

 This work is licensed under a
This work is licensed under a Abstract
Pulmonary arterial hypertension (PAH) is a multifactorial cardiopulmonary disease characterized by an elevation of pulmonary artery pressure (PAP) and pulmonary vascular resistance (PVR), which can lead to right ventricular (RV) failure, multi-organ dysfunction, and ultimately to premature death. Despite the advances in molecular biology, the mechanisms underlying pulmonary hypertension (PH) remain unclear. Nowadays, there is no curative treatment for treating PH. Therefore, it is crucial to identify novel, specific therapeutic targets and to offer more effective treatments against the progression of PH. Increasing amounts of evidence suggest that epigenetic modification may play a critical role in the pathogenesis of PAH. In the presented paper, we provide an overview of the epigenetic mechanisms specifically, DNA methylation, histone acetylation, histone methylation, and ncRNAs. As the recent identification of new pharmacological drugs targeting these epigenetic mechanisms has opened new therapeutic avenues, we also discuss the importance of epigenetic-based therapies in the context of PH.
Keywords: PAH, epigenetics, vascular remodelling
Background
Pulmonary arterial hypertension (PAH) is a vascular remodeling disease characterized by an increase of PVR and an elevation of PAP ≥25 mmHg at rest or 30 mmHg with exercise, leading to RV failure and ultimately death (1, 2, 3, 4). Various biological processes contribute to muscularization, obstruction, and constriction of distal pulmonary arteries (PAs) (5) (Fig. 1). It is now well established that vascular remodeling includes multiple cell types of the vessel wall such as pulmonary artery endothelial cells (PAEC) and pulmonary artery smooth muscle cells (PASMC). Vascular remodeling occurs as a result of pulmonary vascular endothelial dysfunction, PASMC proliferation and migration, medial hypertrophy, inflammation, and thrombosis in situ leading to the formation of plexiform lesions, which progressively increase the vascular resistance in PAH (6). In addition, the imbalance between vasodilators and vasoconstrictors contributes to increase PVR and RV hypertrophy and ultimately to RV dysfunction (7).
Figure 1.
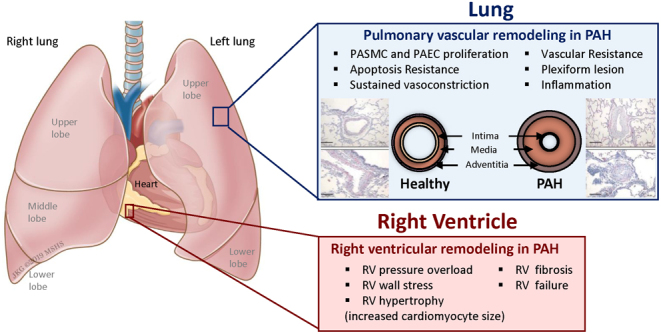
Pathogenesis of pulmonary hypertension. PAH is associated with vascular and cardiac remodeling. In PAH, vascular remodeling is characterized by smooth muscle and endothelial cell dysfunction. Indeed, both cell types contribute to muscularization, obstruction, and constriction of distal pulmonary arteries (PAs) which progressively increase the vascular resistance to induce right ventricle (RV) hypertrophy and ultimately RV dysfunction. Representative pictures of the vascular remodeling in pulmonary arterioles have been previously published by Dr Hadri and collaborators (185). PAH, pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cells; PAEC, pulmonary artery endothelial cell.
The World Health Organization classifies PH into five groups based on the underlying etiology (8) (Fig. 2). Group 1 (PAH) refers to idiopathic or inherited PAH, drug or toxins induced, connective tissue and heart diseases, human immunodeficiency virus infection, portal hypertension, congenital tissue and heart disorders, schistosomiasis, chronic hemolytic anemia, pulmonary capillary hemangiomatosis, pulmonary veno-occlusive disease, and PH of the newborn (8). The term PH is used for all the other groups of the classification: group 2, 3, 4, and 5. Left heart-associated diseases are associated with group 2 (8). Group 3 is associated with lung diseases such as chronic obstructive pulmonary disease and sleep apnea (8). Group 4 develops from thromboembolic diseases (8). Group 5 occurs in response to various disorders related to sarcoid and other rare disorders including blood and metabolic disorders (8). PAH is considered as a rare lung disease and is estimated to affect around 15–60 patients per million population.
Figure 2.
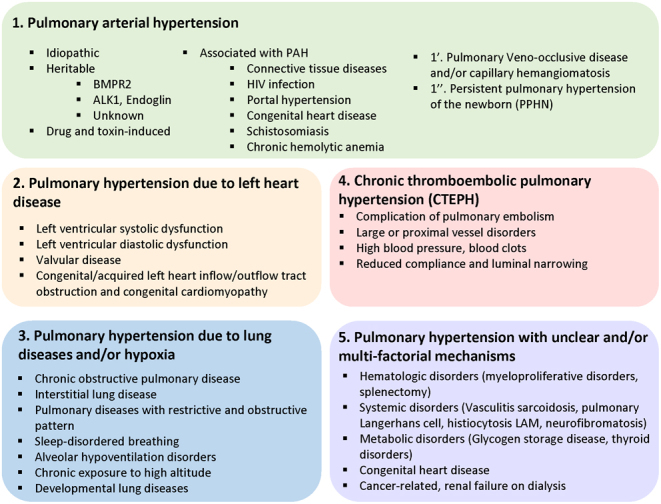
Updated classification of pulmonary hypertension. The World Health Organization classifies PH into five groups based on the underlying etiology. Group I includes PAH. Group II refers to PH from left sided heart disease. Group III refers to PH caused by chronic hypoxia lung disease. Group IV is associated with chronic blood clots, and Group V includes all other forms of PH associated with unclear multifactorial mechanisms such as sarcoidosis and hematological disorders.
Most of the current therapies for treating PH target mostly patients with WHO group I PAH, with a significant pre-capillary component (9). Specific current PAH treatment strategies target mainly three major pathways: Prostacyclin, NO signaling, and Endothelin (ET) receptor; and fall into four categories: Prostacyclin analogues and receptor agonists, Phosphodiesterase 5 inhibitors, ET-receptor antagonists (ERA), and cGMP activators (10). Prostacyclin (also called prostaglandin I2 or PGI2) is a prostaglandin member, which acts as a potent vasodilator and platelet-aggregating inhibitor. Clinical studies have shown that continuous administration of a synthetic prostacyclin analog, named Epoprostenol, increased the survival and exercise capacity in patients with WHO Group I PAH by sustaining the RV function but did not prevent the vascular remodeling (11). Endothelial nitric oxide (NO) is synthesized by endothelial nitric oxide synthase (eNOS) and acts as a powerful vasodilator of pulmonary vessels (12). Inhalation of NO has shown beneficial effects in pediatric cases of PAH. However, inhaled NO remains an expensive approach, and not all patients respond to this therapy. Moreover, rapid withdrawal of inhaled NO therapy can also have deleterious effects and drastically increase pulmonary pressure. Finally, ET-1 is secreted by the vascular endothelial cells and potentiates vasoconstriction and vascular remodeling in lungs (13). While the dual ETA and ETB receptors antagonist bosentan improved exercise capacity and cardiopulmonary hemodynamics in patients with PAH. However, high liver toxicity remains the most prevalent side effect of oral ERA. Increased hepatic aminotransferase levels have been reported in patients under ERA treatment. For example, sitaxentan was withdrawn from the market due to its acute and irreversible liver toxicity. As some ERAs have been associated with liver failure, its use highlighted the critical importance of monitoring hepatic function in patients under ERAs-based therapies.
Unfortunately, our understanding of the PH pathogenesis remains incomplete, and despite advances in molecular biology, there is no curative treatment as of today. Therefore, the identification and characterization of new therapeutic targets for treating PAH are necessary.
Epigenetic mechanisms
Epigenetics is defined as a heritable change occurring in the genome resulting in a change of gene expression without affecting the DNA sequence (14). These heritable changes occur through specific epigenetic modifications called epigenetic marks and have a direct impact on the chromatin packaging, accessibility, and gene expression (15). In mammals, epigenetic mechanisms play a key role during the embryonic development, in genomic imprinting, X-chromosome inactivation, and repression of transposable elements, aging, and carcinogenesis (16, 17, 18). There are three major layers of epigenetic modifications: DNA methylation, histone modifications, and ncRNAs (Fig. 3). These epigenetic modifications can turn the genes ‘off’ and ‘on’. The key mediators associated with DNA and histone modifications are divided into three main groups: writers, erasers, and readers (19). Writers refer to the enzymes that are directly responsible for modifying nucleotide or specific amino acid residue on histones. Erasers refer to a group of enzymes able to remove these marks and readers include all the proteins containing a specialized domain capable of recognizing specific epigenetic marks. ncRNAs also play a critical role in the gene expression as a part of the epigenetic machinery (20). They are RNA molecules transcribed from DNA to RNA, but not translated into proteins. They include miRNA, siRNA, piRNA, and lncRNA. In general, they regulate gene expression at the transcriptional and post-transcriptional level (21). miRNAs have been extensively investigated over the past few decades (22). They belong to a class of small noncoding RNAs of ∼22 nt in length and are involved in the regulation of gene expression at the post-transcriptional level by degrading their target mRNAs and/or inhibiting their translation. There is increasing evidence showing that dysregulation of these epigenetic modifiers plays an important role in cancer and cardiovascular diseases (23, 24, 25, 26). For instance, abnormal hyper-methylation in the promoter region of the tumor suppressor genes has been described as one of the key mediators in tumorigenesis, while hypo-methylation is associated with chromosomal instability and loss of imprinting (27). Recent findings have indicated that targeting DNA or histone methylation/acetylation might be a promising strategy for epigenetics-based therapy (28, 29, 30, 31).
Figure 3.

Major epigenetic mechanisms. There are three major layers of epigenetic modifications: DNA methylation, histone modifications, and non-coding RNAs. These epigenetic marks regulate the expression of genes. DNA methylation is regulated by enzymes called DNA methyltransferases and DNA demethyltransferases. Histone post-translational modifications occur on the N-terminal tails of four core histones (H3, H4, H2A, and H2B). Among the histone modifications, acetylation and methylation remain the most studied mechanisms. ncRNAs also play a critical role in gene expression by degrading their target mRNAs and/or inhibiting their translation. ncRNAs include miRNA, siRNA, Piwi-interacting RNA (piRNA), and lncRNA.
