

Abstract
Purpose:
To estimate the absolute number of adult survivors of childhood cancer in the US population who carry a pathogenic or likely pathogenic variant in a cancer predisposition gene.
Methods:
Using the Surveillance, Epidemiology, and End Results (SEER) Program, we estimated the number of childhood cancer survivors on December 31, 2016 for each childhood cancer diagnosis, multiplied this by the proportion of carriers of pathogenic/likely pathogenic variants in the St. Jude Lifetime Cohort (SJLIFE) study, and projected the resulting number onto the US population.
Results:
Based on genome sequence data, 11.8% of 2,450 SJLIFE participants carry a pathogenic/likely pathogenic variant in one of 156 cancer predisposition genes. Given this information, we estimate that 21,800 adult survivors of childhood cancer in the US carry a pathogenic/likely pathogenic variant in one of these genes. The highest estimated absolute number of variant-carriers are among survivors of central nervous system tumors (n=4,300), particularly astrocytoma (n=1,800) and other gliomas (n=1,700), acute lymphoblastic leukemia (n=4,300), and retinoblastoma (n=3,500). The most frequently mutated genes are RB1 (n=3,000), NF1 (n=2,300), and BRCA2 (n=800).
Conclusion:
Given the increasing number of childhood cancer survivors in the US, clinicians should counsel survivors regarding their potential genetic risk, consider referral for genetic counseling and testing and, as appropriate, implement syndrome-specific cancer surveillance or risk-reducing measures.
INTRODUCTION
Survivors of childhood cancer are at increased risk to develop subsequent neoplasms (SNs) across their lifespan. Indeed, the overall cumulative incidence of invasive SNs among childhood cancer survivors is approximately 8% at 30 years post diagnosis, representing a 6.0-fold increase above rates observed in the general population1. Moreover, approximately 47% of survivors who develop one SN will develop an additional neoplasm within 20 years2. While much of the increase in SN risk is driven by exposure to the radiotherapy and specific chemotherapeutic agents that are administered to treat the primary cancer, some increase in risk may be due to an underlying cancer predisposition.
With recent advances in next generation sequencing technologies, it has become feasible for medical and research centers to analyze samples from large series of patients to identify pathogenic/likely pathogenic (P/LP) germline variants. Among children newly diagnosed with cancer, the frequency of P/LP germline variants in cancer predisposition genes is reported to be 8.5% following analysis of 60 genes with autosomal dominant (AD) inheritance and high penetrance,3 10% following analysis of 112 genes with AD or autosomal recessive (AR) inheritance.4 and 7.6% following analysis of 162 genes with AD or AR inheritance.5 In the St. Jude Lifetime Cohort (SJLIFE6), a longitudinal study of survivors of childhood and adolescent cancers, 5.8% of 3006 survivors harbored a P/LP variant in one of 60 AD cancer predisposition genes with moderate to high penetrance (SJCPG60, see Supplement Table 1) while an additional 5.9% carried a P/LP variant within a further list of 96 genes associated with AD cancer predisposition with moderate to low penetrance, autosomal recessive or X-linked inheritance. When compared to community controls, the prevalence of harboring a SJCPG60 gene variant was approximately 10-fold (95%CI=2.6–39.9) higher among survivors.
Although childhood cancers are considered rare, improving survival rates have resulted in an expanding survivor population with an elevated lifetime risk of developing SNs.7 As this risk is likely driven in part by underlying heritable factors, the aim of this study was to estimate the burden of P/LP cancer predisposing germline variants among childhood cancer survivors in the US using published data from the SJLIFE study. Specifically, we utilized SJLIFE data to estimate the absolute number of adult (≥18 years old) 10-year survivors of childhood cancer in the US population who carry a P/LP variant in a SJCPG60 gene, or in one of an additional 96 cancer predisposition genes (hereafter referred to as SJCPG156, see Supplemental Table 1).
