

Abstract
Modifying the electronic properties of olefins represents the quintessential approach to tuning alkene reactivity. In this context, the exploration of trifluoromethyl groups as divergent electronic modifiers has not been considered. Here, we describe a copper-mediated 1,2-(bis)trifluoromethylation of acetylenes to create—in a single step—E-hexafluorobutenes (E-HFBs). The reaction proceeds with high yield and E/Z selectivity. Since the alkyne captures two trifluoromethyl groups from each molecule of bpyCu(CF3)3, mechanistic studies were conducted to illuminate the role of reactants. Interestingly, E-HFBs exhibit remarkable stability to standard olefin functionalization reactions in spite of the pendant trifluoromethyl groups. This finding has significant implications for medicine, agroscience, and materials.
Keywords: Alkynes, Copper, Difunctionalization, Persulfate, Trifluoromethylation
Graphical Abstract
A copper-mediated 1,2-(bis)trifluoromethylation of acetylenes enables the preparation of E-hexafluorobutenes (E-HFBs)—in a single step. The reaction proceeds with high yield and E/Z selectivity. Significant mechanistic work explains the frontier molecular orbital behavior of the copper complex and the role of persulfate in this and related reactions for the first time.
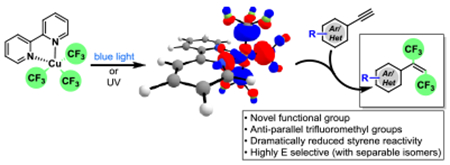
The trifluoromethyl group, with its unique electronegativity, hydrophobicity, metabolic stability, and bioavailability, has permeated organic structures in the context of medicines, agrochemicals, and organic materials.[1] While frequently connected to aromatic systems, the trifluoromethyl group can also be found appended to alkenes in many biologically active molecules and organic functional materials.[2] Interestingly, olefins bearing two trifluoromethyl groups remain rare but should possess unique properties. The ability to introduce trifluoromethyl groups into complex molecular architectures, however, presents a difficult proposition—even with modern organic chemistry. Synthesis of stereochemically defined E-hexafluorobutenes (E-HFBs) requires multi-step synthesis, wherein the difficult-to-append trifluoromethyl groups are added separately or through the difunctionalization of hexafluoro-2-butyne—a gas at 25 °C.[3] For example, the treatment of trans-1,2-diiodoalkenes with copper and FSO2CF2CO2Me leads to the formation of E-HFBs,[4] and the treatment of arynes with a trifluoromethyl-ligated copper produces ortho-bis(trifluoromethyl)benzenes.[5c] Obviously, the direct difunctionalization of alkynes would offer advantages for both step- and atom-economy.[5]
Trifluoromethyl-adorned copper systems have proven robust, and numerous applications for aryl trifluoromethylation have been developed.[6] As early as the 1980s, stable copper complexes containing trifluoromethyl groups were synthesized and studied.[7] In 2015, Grushin et al. reported bpyCu(CF3)3 and related complexes that are bench-stable and easy to prepare.[8] While some reports described similar copper systems as high-valent Cu(III) complexes, theoretical work as early 1995 showed that these structures possess filled 3d orbitals (i.e., d10), in part due to the inversion of the ligand field caused by the CF3 ligands. [9] Consequently, such copper complexes are more accurately described, and spectroscopically observed,[10] as Cu(I)—which has significant implications for reactivity.
Several groups have demonstrated that the unique reactivity of bpyCu(CF3)3 can be used for mono-trifluoromethylation. For example, Li and co-workers developed alkyl trifluoromethylation from halides or carboxylic acids (Scheme 1a)[11] and, later, formed trifluoromethyl ketones from aldehydes with bpyCu(CF3)3 (Scheme 1b).[12] Liu et al. and our group disclosed benzylic C(sp3)−H trifluoromethylation that can be employed for late-stage functionalization (Scheme 1c).[13] Here, we describe the simultaneous introduction of two trifluoromethyl groups across alkynes using bpyCu(CF3)3, persulfate, and blue light to access E-HFBs (Scheme 1d). Under irradiation, bpyCu(CF3)3 liberates one trifluoromethyl radical. We found that the newly formed bpyCu(CF3)2(OSO3H) also liberates a trifluoromethyl radical (Scheme 1e). The conditions provide a tandem radical and inner sphere trifluoromethylation that we harness for the synthesis of E-HFBs.
Scheme 1.