In this review, we focused on different epigenetic mechanisms and emphasized on how they affect various cellular processes. Over the past years, it has become increasingly recognized that epigenetics may play a critical role in the initiation and progression of PAH, as demonstrated by the increasing amount of reviews recently published in the field (32, 33, 34). Here we present the most comprehensive and up-to-date review on the role of epigenetic mechanisms in the context of PAH. This review not only summarized classical histone or DNA modifications, miRNAs or lncRNAs but also critically discussed some controversial findings in the literature. This review also highlights the relationship between ncRNAs and PASMCs contractility. Finally, as recent developments and identification of new pharmacological drugs targeting these epigenetic tools in cancer have opened new avenues for the treatment of PAH, we have also highlighted the importance of epigenetic dysregulation in the progression of this disease.
DNA methylation
DNA methylation is a process by which methyl groups -CH3 are added to the DNA molecules on the 5-carbon of the Cytosine ring to form 5-methylcytosine (35, 36). DNA methylation occurs, almost exclusively, at the Cytosines that are located next to Guanines and bond by a phosphate called CpG dinucleotide sites. Enzymes, known as DNA methyltransferases (DNMTs), tightly regulate this process (37). DNMTs are a family of highly-conserved proteins, including five members in mammals: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L (38). DNMT1 was the first-identified DNMT and remains the most abundant isoform in adult cells. Another two mammalian members, DNMT3A and DNMT3B mediate the de novo methylation. The DNA methylation state is conserved during cell division and transmitted to daughter cells along with the DNA sequence (39). During the replication, DNMT1 is responsible for duplicating the methylation pattern from the parental strand to the daughter strand. This enzyme is described as a maintenance methyltransferase. DNMT1 interacts specifically with hemimethylated DNA. Hemimethylated DNA contains the methylation marks only on one strand. DNMT1 interacts specifically with hemimethylated DNA and contributes to the methylation of the daughter strand to restore fully methylated CpG dinucleotides. Thus, DNMT1 maintains the stability of this epigenetic mark throughout the different generations. Interestingly, recent studies have also associated DNMT1 with methylation of unmethylated human CpG islands in cancer cells. They reported that a majority of the de novo methyltransferase activity was mediated by DNMT1 in a gene-specific manner. DNMT2 enzymes do not methylate DNA but methylate small RNA. However, in 2003, Hermann et al. have demonstrated that DNMT2 may have a de novo CpG methylation activity (40).
DNA methylation at CpG sites represses gene transcription and silences gene expression (41, 42). Methylated CpGs recruit and interact with methylated-CpG-binding proteins known as MeCP1 and MeCP2. These proteins alter the transcription through their transcriptional repression domain. Alternatively, MeCP2 or MeCP1 proteins can form a complex along with other molecular partners to condense the chromatin (43). Repression of gene transcription is also achieved by structural changes directly associated with the methyl group. Addition of a methyl group directly affects the interaction of the transcription factor on its DNA binding sites and will therefore block the transcription of the gene (44). Indeed, DNA methylation induces structural changes that physically obstruct the binding of transcriptional factors onto the gene promoter region (45). In addition to methyltransferase activity, the interaction of DNMT1 with DNMT1-associated proteins, E2F1 transcription factor, histone deacetylases (HDAC), and methyl-CpG binding proteins (MBD) make DNMT1 a central element of transcription repression complex (46). Yet, methylated DNA may recruit additional proteins known as methyl-CpG-binding domain proteins (MBDs), which can interact with other proteins such as HDAC and other chromatin remodeling proteins, resulting in compact and inactive chromatin (47). This relation between DNA methylation and chromatin arrangement may strongly influence disease development. For example, mutations in the MeCP2 gene caused Rett syndrome, a neuro-developmental disorder characterized by mental retardation; and MBD2 mediates the transcriptional repression of hypermethylated genes in cancer (48, 49).
In PAH, DNA methylation plays an important role in the regulation of superoxide dismutase 2 (SOD2) expression – a critical intra-mitochondrial antioxidant enzyme. SOD2 expression is significantly decreased in PASMCs in PAH patients and Fawn-hooded rats (FHR), a rat model which spontaneously develops PH (50, 51). Mechanistically, downregulation of SOD2 activates the hypoxia inducible factor (HIF-1α) which increases the expression of the oxygen-sensitive, voltage-gated K+ channels (Kv1.5). These changes result in impaired oxygen sensing and reduced cytoplasmic and mitochondrial redox state (50). Inhibition of SOD2 expression by siRNA in normal Sprague–Dawley PASMC recapitulates the PAH phenotype. Conversely, SOD2 overexpression inhibits HIF-1α and restores Kv1.5 expression in FHR PASMC. Using genomic bisulfite sequencing, Archer and colleagues (51) identified selective hypermethylation of CpG island in the promoter and an enhancer region of intron 2 of the SOD2 gene (51) (Fig. 4). Of particular interest, increased DNA methylation level correlates with increased expression of the DNMT1 and DNMT3B in FHR lungs and PASMCs. Most importantly, inhibition of DNA methylation by 5-aza-2′-deoxycytidine restores SOD2 expression, reduces proliferation, and promotes apoptosis in FHR PASMC. These data showed that epigenetic attenuation of mitochondrial SOD2 contributes to PAH development.
Figure 4.

Illustration of several underlying epigenetic mechanisms in pulmonary hypertension. DNA methylation plays an important role in the regulation of the expression of superoxide dismutase 2 (SOD2) and bone morphogenetic protein receptor 2 (BMPR2). Low SOD2 and BMPR2 expression is associated with increased proliferation and decreased apoptosis in hPASMC. Several histone deacetylases (HDAC1, 4, 5, and 6) are upregulated in PAH. HDACs inhibitors (VPA, SAHA, MGCD0103, MS-275, and Vorinostat) have been extensively investigated in multiple experimental models of PH. miRNAs (i.e miR-17/29; -21; 20a; -135a; miR-124; mR-204) target key signaling pathways (i.e BMPR2 and PPARγ) to influence the proliferation and migration of PASMCs and PAECs. The aforementioned miRNAs regulate several processes such as BMPR2 and PPARγ and hypoxia and other pro-proliferative signaling pathways implicated in the development of PAH.
Heritable PAH represents (HPAH) approximately 6–10% of all PAH. Genetic studies have identified more than 450 mutations heterozygous germline mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene (52, 53, 54). Accumulated evidence pointed to BMPR2 mutations as being the most important predisposing factors for HPAH, with a penetrance of 27% (55), indicating that a BMPR2 mutation alone is not sufficient to develop HPAH (56). Moreover, Gimelbrant et al. (57) have reported a differential allelic expression using a genome-wide approach. These findings demonstrate that the two copies of many genes are not simultaneously expressed at identical levels (57). This study also suggests that other mechanisms, such as epigenetic marks, may contribute to the diversity in individual cells and clonal descendants. These mechanisms may also affect gene expression of the WT allele and contribute to the early onset of the disease (58).
In 2017, Liu et al. have measured the methylation status of the BMPR2 promoter in DNA samples isolated from peripheral blood mononuclear cells (PBMCs) in 12 patients with HPAH using genomic bisulfite sequencing (59). They have demonstrated that the BMPR2 promoter is hypermethylated in patients with HPAH, resulting in the downregulation of BMPR2 expression. Low BMPR2 expression in lung tissue and endothelial cells increases cell proliferation and promotes the development of PAH. In addition, their results suggest that specific allelic methylation of the WT strand in patients with HPAH may contribute to the penetrance of HPAH and trigger the early onset of disease. However, this study faces several limitations. Only a few patients were enrolled in this study, and the methylation status of the BMPR2 promoter was measured only in PBMCs. Lung tissues or PAEC from patients may be more relevant to decipher the development of HPAH.
On the contrary, a similar study analyzed the methylation status of the upstream region of the BMPR2 gene and its association with BMPR2 mutations in iPAH patients (60). The authors found no significant difference in the methylation of CpG islands of the BMPR2 promoter between iPAH patients and healthy control donors. In another study, Poussada et al. analyzed the methylation pattern of the BMPR2 promoter region in PAH patients and controls from genomic DNA obtained from the peripheral blood using methylation-specific PCR (61). Again, no evidence of methylation was detected in this region in patients and controls. Several differences across these studies may explain the heterogeneity of the previous results. First, only few patients were enrolled in these studies. Second, age, gender, stages of the disease, diversity of PAH group, and intra-individual variability may also affect the methylation pattern. Finally, different regions of the BMPR2 promoter or gene were investigated. Additionally, the genetic profile of the enrolled patients was not reported and may explain the lack of reproducibility. Nonetheless, recent technological advances for epigenetic profiling offer now new methodologies to further investigate the methylation pattern of the BMPR2 gene.
In contrast to DNA methylation, DNA demethylation of a gene promoter is associated with transcriptional activation and gene expression. Ten-eleven translocation methylcytosine dioxygenase 1 (TET1) catalyzes the conversion of the modified DNA base 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) and initiates the first step of active DNA demethylation in mammals (62, 63). Colon, breast, prostate, and liver cancer showed a significant decrease of TET1 expression (64, 65, 66). Additionally, increasing TET1 expression levels in colon cancer cells decreased cell proliferation both in vitro and in vivo through demethylation of promoters of the WNT signaling pathway (67). Degradation of TET1 in hypoxia-induced EMT lung cancer cells decreased metastasis rates and cells (68). By initiating DNA demethylation, TET1 activates gene transcription of tumor suppressor genes and plays a major role in the regulation of several cellular processes including proliferation, apoptosis, and migration (69, 70). Since PAH shows many similarities with cancer such as proliferation, resistance to apoptosis, and dysregulation of tumor suppressor genes, further investigations of DNA demethylation should be considered in future studies in PAH.
Histone post-translational modifications
Histones are highly-evolutionary conserved proteins and constitute the primary components of chromatin (71). The histone octamer is composed of dimers of each of the four central core histones: H2A, H2B, H3, and H4 (72). DNA is wrapped around the histone octamer and forms the nucleosome, which is defined as the repeating unit of chromatin. Histone post-translational modifications modify and affect gene expression patterns and can either activate or repress gene expression. Among the histone modifications, acetylation and methylation remain the most studied mechanisms so far (73). However, other histone marks such as glycosylation, ubiquitination, ADP-ribosylation, biotinylation, phosphorylation, and SUMOylation can also affect the chromatin compacting (73, 74). Histone acetylation and methylation occur on specific amino acids like Lysine (K) and/or Arginine (R) (75). These chemical processes result, respectively, in the addition of acetyl or methyl groups to a specific amino acid located on the histone.