METHODS
Using age-, sex-, calendar year-, and diagnosis-specific incidence and survival data from the Surveillance, Epidemiology and End-Results progam,8 we estimated the number of variant-carriers among adult 10-year survivors of childhood cancer diagnosed under 15 years of age between 1960–2006 in the US. Specifically, for each cancer type, a five-year survivor cohort of SEER cases was created by modeling the annual survival probability for the first five years after diagnosis, using a piecewise-exponential model as a function of year of diagnosis (natural cubic splines with knots at 1980, 1988, 1996, and 2004), the major determinant of the five-year survival, with an extrapolation to diagnosis years of 1960–1972. We then used a multivariable piecewise-exponential model on the five-year survivor cohorts (a cohort from each diagnosis year) to estimate the annual survival probability after five years from diagnosis as a function of sex, age at diagnosis, year of diagnosis, and attained age (natural cubic splines with five knots whose locations differed by diagnosis), and the interaction between age at diagnosis and year of diagnosis. The survival probability on December 31, 2016, for each subject was estimated by the product of annual survival probabilities obtained from the above model and summed over all SEER cases who would be over 18 years old if alive. This number was divided by the proportion of the US population covered by SEER registries to project the number of survivors of a given cancer type in the US.
The estimated number of US survivors was multiplied by the proportion of variant-carriers with the corresponding cancer diagnosis in SJLIFE. For these analyses, we used data from a SJLIFE report by Wang et al. (2016) in which whole genome sequencing (mean coverage per sample 36.8X) was performed on blood samples provided by SJLIFE participants and germline variant pathogenicity was classified according to the American College of Medical Genetics and Genomics guidelines.9,10All survivors who were variant carriers were counted in analyses. regardless of whether their primary, or subsequent, cancer diagnoses were considered concordant with their particular variant. The P/LP variant-carrier proportion in the SJCPG156 gene subset, as well as that in the SJCPG60 subset (AD, with moderate to high penetrance), was calculated. The SJLIFE protocol was approved by the Institutional Review Board.
RESULTS
Prevalence of P/LP germline mutations for cancer predisposition genes was based on 2,450 survivors in the SJLIFE cohort; 52% were male, 84% were white, 15% were black, and 1% of Hispanic ethnicity. The median age at diagnosis was 5.7 (range: 0.0–14.9) years and median attained age 35.6 (range: 18.0–68.6) years. The P/LP variant-carrier proportion was 11.8% (95%CI 10.6% to 13.1%; 309 variants in 289 survivors) and 6.1% (95%CI 5.2% to 7.1%; 149 variants in 149 survivors) for the SJCPG156 and SJCPG60 genes, respectively.
Based on the overall frequency of carriers observed in SLIFE, we estimated that there are approximately 21,800 survivors who harbor (one or more) P/LP variants in a cancer-associated predisposing gene (SJCPG156; Table 1) among 171,300 10-year adult survivors of pediatric cancer in the US as of December 31, 2016 (see Supplemental Table 2). The estimated number of variant carriers for the SJCPG60 subset is 12,500. The estimated number of variant carriers among males and females are 11,100 and 10,700 (SJCPG156), respectively. Within specific cancer diagnoses, the highest estimated absolute number of SJCPG156 variant carriers are survivors of central nervous system tumors (CNS; n=4,300), particulary astrocytoma (n=1,800) and other gliomas (n=1,700), acute lymphoblastic leukemia (n=4,300), and retinoblastoma (n=3,500).