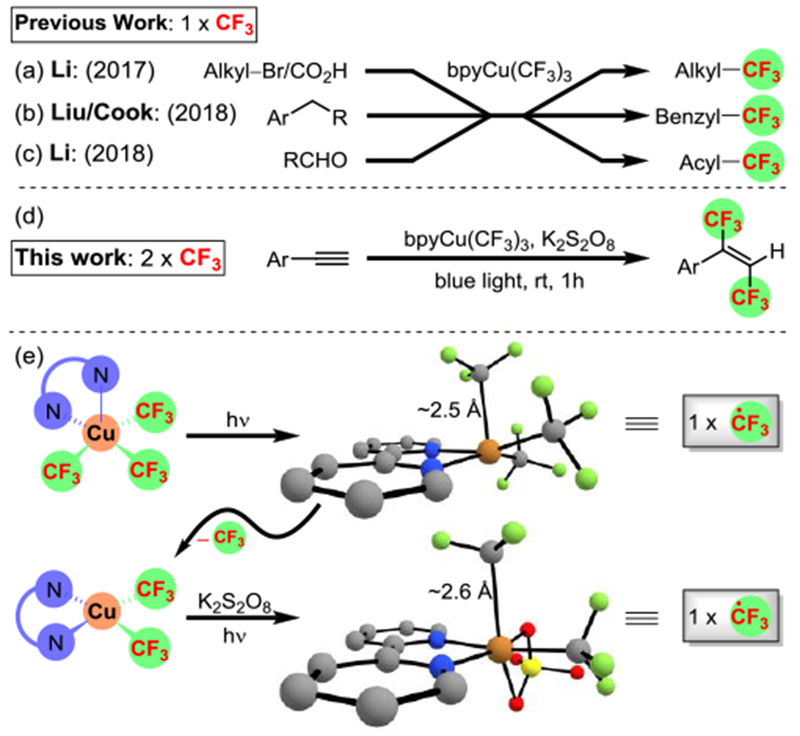
(a-c) Previous studies have established the capture of one trifluoromethyl group from bpyCu(CF3)3; (d-e) the combination of persulfate and light enable the capture of two trifluoromethyl groups from bpyCu(CF3)3.
We have previously shown that UV light can homolyze reagent 2 to form bpyCu(CF3)2 and trifluoromethyl radical.[13b] In that study, the newly formed trifluoromethyl radical was quenched with silane to prevent unwanted side reactions. Here, we envisioned using this radical to add into an alkyne to generate a vinylic radical that could recombine with copper to induce a second trifluoromethylation. Interestingly, visible-light irradiation (447 nm) of bpyCu(CF3)3/K2S2O8 in the presence of 4-bromophenylacetylene (1a) produced the desired 1,2-(bis)trifluoromethylation product (3a) in excellent yield (94%) and stereoselectivity (E/Z = 15:1) (Table 1, entry 1). Since the ratio of 1a to 2 is 1:1, the reaction liberates two trifluoromethyl groups per bpyCu(CF3)3. Surprisingly, lower efficiency was observed under long UV (365 nm) irradiation (Table 1, entry 2).[13b] Interestingly, aliphatic alkynes did not react under these conditions (vide infra). While the optimal solvent system is comprised of acetonitrile and methanol, other common solvent mixtures also work reasonably well for the reaction (Table 1, entry 3, 4, 6-8). Although K2S2O8 is not required for the reaction, the lack of oxidant significantly decreased the yield (Table 1, entry 5). The supporting role of K2S2O8 suggested a critical mechanistic switch that enables the consumption of two trifluoromethyl groups (vida infra).
Table 1.
Effects of Pertinent Reaction Parametersa
 | ||
|---|---|---|
| Entry | Conditons | Yield of 3ab |
| 1 | standard conditions | 94% (E/Z=15:1) |
| 2 | 365 nm instead of 447 nm | 74% (E/Z=13:1) |
| 3 | without MeOH | 80% (E/Z=7:1) |
| 4 | without CH3CN | 31% (E/Z=10:1) |
| 5 | without K2S2O8 | 32% (E/Z=11:1) |
| 6 | DCM instead of MeOH | 70% (E/Z=5:1) |
| 7 | tBuOH instead of MeOH | 82% (E/Z=6:1) |
| 8 | acetone instead of CH3CN | 67% (E/Z=10:1) |
| 9 | performed in the dark | N. D.c |
| 10 | 50 °C intead of blue light | N. D.c |
Unless otherwise noted, all the reactions were run with 1a (0.04 mmol) and 2 (0.04 mmol) in 0.7 mL of solvent for 1 h.