Histone acetylation
Histone acetylation is found at the amino termini of the four core histones and is mediated by the histone acetyltransferases, commonly named HATs or KATs since only lysine residues (K) can be acetylated (76). There are 18 different types of HATs in mice and human. Among the most active HATs in mammals, we can mention cAMP response element-binding protein (CREB) and its binding protein (CBP), p300, p300/CREB-binding protein-associated factor (PCAF), and HIV Tat interactive 60-kDa protein (Tip60) (77, 78). The histone acetylation level is also regulated by histone deacetylases or HDACs. In contrast, HDACs remove acetyl groups from histone proteins and represses DNA transcription by tightening the chromatin (79). They are classified into four different groups: a- class I HDACs (HDAC 1, 2, 3, and 8) are exclusively expressed in nucleus; b- class II HDACs (HDACs 4, 5, 6, 7, 9, and 10) are expressed in nucleus and cytoplasm; c- class III HDACs (Sirtuins 1–7) are located in the nucleus (Sirtuin1–6–7); and d- class IV (HDAC 11) are expressed both in the cytoplasm and nucleus. Histone acetylation is universally associated with transcriptional activation and is not mitotically heritable due to the rapid dynamics of histone acetylation and deacetylation. Interestingly, proteins expressing bromodomain, such as BRD4, recognize specific acetyl groups and may recruit specific proteins to form a molecular complex in order to finely modulate gene transcriptional activity (15). Recruitment of chromatin remodelers might affect the structure and level of condensation of the chromatin.
Over the past decade, many studies have shown that HDACs are major regulators in left ventricular (LV) cardiac hypertrophy in response to stress such as transverse aortic constriction, infusion of isoproterenol, DOCA salt, phenylephrine, or Endothelin-1 (80, 81). Conversely, HDAC inhibitors, such as the small compound Trichostatin A (TSA), prevent pressure overload-induced pathological cardiac hypertrophy and fibrosis and improve ventricular function. With respect to PAH, Zhao et al. have shown that HDAC1, HDAC4, and HDAC5 protein expression are significantly upregulated in human lung isolated from iPAH patients (82) (Fig. 4). Similar results were found in the lungs and RV of hypoxia-induced PH animal models. In this line, increased levels of histone acetylation on H3 and H4 at the promoter region of the endothelial nitric oxide synthase (eNOS) gene have been found in PAEC from Persistent PH of the Newborn (PPHN) (83). Recent studies illustrate the importance of HDACs in RV failure in several PH experimental models. For example, Cho et al. have assessed the therapeutic potential of Valproic Acid (VPA), a histone deacetylase inhibitor, in a rat model of monocrotaline (MCT)-induced PAH and in the rat pulmonary artery banding model (PAB) (84). VPA has been used in the clinic for decades as an anticonvulsant and mood stabilizer. The authors found that VPA administration in drinking water increased histone acetylation in the RV of VPA-treated rats and consequently reduced RV hypertrophy. Echocardiography analysis also revealed that VPA improved RV systolic function in MCT-treated animals (84). In 2015, another study demonstrated that pharmacological inhibition of class IIa HDACs using MC1568 (50 mg/kg; daily intraperitoneal administration) restored MEF2 activity in PAECs, as demonstrated by increased expression of its transcriptional targets such as miR-424 and miR-503 (85). Restoration of MEF2 activity resulted in decreased cell migration and proliferation and rescue of experimental pulmonary hypertension models. These data indicate that HDACs can be considered as promising therapeutic targets in vascular remodeling disease.
Controversially, a separate study from Bogaard and colleagues investigated the effect of a broad-spectrum HDAC inhibitor TSA on RV function and remodeling using the PAB-induced PH model in rats (86). Surprisingly, their results showed that HDAC inhibition with TSA failed to prevent RV remodeling, but instead exacerbated RV fibrosis and promoted RV dysfunction. Their data also showed that isoform-selective HDAC inhibition might be better tolerated than non-selective pan-HDAC inhibition with compounds such as TSA. This was later confirmed by Cavasin et al. who tested two compounds that were highly selective for class I HDACs (HDAC1, HDAC2, and HDAC3): MGCD0103 and MS-275 in the hypoxia-induced PH rat model. MGCD0103 (10 mg/kg) and MS-275 (3 mg/kg) were administrated via intraperitoneal injections for 3 weeks (Fig. 4). Both compounds successfully decreased class I HDAC catalytic activity in the RV and the lungs. Although the ability of HDAC inhibitors to reverse PAH was not explored in this study, they demonstrated that class I HDAC inhibitors impaired hypoxia-induced PH and improved hemodynamics including RV cardiac output, pulmonary vascular resistance PA acceleration time (PAAT) and velocity time integral (VTI), RV end-systolic pressure, and pulmonary artery pressure. Finally, Zhao et al. showed that VPA and SuberoylAnilide Hydroxamic Acid (SAHA), Vorinostat (HDAC class I, II, and IV inhibitor) reversed hypoxia-induced PH in rats and decreased hPASMCs and hPAECs proliferation as well as inflammation in vitro (82, 87). As previously described, there is a controversy surrounding the role of HDAC in PH. PH is also characterized by the heterogeneity and severity of RV remodeling, which may be considered either as adaptive or maladaptive, compensated or decompensated. Therefore, inhibition of adaptive RV response by broad-spectrum HDAC inhibitors may be associated with detrimental outcomes while inhibition of maladaptive RV may be beneficial. It is essential to define the type of RV remodeling and function in PAH patients. This is well illustrated by the electrocardiographic abnormalities, pulmonary embolism, and thrombocytopenia that have been reported after the use of broad-spectrum HDAC inhibitors in patients with lymphoma (88, 89). The use of selective HDAC inhibitors may be an alternative approach to limit side effects on off-target genes. For example, Kim et al. have demonstrated that selective HDAC IIa inhibition protected against RV dilatation (85). Similarly, Cavasin and colleagues showed that selective HDAC I attenuated RV hypertrophy in the chronic hypoxia model of PH (90). The use of different PAH models (Sugen-Hypoxia, MCT, and PAB) and protocols, duration of treatment, frequency of treatment, and selectivity of HDAC inhibitors may explain this controversy surrounding the use of HDAC inhibitors as a therapeutic approach.
Contrary to nuclear HDACs implicated in epigenetic regulation of transcription, HDAC6 is exclusively localized in the cytoplasm and displays a deacetylation activity toward ‘non-histone’ proteins (91). Several studies have demonstrated that HDAC6 is overexpressed in many cancers such as hepatocellular carcinoma, glioblastoma, and prostate cancer. Domain-selective small-molecule inhibitor of HDAC6 inhibits cell growth, cancer progression, and re-sensitizes transformed cells to anticancer agents (92). Boucherat and colleagues reported increased expression levels of HDAC6 in lungs and distal PAs from PAH patients and experimental animal models of PAH induced by MCT and Sugen/Hypoxia (93). They found that HDAC6 was overexpressed only at the protein level in PAH-PASMCs and PAH-PAECs (compared to control cells) as well as RV from PAH patients or Su/Hx and MCT rats. Blockade of HDAC6 activity by either Tubastatin A (TubA) or ACY-775 or siRNA against HDAC6 significantly decreased PAH-PASMC proliferation, resistance to apoptosis, and migration. Pharmacological inhibition of HDAC6 improved pulmonary hypertension in the Sugen/Hypoxia and MCT rat model as demonstrated by decreased RV systolic pressure (RVSP), mean PA pressure (mPAP), cardiac output (CO), total pulmonary vascular resistance (TPR), RV hypertrophy, vascular remodeling, and restoration of the proliferation/apoptosis balance (93). Using the genetically engineered WT (Hdac6 Y/+) and Hdac6-knockout (Hdac6 Y/−) mice models, they showed that the loss of Hdac6 conferred protection against chronic hypoxia-induced PH (93).
Finally, epigenetic readers that recognize histone modification patterns and bind to acetylated lysine residues of histone tails, may also contribute to the development of PAH. This is well illustrated with the BRD4 bromodomain protein, which acts as a cofactor, facilitating transcriptional activation of target genes. BRD4 is upregulated in lungs, distal PAs, and PASMCs of patients with PAH compared with healthy donors (94). Mechanistically, microRNA-204 upregulated BRD4 expression in PAH. Pharmacological inhibition of BRD4 with JQ1 increased p21 expression and induced cycle arrest in PAH-PASMCs. This effect of BRD4 inhibition was accompanied by decreased expression of several oncogenes such as nuclear factor of activated T cells (NFAT), B-cell lymphoma 2 (BCL2), and Survivin, all known to be overexpressed in PAH. Inhibition of this oncogenic signature decreased proliferation and increased apoptosis of PAH-PASMC. Finally, blockade of BRD4 activity by either nebulization of siRNA against BRD4 or JQ1 reversed established PAH in the Sugen/hypoxia rat model in vivo (94).
Histone methylation
In addition to DNA methylation, histone methylation also regulates the gene expression pattern (95). Histone methylation is a process by which methyl groups are transferred to specific amino acids of histone proteins. Methylation of histones tightly influences gene expression. Gene activation or inhibition status depends on which amino acids in the histones are methylated and how many methyl groups are attached (Fig. 3). This is the only post-translational modification that can add up multiple methyl groups onto Lysine (K) residues resulting in mono (me1), di (me2), and tri-methylated (me3) states (96). As acetylated lysines served as docking sites of proteins expressing a Bromodomain, methylated lysines can interact with several different domain types including Chromodomains, Malignant Brain Tumor (MBT) domain, plant homeodomain (PHD) domains, or Tudor domains. Binding of chromodomain-containing proteins has different functional consequences depending on the docking sites. For example, the chromodomain of CHD1 binds to the H3K4 methylation, heterochromatin protein 1 (HP1) chromodomain interacts with the H3K9 methylation, and CBX2 chromodomain recognizes and binds to the H3K27 methylated (97). CHD1, which binds to the active epigenetic methylation H3K4, belongs to the family of ATP-dependent chromatin remodeler (98). CHD1 stimulates DNA unwrapping and makes the nucleosomes more broad. Histone lysine methyltransferases catalyze the transfer of methyl group from adenosyl-methionine. Due to the complexity and heterogeneity of this process, we have highlighted only several well-characterized modifications. These include the mono- and di-methylation of lysine residues 4 and 9 on histone H3 (H3K4me2/3 and H3K9me2/3, respectively) and the tri-methylation of the lysine residues 27 on the histone H3 (H3K27me3).
