

Abstract
Objectives/Hypothesis:
Our objectives were to determine genotype-phenotype correlations in patients with sensorineural hearing loss (SNHL) who undergo testing for GJB2 mutations and to examine the relationship of temporal bone anomalies seen on computed tomography (CT) and GJB2 mutations.
Study design:
We conducted a retrospective review of all children diagnosed with SNHL and who underwent GJB2 testing from 1997 to 2006.
Results:
Of 840 patients, 146 (17.4%) had mutations. Seventy-six (9.1%) had biallelic GJB2 mutations and 70 (8.3%) had heterozygous mutations. When biallelic mutations were categorized as missense or nonsense mutations, the presence of at least one missense mutation was associated with mild or moderate SNHL. Biallelic nonsense mutations were associated with severe to profound SNHL. Among patients with GJB2 mutations, those with heterozygous mutations (n = 14 [20%]) had a higher rate of asymmetric SNHL loss than those with biallelic mutations (n = 6 [7.9%], P = .03). Those with heterozygous mutations were more likely to experience progression than were those with biallelic mutations, though this difference was only marginally significant (26.5% vs. 12.3%, respectively; P = .06). Patients who were wild type for GJB2 were more likely to have an enlarged vestibular aqueduct (EVA) than were those with biallelic and heterozygous mutations (29% vs. 11.9%, respectively; P = .004). Compared to patients who were wild type, those with biallelic mutations had a significantly lower rate of EVA.
Conclusions:
This is the largest single-institution study of pediatric patients with GJB2 mutations and SNHL. The functional consequences of GJB2 mutations correlated with the degree of hearing loss. Patients with M34T mutations and/or mild SNHL had a low risk of progression. Temporal bone anomalies were uncommon in patients with GJB2 mutations.
Keywords: GJB2, mild, progressive, and asymmetric sensorineural hearing loss, temporal bone anomalies
INTRODUCTION
Sensorineural hearing loss (SNHL) is the most common congenital sensory impairment, affecting 1 of every 1000 newborns in the United States.1 In as many as 68% of these cases, SNHL is genetic in origin.1 Autosomal recessive nonsyndromic hearing loss locus 1 (DFNB1) is caused by mutations in the GJB2 gene. Studies have shown that mutations in the GJB2 gene are responsible for approximately 40% of cases attributed to a genetic etiology and that genetic testing for GJB2 is a sensitive, accurate, and cost-effective diagnostic tool.2–4
Early studies of GJB2 identified a high prevalence of the 35delG mutation, which generally leads to severe to profound SNHL.4,5 More recent studies, however, have shown that this particular mutation is far less common than originally reported and have identified multiple novel GJB2 mutations.6–9 The 35delG mutation is a nonsense mutation that results in a truncated protein and thus severe dysfunction or complete loss of protein function. Although estimates vary, this appears to be present in about 50% of mutated alleles.4,6 Many other GJB2 mutations are missense mutations that generate a full-length protein with only a single amino acid substitution. These mutations may lead to milder forms of SNHL.4,10
Despite the importance of the GJB2 gene, there is surprisingly little available data that describes its association with unique audiologic phenotypes (i.e., mild to moderate, asymmetric, and progressive SNHL) and temporal bone anomalies seen on computed tomography (CT). Specifically, Lim et al. found that the prevalence of these phenotypes was relatively low.8 Additionally, whereas Propst et al.11 reported that patients with GJB2 mutations have a very high rate of temporal bone anomalies, results of a study by Madden et al. were less conclusive.12 The purpose of the present study was thus to determine genotype-phenotype correlations in patients with SNHL who undergo genetic testing for GJB2 mutations. To examine the relationship of temporal bone anomalies and SNHL progression with these mutations, we also compared temporal bone CT findings in patients with and without GJB2 mutations. We hypothesized that patients with GJB2 mutations (biallelic vs. heterozygous) would have a low prevalence of these unique audiologic phenotypes, and that patients with biallelic GJB2 mutations would have a lower prevalence of temporal bone anomalies than would those in whom testing showed to be wild type.