Yields of 3a were determined by 19F NMR spectroscopy with 1-fluoro-4-methylbenzene as the internal standard, while the E/Z ratios were determined by 19F NMR spectroscopy of the crude product mixtures.
Not detected by 19F NMR. bpy = 2,2’-bipyridine.
With near quantitative yield of E-HFBs, we sought to examine the scope of various terminal alkynes (Table 2). Two trifluoromethyl groups could be effectively incorporated across a number of electronically and sterically differentiated aromatic alkynes with high E selectivity. Moreover, the pronounced dipole differences between the E and Z isomers allowed the separation of both isomers by silica chromatography in all cases. Aromatic bromides were tolerated under the reaction conditions with positional insignificance (1a-c, Table 2). The reaction tolerated aromatic alkynes bearing electron-donating substituents such as tert-butyl (1i), methylthio (1j), and methoxy (1k) or electron-withdrawing substituents (1a-h, 1p, 1q). Notably, if an aldehyde is present, the reaction proceeds in high yield and produces the acetal (1s). Overall, the reaction proved exceedingly mild, tolerating numerous desirable functional groups such as Bpin (1n), anilines (1r), thioethers (1j), mesyl (1o-p), and a ketone (1q). Using these conditions, the tetra-trifluoromethylation of bis-alkyne (1u) also proceeded with a good yield. Moreover, heteroaryl acetylenes, such as 5-ethynylbenzofuran (1v), 1-(3-ethynylphenyl)imidazole (1w), 4-pyridylacetylene (1x), and 5-ethynyl-2-phenylpyrimidine (1y), function well in the reaction.
Table 2.
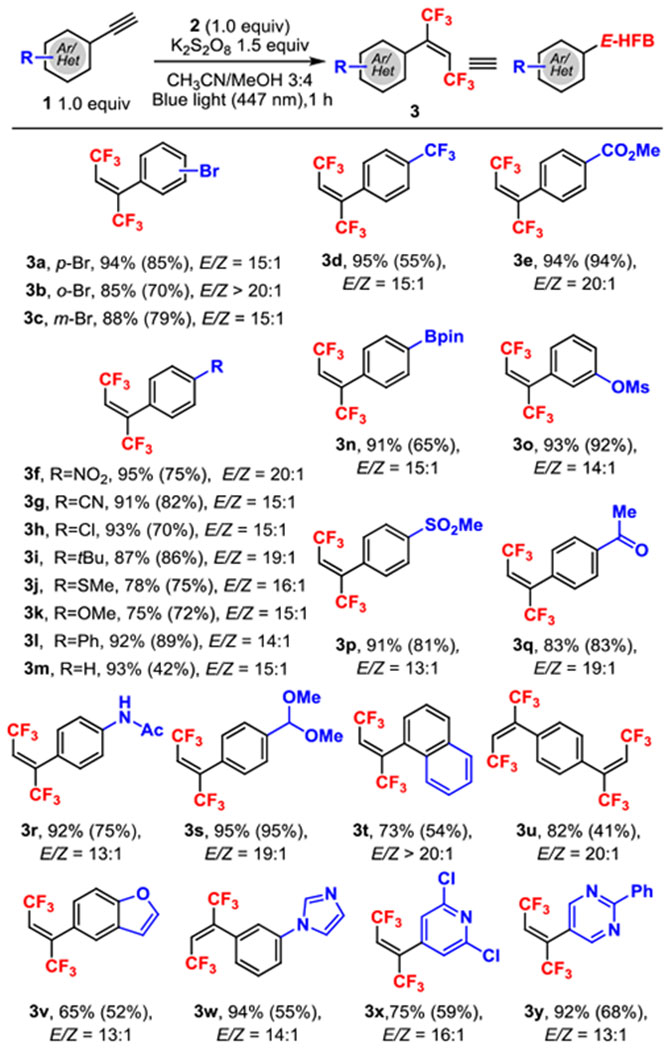 |
All reactions were with 1 (0.16 mmol) and 2 (0.16 mmol) in 2.8 mL of solvent for 1 h.
The 19F NMR yield determined by 19F NMR spectroscopy with 1-fluoro-4-methylbenzene as the internal standard, with the isolated yields given in parentheses. The E/Z ratios were determined by 19F NMR spectroscopy of the crude product mixtures.