H3K4 methylation is restricted to the promoter and enhancer region of genes and is not able to spread over the entire gene (99). H3K4 methylation is associated with an increase of the transcriptional activity while H3K9 methylation represses gene expression and is mostly associated with constitutive heterochromatin (100). Similarly to H3K9me2/3, H3K27 methylation is a repressive methyl mark and can spread over the entire gene locus (101). Heterochromatin is associated with a different type of histone methylation like the H3K9 or H3K27 methylation and is found in regions with inactive gene transcription. A specific class of H3K9 methyltransferases, including SUV39H1/KMT1A, SUV39H2/KMT1B, SETDB1/KMT1E, dimeric G9a/KMT1C-GLP (G9a-like protein)/KMT1D, and PRDM family, mediates the methylation of H3K9. Similarly, H3K27 or H3K4 methylation is mediated by other lysine methyltransferases such as EZH2 and SET1 proteins, which can specifically recognize the H3K27 and H3K4 residues (102). Methyl groups can also be removed from the histone by specific lysine demethylases, which are known as KDMs and express specific activity toward the methylated H3K9, K4, and K27.
Polycomb group proteins are important chromatin-associated factors contributing to the maintenance of transcriptional repression (103). The Polycomb Repressive Complex 2 (PRC2) is one of the most- and best-investigated epigenetic complexes as of today. The PRC2 complex has a very important role in silencing gene expression (104). PRC2-modified histone tails act as docking sites for other chromatin proteins, which can alter the chromatin packaging or recruit other molecular partners (105). EZH2 is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2) and promotes transcriptional gene silencing by trimethylation at lysine 27 of Histone H3 (H3K27me3). EZH2 overexpression has been shown to be important for tumor growth, metastasis and correlates with poor prognosis in several cancer types including melanoma, brain, breast, lung, liver, kidney, prostate, ovarian, gastric, and pancreatic cancer and Diffuse Large B-Cell lymphoma (106, 107). Of particular interest, Aljubran et al. analyzed the role of EZH2 in PASMCs isolated from control and hypoxia-induced PAH mouse model (108). Their data showed that EZH2 overexpression induced proliferation and migration of hPASMCs while decreasing apoptosis (Fig. 4). In 2018, Shi and collaborators have demonstrated that pharmacological inhibition of EZH2 with EPZ005687 protects against TAC-induced PAH by preventing ROS production in the lung (109). At the molecular level, EPZ005687 significantly decreased the H3K27me3 enrichment in the promoter region of SOD1, thereby repressing SOD1 transcription and expression.
Another study has focused on the role of the H3K27me3 mark in pulmonary vascular endothelial cell proliferation, apoptosis, and inflammatory response in PAH (110). It is shown that pharmacological inhibition of JmjC domain-containing protein JMJD3/KDM6B by GSK-J4 significantly decreased proliferation, potentiated apoptosis, and reduced TNF alpha-induced IL-6 release in a concentration-dependent manner in pulmonary vascular endothelial cells. This study strongly suggests that JMJD3/KDM6B may regulate the pulmonary vascular endothelial cell activation in an H3K27me3-dependent manner in PH. Yet, Chen and collaborators investigated the role of the transcriptional modulator megakaryocytic leukemia 1 (MKL1), also known as myocardin-related transcription factor A (MRTF-A) in hypoxia-induced PAH. Loss of MKL1/MRTF-A protected against the development of hypoxia-induced PH (111, 112). Specifically, it is shown that MKL1 coordinates epigenetic alterations on cell adhesion molecules (CAMs) promoters in human umbilical vein endothelial cells. Indeed, MKL1/MRTF-A mediates the recruitment of the H3K4 complex to the CAM promoters (113). In addition, silencing of ASH2 and WDR5, two key components of the histone H3K4 methyltransferase complex, reduced hypoxia-induced PH in mice (113).
Non-coding RNA (ncRNA)
Another layer of epigenetic regulation of gene expression, not taking place on DNA, is ncRNAs (Fig. 3). There are several groups of ncRNA clustered based on their function or length as previously mentioned, but here, we will only review the contribution of micro RNA (miRNA) and long ncRNA (lncRNA) to the development of PAH.
micro RNA
miRNAs are short non-coding transcripts with the length 21–25 nucleotides. Single miRNA can regulate the expression of multiple genes and inversely the expression of a single gene can be regulated by multiple miRNAs (114). miRNAs are transcribed as pre-miRNAs by RNA polymerase II from either intronic or exonic regions (115). Pre-miRNAs are several hundreds of bases long and contain 5′ m7G cap and 3′ poly-A tail (116) and are then processed in the nucleus by Drosha to yield 70 nt transcript in a form of hairpin (116), which is exported to cytoplasm (117). The pre-miRNA is then spliced by DICER to produce double-strand miRNA, then separated and loaded onto the RNA-induced silencing complex (RISC) (118). miRNAs within the RISC complex then regulate cellular processes such as proliferation, differentiation, or apoptosis, and therefore miRNAs are also implicated in the pathogenesis of PAH (119). miRNAs play an important regulatory role in the proliferation and migration of both PASMCs and PAECs. These miRNAs regulate several pathways such as BMPR2, PPARγ, Hypoxia, and other pro-proliferative signaling pathways implicated in the development of PAH.
One of the key signaling pathways involved in the regulation of many miRNAs is the hypoxia-regulated pathways through the hypoxia inducible factor (HIF). It has been shown that hypoxia stimulates the expression of miR-1; -9; -17/92; -21; -27a; -27b; -138; -190; -199-5p; -210; -322; -361-5p; and -23a (120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132). Expression of most of these miRNAs is upregulated by the action of HIF-1α. Several of these miRNAs affect multiple signaling modules involved in the development of PAH such as the BMP/SMAD signaling or expression of transmembrane ion channels. Among these miRNAs, miR-17/92 downregulates prolyl hydroxylase 2 which leads to HIF-1α stabilization and stimulation of proliferation of PASMC after hypoxia (133). miR-17/92 also targets proteins such as BMPR2, p21, PTEN, MFN2, and promotes cell survival in PASMCs (122, 134, 135) (Fig. 4). Moreover, miR-17/92 is stimulated by inflammation and activates NF-ΚB, thus upregulating the retinoblastoma protein (136). Similar to miR-17/92, miR-138 exerts pleiotropic effects and stimulates cell survival in PASMCs via AKT and Mst1 kinase (137). Additionally, it exerts a profound effect on nitric oxide (NO) production. Indeed, miR-138 targets S100A1 to promote downregulation of eNOS activation and subsequent inhibition of NO production (126, 138) (Fig. 4). miR-27b and miR-195a-5p also modulate NO production. miR-199a-5p inhibits NO production by targeting SMAD3 in PASMCs and PAECs (128). miR-27b targets PPARγ, thus altering the stability of eNOS/HSP90 complex important for NO production (125).
PPARγ is another key player in the miRNA network – many miRNAs target PPARγ and vice versa PPARγ downregulates several miRNAs. miR-21 is one of the miRNAs that is negatively regulated by PPARγ. Downstream, multiple targets of miR-21 have been identified. Indeed, miRNA-21 downregulates BMPR2, RhoB, PDCD4, SPRY2, PPARα, PTEN or SATB1, leading to the stimulation of PASMCs and PAECs proliferation, migration and contraction and therefore to the development of PAH (123, 139, 140, 141). Moreover, PPARγ also downregulates miR-27a or miR-130/301 (124, 142, 143). One of the hallmarks of PAH is the alteration of the bone morphogenetic proteins/SMAD signaling (144). A large body of evidence indicates that miRNAs play an important role in regulating BMP/SMAD signaling. One of the proteins, whose expression is tightly regulated by miRNAs, is BMPR2. Indeed, miR-17/29; -21; -20a; -135a; -23a targets BMPR2 and regulates proliferation and differentiation of PASMC and PAECs (122, 123, 132, 145, 146). Additionally, miR-322 targets other members of the cascade, namely BMPR1 and Smad5, thus stimulating the proliferation of PASMCs (130).
The development of PAH is also characterized by increased contractility of PASMCs (147, 148). Contractility is dependent on the activation of the Rho/ROCK pathway, which stimulates actomyosin cytoskeleton and the expression of transmembrane transporters (149). It has been shown that miRNA targets several unrelated transporters. One of them is the ATP-binding cassette transporter A1 (ABCA1). ABCA1 is a c-AMP dependent transporter exporting intracellular cholesterol and phospholipids (150). Two miRNAs – miR-143/145 and miR-361-5p – have been shown to downregulate the expression of ABCA1 in PASMCs in hypoxic conditions, therefore stimulating proliferation and survival (131, 151). Moreover, voltage-dependent potassium channels are also targeted by miRNAs – KCNA5 as a target of miR-1, KCNQ5 as a target of miR-190 and TASK1 as a target of miR-138 (120, 127, 152). Downregulation of these channels alters the membrane potential of PASMCs and stimulates Ca2+ influx, thus stimulating the contractility of PASMCs (153).
On the other side, multiple miRNAs inhibit PASMCs and PAECs proliferation and are downregulated in hypoxic and PAH conditions. One of the most studied is miR-124. It has been shown that miR-124 is downregulated after hypoxia in PASMCs (Fig. 4). miR-124 inhibits the proliferation of PASMCs by inhibiting NFAT and Notch-FoxO3 pathways via stimulation of PTBP1 expression (154, 155). Similarly, in pulmonary adventitial fibroblasts, miR-124 normalizes the ratio of PKM2/PKM1 via PTBP1, consequently inhibiting the proliferation and the migration of the fibroblast (156). miR-204 is also downregulated in hypoxia and targets SHP2 phosphatase, thus inhibiting NFAT and Src pathways, PASMCs proliferation and survival (157) (Fig. 4). Moreover, miR-204 downregulates RUNX2 and inhibits HIF1α, which inhibits the proliferation and promotes the apoptosis in PASMCs under physiological conditions (158).
