalai M, Saelens X & Vandenabeele P (2002) Alice in caspase land. A phylogenetic analysis of caspases from worm to man, Cell Death Differ. 9, 358–61. [DOI] [PubMed] [Google Scholar]
- 2.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J & et al. (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes, Nature. 356, 768–74. [DOI] [PubMed] [Google Scholar]
- 3.Ayala JM, Yamin TT, Egger LA, Chin J, Kostura MJ & Miller DK (1994) IL-1 beta-converting enzyme is present in monocytic cells as an inactive 45-kDa precursor, J Immunol. 153, 2592–9. [PubMed] [Google Scholar]
- 4.Martinon F, Burns K & Tschopp J (2002) The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β, Molecular Cell. 10, 417–426. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, Chang HY & Baltimore D (1998) Autoproteolytic activation of pro-caspases by oligomerization, Mol Cell. 1, 319–25. [DOI] [PubMed] [Google Scholar]
- 6.Elliott JM, Rouge L, Wiesmann C & Scheer JM (2009) Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation, J Biol Chem. 284, 6546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR & Salvesen GS (2003) A unified model for apical caspase activation, Mol Cell. 11, 529–41. [DOI] [PubMed] [Google Scholar]
- 8.Baliga BC, Read SH & Kumar S (2004) The biochemical mechanism of caspase-2 activation, Cell Death Differ. 11, 1234–41. [DOI] [PubMed] [Google Scholar]
- 9.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, Wong W, Kamen R, Tracey D & Allen H (1997) Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production, Nature. 386, 619–23. [DOI] [PubMed] [Google Scholar]
- 10.Dinarello CA (1998) Interleukin-1β, Interleukin-18, and the Interleukin-1β Converting Enzymea, Annals of the New York Academy of Sciences. 856, 1–11. [DOI] [PubMed] [Google Scholar]
- 11.Dinarello CA, Gatti S & Bartfai T (1999) Fever: Links with an ancient receptor, Current Biology. 9, R143–R146. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura K, Okamura H, Wada M, Nagata K & Tamura T (1989) Endotoxin-induced serum factor that stimulates gamma interferon production, Infect Immun. 57, 590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamura H, Tsutsui H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattori K, Akita K, Namba M, Tanabe F, Konishi K, Fukuda S & Kurimoto M (1995) Cloning of a new cytokine that induces IFN-γ production by T cells, Nature. 378, 88. [DOI] [PubMed] [Google Scholar]
- 14.Dinarello CA & Fantuzzi G (2003) Interleukin-18 and host defense against infection, The Journal of infectious diseases. 187 Suppl 2, S370–84. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, Kurimoto M, Tanimoto T, Flavell RA, Sato V, Harding MW, Livingston DJ & Su MS (1997) Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme, Science. 275, 206–9. [DOI] [PubMed] [Google Scholar]
- 16.Wang J & Lenardo MJ (2000) Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies, J Cell Sci. 113 ( Pt 5), 753–7. [DOI] [PubMed] [Google Scholar]
- 17.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J & et al. (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock, Cell. 80, 401–11. [DOI] [PubMed] [Google Scholar]
- 18.Lamkanfi M, Moreira LO, Makena P, Spierings DC, Boyd K, Murray PJ, Green DR & Kanneganti TD (2009) Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival, Blood. 113, 2742–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cookson BT & Brennan MA (2001) Pro-inflammatory programmed cell death, Trends in microbiology. 9, 113–4. [DOI] [PubMed] [Google Scholar]
- 20.Brennan MA & Cookson BT (2000) Salmonella induces macrophage death by caspase-1-dependent necrosis, Molecular microbiology. 38, 31–40. [DOI] [PubMed] [Google Scholar]
- 21.Fink SL & Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages, Cellular microbiology. 8, 1812–25. [DOI] [PubMed] [Google Scholar]
- 22.Sun GW, Lu J, Pervaiz S, Cao WP & Gan YH (2005) Caspase-1 dependent macrophage death induced by Burkholderia pseudomallei, Cellular microbiology. 7, 1447–58. [DOI] [PubMed] [Google Scholar]
- 23.Rubartelli A, Cozzolino F, Talio M & Sitia R (1990) A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence, EMBO J. 9, 1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullen SP, Kearney CJ, Clancy DM & Martin SJ (2015) Diverse Activators of the NLRP3 Inflammasome Promote IL-1beta Secretion by Triggering Necrosis, Cell Rep. 11, 1535–48. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, Kaisho T, Takemoto K, Suzuki T, Kuranaga E, Ohara O & Miura M (2014) Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response, Cell Rep. 8, 974–82. [DOI] [PubMed] [Google Scholar]