Table 1:
Prevalence of pathogenic/likely pathogenic variants in cancer predisposition genes among adult survivors of childhood cancer participating in the SJLIFE study and in the US populationa
| CHARACTERISTICb | US | SJLIFE | Cancer-related genes (SJCPG156)c | Cancer predisposition genes (SJCPG60)d | ||||
|---|---|---|---|---|---|---|---|---|
| SJLIFE | SJLIFE | US | SJLIFE | SJLIFE | US | |||
| No. of survivors | No. of survivorse | No. of survivors with P/LP variants | % P/LP variant carriers | No. of survivors with P/LP variantsg | No. of survivors with P/LP variants | % P/LP variant carriers | No. of survivors with P/LP variantsg | |
| SEX | ||||||||
| Male | 89100 | 1276 | 144 | 11.3 | 11100 | 79 | 6.7 | 6300 |
| Female | 82200 | 1174 | 145 | 12.4 | 10700 | 70 | 5.5 | 6200 |
| AGE AT DIAGNOSIS | ||||||||
| 0–4 years | 72300 | 1104 | 159 | 14.4 | 10500 | 96 | 8.7 | 6800 |
| 5–9 years | 45300 | 674 | 67 | 9.9 | 5200 | 25 | 3.7 | 2700 |
| 10–14 years | 53700 | 672 | 63 | 9.4 | 6100 | 28 | 4.2 | 3000 |
| CURRENT AGE | ||||||||
| 18–29 years | 68900 | 797 | 98 | 12.3 | 8600 | 59 | 7.4 | 4900 |
| 30–39 years | 49200 | 936 | 102 | 10.9 | 6200 | 54 | 5.8 | 3500 |
| 40–49 years | 29200 | 552 | 70 | 12.7 | 3700 | 33 | 6.0 | 2100 |
| 50–59 years | 18000 | 158 | 18 | 11.4 | 2500 | 3 | 1.0 | 1600 |
| ≥60 years | 6000 | 7 | 1 | 14.3 | 730 | 0 | 0 | 400 |
| DIAGNOSIS | ||||||||
| Leukemias, & myeloproliferative & myelodysplastic diseases | 49500 | 934 | 92 | 9.93 | 52200 | 31 | 3.3 | 2200 |
| Lymphoid leukemia | 42800 | 874 | 87 | 10.0 | 4300 | 28 | 3.2 | 1400 |
| Acute myeloid leukemia | 4000 | 56 | 4 | 7.1 | 300 | 2 | 3.6 | 100 |
| Other leukemia | 2700 | 4 | 1 | 25.0 | 700 | 1 | 25.0 | 700 |
| Lymphoma & reticuloendothelial neoplasms | 21900 | 385 | 35 | 9.1 | 2000 | 12 | 3.1 | 700 |
| Hodgkin lymphoma | 10200 | 200 | 17 | 8.5 | 900 | 4 | 2.0 | 200 |
| Non-Hodgkin lymphomaf | 11700 | 185 | 18 | 9.7 | 1100 | 8 | 4.3 | 500 |
| CNS & intraspinal neoplasms | 30200 | 285 | 42 | 14.7 | 4300 | 28 | 9.8 | 2900 |
| Ependymoma & choroid plexus | 1900 | 39 | 5 | 12.8 | 200 | 2 | 5.1 | 100 |
| Astrocytoma | 18000 | 89 | 9 | 10.1 | 1800 | 4 | 4.5 | 800 |
| Medulloblastoma/PNET | 4800 | 76 | 8 | 10.5 | 500 | 5 | 6.6 | 300 |
| Other glioma | 4700 | 45 | 16 | 35.6 | 1700 | 16 | 35.6 | 1700 |
| Other CNS | 600 | 36 | 4 | 11.1 | <100 | 1 | 2.8 | <100 |
| Neuroblastoma & peripheral nervous system tumors | 11000 | 152 | 11 | 7.2 | 800 | 6 | 3.9 | 400 |
| Retinoblastoma | 7100 | 90 | 44 | 48.9 | 3500 | 42 | 46.7 | 3300 |
| Renal tumors | 13000 | 209 | 26 | 12.4 | 1600 | 12 | 5.7 | 700 |
| Hepatic tumors | 1400 | 17 | 2 | 11.8 | 200 | 2 | 11.8 | 200 |
| Bone tumors | 6700 | 119 | 11 | 9.2 | 600 | 3 | 2.5 | 200 |
| Osteosarcoma | 3700 | 72 | 9 | 12.5 | 500 | 2 | 2.8 | 100 |
| Ewings sarcoma | 2200 | 43 | 2 | 4.7 | 100 | 1 | 2.3 | 100 |