MATERIALS AND METHODS
Patients
We conducted a retrospective study of all children diagnosed with SNHL and who underwent GJB2 genetic testing at our institution from 1997 to 2006. Children were excluded from the study if their audiologic data from at least one visit could not be obtained. This study was approved by our institutional review board.
Audiologic Evaluation
Age-appropriate audiologic evaluation was performed either by pure-tone audiometry (PTA) in a standard audiologic booth or by auditory brainstem-evoked response. Severity of SNHL, asymmetry, and SNHL progression were documented. Pure tone averages of 500 Hz, 1 kHz, 2 kHz, and 4 kHz were used to categorize the severity of SNHL. Severity levels were classified as mild (20–40 dB), moderate to moderately severe (41–69 dB), or severe to profound (70dB or greater). High-frequency SNHL was also noted. Asymmetric hearing loss was defined as a 15 dB or greater difference between the two ears in at least two frequencies. Children with bilateral profound losses, but with differences of 15 dB or greater between ears were not considered to have asymmetric hearing loss. SNHL progression was defined as an elevation of the 4-tone PTA by more than 15 dB in one or both ears between audiograms. Only patients with multiple audiograms and at least a 4-month gap between audiograms were included in the analysis for progressive SNHL.
Genetic Testing
DNA was purified by an alcohol extraction method (Qiagen Inc., Valencia, CA). The entire 732 base-pair coding region of exon 2 and the splice site in exon 1 of the GJB2 gene was amplified by polymerase chain reaction (PCR).7 The PCR products were sequenced with a 3700 Fluorescent Automated Sequencer (Applied Biosystems Division of Perkin-Elmer, Foster City, CA). The sequences of the entire coding and splice sites were then analyzed for mutations using the Sequencher 4.0.5 software package (Gene Codes, Ann Arbor, MI). Mutations were identified by comparison of the identified sequences with the wild-type GJB2 sequence (Genebank: XM_007169) and with previously characterized mutations.13
Imaging
High-resolution temporal bone CT scans were performed with our standard temporal bone protocol of contiguous 1.0 to 1.25 mm scans. Measurements were made with images enlarged 10× to 15× using current workstation software (Centricity GE Medical Systems, Milwaukee, WI). These scans were visualized using the Centricity Picture Archiving Communication System (PACS, GE Healthcare, Milwaukee, WI).
Based on published norms, enlarged vestibular aqueduct (EVA) was defined as ≥ 2 mm at the operculum and/or ≥ 1 mm at the midpoint.14 These measurements were performed by two board-certified radiologists (CB and MH).
Statistical Analysis
Data distributions were reported as either means (standard deviations) or medians (ranges) as appropriate for continuous variables and proportions with 95% confidence intervals (CI) for categorical variables. Patients with GJB2 mutations were compared to those who were wild-type for GJB2 in regard to associations with mild, asymmetric, and progressive SNHL and with EVA. Additionally, patients with GJB2 biallelic mutations were compared to those with GJB2 heterozygous mutations in regard to these same parameters. Associations between the GJB2- positive and wild-type groups were analyzed using the Pearson’s chi-square or Fisher’s exact test, as appropriate. A Bonferroni correction was utilized when comparing values across the three groups (wild type, GJB2 biallelic mutation, GJB2 heterozygous mutation).
RESULTS
We identified 919 patients with SNHL who underwent GJB2 genetic testing over the study duration; 79 patients had either missing or incomplete audiologic data and were thus excluded from further analyses. Of the remaining 840 patients, 146 had mutations, yielding a mutation rate of 17.4% (95% CI 14.8, 19.9). Seventy-six (9.1%) patients had biallelic GJB2 mutations and 70 (8.3%) had heterozygous mutations. The mean follow-up time between initial and final audiologic testing was 36 months (SD 29 months), with no difference between the two GJB2 groups (P = .96).