- 26.Evavold CL, Ruan J, Tan Y, Xia S, Wu H & Kagan JC (2018) The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages, Immunity. 48, 35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conos SA, Lawlor KE, Vaux DL, Vince JE & Lindqvist LM (2016) Cell death is not essential for caspase-1-mediated interleukin-1beta activation and secretion, Cell Death Differ. 23, 1827–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monack DM, Detweiler CS & Falkow S (2001) Salmonella pathogenicity island 2-dependent macrophage death is mediated in part by the host cysteine protease caspase-1, Cellular microbiology. 3, 825–37. [DOI] [PubMed] [Google Scholar]
- 29.Van de Craen M, Vandenabeele P, Declercq W, Van den Brande I, Van Loo G, Molemans F, Schotte P, Van Criekinge W, Beyaert R & Fiers W (1997) Characterization of seven murine caspase family members, FEBS Lett. 403, 61–9. [DOI] [PubMed] [Google Scholar]
- 30.Lin XY, Choi MS & Porter AG (2000) Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma, J Biol Chem. 275, 39920–6. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Miura M, Jung YK, Zhu H, Li E & Yuan J (1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE, Cell. 92, 501–9. [DOI] [PubMed] [Google Scholar]
- 32.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M & Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11, Nature. 479, 117–21. [DOI] [PubMed] [Google Scholar]
- 33.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti T-D & Dixit VM (2010) Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia, The Journal of Immunology. 185, 4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW & Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4, Science. 341, 1246–9. [DOI] [PubMed] [Google Scholar]
- 35.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L & Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS, Nature. 514, 187–92. [DOI] [PubMed] [Google Scholar]
- 36.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM & Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1, Nature. 490, 288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC & Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling, Nature. 526, 666–71. [DOI] [PubMed] [Google Scholar]
- 38.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F & Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, Nature. 526, 660–5. [DOI] [PubMed] [Google Scholar]
- 39.Feng S, Fox D & Man SM (2018) Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death, Journal of Molecular Biology. 430, 3068–3080. [DOI] [PubMed] [Google Scholar]
- 40.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM & Dueber EC (2016) GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes, Proc Natl Acad Sci U S A. 113, 7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H & Lieberman J (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores, Nature. 535, 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruan J, Xia S, Liu X, Lieberman J & Wu H (2018) Cryo-EM structure of the gasdermin A3 membrane pore, Nature. 557, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA & Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta, Nature. 403, 98–103. [DOI] [PubMed] [Google Scholar]
- 44.Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, Meeus A, Declercq W & Vandenabeele P (2003) Regulation of the expression and processing of caspase-12, J Cell Biol. 162, 457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M & Cecconi F (2006) Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism, J Biol Chem. 281, 2693–700. [DOI] [PubMed] [Google Scholar]
- 46.Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, Ulevitch RJ, Green DR & Nicholson DW (2006) Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice, Nature. 440, 1064–8. [DOI] [PubMed] [Google Scholar]
- 47.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS, Buchman TG, Zehnbauer BA, Hayden MR, Farrer LA, Roy S & Nicholson DW (2004) Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms, Nature. 429, 75–9. [DOI] [PubMed] [Google Scholar]
- 48.Park HH, Lo YC, Lin SC, Wang L, Yang JK & Wu H (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation, Annual review of immunology. 25, 561–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V & Ward PA (2008) The NLR gene family: a standard nomenclature, Immunity. 28, 285–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S & Dixit VM (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf, Nature. 430, 213. [DOI] [PubMed] [Google Scholar]