With general access to a wide range of E-HFBs, we sought to explore the reactivity of this unusual olefin and add to the dearth of known reactions of E-HFBs. With two antiparallel trifluoromethyl groups, E-HFBs are expected to be rather electron-deficient. Conversely, near cancellation of their dipoles might confer added stability. Remarkably, various attempts to leverage the electronic deficiency of the E-HFB in 3c failed. For example, E-HFBs fail to engage with Danishefsky diene[14] or furan[15] but are also inert to α-diazo esters,[16] catalytic Rh carbenes,[17] mCPBA,[18] and bromine—demonstrating that E-HFBs are much more stable than the parent styrene. Counterintuitively, the Os-catalyzed dihydroxylation of the E-HFB proceeded in 78% yield to produce the vicinal diol (see SI). This stability has significant implications for fields ranging from medicinal chemistry to materials.
Based on the remarkable scope developed in Table 2, we thought this reaction might serve in the late-stage functionalization of bioactive molecules. To that end, a variety of alkyne-containing molecules were used to test whether this chemistry can offer direct access to E-HFBs as a potentially new functional group. We found the E-HFB functional group could be easily installed in natural product scaffolds such as unnatural amino acids (4), steroid structures (6), and tocopherol (7). An alkyne-containing medicine with sensitive functionalities (5) could also be converted to novel E-HFB structures.
To explore the role of oxidant and copper, a series of simple mechanistic reactions were conducted. First, deuterated solvent led to the detection of deuterated fluoroform (18% yield) under the standard reaction conditions using 4-bromophenylacetylene (1a) as substrate (Scheme 3A)—implying that quenching of trifluoromethyl radical by solvent competes with our desired reaction.[19] To further investigate the presence of trifluoromethyl radicals, complex 2 was irradiated with blue light in the presence of TEMPO (Scheme 3B). Interestingly, the trifluoromethyl ether of TEMPO could be obtained in 300% yield with K2S2O8, but only 60% yield without—suggesting that K2S2O8 interacts directly with copper to facilitate homolysis and generate trifluoromethyl radical. This was further investigated under the standard reaction conditions by varying the ratio of substrate 1a to Grushin’s reagent 2 with and without the oxidant (Scheme 3C). While a ratio of 1:1 without K2S2O8 provided low yield (32%), the yield could be rescued (to 82%) by increasing the ratio to 1:3 (Scheme 3C). These reactions suggest that copper complex 2 provides one trifluoromethyl radical under blue light irradiation without K2S2O8 and, with K2S2O8, 2 offers all three trifluoromethyl groups. Since these reactions revealed the critical role of K2S2O8 for the efficient release of trifluoromethyl groups from 2, we turned to DFT to better clarify these observations.