| Other bone tumors | 800 | 4 | 0 | 0.0 | 0 | 0 | 0.0 | 0 |
| Soft tissue & extraosseous sarcomas | 12600 | 153 | 13 | 8.5 | 1000 | 8 | 5.2 | 700 |
| Germ cell tumors, trophoblastic tumors & neoplasms of the gonads | 6900 | 66 | 7 | 10.6 | 700 | 2 | 3.0 | 200 |
| Epithelial neoplasms & melanomas | 10600 | 39 | 5 | 12.8 | 1400 | 2 | 5.1 | 500 |
| Other & unspecified malignant neoplasmsh | 500 | 1 | 1 | 100.0 | 500 | 1 | 100.0 | 500 |
| TOTAL | 171300 | 2450 | 289 | 11.8 | 21800 | 149 | 6.1 | 12500 |
The US childhood cancer survivor population was estimated from SEER data for adult survivors (≥18 years of age) diagnosed between 1960 and 2006 before 15 years of age, who were more than 10 years post diagnosis, and alive on December 31, 2016.
Cancer diagnoses were categorized according to the International Classification of Childhood Cancer (ICCC) scheme5
SJCPG156 - 156 genes related to cancer predisposition were selected for analysis and included genes with well-established associations with monogenic cancer risk inherited in an autosomal dominant fashion, genes with autosomal recessive or X-linked inheritance, somatic mosaicism and genes in which common variants increase cancer risk
SJCPG60 - 60 genes with well-established associations with monogenic cancer risk inherited in an autosomal dominant fashion at moderate to high penetrance were included in analyses. These 60 genes are a subset of the SJCPG156
All survivors who were variant carriers were counted in analyses regardless of whether their primary (or subsequent) cancer diagnosis was considered concordant with their particular gene variant.
Includes ICD-O categories of non-Hodgkin lymphoma, Burkitt lymphoma, miscellaneous lymphoreticular neoplasms and unspecified lymphoma.
The P/LP carrier percentages were from SJLIFE and were multiplied to the estimates of US survivor counts within each diagnosis subgroup to obtain the estimates of US survivors in the subgroup who carry a P/LP mutation. Therefore, their values stratified by sex, age at diagnosis current age, and group of diagnosis subgroups do not necessarily match with those of the US survivors.
The single SJLIFE survivor had a diagnosis of pleuropulmonary blastoma and a mutation in DICER1
No.=numbers
Figure 1 shows the estimated number of US survivors with P/LP variants according to specific genes by cancer diagnosis. Genes with the highest estimated numbers of survivors are RB1 (n=3,000), NF1 (n=2,300), and BRCA2 (n=800). Cancer predisposition syndromes and available family history information for among SJLIFE participants who carry a SJCPG60 P/LP variant are summarized in Supplementary Table 3.
Figure 1: The estimated number of US survivors with pathogenic/likely pathogenic SJCPG60 variants according to specific genes and by initial cancer diagnosis.
