

Abstract
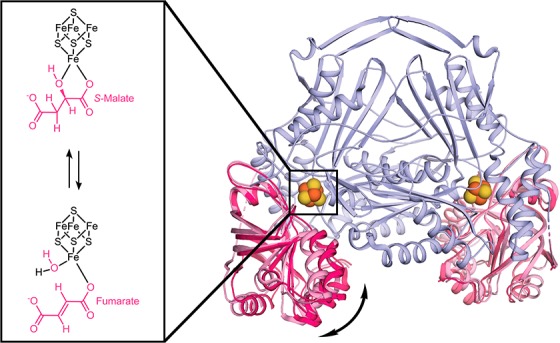
Class I fumarate hydratases (FHs) are central metabolic enzymes that use a [4Fe-4S] cluster to catalyze the reversible conversion of fumarate to S-malate. The parasite Leishmania major, which is responsible for leishmaniasis, expresses two class I FH isoforms: mitochondrial LmFH-1 and cytosolic LmFH-2. In this study, we present kinetic characterizations of both LmFH isoforms, present 13 crystal structures of LmFH-2 variants, and employ site-directed mutagenesis to investigate the enzyme’s mechanism. Our kinetic data confirm that both LmFH-1 and LmFH-2 are susceptible to oxygen-dependent inhibition, with data from crystallography and electron paramagnetic resonance spectroscopy showing that oxygen exposure converts an active [4Fe-4S] cluster to an inactive [3Fe-4S] cluster. Our anaerobically conducted kinetic studies reveal a preference for fumarate over S-malate. Our data further reveal that single alanine substitutions of T467, R421, R471, D135, and H334 decrease kcat values 9–16000-fold without substantially affecting Km values, suggesting that these residues function in catalytic roles. Crystal structures of LmFH-2 variants are consistent with this idea, showing similar bidentate binding to the unique iron of the [4Fe-4S] cluster for substrate S-malate as observed in wild type FH. We further present LmFH-2 structures with substrate fumarate and weak inhibitors succinate and malonate bound in the active site and the first structure of an LmFH that is substrate-free and inhibitor-free, the latter showing increased mobility in the C-terminal domain. Collectively, these data provide insight into the molecular basis for the reaction catalyzed by LmFHs, enzymes that are potential drug targets against leishmaniasis.
Fumarate hydratase (FH, fumarase, EC 4.2.1.2) catalyzes the stereospecific reversible conversion of S-malate to fumarate (Scheme 1). The reactions involve the anti-elimination of a water molecule from S-malate to form fumarate and the anti-addition of water to the carbon–carbon double bond of fumarate to form S-malate.1 FH participates in the tricarboxylic acid (TCA) cycle, the succinic fermentation pathway,2 and DNA repair3 and is thought to provide fumarate for the de novo pyrimidine biosynthetic pathway.4 There are two classes of FHs with ∼20% amino acid sequence identity and distinct protein structures.5,6 Class I FHs are homodimeric enzymes that contain an oxygen-sensitive [4Fe-4S] cluster as a cofactor and are found in archaea,7 prokaryotes,8 and unicellular eukaryotes.9−11 Class II FHs are homotetrameric enzymes that do not require iron for catalysis and are found in prokaryotes6 and eukaryotes.12
Scheme 1. FH Reversible Reaction.

The parasite Leishmania major, which is responsible for the neglected tropical disease cutaneous leishmaniasis, expresses two isoforms of class I FHs that share 59% sequence identity and are localized in the mitochondria (LmFH-1) and cytosol/glycosome (LmFH-2).9 The mitochondrial LmFH-1 catalyzes the hydration of fumarate to S-malate (Scheme 1) in the TCA cycle as well as the dehydration of S-malate to fumarate (Scheme 1) in the succinic acid fermentation pathway. The cytosolic/glycosomal LmFH-2 catalyzes the dehydration of S-malate to fumarate as part of the glycosomal succinic acid fermentation pathway and for use by dihydroorotate dehydrogenase,13 an enzyme that is part of the de novo pyrimidine biosynthetic pathway. The role of LmFHs in these central metabolic pathways together with the lack of structural similarity between class I parasitic FHs and the class II human FH make LmFHs potential drug targets against leishmaniasis. Leishmaniasis affects 1 million people in low-income countries and has no effective treatment, according to the World Health Organization.