Analysis of the frequency of alleles in patients with biallelic mutations (Table I) indicated that the 35delG allele was by far the most common, accounting for 82 (53.9%) of the 152 alleles. The V27I sequence change is considered nonpathogenic. For our analysis, the patient with the V27I/V27I genotype was considered not to have GJB2-related hearing loss. This was followed by M34T (12.5%), V37I (7.9%), L90P (5.3%), and 167delT (3.3%). The percentage of patients with GJB2 mutations in each of the three SNHL severity classifications as well as in each genotypic subgroup are shown in Figure 1A. The 35delG mutation was associated with severe to profound SNHL, whereas M34T and V37I mutations were associated with mild or moderate SNHL. When biallelic mutations were categorized as missense (M) or nonsense (N) mutations, the presence of at least one M mutation was associated with mild or moderate SNHL (Figure 1B). In contrast, biallelic N mutations were associated with severe to profound SNHL. Patients with the M/M genotype had a slightly higher rate of mild and moderate SNHL (88.9%) as compared to those with the M/N genotype (72.7%), although this finding was not statistically significant (P = .26). There was a more striking difference in the rate of moderate SNHL in these two groups (26.2% vs. 9.1%, respectively; P = .21); however, this too was not significant.
TABLE I.
Analysis of the Frequency of Alleles in Patients With Biallelic Mutations.
| Mutation | Prevalence |
|---|---|
| 35delG | 82 (53.9%) |
| M34T | 19 (12.5%) |
| V37I | 12 (7.9%) |
| L90P | 8 (5.3%) |
| 167del T | 5 (3.3%) |