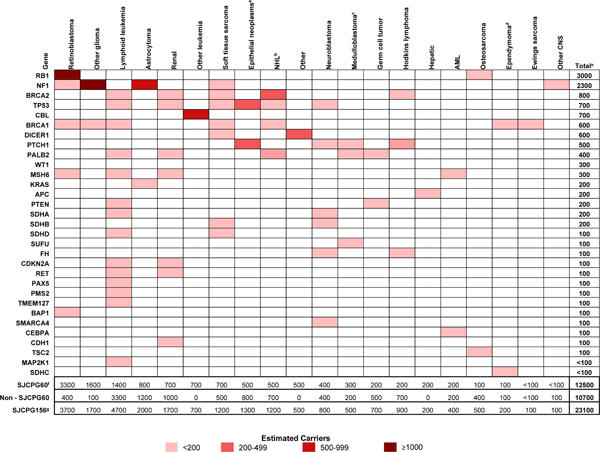
Genes are ranked in the order from highest to lowest estimated number of survivors. a) Epithelial neoplasms and melanomas; b) Non-Hodgkin lymphoma, Burkitt lymphoma, miscellaneous lymphoreticular neoplasms and unspecified lymphoma; c) Medulloblastoma and primitive neuroectodermal tumor; d) Ependymoma and choroid plexus tumors; e) Several individuals carried two P/LP variants, accordingly, the estimated number of affected genes among the US survivor population is higher than the estimated number of survivors who carry P/LP variants in the US; f) SJCPG60 - 60 genes with well-established associations with monogenic cancer risk inherited in an autosomal dominant fashion at moderate to high penetrance. These 60 genes are a subset of the SJCPG156; g) SJCPG156 - 156 genes with well-established associations with cancer risk inherited in an autosomal dominant, autosomal recessive or X-linked fashion.
DISCUSSION
Although childhood cancers are considered rare, survival rates have steadily improved over the past several decades, with an overall survival rate near 85%.11 Accordingly, there is a growing population of childhood cancer survivors who have elevated risks of developing additional cancers relative to general population, and who require changes in medical management to maximize their own health outcomes as well as the health outcomes of their offspring. Our data suggest that more than 20,000 10-year survivors of pediatric cancer carry a P/LP variant in a cancer-associated predisposing gene. Our findings are medically relevant as they provide an estimate of the absolute number childhood cancer survivors, who if identified through appropriate genetic counseling and testing, could potentially benefit from long term cancer surveillance. Identification of childhood cancer survivors who harbor cancer-predisposing variants is important for multiple reasons. First, while increased cancer surveillance will not prevent SNs from occurring, it will enable their early detection and treatment of specific cancers, thereby enhancing the chances for cure. Knowledge of a cancer-predisposing variant can inform the clinical care and choice of therapeutic agents used to treat a SN, such as avoidance of radiation therapy, to minimize toxicities and further enhance long-term outcome. Second, some predisposed survivors may benefit from preventative measures, such as prophylactic removal of at-risk organs. Third, early detection of SNs that reduces the need for intensive treatments may lower healthcare costs associated with treatment of SNs. Fourth, as germline variants are inherited in the vast majority of cases, it is likely that for most survivors who harbor a P/LP variant, one parent, as well as additional close relatives such as a sibling or offspring might also be affected and could also benefit from additional cancer surveillance and/or interventions. Finally, quantification of the burden of cancer predisposition among pediatric cancer survivors can assist health care providers, researchers and policy makers to develop the health services and resources needed to provide genetic counselling and cancer surveillance for this high-risk population.
In this study, the highest estimated absolute number of variant carriers, stratified according to primary childhood cancer diagnosis, occurred among survivors of CNS and intraspinal neoplasms and lymphoid leukemia. The estimated number of US survivors of CNS and intraspinal neoplasms carrying a P/LP variant in a SJCPG156 and SJCPG60 gene were approximately 4,300 and 2,900, respectively. Our finding is consistent with recent genome sequencing efforts among newly-diagnosed children which suggest that variants within predisposition genes are common within certain CNS subtypes, including choroid plexus carcinoma (25.0%), medulloblastoma (13.5%), and high-grade -(7.9%−9.1%), and low-grade (7.9%) glioma.3 Lymphoid leukemia predominantly consisting of ALL, represented the second largest group in absolute number of SJCPG156 variant carriers (N=4,000) despite a modest frequency of variant carriers (10.4%). This observation is likely due to a combination of factors including the high prevalence of ALL, the most common cancer among children, and that survival rates for this cancer have improved dramatically over time.12
A high frequency of variant carriers for either gene list were observed among retinoblastoma survivors, consistent with the known biology of this tumor where approximately 40–50% of cases arise because of a dominantly inherited pathogenic variant in the RB1 gene.13,14 Although a high frequency of variant carriers were observed among survivors of epithelial neoplasms and melanomas (SJCPG156, 12.8%) and hepatic tumors (SJCPG60, 11.8%), , they reflected a relatively small number of variant carriers (1,400 and 200, respectively) because of the low incidence rate in the pediatric age range.