Many miRNAs targeting the BMP/Smad signaling are downregulated in hypoxic and PAH conditions. miR-140-5p targets the SMAD-specific E3 ubiquitin-protein ligase 1 (SMURF1), which leads to the stimulation of BMP/SMAD signaling and inhibition of PASMCs proliferation (159). Several miRNAs target tyrosine kinase receptors, resulting in the activation of the downstream signaling and inhibition of proliferation and survival. For example miR-193-3p and miRNA-328 downregulate the IGF1R receptor (160, 161). miR-424/503 and miR-339 inhibit FGF-stimulated signaling via targeting FGF2, FGFR1, or FRS2 (162, 163). PDGFRA expression is downregulated by miR-34a in PASMCs (164). Finally, contractility of PASMCs is inhibited by several miRNAs. Especially miR-223 is especially important in the regulation of actomyosin contractility by targeting upstream regulators such as ITGB3, RhoB, and MLC2 (165, 166). Furthermore, calcium levels are important for contractility and are regulated by CaSR, which is targeted by two miRNAs – miR-429 and miR-424-5p (167). An interesting connection between hypoxia, PAH, miRNAs, and DNA damage-stimulated pathways is illustrated by miR-223. Hypoxia-stimulated HIF1α downregulates miR-223, which leads to upregulation of miR-223 target – PARP-1. Increased PARP-1 levels subsequently stimulate PASMCs proliferation and resistance to apoptosis, promoting the development of PAH (168).
Long non-coding RNAs
Long ncRNAs are RNA molecules which are longer than 200 nt but do not encode for any specific and functional protein (169). Based on the DNA element that is being transcribed from, they are classified into sense lncRNAs, antisense lncRNAs, bidirectional lncRNAs, intronic lncRNAs, and intergenic lncRNAs (170). LncRNAs are usually transcribed by RNA polymerase II or III and processed similarly to mRNAs – spliced, 3′-polyadenylated, or 5′-capped (171, 172). LncRNAs can have multiple functions: signal lncRNAs are expressed in specific cellular compartments or at specific times and usually regulate the transcription of other genes. Decoy lncRNAs are used to sequester regulatory proteins or miRNAs to regulate gene expression at transcriptional or translational levels. Guide lncRNAs can recruit transcription activators or repressors to specific genome locations. Scaffold lncRNAs act as a platform for the assembly of various ribonucleoprotein complexes to regulate the chromatin landscape. The most recently discovered class is a circular lncRNAs and acts as transcriptional regulators (173).
Because lncRNAs play an important role in gene expression, they are prime candidates to be involved in the development of PAH. Similar to miRNA regulation, hypoxia is an important stimulus altering the expression of lncRNAs. One of the important pleiotropic lncRNA stimulated by hypoxia and shared with cancer cells is MALAT1. This lncRNA controls the phenotypic switch and stimulates the proliferation of PAECs (174). H19 lncRNA is stimulated by hypoxia via increased PDGF-BB expression (Fig. 4). H19 then increases AT1R expression via sponging let-7b miRNA, which targets AT1R. Increased AT1R expression then leads to the stimulation of PASMCs proliferation (175). Similarly, hypoxia promotes the expression of lncRNA PAXIP1-AS1. PAXIP1-AS1 alters the focal adhesion signaling and upregulates paxillin, which leads to increased proliferation and migration of PASMCs (176). Another hypoxia-induced lncRNA is UCA1 (urothelial carcinoma-associated-1). The mechanism of UCA1 action is sequestering ING5 from hnRNP I, which induces the stimulation of PASMCs proliferation and apoptosis resistance (177). Finally, hypoxia also induced the expression of the lncRNA Hoxaas3, located within the HoxA gene cluster. LncRNA Hoxaas3 was shown to stimulate the expression of HoxA3, which increases the expression of Cyclins A, D, and E, thus stimulating the cell cycle progression and proliferation (178).
Several other lncRNAs are downregulated under hypoxic stress. One of them is lncRNA MEG3 and has been shown to be downregulated in the lungs of patients with PAH (Fig. 4). Inhibition of MEG3 in PASMCs results in the induction of PCNA, cyclin A, and cyclin E expression, thus leading to faster progression through the cell cycle (179). It has been shown that MEG3 is important for p53 activation and therefore cell cycle inhibition (179). Moreover, hypoxia-mediated downregulation of MEG3 promotes miR-21 expression and downregulates PTEN expression, leading to the stimulation of PASMC proliferation (180). LincRNA-p21, a transcriptional target of p53, feeds back to enhance p53 transcriptional activity by physically interacting with the E3 ubiquitin-protein ligase called mouse double minute 2 (MDM2). The physical interaction of lincRNA-p21 with MDM2 releases MDM2 repression of p53, which then allows p53 to interact with p300 and to bind the promoters/enhancers of its target genes (181) (Fig. 4).
Hypoxia also represses the expression of lncRNAs TCONS_00034812 and CASC2. TCONS_00034812 stimulates the expression of the Stox1 transcription factor, which inhibits the activity of major effectors of the MAPK pathway – ERK, JNK, and p38. After hypoxia, downregulation of TCONS_00034812 results in the stimulation of MAPK pathway activity, PASMCs proliferation, and apoptosis resistance (182). LncRNA CASC2 inhibits proliferation, migration, and stimulates apoptosis of PASMCs. It has been shown that CASC2 inhibits the expression of phenotype switch-related marker alpha smooth muscle actin (α-SMA) (183) in PASMCs.
Finally, it is now well-known that altered PDGF signaling contributes to the PAH pathogenesis. However, the underlying mechanism remains not fully understood. Chen and colleagues have shown that PDGF-BB exerts its function partially by repressing the lncRNA LnRPT. LnRPT inhibits the proliferation of PASMCs by decreasing the expression of Cyclin A and by inhibiting the Notch signaling via the downregulation of Notch3 and Jag1 (184).
Conclusion
Considerable progresses have been made in our understanding of gene expression regulation. In the past decades, numerous studies have demonstrated the critical role of epigenetics in cancer and other diseases. New advances in epigenetic drugs have led to the development of new pharmacological drugs for treating several cancers and are now under clinical investigation in phases II and III. Since PAH shows many common features with cancer, new opportunities to investigate the current therapeutic strategies used in cancer should be considered to treat PAH. Indeed, increasing evidence have identified DNA methylation, histone post-translational modifications (acetylation, methylation), and ncRNAs as critical in the development, severity, and progression of PAH. Although several unknown epigenetic mechanisms in PAH are yet to be elucidated, strong evidence suggests that targeting these epigenetic mechanisms may represent an emerging and very promising therapeutic strategy in PAH. Moreover, recent technological advances in epigenetic profiling may help to further investigate and decipher the underlying molecular mechanisms in PAH to identify new therapeutic targets.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This study was supported by the US NIH R01 HL133554 (to L H).
Author contribution statement
M B, R J, F L, and L H contributed to the writing and editing of the manuscript and read and approved the final version.
Acknowledgements
Not applicable. Only the authors listed on the manuscript contributed toward the article.
References
- 1.Montani D, Gunther S, Dorfmuller P, Perros F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud-Macari E, Price LC, et al Pulmonary arterial hypertension. Orphanet Journal of Rare Diseases 2013. 97 ( 10.1186/1750-1172-8-97) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circulation Research 2014. 115–130. ( 10.1161/CIRCRESAHA.115.301146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, et al Diagnosis and assessment of pulmonary arterial hypertension. Journal of the American College of Cardiology 2009. S55–S66. ( 10.1016/j.jacc.2009.04.011) [DOI] [PubMed] [Google Scholar]
- 4.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, et al Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). European Heart Journal 2009. 2493–2537. ( 10.1093/eurheartj/ehp297) [DOI] [PubMed] [Google Scholar]
- 5.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. Journal of Molecular Medicine 2013. 297–309. ( 10.1007/s00109-013-0998-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovascular Research 2018. 551–564. ( 10.1093/cvr/cvy004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coronary Artery Disease 2005. 13–18. ( 10.1097/00019501-200502000-00003) [DOI] [PubMed] [Google Scholar]
- 8.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. European Respiratory Journal 2019. 1801913 ( 10.1183/13993003.01913-2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenmark KR, Rabinovitch M. Emerging therapies for the treatment of pulmonary hypertension. Pediatric Critical Care Medicine 2010. S85–S90. ( 10.1097/PCC.0b013e3181c76db3) [DOI] [PubMed] [Google Scholar]
- 10.Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016. 73–83. ( 10.1136/thoraxjnl-2015-207170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M, Simonneau G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. Journal of the American College of Cardiology 2002. 780–788. ( 10.1016/S0735-1097(02)02012-0) [DOI] [PubMed] [Google Scholar]
- 12.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. European Heart Journal 2012. 829–837, 837a–837d. ( 10.1093/eurheartj/ehr304) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hynynen MM, Khalil RA. The vascular endothelin system in hypertension – recent patents and discoveries. Recent Advances in Cardiovascular Drug Discovery 2006. 95–108. ( 10.2174/157489006775244263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinhold B. Epigenetics: the science of change. Environ Health Perspect 2006. A160–A167. ( 10.1289/ehp.114-a160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 2011. 2145–2156. ( 10.1161/CIRCULATIONAHA.110.956839) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Reviews in Endocrine and Metabolic Disorders 2007. 173–182. ( 10.1007/s11154-007-9042-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim KC, Friso S, Choi SW. DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. Journal of Nutritional Biochemistry 2009. 917–926. ( 10.1016/j.jnutbio.2009.06.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. Journal of Dental Research 2009. 400–408. ( 10.1177/0022034509335868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillette TG, Hill JA. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circulation Research 2015. 1245–1253. ( 10.1161/CIRCRESAHA.116.303630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular Cell 2011. 904–914. ( 10.1016/j.molcel.2011.08.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaikkonen MU, Lam MT, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovascular Research 2011. 430–440. ( 10.1093/cvr/cvr097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Current Genomics 2010. 537–561. ( 10.2174/138920210793175895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Frontiers in Cell and Developmental Biology 2014. 