Scheme 3.
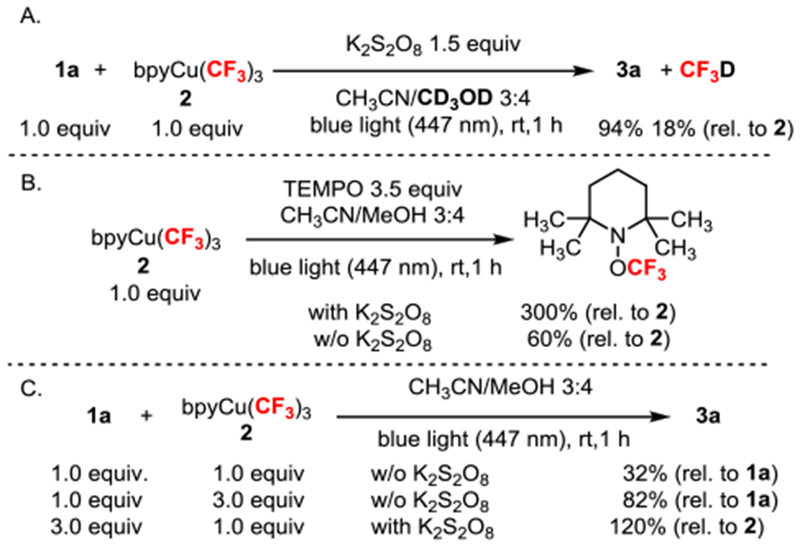
Control Experiments
We chose phenylacetylene and employed DFT to calculate the relative Gibbs free energies for all intermediates and transition states (see SI for computational details). The absorption of light by 2 results in the formation of the triplet complex 32 that elongates the axial Cu–CF3 bond (Figure 1). This system decomposes into a trifluoromethyl radical and bpyCu(CF3)2 (I) —which we used as our starting point for relative energies (Figure 1). The addition of the trifluoromethyl radical into the alkyne proceeds with a transition state (I-TS) of 11.3 kcal/mol to produce an allenyl radical II, which is −28.7 kcal/mol more stable than the trifluoromethyl radical (Figure 2). The conjugated allenyl radical explains the lack of reactivity for aliphatic alkynes. Allenyl radical II recombines with the copper(II) species to form intermediate III through a barrier (II-TS) of 12.8 kcal/mol, with III lower in energy (−43.6 kcal/mol) than I. Intermediate III then undergoes reductive elimination to form the 1,2-(bis)trifluoromethylated product IV and Cu(I). This step (III-TS) has an energetic barrier of 15.9 kcal/mol and was deemed rate-determining (Figure 2). Under these reaction conditions, the homolysis of persulfate into two hydrogen sulfate (or potassium sulfate) radicals was assumed. Consistent with the reactions in Scheme 3, the binding of sulfate to copper results in a 9.8 kcal/mol reduction in the Cu-CF3 bond-dissociation energy (see SI for details). Moreover, the reductive elimination now proceeds with a lower barrier (4.3 kcal/mol), thereby changing the rate-determining step (RDS) to the radical addition III-TS (11.3 kcal/mol). The change in RDS and lower overall transition state energy explains the higher yield observed with persulfate since it allows radical addition to vastly outcompete the quench of the trifluoromethyl radical by solvent (see SI for more details).
Figure 1.
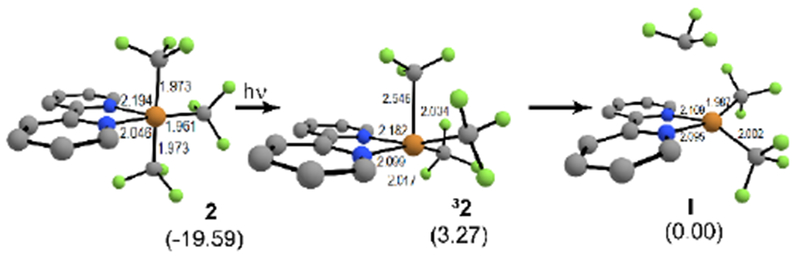
Computed geometries (uM06/LACVP**) and ΔGsol energies for 2, 32, and bipyCu (CF3)2. Bond lengths are measured in angstroms (Å).
Figure 2.
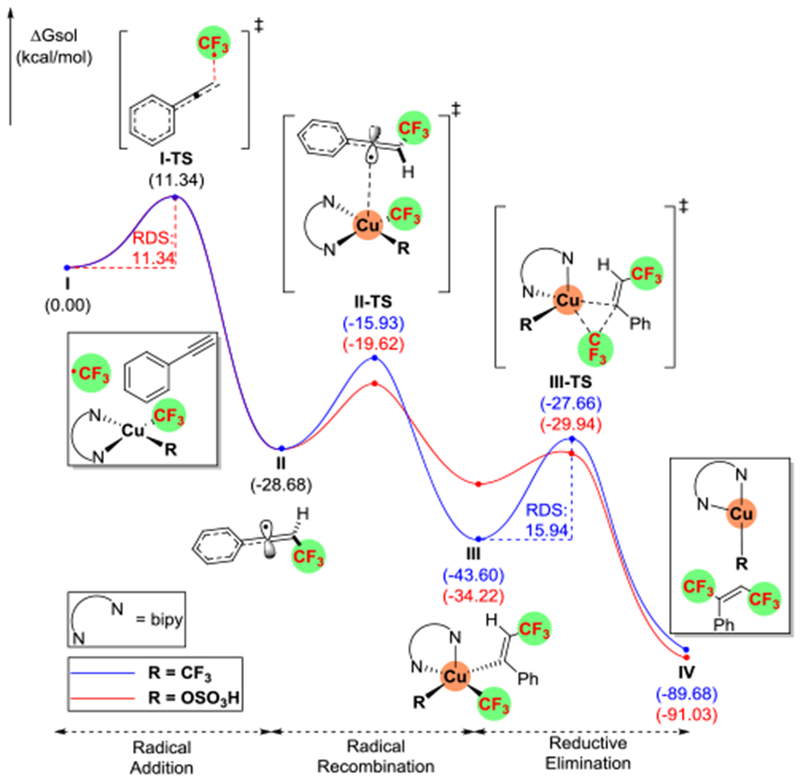
Computed ΔGsol energies (with uM06/cc-pVTZ(-f)–LACV3P**//uM06/LACVP**) for all intermediates and transition states along the reaction pathway from I to IV.