In these analyses, we estimated that the most frequently mutated SJCPG60 genes (in absolute numbers) among survivors of childhood cancer included RB1, NF1, and TP53, as well as BRCA1 and BRCA2. It is perhaps not surprising that RB1, NF1, and TP53 accounted for a substantial proportion of mutated genes (~50% of SJCPG60 genes and ~25% of SJCPG156 genes) in the US childhood cancer survivor population given that these genes have well-established etiological links with specific childhood cancers. In contrast, the occurrence of BRCA1 and BRCA2 among the most frequently mutated SJCPG60 genes was unexpected as these genes are generally not considered to predispose to childhood cancers. Currently it remains unknown whether these unexpected variants are causal for the primary cancer or SN diagnoses, or simply incidental findings. A recent report found that pediatric.adolescent non-Hodgkin lymphoma survivors had a five-fold excess risk of BRCA2 mutations.15 Nevertheless, the current analysis remains important because it estimates the size of a high-risk population of survivors with potential implications for cancer surveillance and/or preventive interventions. Beyond the implications for survivors, their first degree relatives may also benefit from genetic counselling. Given that the genes considered in our analyses have well-established associations with cancer risk, and that the variants within these genes were rigorously assessed using a widely accepted framework for variant classification, we believe our results to be a reasonable estimate of the burden of cancer predisposition among the 10-year survivors of childhood cancer in the US.
Limitations of this study include potential bias in the modeling of survival probabilities, including the use of SEER to represent the US population and extrapolation to diagnosis years of 1960–1972 when SEER data were not available. However, SEER data had 8.3% censoring: thus, survival was estimated for a small portion of the SEER survivor data. Second, our analyses are based on the assumption that the SJLIFE population was representative of all survivors in the US. While the SJLIFE population is similar to other survivor groups,16 the frequency of gene variants in the SJLIFE cohort may be biased as it reflects individuals treated at a single institution. Third, for rare cancers such as chronic leukemias, estimates may have been unstable given the low numbers of survivors treated for these cancers in SJLIFE. Fourth, we could not assess potential differences of variant carrier rates by race due to the small numbers of non-Caucasian survivors with variants in each childhood cancer diagnosis group. Finally, because the variant-carrier rates we applied are from adult 10-year survivors, we cannot estimate the prevalence for younger survivors with shorter time since diagnosis. However, had we removed our restrictions (≥18 years of age, 10-year survivor) we would have estimated there to be approximately 276,000 individuals currently living in the US diagnosed with cancer before the age of 15 years since 1960. From this, we conservatively estimate that approximately 35,000 survivors carry a P/LP variant in a SJCPG156 gene and 20,000 survivors carry a variant in a SJCPG60 gene.
With the substantial number of childhood cancer survivors in the US estimated to carry a P/LP variant in a cancer-predisposing gene, clinicians should monitor for signs (e.g., phenotypic characteristics or tumor types suggestive of an underlying syndrome and/or family history of cancer) indicating an increased predisposition to cancer, as these individuals may benefit from referral to cancer predisposition programs or providers for genetic counselling services and genetic testing.17,18 Having estimates of the total number of survivors with a P/LP variant provide an important context for consideration of the potential demands on health care providers and services relative to genetic counselling and surveillance, as well as the broader public health perspective. Implementation of appropriate cancer surveillance and risk-reducing strategies may improve health outcomes among variant-carriers and provide important information regarding risk for family members.