Recently, we found that LmFH isoforms have very similar homodimeric structures, with each monomer comprised of an N-terminal domain and a C-terminal domain, connected by a flexible linker, that are arranged around the catalytic [4Fe-4S] cluster.5,14 An LmFH-2 co-crystal structure revealed substrate S-malate bound in a bidentate fashion to the [4Fe-4S] cluster in the active site as well as at a cavity located between the N- and C-terminal domains that leads to the active site.5 This structure suggested a key catalytic role for the conserved active site residues T467, R421, R471, D135, H334, and R173, as well as C-terminal domain mobility that could regulate access of the substrate to the active site.
Motivated by these discoveries, we applied site-directed mutagenesis, biochemical, and structural approaches to elucidate the molecular basis for the reaction catalyzed by class I FHs. Here, we report a total of 13 crystal structures of LmFH-2, both of the wild type protein and of five protein variants, and both in the presence and in the absence of substrates and inhibitors. We further report kinetic data for wild type LmFH-2 and its variants. Our findings shed light on the mechanism of action of class I FHs and contribute to further evaluation of LmFHs as valuable drug targets for the development of new therapies against leishmaniasis.
Materials and Methods
Site-Directed Mutagenesis, Expression, and Purification
LmFH-2 variants were constructed using a pET28a(+) vector containing the LmFH-2 gene (GeneDB accession code LmjF.29.1960) as a template.9 LmFH-2-D135A, LmFH-2-R173A, LmFH-2-H334A, LmFH-2-R421A, LmFH-2-T467A, and LmFH-2-R471A were constructed using the protocol of the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies).15 The primers used for site-directed mutagenesis are listed in Table S1. Recombinant LmFH-1, LmFH-2, and variants were expressed in Escherichia coli T7 express and purified by nickel affinity chromatography as described previously9 at 4 °C in an MBraun anaerobic glovebox. For crystallization assays, the purifications were performed with 1 mM dithiothreitol (DTT) in all buffers.
Steady-State Enzyme Kinetics
The activity measurements of LmFH-1, LmFH-2, and the variants LmFH-2-D135A, LmFH-2-R173A, LmFH-2-R421A, LmFH-2-T467A, LmFH-2-R471A, and LmFH-2-H334A were carried out in an MBraun anaerobic glovebox (<0.1 ppm O2). Fumarate production or consumption was measured at 250 nm (ε250 nmfumarate = 1450 M–1 cm–1) and 300 nm (ε300 nm = 36.6 M–1 cm–1) at 23 °C using a USB 4000 fiber optic spectrometer (Ocean Optics). Fumarate consumption was measured at 250 nm for 0.125–1 mM fumarate and 300 nm for 2–32 mM fumarate. Fumarate production was measured at 250 nm for the LmFH-2 variants and 250 nm (0.125–4 mM S-malate) and 300 nm (8–100 mM S-malate) for wild type LmFH-1 and LmFH-2. Activity assays were performed in a reaction mixture containing 50 mM Tris (pH 9) and 150 mM NaCl with the substrate, S-malate or fumarate, in a total volume of 1 mL. The reaction was started by adding the enzyme [the molecular weight (MW) of LmFH-1 is 60.8 kDa, and that of LmFH-2 is 62.6 kDa]: ∼9 μg mL–1 LmFH-1, ∼7 μg mL–1 LmFH-2, ∼8 μg mL–1 LmFH-2-H334A, ∼100 μg mL–1 LmFH-2-D135A, ∼160 μg mL–1 LmFH-2-R173A and LmFH-2-R471A, and ∼170 μg mL–1 LmFH-2-R421A and LmFH-2-T467A, all in 50 mM Tris (pH 8.5) and 150 mM NaCl. The kinetic parameters were determined from the Michaelis–Menten equation (eq 1) fitted to the experimental data obtained by varying the concentration of S-malate (from 0.125 to 100 mM) and fumarate (from 0.125 to 32 mM).
| 1 |
where v is the reaction velocity, Vmax is the maximum velocity of the reaction, [S] is the substrate concentration, and Km is the concentration of the substrate when the reaction velocity is half of the Vmax. Data were fit using Prism 8 software. The protein concentration was corrected on the basis of the extinction coefficient at 410 nm (ε410 nm[4Fe-4S] = 15000 M–1 cm–1),16 which reports on the amount of intact [4Fe-4S] cluster, i.e., the type of cluster that makes active protein. Approximately 60% of LmFH had an intact [4Fe-4S] cluster based on this analysis. Notably, although the cluster does not play a redox role in catalysis, the presence of an intact cluster is required for catalytic activity.