Finally, we employed TDDFT[20] to examine the requisite wavelength of light for the excitation of this system (see SI for computational details). Interestingly, there exist several transitions to the first excited state during the singlet-singlet excitation (see SI), but the major transition promotes an electron from the HOMO −1 to the LUMO +1 (Figure 3). This transition had a calculated wavelength of 362 nm and results in the formation of 3I after intersystem crossing. We found that the first excited state for the singlet-triplet excitation, however, results from promoting an electron from the HOMO to the LUMO +1 with a calculated wavelength of 486 nm. While this transition is spin-forbidden, there exist several examples of populating a triplet system from a singlet ground state.[21] Calculations suggest that UV irradiation proceeds through a singlet-to-singlet excitation, followed by intersystem crossing to a triplet prior to Cu–CF3 bond scission, whereas blue light irradiation leads directly to a singlet-to-triplet excitation. Further experiments will be needed to better understand these properties.
Figure 3.
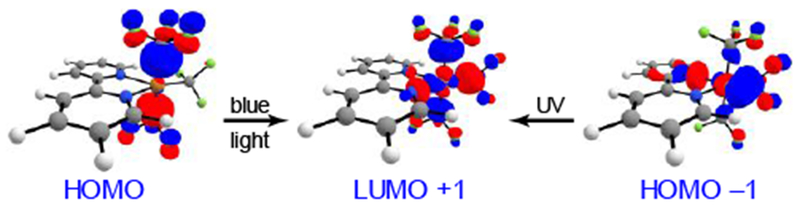
Both UV and blue light excite to the same σ* orbital (LUMO +1). This results in the formation of 32 leading to CF3 bond homolysis.
In conclusion, we have developed a method for the difunctionalization of alkynes with two trifluoromethyl groups to produce E-hexafluorobutenes—E-HFBs. Both trifluoromethyl groups come from a single equivalent of bpyCu(CF3)3 through a combination of persulfate and blue light (447 nm). Our mechanistic studies explore the photochemical properties and subtle changes in the reaction coordinate. We propose, for the first time, a role for persulfate in bpyCu(CF3)3-based reactions wherein persulfate serves as an oxidant and offers a critical replacement ligand in the form of sulfate. The sulfate ligand changes the RDS of these transformations. Such insights should aid in the future design of reaction manifolds that employ bpyCu(CF3)3. The reaction has excellent substrate scope and functional-group compatibility providing the desired products in high yields. The chemistry functions well for the late-stage functionalization of alkynes in the presence of various functional groups and heterocycles. Moreover, the E-HFBs demonstrate remarkable chemical stability toward a range of olefin functionalization reactions. Simple access to aromatic E-HFBs provides exciting opportunities in medicinal chemistry, agroscience and materials.
Supplementary Material
Scheme 2.
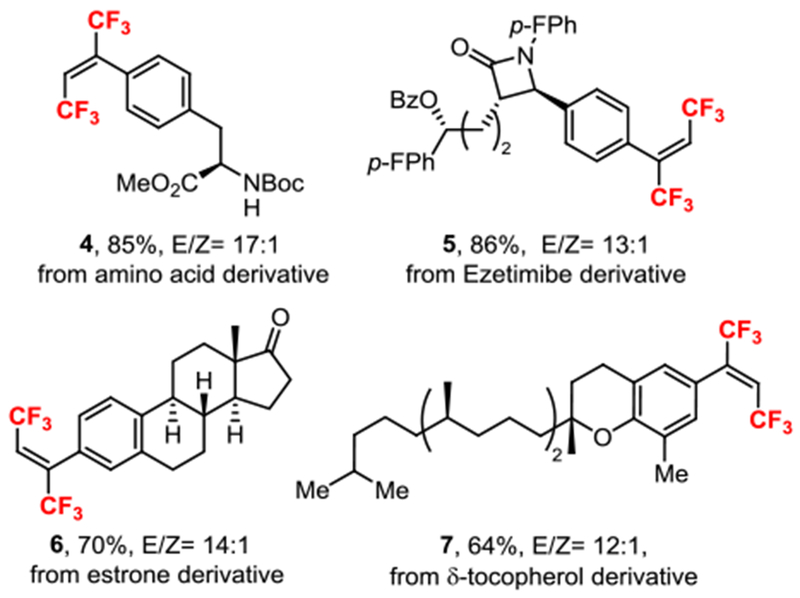
Late-Stage 1,2-(Bis)trifluoromethylation of Bioactive Moleculesa
a All reactions were with alkyne substrate (0.16 mmol) and 2 (0.16 mmol) in 2.8 mL of solvent for 1 h.