Supplementary Material
Acknowledgments
Funding: This work was supported by the American Lebanese Syrian Associated Charities and the National Institutes of Health (P30 CA021765, U01 CA195547). The funders had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the communication; or the decision to submit the communication for publication.
Conflict of Interest Disclosures: Dr. Nichols has received research funding (unrelated to the current research) from Incyte, Apline Immune Sciences, and Imago Pharmaceuticals. Dr. S. Wang has been previously employed by Bristol-Myers Squibb. Dr. Lanctot has a family member who has been employed by Covidien and Medtronic and who owns stocks in these companies. The remaining authors declare no conflicts of interest.
Abbreviation
- ~
approximately more than or equal to
- ≥
more than or equal to
- %
percent
- AD
autosomal dominant
- ALL
acute lymphocytic leukemia
- AR
autosomal recessive
- BAM
compressed binary SAM file
- BRCA1
Breast cancer 1, early onset
- BRCA2
Breast cancer 2, early onset
- CI
confidence interval
- CNS
central nervous system tumors
- et al
and others
- gVCF
joint genotype calls
- n
number
- NF1
neurofibromin 1
- P/LP
pathogenic/likely pathogenic
- RB1
RB transcriptional corepressor 1
- SEER
Surveillance, Epidemiology, and End Results
- SJLIFE
St. Jude Life Cohort
- SNs
Subsequent neoplasms
- TP53
tumor protein 53
- US
United States
Footnotes
Data Availability: Aligned BAM files for 2450 survivors and the joint genotype calls (gVCFs) are accessible through St. Jude Cloud (https://stjude.cloud).
REFERENCES
- 1.Friedman DL, Whitton J, Leisenring W, et al. Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 2010;102(14):1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong GT, Liu W, Leisenring W, et al. Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 2011;29(22):3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Walsh MF, Wu G, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 2015;373(24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons DW, Roy A, Yang Y, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors [published online ahead of print Jan 28, 2016]. JAMA Oncol 2016; 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–327. [DOI] [PubMed] [Google Scholar]
- 6.Hudson MM, Ehrhardt MJ, Bhakta N, et al. Approach for Classification and Severity Grading of Long-term and Late-Onset Health Events among Childhood Cancer Survivors in the St. Jude Lifetime Cohort. Cancer Epidemiol Biomarkers Prev 2017;26(5):666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibson TM, Mostoufi-Moab S, Stratton KL, et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970–99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol 2018;19(12):1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.National Institutes of Health, National Cancer Institute, Surveillance E, and End Results Program. ICCC Recode ICD-O-3/WHO 2008; 2008; https://seer.cancer.gov/iccc/iccc-who2008.html. [Google Scholar]
- 9.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19(2):249–255. [DOI] [PubMed] [Google Scholar]
- 11.Childhood Cancer Statistics, 5-Year Survival Rate. 2019; https://curesearch.org/5-Year-Survival-Rate. Accessed Aug 20, 2019.
- 12.Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood 2012;120(6):1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet C Semin Med Genet 2004;129C(1):23–28. [DOI] [PubMed] [Google Scholar]
- 14.Ripperger T, Bielack SS, Borkhardt A, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A 2017;173(4):1017–1037. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Wilson CL, Armstrong GT, et al. Association of Germline BRCA2 Mutations With the Risk of Pediatric or Adolescent Non-Hodgkin Lymphoma. JAMA Oncol 2019; 10.1001/jamaoncol.2019.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips SM, Padgett LS, Leisenring WM, et al. Survivors of childhood cancer in the United States: prevalence and burden of morbidity. Cancer Epidemiol Biomarkers Prev 2015;24(4):653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballinger ML, Best A, Mai PL, et al. Baseline Surveillance in Li-Fraumeni Syndrome Using Whole-Body Magnetic Resonance Imaging: A Meta-analysis. JAMA Oncol 2017;3(12):1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villani A, Shore A, Wasserman JD, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016;17(9):1295–1305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.