- 51.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J & Alnemri ES (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation, Cell death and differentiation. 14, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H & Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes, Cell. 156, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanders MG, Parsons MJ, Howard AG, Liu J, Fassio SR, Martinez JA & Bouchier-Hayes L (2015) Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes, Cell Death Dis. 6, e1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM & Monack DM (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella, The Journal of experimental medicine. 207, 1745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Broz P, von Moltke J, Jones JW, Vance RE & Monack DM (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing, Cell host & microbe. 8, 471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai X, Chen J, Xu H, Liu S, Jiang Q-X, Halfmann R & Chen ZJ (2014) Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation, Cell. 156, 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C & Wu H (2018) Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1, Proc Natl Acad Sci U S A. 115, 10845–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, Tong AB, Ploegh HL, Mao Y & Wu H (2016) Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism, Nature structural & molecular biology. 23, 416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu A, Magupalli Venkat G., Ruan J, Yin Q, Atianand Maninjay K., Vos MR, Schröder Gunnar F., Fitzgerald Katherine A., Wu H & Egelman Edward H. (2014) Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes, Cell. 156, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gurcel L, Abrami L, Girardin S, Tschopp J & van der Goot FG (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival, Cell. 126, 1135–45. [DOI] [PubMed] [Google Scholar]
- 61.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM & Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP, Nature. 440, 228–32. [DOI] [PubMed] [Google Scholar]
- 62.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT & Tschopp J (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica, Science. 320, 674–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS & Flavell RA (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants, Nature. 453, 1122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinon F, Petrilli V, Mayor A, Tardivel A & Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome, Nature. 440, 237–41. [DOI] [PubMed] [Google Scholar]
- 65.Perregaux D, Barberia J, Lanzetti AJ, Geoghegan KF, Carty TJ & Gabel CA (1992) IL-1 beta maturation: evidence that mature cytokine formation can be induced specifically by nigericin, J Immunol. 149, 1294–303. [PubMed] [Google Scholar]
- 66.Pelegrin P & Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor, EMBO J. 25, 5071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW & Flavell RA (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1, Immunity. 24, 317–27. [DOI] [PubMed] [Google Scholar]
- 68.Pelegrin P & Surprenant A (2007) Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway, J Biol Chem. 282, 2386–94. [DOI] [PubMed] [Google Scholar]
- 69.Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti T-D, Rehman J & Malik AB (2018) The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation, Immunity. 49, 56–65.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He Y, Zeng MY, Yang D, Motro B & Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux, Nature. 530, 354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, Kravchenko VV, Mathison JC, Moresco EM, Monson NL, Ulevitch RJ & Beutler B (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component, Nat Immunol. 17, 250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou R, Yazdi AS, Menu P & Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation, Nature. 469, 221–5. [DOI] [PubMed] [Google Scholar]
- 73.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J & Zychlinsky A (2010) Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease, Blood. 116, 1570–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Bruggen R, Koker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW & van den Berg TK (2010) Human NLRP3 inflammasome activation is Nox1–4 independent, Blood. 115, 5398–400. [DOI] [PubMed] [Google Scholar]
- 75.van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW & Netea MG (2010) Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease, Proc Natl Acad Sci U S A. 107, 3030–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V & Latz E (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression, J Immunol. 183, 787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Franchi L, Eigenbrod T & Nunez G (2009) Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation, J Immunol. 183, 792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G & Hornung V (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome, J Immunol. 187, 613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T & Karin M (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation, Nature. 560, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T & Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis, Immunity. 36, 401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI & Flavell RA (2007) Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome, The Journal of experimental medicine. 204, 3235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C & Nunez G (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages, PLoS pathogens. 3, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T & Alnemri ES (2001) Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1, J Biol Chem. 276, 28309–13. [DOI] [PubMed] [Google Scholar]