| E47X | 3 (2.0%) |
| W24X | 2 (1.3%) |
| F83I | 2 (1.3%) |
| K15T | 2 (1.3%) |
| 312del14 | 2 (1.3%) |
| V84L | 1 (0.6%)* |
| M195T | |
| 235delC | |
| R143W | |
| K122I | |
| V95M | |
| S139N | |
| R184P | |
| G160S | |
| 269insT | |
| 120delE |
One mutation was present for each of these alleles.
Fig. 1.
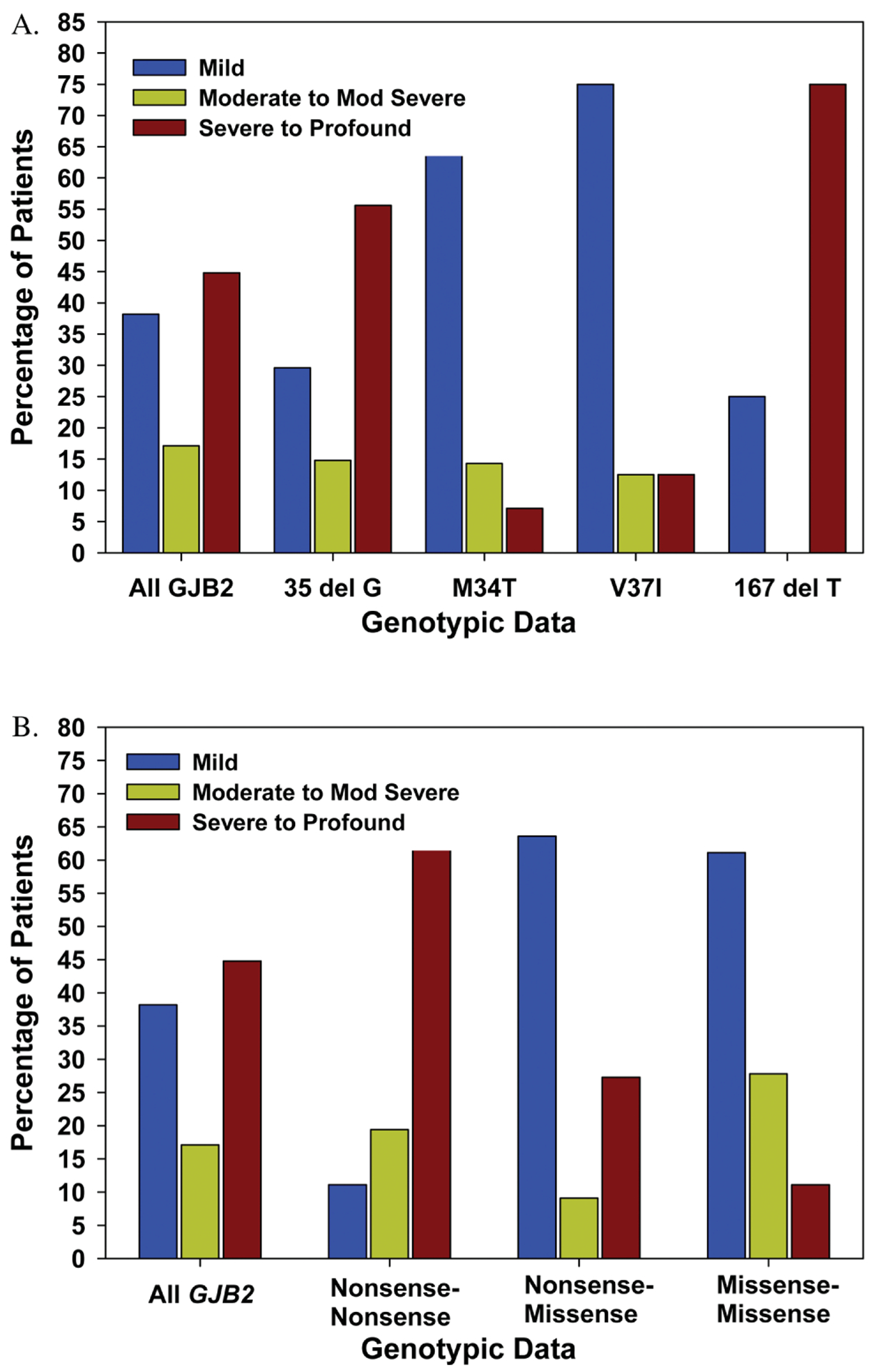
(A) Percentage of mild (20–40 dB), moderate to moderately severe (41–69 dB), and severe to profound (>70 dB) sensorineural hearing loss in the better hearing ear compared with different alleles (n = 76); (B) compared with the genotypes 1) nonsense/nonsense; 2) missense/nonsense; and 3) missense/missense. Percentages are values within each subgroup. Mod = moderate.
Initial audiologic testing indicated that 226 (26.9%) patients had mild, bilateral, symmetric SNHL; 39 additional patients showed a “slight’” bilateral deficit, ≤ the 20 dB criteria for classification as mild. Comparison of initial and final testing indicated that 363 (43.3%) vs. 369 (44%) patients respectively had mild SNHL in the better hearing ear. Patients with GJB2 mutations (57 of 146 [39.3%]) were less likely to have mild or slight hearing loss in the better hearing ear at initial testing than those who were wild type for GJB2 (374 of 694 [54%]; P = .001). There was no difference in the rate of mild or slight SNHL in the better hearing ear between patients with biallelic mutations and those with heterozygous mutations (38.2% vs. 40.6%, respectively; P = .77). In patients with slight or mild bilateral SNHL, 20 (32%) of the 62 alleles genotyped were M34T.