Inhibition Assays with Succinate
Inhibition analyses of LmFH-2 by succinate were carried out in an MBraun anaerobic glovebox. Fumarate production and consumption were measured at 250 nm (ε250 nmfumarate = 1450 M–1 cm–1) at 23 °C in a USB 4000 fiber optic spectrometer (Ocean Optics). Inhibition assays were performed in a reaction mixture containing 50 mM Tris (pH 9) and 150 mM NaCl with the substrate (2 mM S-malate or 0.5 mM fumarate) and inhibitor (0–120 mM succinate) in a total volume of 1 mL. The reaction was started by adding ∼10 μg mL–1 LmFH-2 in 50 mM Tris (pH 8.5) and 150 mM NaCl. The IC50 was determined from the dose–response equation (eq 2) fitted to the experimental data obtained by varying the concentration of succinate.
| 2 |
where inhibition(%) is written as a function of log[I], the log of inhibitor concentration, Imin(%) is the minimum inhibition, Imax(%) is the maximum inhibition, IC50 is the half-maximal inhibitory concentration, and h is the Hill coefficient. Data were fitted using Origin software (http://www.originlab.com).
Electron Paramagnetic Resonance (EPR) Spectroscopy
EPR measurements were carried out on a Bruker ELEXSYS E580 spectrometer operating at X-band (9.5 GHz) frequency. The experiment was performed at liquid helium temperature (∼4 K), with a microwave power of 10 mW, a modulation amplitude of 2 G, and a modulation frequency of 100 kHz. A sample containing ∼4 mg mL–1 LmFH-2 was prepared aerobically and frozen in liquid nitrogen.
Crystallization of LmFH-2 Variants
Crystals of wild type LmFH-2 and variants LmFH-2-R173A, LmFH-2-H334A, LmFH-2-R421A, LmFH-2-T467A, and LmFH-2-R471A were obtained by the hanging drop vapor diffusion at 23 °C in a Coy anaerobic chamber (<6 ppm O2) as described previously.5 The crystallization condition consists of polyethylene glycol (PEG) 3350 [12–14% (v/v)] and tacsimate [2–4% (v/v), pH 5]. Tacsimate has in its composition the substrate malate and inhibitors malonate and succinate,17 which results in LmFH-2 structures that have both malate and malonate molecules bound without further addition of these molecules to the crystallization buffer.5 To obtain crystals only in the presence of a substrate (S-malate or fumarate) or an inhibitor (malonate or succinate), different formulations of tacsimate were prepared to contain the ligand of interest (Table S2). Another formulation of tacsimate was also prepared without malate, malonate, and succinate (Table S2) to obtain crystals in the absence of ligands. In addition, the proteins were previously incubated with 5–10 mM S-malate or fumarate. Drops were prepared by mixing 1 μL of a protein solution [5–10 mg mL–1 in 50 mM Tris (pH 8.5), 150 mM NaCl, and 1 mM DTT], 1 μL of a reservoir solution [12% (v/v) PEG 3350, 4% (v/v) tacsimate (or its variations, Table S2) (pH 5)], and 0.2 μL of additive [30% (v/v) ethanol] and equilibrating against 400 μL of a reservoir solution. After 1 day, brownish-yellow needle cluster-like crystals were obtained. The optimization of the crystals was performed using microseeding techniques.18 The crystals were transferred to a cryoprotectant solution [25% (v/v) glycerol, 18% (v/v) PEG 3350, and 8% (v/v) tacsimate (or its variations, Table S2) (pH 5)] and flash-cooled in liquid nitrogen in the Coy chamber.