- 84.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S & Dixit VM (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf, Nature. 430, 213–8. [DOI] [PubMed] [Google Scholar]
- 85.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM & Vance RE (2008) Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin, Nat Immunol. 9, 1171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kofoed EM & Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity, Nature. 477, 592–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L & Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus, Nature. 477, 596–600. [DOI] [PubMed] [Google Scholar]
- 88.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, Shao F, Wang HW, Sui SF & Chai J (2015) Structural and biochemical basis for induced self-propagation of NLRC4, Science. 350, 399–404. [DOI] [PubMed] [Google Scholar]
- 89.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, Marks C, Ouyang Q, Zhang X, Mao Y & Wu H (2015) Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization, Science. 350, 404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA & Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production, J Immunol. 191, 5239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, Philip NH, Ayres JS, Brodsky IE, Gronert K & Vance RE (2017) NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and −8, Immunity. 46, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, Manin GZ, Cunha LD & Zamboni DS (2017) Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome, PLoS pathogens. 13, e1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, Cuellar TL, Reichelt M & Kayagaki N (2018) ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages, Scientific reports. 8, 3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Goncalves A, Vande Walle L, Demon D, Matusiak M, Van Hauwermeiren F, D’Hont J, Hochepied T, Krautwald S, Kanneganti TD & Lamkanfi M (2017) Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4, Cell Rep. 21, 3427–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS & Henry T (2012) AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages, Cell Death Differ. 19, 1709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP & Koller BH (2012) NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice, J Immunol. 189, 2006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S, Godzik A & Reed JC (2001) A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis, J Biol Chem. 276, 9239–45. [DOI] [PubMed] [Google Scholar]
- 98.Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM, Vincenz C & Ward PA (2001) Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins, J Biol Chem. 276, 9230–8. [DOI] [PubMed] [Google Scholar]
- 99.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I & Reed JC (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation, Mol Cell. 25, 713–24. [DOI] [PubMed] [Google Scholar]
- 100.Boyden ED & Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin, Nat Genet. 38, 240–4. [DOI] [PubMed] [Google Scholar]
- 101.Chavarria-Smith J & Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor, PLoS pathogens. 9, e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guey B, Bodnar M, Manie SN, Tardivel A & Petrilli V (2014) Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function, Proc Natl Acad Sci U S A. 111, 17254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, Meltzer PS & Trent JM (1997) Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma, Oncogene. 15, 453. [DOI] [PubMed] [Google Scholar]
- 104.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E & Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC, Nature. 458, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL & Superti-Furga G (2009) An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome, Nature Immunology. 10, 266. [DOI] [PubMed] [Google Scholar]