Of the 840 patients in our study, 211 (25.1%) had asymmetric SNHL. In 54 (26%) patients, SNHL was unilateral. Twenty of the 146 (13.7%) patients with mutations had asymmetric SNHL, whereas 191 of the 694 (27.5%) who were wild type for GJB2 had asymmetric SNHL (P = .0005). Among patients with GJB2 mutations, those with heterozygous mutations (n = 14) had a higher rate of asymmetric SNHL than those with biallelic mutations (n = 6) (20% vs. 7.9%, respectively; P = .03). Table II shows the asymmetry on the last available audiogram and specifies the genotypes of patients with GJB2 mutations. Three of the 146 (2%) patients with GJB2 mutations had unilateral SNHL as compared to 51 of the 694 (7%) patients with unilateral SNHL who were wild type for GJB2 (P = .02). All three of the patients with GJB2 mutations and unilateral SNHL had a heterozygous genotype.
TABLE II.
Asymmetric Hearing Loss With Corresponding Genotype in Patients With GJB2 Mutations*.
| PTA (dB) Left | PTA (dB) Right | Genotype | |
|---|---|---|---|
| 1 | 26 | 109 | 167delT and wild type |
| 2 | 91 | 9 | 35delG and wild type |
| 3 | 6 | 83 | M34T and wild type |
| 4 | 130 | 55 | 35delG and L90P |
| 5 | 119 | 55 | 35delG and wild type |
| 6 | 90 | 26 | R32C and wild type |
| 7 | 109 | 55 | 35delG and 35delG |
| 8 | 16 | 69 | M34T and wild type |
| 9 | 11 | 60 | 499G and wild type |
| 10 | 31 | 68 | M34T and wild type |
| 11 | 91 | 59 | F83L and wild type |
| 12 | 48 | 71 | 235delC and wild type |
| 13 | 43 | 65 | M34T and K122I |
| 14 | 40 | 19 | 35delG and M34T |
| 15 | 49 | 70 | 235delC and wild type |
| 16 | 20 | 41 | 35delG and wild type |
| 17 | 34 | 14 | V37I and V37I |
| 18 | 10 | 30 | V27I, EI14G, and wild type |
| 19 | 41 | 22 | G4D and wild type |
| 20 | 46 | 29 | V37I and 235delC |
PTA = pure tone average.
From greatest to least asymmetry.
Only 570 patients met the criteria for inclusion in analysis of progressive SNHL; 99 had progressive SNHL, yielding an overall rate of 17.4%. Of these 99 patients, 20 had GJB2 mutations. There was no difference between the rate of progressive SNHL in patients with and without GJB2 mutations (18.9% vs. 17%, respectively; P = .7). Those with heterozygous mutations were more likely to experience progressive SNHL than were those with biallelic mutations, though this difference was only marginally significant (26.5% vs. 12.3%, respectively; P = .06). Patients with progressive SNHL and a GJB2 mutation were more likely to exhibit a bilateral progression than were those who were wild type for GJB2 (11 of 20 [55%] vs. 18 of 79 [23%], respectively; P = .005). There was no difference in the rate of bilateral progression in patients with heterozygous mutations (53.9%) and those with biallelic mutations (57%) (P = .99). Patients with mild SNHL in at least one ear and a heterozygous GJB2 mutation were more likely to experience a worsening of hearing in that ear than were those with a biallelic mutation, though this finding was not statistically significant (18.8% vs. 5%, respectively; P = .31). There was no difference between patients a GJB2 mutation and those who were wild type for GJB2 regarding progression of mild SNHL.
Temporal bone CT scans were available on 412 (49%) of the 840 patients in our study. In the remaining patients, CT scans were either not performed or not available for review. Imaging revealed an EVA in 108 (26%) patients. Patients who were wild type for GJB2 were more likely to have an EVA than were those with GJB2 biallelic and heterozygous mutations (29% vs. 11.9%, respectively; P = .004). Although patients with biallelic mutations (n = 2) had a lower rate of EVA than those with heterozygous mutations (n = 6), this difference was not statistically significant (5.7% vs. 18.8%, respectively; Fisher’s exact P = .14). Compared to patients who were wild type for GJB2, those with biallelic mutations had a significantly lower rate of EVA (Bonferroni adjusted P = .009). There was, however, no significant difference in the rate of patients with heterozygous mutations who had an EVA as compared to those who were wild type (Bonferroni adjusted P = .6).