49 ( 10.3389/fcell.2014.00049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ha TY. MicroRNAs in human diseases: from cancer to cardiovascular disease. Immune Netwowk 2011. 135–154. ( 10.4110/in.2011.11.3.135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosutti A, Zanconati F, Grassi G, Dapas B, Passamonti S, Scaggiante B. Epigenetic and miRNAs dysregulation in prostate cancer: the role of nutraceuticals. Anti-Cancer Agents in Medicinal Chemistry 2016. 1385–1402. ( 10.2174/1871520616666160425105257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochberg Z, Feil R, Constancia M, Fraga M, Junien C, Carel JC, Boileau P, Le Bouc Y, Deal CL, Lillycrop K, et al Child health, developmental plasticity, and epigenetic programming. Endocrine Reviews 2011. 159–224. ( 10.1210/er.2009-0039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010. 27–36. ( 10.1093/carcin/bgp220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends in Pharmacological Sciences 2010. 536–546. ( 10.1016/j.tips.2010.08.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annual Review of Medicine 2016. 73–89. ( 10.1146/annurev-med-111314-035900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. Journal of Clinical Investigation 2014. 56–63. ( 10.1172/JCI69736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. British Journal of Cancer 2004. 761–769. ( 10.1038/sj.bjc.6601575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luna RCP, de Oliveira Y, Lisboa JVC, Chaves TR, de Araújo TAM, de Sousa EE, Miranda Neto M, Pirola L, Braga VA, de Brito Alves JL. Insights on the epigenetic mechanisms underlying pulmonary arterial hypertension. Brazilian Journal of Medical and Biological Research 2018. e7437 ( 10.1590/1414-431X20187437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng X, Wang Y, Du L. Epigenetic modulation in the initiation and progression of pulmonary hypertension. Hypertension 2019. 733–739. ( 10.1161/HYPERTENSIONAHA.119.13458) [DOI] [PubMed] [Google Scholar]
- 34.Bourgeois A, Omura J, Habbout K, Bonnet S, Boucherat O. Pulmonary arterial hypertension: new pathophysiological insights and emerging therapeutic targets. International Journal of Biochemistry and Cell Biology 2018. 9–13. ( 10.1016/j.biocel.2018.08.015) [DOI] [PubMed] [Google Scholar]
- 35.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harbor Perspectives in Biology 2014. a019133 ( 10.1101/cshperspect.a019133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science 2001. 1068–1070. ( 10.1126/science.1063852) [DOI] [PubMed] [Google Scholar]
- 37.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013. 23–38. ( 10.1038/npp.2012.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bestor TH. The DNA methyltransferases of mammals. Human Molecular Genetics 2000. 2395–2402. ( 10.1093/hmg/9.16.2395) [DOI] [PubMed] [Google Scholar]
- 39.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nature Reviews Genetics 2009. 805–811. ( 10.1038/nrg2651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermann A, Schmitt S, Jeltsch A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. Journal of Biological Chemistry 2003. 31717–31721. ( 10.1074/jbc.M305448200) [DOI] [PubMed] [Google Scholar]
- 41.Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Molecular and Cellular Biology 2002. 3157–3173. ( 10.1128/MCB.22.9.3157-3173.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. Journal of Cellular Physiology 2007. 384–390. ( 10.1002/jcp.21224) [DOI] [PubMed] [Google Scholar]
- 43.Clouaire T, Stancheva I. Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin? Cellular and Molecular Life Sciences 2008. 1509–1522. ( 10.1007/s00018-008-7324-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MS, Kawaji H, Lassmann T, Harbers M, Forrest AR, Bajic VB, et al Effects of cytosine methylation on transcription factor binding sites. BMC Genomics 2014. 119 ( 10.1186/1471-2164-15-119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JY, Lee TH. Effects of DNA methylation on the structure of nucleosomes. Journal of the American Chemical Society 2012. 173–175. ( 10.1021/ja210273w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, Cigudosa JC, Huang TH, Esteller M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO Journal 2003. 6335–6345. ( 10.1093/emboj/cdg604) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 2009. 549–565. ( 10.1007/s00412-009-0221-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. British Journal of Cancer 2008. 1881–1885. ( 10.1038/sj.bjc.6604374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huppke P, Laccone F, Kramer N, Engel W, Hanefeld F. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Human Molecular Genetics 2000. 1369–1375. ( 10.1093/hmg/9.9.1369) [DOI] [PubMed] [Google Scholar]
- 50.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, et al An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006. 2630–2641. ( 10.1161/CIRCULATIONAHA.105.609008) [DOI] [PubMed] [Google Scholar]
- 51.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thébaud B, Husain AN, et al Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010. 2661–2671. ( 10.1161/CIRCULATIONAHA.109.916098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulmonary Circulation 2011. 305–319. ( 10.4103/2045-8932.87293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proceedings of the American Thoracic Society 2006. 680–686. ( 10.1513/pats.200605-118SF) [DOI] [PubMed] [Google Scholar]
- 54.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circulation Research 2014. 189–202. ( 10.1161/CIRCRESAHA.115.303404) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. European Respiratory Journal 2019. 1801899 ( 10.1183/13993003.01899-2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quarck R, Perros F. Rescuing BMPR2-driven endothelial dysfunction in PAH: a novel treatment strategy for the future? Stem Cell Investigation 2017. 56 ( 10.21037/sci.2017.05.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science 2007. 1136–1140. ( 10.1126/science.1148910) [DOI] [PubMed] [Google Scholar]
- 58.Raslova H, Komura E, Le Couedic JP, Larbret F, Debili N, Feunteun J, Danos O, Albagli O, Vainchenker W, Favier R. FLI1 monoallelic expression combined with its hemizygous loss underlies Paris-Trousseau/Jacobsen thrombopenia. Journal of Clinical Investigation 2004. 77–84. ( 10.1172/JCI21197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu D, Yan Y, Chen JW, Yuan P, Wang XJ, Jiang R, Wang L, Zhao QH, Wu WH, Simonneau G, et al Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. American Journal of Respiratory and Critical Care Medicine 2017. 925–928. ( 10.1164/rccm.201611-2273LE) [DOI] [PubMed] [Google Scholar]
- 60.Viales RR, Eichstaedt CA, Ehlken N, Fischer C, Lichtblau M, Grünig E, Hinderhofer K. Mutation in BMPR2 promoter: a ‘second hit’ for manifestation of pulmonary arterial hypertension? PLoS ONE 2015. e0133042 ( 10.1371/journal.pone.0133042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pousada G, Baloira A, Valverde D. Methylation analysis of the BMPR2 gene promoter region in patients with pulmonary arterial hypertension. Archivos de Bronconeumología 2016. 293–298. ( 10.1016/j.arbres.2015.10.006) [DOI] [PubMed] [Google Scholar]
- 62.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes and Development 2016. 733–750. ( 10.1101/gad.276568.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao H, Chen T. Tet family of 5-methylcytosine dioxygenases in mammalian development. Journal of Human Genetics 2013. 421–427. ( 10.1038/jhg.2013.63) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu C, Liu L, Chen X, Shen J, Shan J, Xu Y, Yang Z, Wu L, Xia F, Bie P, et al Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS ONE 2013. e62828 ( 10.1371/journal.pone.0062828) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang L, Yu SJ, Hong Q, Yang Y, Shao ZM. Reduced expression of TET1, TET2, TET3 and TDG mRNAs are associated with poor prognosis of patients with early breast cancer. PLoS ONE 2015. e0133896 ( 10.1371/journal.pone.0133896) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li L, Li C, Mao H, Du Z, Chan WY, Murray P, Luo B, Chan AT, Mok TS, Chan FK, et al Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Scientific Reports 2016. 26591 ( 10.1038/srep26591) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, Maldotti M, Parlato C, Paliogiannis P, Oliviero S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015. 4168–4176. ( 10.1038/onc.2014.356) [DOI] [PubMed] [Google Scholar]
- 68.Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, Song C, Teng SC, He C, Wu KJ. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biology 2014. 513 ( 10.1186/s13059-014-0513-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Good CR, Panjarian S, Kelly AD, Madzo J, Patel B, Jelinek J, Issa JJ. TET1-mediated hypomethylation activates oncogenic signaling in triple-negative breast cancer. Cancer Research 2018. 4126–4137. ( 10.1158/0008-5472.CAN-17-2082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fan M, He X, Xu X. Restored expression levels of TET1 decrease the proliferation and migration of renal carcinoma cells. Molecular Medicine Reports 2015. 4837–4842. ( 10.3892/mmr.2015.4058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cutter AR, Hayes JJ. A brief review of nucleosome structure. FEBS Letters 2015. 2914–2922. ( 10.1016/j.febslet.2015.05.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Review of Proteomics 2005. 719–729. ( 10.1586/14789450.2.5.719) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Research 2011. 381–395. ( 10.1038/cr.2011.22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. PNAS 2003. 13225–13230. ( 10.1073/pnas.1735528100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Reviews Genetics 2012. 343–357. ( 10.1038/nrg3173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harbor Perspectives in Biology 2014. a018762 ( 10.1101/cshperspect.a018762) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clinical Epigenetics 2016. 59 ( 10.1186/s13148-016-0225-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chemical Reviews 2015. 2419–2452. ( 10.1021/cr500452k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor Perspectives in Biology 2014. a018713 ( 10.1101/cshperspect.a018713) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, Irwin DC, McCune SA, Chung E, Leinwand LA, et al Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. Journal of Molecular and Cellular Cardiology 2011. 41–50. ( 10.1016/j.yjmcc.2011.04.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie M, Hill JA. HDAC-dependent ventricular remodeling. Trends in Cardiovascular Medicine 2013. 229–235. ( 10.1016/j.tcm.2012.12.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, et al Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012. 455–467. ( 10.1161/CIRCULATIONAHA.112.103176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu XF, Ma XL, Shen Z, Wu XL, Cheng F, Du LZ. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. Journal of Hypertension 2010. 2227–2235. ( 10.1097/HJH.0b013e32833e08f1) [DOI] [PubMed] [Google Scholar]
- 84.Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI, Ma JS, Kook H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circulation Journal 2010. 760–770. ( 10.1253/circj.CJ-09-0580) [DOI] [PubMed] [Google Scholar]