Data Collection and Structure Determination of LmFH-2 Variants
Data collection was performed at beamline 24-ID-C of the Advanced Photon Source. Diffraction data were processed and scaled using HKL200019 with the CC1/2 value used to determine the resolution cutoff. The crystal structures of LmFH-2 and variants LmFH-2-T467A, LmFH-2-H334A, LmFH-2-R173A, LmFH-2-R471A, and LmFH-2-R421A in a complex with the substrate and/or inhibitor or without a ligand were determined by molecular replacement techniques implemented in Phaser.20 The coordinates of the LmFH-2 structure [Protein Data Bank (PDB) entry 5L2R(5)] were used to obtain the initial phases. Following molecular replacement, simulated annealing was performed in phenix.refine21 to remove model bias. The models were refined by iterative rounds of model building and addition of water molecules using Coot.22 Refinement in phenix.refine21 used noncrystallographic symmetry restraints, TLS (translation, libration, and screw), and positional and individual B-factor refinement, with S-malate, fumarate, succinate, and malonate geometry restraints generated by phenix.elbow.21 The data collection and refinement statistics are summarized in Tables S3 and S4. The residues visualized in each structure, of 568 residues, are listed in Table S5. Figures were created with PyMol Software.23 Crystallographic software packages were compiled by SBGrid.24
Results
Steady-State Kinetics of LmFH Isoforms Indicate a Preference for S-Malate Formation
We have previously determined the kinetic parameters for LmFH isoforms, but these studies were performed using a glovebag (>20 ppm O2), conditions under which the cluster was being degraded over the course of the experiment.9 Here, we measured the initial velocities of mitochondrial LmFH-1 and cytosolic LmFH-2 as a function of S-malate and fumarate concentration in an MBraun anaerobic glovebox (<0.1 ppm O2). For the concentration ranges investigated for both S-malate and fumarate, LmFH-1 and LmFH-2 data show hyperbolic curves and a good fit to the Michaelis–Menten equation (eq 1) (Figure 1A,B).
Figure 1.

Initial rates of LmFH isoforms and LmFH-2 variants as a function of S-malate and fumarate concentration. (A) Initial velocities of LmFH isoforms vs S-malate concentration. (B) Initial velocities of LmFH isoforms vs fumarate concentration. (C) Initial velocities of LmFH-2 variants vs S-malate concentration. (D) Initial velocities of LmFH-2 variants vs fumarate concentration. The plots were fit with the Michaelis–Menten equation (eq 1). The Y-axis on the right corresponds to the initial velocities of the LmFH-2-H334A variant, which are colored gray. The error bars were calculated from triplicate reactions. Error bars are given for all measurements but are not easily visible in all cases. (E) Catalytic efficiency (kcat/Km) of LmFH isoforms and LmFH-2 variants for S-malate and fumarate.
The kinetic parameters of LmFH isoforms are listed in Table 1. The Km values for LmFH-1 and LmFH-2 are approximately 3-fold higher for S-malate than for fumarate. The kcat values for LmFHs are similar for fumarate but approximately 1.5-fold higher for LmFH-2 than for LmFH-1 for S-malate. Additionally, the catalytic efficiency (kcat/Km) is approximately 10- and 7-fold higher for fumarate than for S-malate for LmFH-1 and LmFH-2, respectively, indicating that both isoforms have a preference for S-malate formation (Scheme 1 and Figure 1E).
Table 1. Kinetic Parameters of LmFH Enzymes.