- 106.Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, Hornung V, Latz E, Bowie AG, Fitzgerald KA & Xiao TS (2012) Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor, Immunity. 36, 561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin T, Perry A, Smith P, Jiang J & Xiao TS (2013) Structure of the Absent in Melanoma 2 (AIM2) Pyrin Domain Provides Insights into the Mechanisms of AIM2 Autoinhibition and Inflammasome Assembly, Journal of Biological Chemistry. 288, 13225–13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E & Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC, Nature. 458, 514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP & Alnemri ES (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis, Nat Immunol. 11, 385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E & Fitzgerald KA (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses, Nat Immunol. 11, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM & Monack DM (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis, Proc Natl Acad Sci U S A. 107, 9771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P & Portnoy DA (2010) Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol, Cell host & microbe. 7, 412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E & Hornung V (2010) Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome, Eur J Immunol. 40, 1545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R & Mitsuyama M (2010) Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes, J Immunol. 185, 1186–95. [DOI] [PubMed] [Google Scholar]
- 115.Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA & Aderem A (2010) Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2, J Immunol. 185, 818–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu J, Fernandes-Alnemri T & Alnemri ES (2010) Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes, Journal of clinical immunology. 30, 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Peng K, Broz P, Jones J, Joubert LM & Monack D (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis, Cellular microbiology. 13, 1586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Komada T, Chung H, Lau A, Platnich JM, Beck PL, Benediktsson H, Duff HJ, Jenne CN & Muruve DA (2018) Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD, Journal of the American Society of Nephrology. 29, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, Fitzgerald KA & Rathinam VAK (2018) Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis, Immunity. 49, 413–426 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, Wittmann G, Latz E, Subklewe M & Hornung V (2017) The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3, Cell. 171, 1110–1124 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P & Chandran B (2011) IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection, Cell host & microbe. 9, 363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M & Kanneganti T-D (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens, Nature. 488, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ & Ting JP-Y (2007) Cutting Edge: Monarch-1 Suppresses Non-Canonical NF-κB Activation and p52-Dependent Chemokine Expression in Monocytes, The Journal of Immunology. 178, 1256–1260. [DOI] [PubMed] [Google Scholar]
- 124.Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, Vogel SN, Braunstein M & Ting JPY (2005) The CATERPILLER Protein Monarch-1 Is an Antagonist of Toll-like Receptor-, Tumor Necrosis Factor α-, and Mycobacterium tuberculosis-induced Pro-inflammatory Signals, Journal of Biological Chemistry. 280, 39914–39924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI & Flavell RA (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis, Cell. 145, 745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY & Nunez G (2018) The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection, Cell. 175, 1651–1664 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA & Lien E (2012) The NLRP12 inflammasome recognizes Yersinia pestis, Immunity. 37, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, Bagga R, Kircheisen R, Ao A, Ratti B, Hanash S, Rouleau GA & Slim R (2006) Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans, Nat Genet. 38, 300–2. [DOI] [PubMed] [Google Scholar]
- 129.Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA & Ting JP (2011) Cutting edge: NLRC5-dependent activation of the inflammasome, J Immunol. 186, 1333–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heneka MT, McManus RM & Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease, Nature reviews Neuroscience. 19, 610–621. [DOI] [PubMed] [Google Scholar]
- 131.Toldo S & Abbate A (2018) The NLRP3 inflammasome in acute myocardial infarction, Nature reviews Cardiology. 15, 203–214. [DOI] [PubMed] [Google Scholar]
- 132.Aziz M, Jacob A & Wang P (2014) Revisiting caspases in sepsis, Cell Death Dis. 5, e1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ting JP, Kastner DL & Hoffman HM (2006) CATERPILLERs, pyrin and hereditary immunological disorders, Nature reviews Immunology. 6, 183–95. [DOI] [PubMed] [Google Scholar]
- 134.Hoffman HM, Mueller JL, Broide DH, Wanderer AA & Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome, Nat Genet. 29, 301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A & de Saint Basile G (2002) Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes, American journal of human genetics. 71, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, Meyrier A, Watts RA, Scott DG, Nicholls A, Granel B, Frances C, Garcier F, Edery P, Boulinguez S, Domergues JP, Delpech M & Grateau G (2002) New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes, American journal of human genetics. 70, 1498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hawkins PN, Lachmann HJ & McDermott MF (2003) Interleukin-1-receptor antagonist in the Muckle-Wells syndrome, The New England journal of medicine. 348, 2583–4. [DOI] [PubMed] [Google Scholar]