DISCUSSION
Our study analyzed genotypic and phenotypic findings in patients with SNHL and GJB2 mutations and also determined the prevalence of EVA in these patients. To our knowledge, it is the largest single-institution study of this specific pediatric population. Our audiologic focus was SNHL severity. Overall, we found a lower percentage of patients with SNHL related to 35delG than previously reported3,4,6,8,9; however, the mix of other mutations was consistent with earlier publications. We also found a higher prevalence of patients with mild SNHL than previously reported. This finding can likely be attributed to the inclusion of all patients with SNHL (regardless of hearing loss severity) in our study. Genetic testing revealed that many with mild hearing loss were GJB2 positive.
We confirmed important genotype/phenotype correlations described in our earlier studies4,8 and in a study reported by Snoeckx et al.15 Specifically, M mutations are associated with milder forms of SNHL, whereas N mutations are associated with more severe hearing loss. Our study showed a higher prevalence of milder forms of hearing loss associated with M mutations than previously reported.15 In addition to these findings, we now report that for patients with at least one M mutation, the type of mutation in the second allele does not appear to influence the audiometric finding. This may be explained by cases in which the second allele has an N mutation. Thus, no functional protein is produced and the patient is functionally hemizygous for the M allele.
Patients who were wild type for GJB2 had an increased likelihood of having slight/mild hearing loss as compared to those with GJB2 mutations. In the patients with GJB2 mutations, there was no difference in the rate of mild/slight hearing loss between those with biallelic or heterozygous mutations. These findings suggest that heterozygous mutations in patients with GJB2 may be a contributing factor in hearing loss.
Liu et al.16 reported a low prevalence of asymmetric SNHL in patients with biallelic N GJB2 mutations. Our study showed that asymmetric hearing loss was significantly less common in patients with GJB2 mutations than in those who were wild type for GJB2. Additionally, there was a significant decrease in the prevalence of biallelic vs heterozygous mutations in patients with asymmetric hearing loss. Overall, patients with biallelic mutations presented with symmetric SNHL.
M34T was the most common M allele in patients with mild SNHL. Although the pathogenicity of this mutation has been controversial, recent data support that M34T contributes to mild SNHL.4,6,15,17 Only 5 patients with mild SNHL showed hearing loss progression; these patients all had different genotypes. We conclude that progression of GJB2-related SNHL is rare and may be attributed to nongenotypic etiologies. Moreover, no patient with the M34T allele showed hearing loss progression. This mutation appears to cause a mild, stable hearing impairment in most patients. Overall, when comparing initial and final audiologic testing, we found no difference in the number of patients with mild SNHL. We may be observing a “threshold effect” in which beyond a certain degree of hearing loss, progression may be more likely to occur. Further analysis is required to support this observation.
Analysis of temporal bone anomalies in our DFNB1 patient population revealed that only two patients (2.6%) had an EVA, the most common temporal bone anomaly in our study. This result is in sharp contrast to the finding by Propst and colleagues that 72% of patients with DFNB1 had temporal bone anomalies.11 These authors identified dilated endolymphatic fossa and hypoplastic modiolus as the two most common temporal bone anomalies (28% and 25% respectively). Our study did not quantitatively evaluate for the latter anomaly. Nonetheless, our cohort appears to have a much lower rate of vestibular aqueduct anomalies. Of patients with EVA, we found that only 1.9% had biallelic mutations. Patients who were wild type for GJB2 were far more likely to have a temporal bone anomaly than those who were GJB2 positive. Collectively, these data suggest that there is a poor correlation between GJB2 and temporal bone anomalies.
We concur with Propst et al.11 that perturbations in inner ear function may affect the development of the inner ear and lead to temporal bone anomalies. Other genetic causes of deafness such as Waardenburg syndrome may also present with EVA.18 Further investigation of temporal bone anomalies in patients with genetic deafness is ongoing.