- 85.Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, et al Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015. 190–199. ( 10.1161/CIRCULATIONAHA.114.013339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. American Journal of Respiratory and Critical Care Medicine 2011. 1402–1410. ( 10.1164/rccm.201007-1106OC) [DOI] [PubMed] [Google Scholar]
- 87.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wilkins MR, Wang D, Li M, Stenmark KR, et al Response to letter regarding article, ‘histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid’. Circulation 2013. e540 ( 10.1161/circulationaha.112.154757) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, et al Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology 2007. 3109–3115. ( 10.1200/JCO.2006.10.2434) [DOI] [PubMed] [Google Scholar]
- 89.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, et al Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology 2010. 4485–4491. ( 10.1200/JCO.2010.28.9066) [DOI] [PubMed] [Google Scholar]
- 90.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, et al Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circulation Research 2012. 739–748. ( 10.1161/CIRCRESAHA.111.258426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Review of Anticancer Therapy 2010. 935–954. ( 10.1586/era.10.62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. PNAS 2003. 4389–4394. ( 10.1073/pnas.0430973100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boucherat O, Chabot S, Paulin R, Trinh I, Bourgeois A, Potus F, Lampron MC, Lambert C, Breuils-Bonnet S, Nadeau V, et al HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Scientific Reports 2017. 4546 ( 10.1038/s41598-017-04874-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, et al Bromodomain-containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circulation Research 2015. 525–535. ( 10.1161/CIRCRESAHA.115.307004) [DOI] [PubMed] [Google Scholar]
- 95.Dong X, Weng Z. The correlation between histone modifications and gene expression. Epigenomics 2013. 113–116. ( 10.2217/epi.13.13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res 2011. 564–578. ( 10.1038/cr.2011.42) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yap KL, Zhou MM. Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 2011. 1966–1980. ( 10.1021/bi101885m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee Y, Park D, Iyer VR. The ATP-dependent chromatin remodeler Chd1 is recruited by transcription elongation factors and maintains H3K4me3/H3K36me3 domains at actively transcribed and spliced genes. Nucleic Acids Research 2017. 7180–7190. ( 10.1093/nar/gkx321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng J, Blum R, Bowman C, Hu D, Shilatifard A, Shen S, Dynlacht BD. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Molecular Cell 2014. 979–992. ( 10.1016/j.molcel.2014.02.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications – writers that read. EMBO Reports 2015. 1467–1481. ( 10.15252/embr.201540945) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by polycomb proteins. Science Advances 2015. e1500737 ( 10.1126/sciadv.1500737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Experimental and Molecular Medicine 2017. e324 ( 10.1038/emm.2017.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simon JA, Kingston RE. Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Molecular Cell 2013. 808–824. ( 10.1016/j.molcel.2013.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grossniklaus U, Paro R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harbor Perspectives in Biology 2014. a019331 ( 10.1101/cshperspect.a019331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nature Structural and Molecular Biology 2012. 1218–1227. ( 10.1038/nsmb.2436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018. 2125–2137. ( 10.1182/blood-2017-08-804344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yamaguchi H, Hung MC. Regulation and role of EZH2 in cancer. Cancer Research and Treatment 2014. 209–222. ( 10.4143/crt.2014.46.3.209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aljubran SA, Cox R, Jr, Tamarapu Parthasarathy P, Kollongod Ramanathan G, Rajanbabu V, Bao H, Mohapatra SS, Lockey R, Kolliputi N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS ONE 2012. e37712 ( 10.1371/journal.pone.0037712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shi ZL, Fang K, Li ZH, Ren DH, Zhang JY, Sun J. EZH2 inhibition ameliorates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Canadian Respiratory Journal 2018. 9174926 ( 10.1155/2018/9174926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gambaryan N, Meng C, Humbert M, Adcock I, Wort S. H3K27 histone lysine methylation as potential therapeutic target in pulmonary arterial hypertension. European Respiratory Journal 2013. P5157. [Google Scholar]
- 111.Chen D, Yang Y, Cheng X, Fang F, Xu G, Yuan Z, Xia J, Kong H, Xie W, Wang H, et al Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 2015. 821–833. ( 10.1161/HYPERTENSIONAHA.114.04585) [DOI] [PubMed] [Google Scholar]
- 112.Yuan Z, Chen J, Chen D, Xu G, Xia M, Xu Y, Gao Y. Megakaryocytic leukemia 1 (MKL1) regulates hypoxia induced pulmonary hypertension in rats. PLoS ONE 2014. e83895 ( 10.1371/journal.pone.0083895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu L, Fang F, Dai X, Xu H, Qi X, Fang M, Xu Y. MKL1 defines the H3K4Me3 landscape for NF-kappaB dependent inflammatory response. Scientific Reports 2017. 191 ( 10.1038/s41598-017-00301-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Reviews Genetics 2008. 102–114. ( 10.1038/nrg2290) [DOI] [PubMed] [Google Scholar]
- 115.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO Journal 2004. 4051–4060. ( 10.1038/sj.emboj.7600385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004. 1957–1966. ( 10.1261/rna.7135204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, et al The nuclear RNase III Drosha initiates microRNA processing. Nature 2003. 415–419. ( 10.1038/nature01957) [DOI] [PubMed] [Google Scholar]
- 118.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001. 363–366. ( 10.1038/35053110) [DOI] [PubMed] [Google Scholar]
- 119.Babashah S, Soleimani M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. European Journal of Cancer 2011. 1127–1137. ( 10.1016/j.ejca.2011.02.008) [DOI] [PubMed] [Google Scholar]
- 120.Mondejar-Parreno G, Callejo M, Barreira B, Morales-Cano D, Esquivel-Ruiz S, Moreno L, Cogolludo A, Perez-Vizcaino F. miR-1 is increased in pulmonary hypertension and downregulates Kv1.5 channels in rat pulmonary arteries. Journal of Physiology 2019. 1185–1197. ( 10.1113/JP276054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shan F, Li J, Huang QY. HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. Journal of Cellular Physiology 2014. 1511–1520. ( 10.1002/jcp.24593) [DOI] [PubMed] [Google Scholar]
- 122.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circulation Research 2009. 1184–1191. ( 10.1161/CIRCRESAHA.109.197491) [DOI] [PubMed] [Google Scholar]
- 123.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, et al MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012. 1520–1532. ( 10.1161/CIRCULATIONAHA.111.060269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kang BY, Park KK, Green DE, Bijli KM, Searles CD, Sutliff RL, Hart CM. Hypoxia mediates mutual repression between microRNA-27a and PPARgamma in the pulmonary vasculature. PLoS ONE 2013. e79503 ( 10.1371/journal.pone.0079503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bi R, Bao C, Jiang L, Liu H, Yang Y, Mei J, Ding F. MicroRNA-27b plays a role in pulmonary arterial hypertension by modulating peroxisome proliferator-activated receptor gamma dependent Hsp90-eNOS signaling and nitric oxide production. Biochemical and Biophysical Research Communications 2015. 469–475. ( 10.1016/j.bbrc.2015.03.057) [DOI] [PubMed] [Google Scholar]
- 126.Sen A, Ren S, Lerchenmuller C, Sun J, Weiss N, Most P, Peppel K. MicroRNA-138 regulates hypoxia-induced endothelial cell dysfunction by targeting S100A1. PLoS ONE 2013. e78684 ( 10.1371/journal.pone.0078684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D. MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K(+) channel in arterial smooth muscle cells. Journal of Cellular Biochemistry 2014. 1196–1205. ( 10.1002/jcb.24771) [DOI] [PubMed] [Google Scholar]
- 128.Liu Y, Liu G, Zhang H, Wang J. MiRNA-199a-5p influences pulmonary artery hypertension via downregulating Smad3. Biochemical and Biophysical Research Communications 2016. 859–866. ( 10.1016/j.bbrc.2016.03.140) [DOI] [PubMed] [Google Scholar]
- 129.Gou D, Ramchandran R, Peng X, Yao L, Kang K, Sarkar J, Wang Z, Zhou G, Raj JU. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. American Journal of Physiology: Lung Cellular and Molecular Physiology 2012. L682–L691. ( 10.1152/ajplung.00344.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zeng Y, Liu H, Kang K, Wang Z, Hui G, Zhang X, Zhong J, Peng W, Ramchandran R, Raj JU, et al Hypoxia inducible factor-1 mediates expression of miR-322: potential role in proliferation and migration of pulmonary arterial smooth muscle cells. Scientific Reports 2015. 12098 ( 10.1038/srep12098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang X, Shao R, Gao W, Sun G, Liu Y, Fa X. Inhibition of miR-361-5p suppressed pulmonary artery smooth muscle cell survival and migration by targeting ABCA1 and inhibiting the JAK2/STAT3 pathway. Experimental Cell Research 2018. 255–261. ( 10.1016/j.yexcr.2018.01.015) [DOI] [PubMed] [Google Scholar]
- 132.Zhang Y, Peng B, Han Y. MiR-23a regulates the proliferation and migration of human pulmonary artery smooth muscle cells (HPASMCs) through targeting BMPR2/Smad1 signaling. Biomed Pharmacother 2018. 1279–1286. ( 10.1016/j.biopha.2018.04.172) [DOI] [PubMed] [Google Scholar]
- 133.Chen T, Zhou Q, Tang H, Bozkanat M, Yuan JX, Raj JU, Zhou G. miR-17/20 controls prolyl hydroxylase 2 (PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary artery smooth muscle cell proliferation. Journal of the American Heart Association 2016. e004510 ( 10.1161/JAHA.116.004510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK, Ghofrani HA, Weissmann N, Grimminger F, Bonauer A, et al Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine 2012. 409–419. ( 10.1164/rccm.201106-1093OC) [DOI] [PubMed] [Google Scholar]
- 135.Lu Z, Li S, Zhao S, Fa X. Upregulated miR-17 regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation and apoptosis by targeting mitofusin 2. Medical Science Monitor 2016. 3301–3308. ( 10.12659/MSM.900487) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang D, Sun C, Zhang J, Lin S, Zhao L, Wang L, Lin R, Lv J, Xin S. Proliferation of vascular smooth muscle cells under inflammation is regulated by NF-kappaB p65/microRNA-17/RB pathway activation. International Journal of Molecular Medicine 2018. 43–50. ( 10.3892/ijmm.2017.3212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li S, Ran Y, Zhang D, Chen J, Li S, Zhu D. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochemical Journal 2013. 281–291. ( 10.1042/BJ20120680) [DOI] [PubMed] [Google Scholar]