Acknowledgements
We acknowledge the NIH (GM121840). Eli Lilly & Co., and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award. We would like to acknowledge Mackenzie Fahey, Prof. Jeffrey Zaleski, and Prof. Jeremy Smith for insightful discussions about the oxidation state and photophysical properties of these copper complexes.
Footnotes
Supporting information for this article is given via a link at the end of the document
Conflicts of Interest
The authors declair no conflicts of interest.
References
- [1].a) Muller K, Faeh C, Diederich F, Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]; b) Hagmann WK, J. Med. Chem. 2008, 51, 4359–4369; [DOI] [PubMed] [Google Scholar]; c) Ilardi EA, Vitaku E, Njardarson JT, J. Med. Chem. 2013, 57, 2832–2842; [DOI] [PubMed] [Google Scholar]; d) Fujiwara T, O’Hagan D, J. Fluor. Chem. 2014, 167, 16–29; [Google Scholar]; e) Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA, J. Med. Chem. 2015, 58, 8315–8359; [DOI] [PubMed] [Google Scholar]; f) Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev. 2016, 116, 422–518; [DOI] [PubMed] [Google Scholar]; g) Meanwell NA, J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- [2].a) Shimizu M, Hiyama T, Angew. Chem., Int. Ed. 2005, 44, 214–231; [DOI] [PubMed] [Google Scholar]; b) Rivkin A, Chou T-C, Danishefsky SJ, Angew. Chem., Int. Ed. 2005, 44, 2838–2850; [DOI] [PubMed] [Google Scholar]; c) Lu H, Silverman RB, J. Med. Chem. 2006, 49, 7404–7412; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shimizu M, Takeda Y, Higashi M, Hiyama T, Angew. Chem., Int. Ed. 2009, 48, 3653–3656; [DOI] [PubMed] [Google Scholar]; e) Innocenti P, Cheung K-MJ, Solanki S, Mas-Droux C, Rowan F, Yeoh S, Boxall K, Westlake M, Pickard L, Hardy T, Baxter JE, Aherne GW, Bayliss R, Fry AM, Hoelder S, J. Med. Chem 2012, 55, 3228–3241; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ma J-J, Yi W-B, Lu G-P, Cai C, Adv. Synth. Catal. 2015, 357, 3447–3452. [Google Scholar]
- [3].a) Chambers RD, Jones CGP, Silvester MJ, J. Fluor. Chem. 1986, 32, 309–317; [Google Scholar]; b) MacNeil KJ, Burton DJ, J. Org. Chem. 1993, 58, 4411–4417; [Google Scholar]; c) Yamamoto M, Burton DJ, Swenson DC, J. Fluor. Chem. 1995, 72, 49–54; [Google Scholar]; d) Jia X, Zhou X, Quan H, Tamura M, Sekiya A, J. Fluor. Chem. 2011, 132, 1188–1193; [Google Scholar]; e) Zhao B, Li Y, Tu D-H, Zhang W, Liu Z-T, Lu J, Tetrahedron Lett. 2016, 57, 4345–4347; [Google Scholar]; f) Yamamoto M, Swenson DC, Burton DJ, J. Fluor. Chem. 2016, 185, 213–223. [Google Scholar]
- [4].Duan J, Dolbier WR, Chen Q-Y, J. Org. Chem. 1998, 63, 9486–9489. [Google Scholar]
- [5].a) Besset T, Poisson T, Pannecoucke X, Eur. J. Org. Chem. 2015, 2015, 2765–2789; [Google Scholar]; b) Gao P, Song X-R, Liu X-Y, Liang Y-M, Chem. Eur. J 2015, 21, 7648–7661. [DOI] [PubMed] [Google Scholar]; c) Yang X-K, Tsui GC, Chem. Sci., 2018, 9, 8871–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) McLoughlin VCR, Thrower J, Tetrahedron 1969, 25, 5921–5940; [Google Scholar]; b) Tomashenko OA, Escudero-Adán EC, Belmonte MM, Grushin VV, Angew. Chem., Int. Ed. 2011, 50, 7655–7659; [DOI] [PubMed] [Google Scholar]; c) Tomashenko OA, Grushin VV, Chem. Rev. 2011, 111, 4475–4521; [DOI] [PubMed] [Google Scholar]; d) Hickman AJ, Sanford MS, Nature 2012, 484, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Willert-Porada MA, Burton DJ, Baenziger NC, J. Chem. Soc., Chem. Commun. 1989, 10.1039/c39890001633, 1633–1634; [DOI] [Google Scholar]; b) Naumann D, Roy T, Tebbe K-F, Crump W, Angew. Chem. Int. Ed. Engl. 1993, 32, 1482–1483. [Google Scholar]