CONCLUSION
This study represents the largest single-institution study of pediatric patients with GJB2 mutations and SNHL. The functional consequences of GJB2 mutations correlated with the degree of hearing loss. Genetic testing revealed that many patients with mild hearing loss were GJB2 positive. Patients with biallelic mutations presented with symmetric SNHL. Patients with M34T mutations and/or mild SNHL had a low risk of progression. In our study, temporal bone anomalies were uncommon in patients with GJB2 mutations.
BIBLIOGRAPHY
- 1.Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med 2006;354:2151–2164. [DOI] [PubMed] [Google Scholar]
- 2.Van Camp G, Smith R. [Hereditary Hearing Loss homepage]. Available at: http://webh01.ua.ac.be/hhh/. Accessed February 25, 2008.
- 3.Green GE, Scott DA, McDonald J, Woodworth G, Sheffield V, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA 1999;281:2211–2216. [DOI] [PubMed] [Google Scholar]
- 4.Preciado DA, Lawson L, Madden C, et al. Improved diagnostic effectiveness with a sequential diagnostic paradigm in idiopathic pediatric sensorineural hearing loss. Otol Neurotol 2005;26:610–615. [DOI] [PubMed] [Google Scholar]
- 5.Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patients with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 2001;127: 1037–1042. [DOI] [PubMed] [Google Scholar]
- 6.Putcha GV, Bejjani BA, Bleoo S, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med 2007;9:413–426. [DOI] [PubMed] [Google Scholar]
- 7.Estivill X, Fortina P, Surrey S, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 1998;351:394–398. [DOI] [PubMed] [Google Scholar]
- 8.Lim LH, Bradshaw JK, Guo Y, et al. Genotypic and phenotypic correlations of DFNB1-related hearing impairment in the Midwestern United States. Arch Otolaryngol Head Neck Surg 2003;129:836–840. [DOI] [PubMed] [Google Scholar]
- 9.Yaeger D, McCallum J, Lewis K, et al. Outcomes of clinical examination and genetic testing of 500 individuals with hearing loss evaluated through a genetics of hearing loss clinic. Am J Med Genet A 2006;140:827–836. [DOI] [PubMed] [Google Scholar]
- 10.Cryns K, Orzan E, Murgia A, et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J Med Genet 2004;41:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Propst EJ, Blaser S, Stockley TL, Harrison RV, Gordon KA, Papsin BC. Temporal bone imaging in GJB2 deafness. Laryngoscope 2006;116:2178–2186. [DOI] [PubMed] [Google Scholar]
- 12.Madden C, Halsted M, Meinzen-Derr J, et al. The influence of mutations in the SLC26A4 gene on the temporal bone in a population with enlarged vestibular aqueduct. Arch Otolaryngol Head Neck Surg 2007;133:162–168. [DOI] [PubMed] [Google Scholar]
- 13.Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80–83. [DOI] [PubMed] [Google Scholar]
- 14.Vijayasekaran S, Halsted MJ, Boston M, et al. When is the vestibular aqueduct enlarged? A statistical analysis of the normative distribution of vestibular aqueduct size. Am J Neuroradiol 2007;28:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snoeckx R, Huygen P, Feldmann D, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 2005;77:945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Pandya A, Angeli S, et al. Audiological features ofGJB2 (connexin 26) deafness. Ear Hear 2005;26:361–369. [DOI] [PubMed] [Google Scholar]
- 17.Pollak A, Skorka A, Mueller-Malesinska M, et al. M34T and V37I mutations in GJB2 associated hearing impairment: evidence for pathogenicity and reduced penetrance. Am J Med Genet A 2007;143:2534–2543. [DOI] [PubMed] [Google Scholar]
- 18.Madden C, Halstead MJ, Hopkin RJ, Choo DI, Benton C, Greinwald JH. Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope 2003;113:2035–2041. [DOI] [PubMed] [Google Scholar]