- 138.Sen A, Most P, Peppel K. Induction of microRNA-138 by pro-inflammatory cytokines causes endothelial cell dysfunction. FEBS Letters 2014. 906–914. ( 10.1016/j.febslet.2014.01.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. American Journal of Physiology: Lung Cellular and Molecular Physiology 2010. L861–L871. ( 10.1152/ajplung.00201.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang S, Banerjee S, Freitas A, Cui H, Xie N, Abraham E, Liu G. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. American Journal of Physiology: Lung Cellular and Molecular Physiology 2012. L521–L529. ( 10.1152/ajplung.00316.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Green DE, Murphy TC, Kang BY, Searles CD, Hart CM. PPARgamma ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of microRNA-21. PLoS ONE 2015. e0133391 ( 10.1371/journal.pone.0133391) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, et al Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. Journal of Clinical Investigation 2014. 3514–3528. ( 10.1172/JCI74773) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, et al The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. Journal of Biological Chemistry 2015. 2069–2085. ( 10.1074/jbc.M114.617845) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Upton PD, Morrell NW. The transforming growth factor-beta-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Experimental Physiology 2013. 1262–1266. ( 10.1113/expphysiol.2012.069104) [DOI] [PubMed] [Google Scholar]
- 145.Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE, Gassmann M, Ostergaard L, Gay S, Speich R, et al AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. European Heart Journal 2014. 3203–3211. ( 10.1093/eurheartj/ehs060) [DOI] [PubMed] [Google Scholar]
- 146.Lee HW, Park SH. Elevated microRNA-135a is associated with pulmonary arterial hypertension in experimental mouse model. Oncotarget 2017. 35609–35618. ( 10.18632/oncotarget.16011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, Chung SW, Schissel SL, Ingber DE, Mitsialis SA, et al Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation 2008. 2114–2122. ( 10.1161/CIRCULATIONAHA.107.716241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010. 2045–2066. ( 10.1161/CIRCULATIONAHA.108.847707) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacological Reviews 2016. 476–532. ( 10.1124/pr.115.010652) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Krimbou L, Denis M, Haidar B, Carrier M, Marcil M, Genest J., Jr Molecular interactions between apoE and ABCA1: impact on apoE lipidation. Journal of Lipid Research 2004. 839–848. ( 10.1194/jlr.M300418-JLR200) [DOI] [PubMed] [Google Scholar]
- 151.Yue Y, Zhang Z, Zhang L, Chen S, Guo Y, Hong Y. miR-143 and miR-145 promote hypoxia-induced proliferation and migration of pulmonary arterial smooth muscle cells through regulating ABCA1 expression. Cardiovascular Pathology 2018. 15–25. ( 10.1016/j.carpath.2018.08.003) [DOI] [PubMed] [Google Scholar]
- 152.Liu JJ, Zhang H, Xing F, Tang B, Wu SL, Xuan L, Kang PF, Xu Q, Wang HJ, Zhang NR, et al MicroRNA138 promotes proliferation and suppresses mitochondrial depolarization in human pulmonary artery smooth muscle cells through targeting TASK1. Molecular Medicine Reports 2018. 3021–3027. ( 10.3892/mmr.2017.8200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mandegar M, Yuan JX. Role of K+ channels in pulmonary hypertension. Vascular Pharmacology 2002. 25–33. ( 10.1016/S1537-1891(02)00123-4) [DOI] [PubMed] [Google Scholar]
- 154.Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L, Weng T, Zhang H, Ramchandran R, Raj JU, et al MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. Journal of Biological Chemistry 2013. 25414–25427. ( 10.1074/jbc.M113.460287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, et al MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circulation Research 2014. 67–78. ( 10.1161/CIRCRESAHA.114.301633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang H, Wang D, Li M, Plecitá-Hlavatá L, D’Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, et al Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a microRNA-124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation 2017. 2468–2485. ( 10.1161/CIRCULATIONAHA.117.028069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J, et al Role for miR-204 in human pulmonary arterial hypertension. Journal of Experimental Medicine 2011. 535–548. ( 10.1084/jem.20101812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ruffenach G, Chabot S, Tanguay VF, Courboulin A, Boucherat O, Potus F, Meloche J, Pflieger A, Breuils-Bonnet S, Nadeau V, et al Role for runt-related transcription factor 2 in proliferative and calcified vascular lesions in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine 2016. 1273–1285. ( 10.1164/rccm.201512-2380OC) [DOI] [PubMed] [Google Scholar]
- 159.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, et al MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. Journal of Clinical Investigation 2016. 2495–2508. ( 10.1172/JCI83361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sharma S, Umar S, Potus F, Iorga A, Wong G, Meriwether D, Breuils-Bonnet S, Mai D, Navab K, Ross D, et al Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 2014. 776–785. ( 10.1161/CIRCULATIONAHA.114.007405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S, Tian H, Jiang C, Zhu D. The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-alpha1C. Hypertension 2012. 1006–1013. ( 10.1161/HYPERTENSIONAHA.111.185413) [DOI] [PubMed] [Google Scholar]
- 162.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, et al An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nature Medicine 2013. 74–82. ( 10.1038/nm.3040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen J, Cui X, Li L, Li L, Qu J, Raj JU, Gou D. MiR-339 inhibits proliferation of pulmonary artery smooth muscle cell by targeting FGF signaling. Physiological Reports 2017. e13441 ( 10.14814/phy2.13441) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang P, Xu J, Hou Z, Wang F, Song Y, Wang J, Zhu H, Jin H. miRNA-34a promotes proliferation of human pulmonary artery smooth muscle cells by targeting PDGFRA. Cell Proliferation 2016. 484–493. ( 10.1111/cpr.12265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu A, Liu Y, Li B, Yang M, Liu Y, Su J. Role of miR-223-3p in pulmonary arterial hypertension via targeting ITGB3 in the ECM pathway. Cell Proliferation 2019. e12550 ( 10.1111/cpr.12550) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zeng Y, Zhang X, Kang K, Chen J, Wu Z, Huang J, Lu W, Chen Y, Zhang J, Wang Z, et al MicroRNA-223 attenuates hypoxia-induced vascular remodeling by targeting RhoB/MLC2 in pulmonary arterial smooth muscle cells. Scientific Reports 2016. 24900 ( 10.1038/srep24900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li C, Qin F, Xue M, Lei Y, Hu F, Xu H, Sun G, Wang T, Guo M. miR-429 and miR-424-5p inhibit cell proliferation and Ca(2+) influx by downregulating CaSR in pulmonary artery smooth muscle cells. American Journal of Physiology: Cell Physiology 2019. C111–C120. ( 10.1152/ajpcell.00219.2018) [DOI] [PubMed] [Google Scholar]
- 168.Meloche J, Le Guen M, Potus F, Vinck J, Ranchoux B, Johnson I, Antigny F, Tremblay E, Breuils-Bonnet S, Perros F, et al miR-223 reverses experimental pulmonary arterial hypertension. American Journal of Physiology: Cell Physiology 2015. C363–C372. ( 10.1152/ajpcell.00149.2015) [DOI] [PubMed] [Google Scholar]
- 169.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annual Review of Biochemistry 2012. 145–166. ( 10.1146/annurev-biochem-051410-092902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circulation Research 2015. 751–762. ( 10.1161/CIRCRESAHA.116.303549) [DOI] [PubMed] [Google Scholar]
- 171.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al Landscape of transcription in human cells. Nature 2012. 101–108. ( 10.1038/nature11233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009. 223–227. ( 10.1038/nature07672) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bar C, Chatterjee S, Thum T. Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation 2016. 1484–1499. ( 10.1161/CIRCULATIONAHA.116.023686) [DOI] [PubMed] [Google Scholar]
- 174.Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, et al Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circulation Research 2014. 1389–1397. ( 10.1161/CIRCRESAHA.114.303265) [DOI] [PubMed] [Google Scholar]
- 175.Su H, Xu X, Yan C, Shi Y, Hu Y, Dong L, Ying S, Ying K, Zhang R. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respiratory Research 2018. 254 ( 10.1186/s12931-018-0956-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jandl K, Thekkekara Puthenparampil H, Marsh LM, Hoffmann J, Wilhelm J, Veith C, Sinn K, Klepetko W, Olschewski H, Olschewski A, et al Long non-coding RNAs influence the transcriptome in pulmonary arterial hypertension: the role of PAXIP1-AS1. Journal of Pathology 2019. 357–370. ( 10.1002/path.5195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhu TT, Sun RL, Yin YL, Quan JP, Song P, Xu J, Zhang MX, Li P. Long noncoding RNA UCA1 promotes the proliferation of hypoxic human pulmonary artery smooth muscle cells. Pflügers Archiv 2019. 347–355. ( 10.1007/s00424-018-2219-8) [DOI] [PubMed] [Google Scholar]
- 178.Zhang H, Liu Y, Yan L, Wang S, Zhang M, Ma C, Zheng X, Chen H, Zhu D. Long noncoding RNA Hoxaas3 contributes to hypoxia-induced pulmonary artery smooth muscle cell proliferation. Cardiovascular Research 2019. 647–657. ( 10.1093/cvr/cvy250) [DOI] [PubMed] [Google Scholar]
- 179.Sun Z, Nie X, Sun S, Dong S, Yuan C, Li Y, Xiao B, Jie D, Liu Y. Long non-coding RNA MEG3 downregulation triggers human pulmonary artery smooth muscle cell proliferation and migration via the p53 signaling pathway. Cellular Physiology and Biochemistry 2017. 2569–2581. ( 10.1159/000480218) [DOI] [PubMed] [Google Scholar]
- 180.Zhu B, Gong Y, Yan G, Wang D, Qiao Y, Wang Q, Liu B, Hou J, Li R, Tang C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochemical and Biophysical Research Communications 2018. 2125–2132. ( 10.1016/j.bbrc.2017.11.185) [DOI] [PubMed] [Google Scholar]
- 181.Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, et al LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 2014. 1452–1465. ( 10.1161/CIRCULATIONAHA.114.011675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liu Y, Sun Z, Zhu J, Xiao B, Dong J, Li X. LncRNA-TCONS_00034812 in cell proliferation and apoptosis of pulmonary artery smooth muscle cells and its mechanism. Journal of Cellular Physiology 2018. 4801–4814. ( 10.1002/jcp.26279) [DOI] [PubMed] [Google Scholar]
- 183.Gong J, Chen Z, Chen Y, Lv H, Lu H, Yan F, Li L, Zhang W, Shi J. Long non-coding RNA CASC2 suppresses pulmonary artery smooth muscle cell proliferation and phenotypic switch in hypoxia-induced pulmonary hypertension. Respiratory Research 2019. 53 ( 10.1186/s12931-019-1018-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chen J, Guo J, Cui X, Dai Y, Tang Z, Qu J, Raj JU, Hu Q, Gou D. The long noncoding RNA LnRPT is regulated by PDGF-BB and modulates the proliferation of pulmonary artery smooth muscle cells. American Journal of Respiratory Cell and Molecular Biology 2018. 181–193. ( 10.1165/rcmb.2017-0111OC) [DOI] [PubMed] [Google Scholar]
- 185.Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmüller P, Ladage D, Guignabert C, Ishikawa K, Aguero J, Ibanez B, et al Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013. 512–523. ( 10.1161/CIRCULATIONAHA.113.001585) [DOI] [PMC free article] [PubMed] [Google Scholar]