- [8].Romine AM, Nebra N, Konovalov AI, Martin E, Benet-Buchholz J, Grushin VV, Angew. Chem., Int. Ed. 2015, 54, 2745–2749. [DOI] [PubMed] [Google Scholar]
- [9].a) Snyder JP, Angew. Chem. Int. Ed. Engl. 1995, 34, 986–987; [Google Scholar]; b) Snyder JP, Angew. Chem. Int. Ed. Engl. 1995, 34, 80–81; [Google Scholar]; c) Kaupp M, von Schnering HG, Angew. Chem. Int. Ed. Engl. 1995, 34, 986–986. [Google Scholar]
- [10].Walroth RC, Lukens JT, MacMillan SN, Finkelstein KD, Lancaster KM, J. Am. Chem. Soc. 2016, 138, 1922–1931. [DOI] [PubMed] [Google Scholar]
- [11].a) Shen H, Liu Z, Zhang P, Tan X, Zhang Z, Li C, J. Am. Chem. Soc. 2017, 139, 9843–9846; [DOI] [PubMed] [Google Scholar]; b) Tan X, Liu Z, Shen H, Zhang P, Zhang Z, Li C, J. Am. Chem. Soc. 2017, 139, 12430–12433. [DOI] [PubMed] [Google Scholar]
- [12].Zhang P, Shen H, Zhu L, Cao W, Li C, Org. Lett. 2018, 20, 7062–7065. [DOI] [PubMed] [Google Scholar]
- [13].a) Paeth M, Carson W, Luo J-H, Tierney D, Cao Z, Cheng M-J, Liu W, Chem. Eur. J. 2018, 24, 11559–11563; [DOI] [PubMed] [Google Scholar]; b) Guo S, AbuSalim DI, Cook SP, J. Am. Chem. Soc. 2018, 140, 12378–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Danishefsky S, Kitahara T, J. Am. Chem. Soc. 1974, 96, 7807–7808. [Google Scholar]
- [15].a) Ashton PR, Brown GR, Isaacs NS, Giuffrida D, Kohnke FH, Mathias JP, Slawin AMZ, Smith DR, Stoddart JF, Williams DJ, J. Am. Chem. Soc. 1992, 114, 6330–6353; [Google Scholar]; b) Oliver Kappe C, Shaun Murphree S, Padwa A, Tetrahedron 1997, 53, 14179–14233; [Google Scholar]; c) Vogel P, Cossy J, Plumet J, Arjona O, Tetrahedron 1999, 55, 13521–13642. [Google Scholar]
- [16].Huisgen R, Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar]
- [17].Brookhart M, Studabaker WB, Chem. Rev. 1987, 87, 411–432. [Google Scholar]
- [18].Prileschajew N, Berichte der deutschen chemischen Gesellschaft 1909, 42, 4811–4815. [Google Scholar]
- [19].Speight JG, Lange’s Handbook of Chemistry, Seventeenth Edition 2017 ed., McGraw-Hill Education: New York, Chicago, San Francisco, Athens, London, Madrid, Mexico City, Milan, New Delhi, Singapore, Sydney, Toronto, 2017. [Google Scholar]
- [20].Bauernschmitt R, Ahlrichs R, Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar]
- [21].a) Zheng GY, Rillema DP, DePriest J, Woods C, Inorg. Chem. 1998, 37, 3588–3592; [DOI] [PubMed] [Google Scholar]; b) Amemori S, Sasaki Y, Yanai N, Kimizuka N, J. Am. Chem. Soc. 2016, 138, 8702–8705; [DOI] [PubMed] [Google Scholar]; c) Kianfar E, Apaydin DH, Knör G, Chemphotochem 2017, 1, 378–382; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yuan J, Chen R-F, Tang X, Tao Y, Xu S, Jin L, Chen C, Zhou X, Zheng C, Huang W, Chem. Sci. 2019, DOI: 10.1039/c8sc05198d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.