- 138.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA & Firestein GS (2004) Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist, Lancet (London, England). 364, 1779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O’Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM & Goldbach-Mansky R (2014) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome, Nat Genet. 46, 1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Romberg N, Vogel TP & Canna SW (2017) NLRC4 inflammasomopathies, Current opinion in allergy and clinical immunology. 17, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, Schiffrin EJ, Foell D, Grom AA, Ammann S, Ehl S, Hoshino T, Goldbach-Mansky R, Gabay C & Canna SW (2018) Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome, Blood. 131, 1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverenyi I, Takeichi T, Balaji R, Lau A, Tye H, Roy K, Bonnard C, Ahl PJ, Jones LA, Baker PJ, Lacina L, Otsuka A, Fournie PR, Malecaze F, Lane EB, Akiyama M, Kabashima K, Connolly JE, Masters SL, Soler VJ, Omar SS, McGrath JA, Nedelcu R, Gribaa M, Denguezli M, Saad A, Hiller S & Reversade B (2016) Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation, Cell. 167, 187–202 e17. [DOI] [PubMed] [Google Scholar]
- 143.Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR & Spritz RA (2007) NALP1 in vitiligo-associated multiple autoimmune disease, The New England journal of medicine. 356, 1216–25. [DOI] [PubMed] [Google Scholar]
- 144.Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, Frouin E, Seyger M, Girard M, Puechberty J, Costes V, Rodiere M, Carbasse A, Jeziorski E, Portales P, Sarrabay G, Mondain M, Jorgensen C, Apparailly F, Hoppenreijs E, Touitou I & Genevieve D (2017) A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis), Annals of the rheumatic diseases. 76, 1191–1198. [DOI] [PubMed] [Google Scholar]
- 145.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, Xiao TS, Li X & Abbott DW (2018) Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis, Science immunology. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang J, Liu Z, Wang C, Yang R, Rathkey JK, Pinkard OW, Shi W, Chen Y, Dubyak GR, Abbott DW & Xiao TS (2018) Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor, Proc Natl Acad Sci U S A. 115, 6792–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Spritz RA, Gowan K, Bennett DC & Fain PR (2004) Novel vitiligo susceptibility loci on chromosomes 7 (AIS2) and 8 (AIS3), confirmation of SLEV1 on chromosome 17, and their roles in an autoimmune diathesis, American journal of human genetics. 74, 188–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Żurawek M, Fichna M, Januszkiewicz-Lewandowska D, Gryczyńska M, Fichna P & Nowak J (2010) A coding variant in NLRP1 is associated with autoimmune Addison’s disease, Human Immunology. 71, 530–534. [DOI] [PubMed] [Google Scholar]
- 149.Magitta NF, Bøe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, Undlien DE, Joner G, Njølstad PR, Kvien TK, Førre Ø, Knappskog PM & Husebye ES (2008) A coding polymorphism in NALP1 confers risk for autoimmune Addison's disease and type 1 diabetes, Genes And Immunity. 10, 120. [DOI] [PubMed] [Google Scholar]
- 150.Pontillo A, Girardelli M, Kamada AJ, Pancotto J. A. t., Donadi EA, Crovella S & Sandrin-Garcia P (2012) Polimorphisms in Inflammasome Genes Are Involved in the Predisposition to Systemic Lupus Erythematosus, Autoimmunity. 45, 271–278. [DOI] [PubMed] [Google Scholar]
- 151.Onoyama S, Ihara K, Yamaguchi Y, Ikeda K, Yamaguchi K, Yamamura K, Hoshina T, Mizuno Y & Hara T (2012) Genetic susceptibility to Kawasaki disease: Analysis of pattern recognition receptor genes, Human Immunology. 73, 654–660. [DOI] [PubMed] [Google Scholar]
- 152.Pontillo A, Vendramin A, Catamo E, Fabris A & Crovella S (2011) The Missense Variation Q705K in CIAS1/NALP3/NLRP3 Gene and an NLRP1 Haplotype Are Associated With Celiac Disease, The American Journal Of Gastroenterology. 106, 539. [DOI] [PubMed] [Google Scholar]
- 153.Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, West B, Podoltsev NA, Boggon TJ, Kazmierczak BI & Lifton RP (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation, Nature Genetics. 46, 1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Han Y, Chen Z, Hou R, Yan D, Liu C, Chen S, Li X & Du W (2015) Expression of AIM2 is correlated with increased inflammation in chronic hepatitis B patients, Virology journal. 12, 129–129. [DOI] [PMC free article] [PubMed] [Google Scholar]