| enzyme | substrate | Km (mM) | Vmax (μmol min–1 mg–1) | kcat (s–1) | kcat/Km (×103 M–1 s–1) | reaction condition | ref |
|---|---|---|---|---|---|---|---|
| LmFH-1 | S-malate | 1.2 ± 0.3 | 0.50 ± 0.03 | 0.55 ± 0.03 | 0.5 ± 0.1 | aerobic | (9) |
| LmFH-1 | S-malate | 13.0 ± 0.8 | 338 ± 6 | 364 ± 6 | 28 ± 2 | anaerobic | this study |
| LmFH-2 | S-malate | 5.7 ± 0.2 | 22.7 ± 0.5 | 24.9 ± 0.5 | 4.4 ± 0.2 | aerobic | (9) |
| LmFH-2 | S-malate | 11.5 ± 0.5 | 485 ± 6 | 537 ± 7 | 47 ± 3 | anaerobic | this study |
| LmFH-2-H334A | S-malate | 4.6 ± 0.1 | 52.0 ± 0.4 | 57.6 ± 0.4 | 12.5 ± 0.4 | anaerobic | this study |
| LmFH-2-R421A | S-malate | 4.1 ± 0.3 | 0.130 ± 0.002 | 0.144 ± 0.002 | 0.035 ± 0.003 | anaerobic | this study |
| LmFH-2-D135A | S-malate | 5.3 ± 0.3 | 0.068 ± 0.001 | 0.075 ± 0.001 | 0.014 ± 0.001 | anaerobic | this study |
| LmFH-2-T467A | S-malate | 4.4 ± 0.2 | 0.047 ± 0.001 | 0.052 ± 0.001 | 0.012 ± 0.001 | anaerobic | this study |
| LmFH-2-R471A | S-malate | 4.3 ± 0.2 | 0.034 ± 0.001 | 0.038 ± 0.001 | 0.009 ± 0.001 | anaerobic | this study |
| LmFH-2-R173A | S-malate | 4.7 ± 0.4 | 0.030 ± 0.001 | 0.033 ± 0.001 | 0.007 ± 0.001 | anaerobic | this study |
| LmFH-1 | fumarate | 1.3 ± 0.3 | 1.8 ± 0.3 | 1.9 ± 0.3 | 1.5 ± 0.6 | aerobic | (9) |
| LmFH-1 | fumarate | 4.2 ± 0.6 | 1,076 ± 50 | 1,159 ± 54 | 280 ± 50 | anaerobic | this study |
| LmFH-2 | fumarate | 1.9 ± 0.3 | 44 ± 5 | 49 ± 5 | 26 ± 7 | aerobic | (9) |
| LmFH-2 | fumarate | 4.6 ± 0.5 | 1,355 ± 49 | 1,500 ± 54 | 330 ± 50 | anaerobic | this study |
| LmFH-2-H334A | fumarate | 4.2 ± 0.2 | 177 ± 4 | 196 ± 4 | 47 ± 3 | anaerobic | this study |
| LmFH-2-R421A | fumarate | 4.9 ± 0.7 | 0.65 ± 0.03 | 0.72 ± 0.03 | 0.15 ± 0.03 | anaerobic | this study |
| LmFH-2-D135A | fumarate | 1.8 ± 0.3 | 0.21 ± 0.01 | 0.23 ± 0.01 | 0.13 ± 0.03 | anaerobic | this study |
| LmFH-2-T467A | fumarate | 3.1 ± 0.5 | 0.18 ± 0.01 | 0.20 ± 0.01 | 0.06 ± 0.01 | anaerobic | this study |
| LmFH-2-R471A | fumarate | 2.3 ± 0.7 | 0.11 ± 0.01 | 0.12 ± 0.01 | 0.05 ± 0.02 | anaerobic | this study |
| LmFH-2-R173A | fumarate | 2.6 ± 0.6 | 0.14 ± 0.01 | 0.15 ± 0.01 | 0.06 ± 0.02 | anaerobic | this study |
Substitutions of LmFH-2 Active Site Residues Affect the kcat More Than the Km
To investigate the role of active site residues in the catalytic mechanism of class I FHs, LmFH-2 residues D135, R173, H334, R421, T467, and R471 were changed to alanine and the kinetics of the enzyme variants were investigated. As observed for wild type LmFH-2, the variants follow the Michaelis–Menten equation (eq 1) for S-malate (Figure 1C) and fumarate (Figure 1D). The kinetic results (Table 1) indicate that D135A, R173A, H334A, R421A, T467A, and R471A substitutions decrease Km and kcat values by approximately 2–3-fold and 9–16000-fold, respectively, for both substrates in comparison with those of the wild type enzyme. The catalytic efficiency (kcat/Km) for both substrates is drastically reduced (∼1000- to >6000-fold) for LmFH-2 variants in comparison with that of the wild type, except for the variant LmFH-2-H334A, which has a kcat/Km that is approximately 4–7-fold lower (Figure 1E).
LmFH-2 Variants Show Modes of S-Malate Binding Similar to That of the Wild Type Enzyme
We have previously determined the crystal structure of cytosolic LmFH-2 in a complex with substrate S-malate in the active site and inhibitor malonate both in the dimer interface and on the top of the protein at 2.05 Å resolution.5 Here, we determined the crystal structure of LmFH-2 with only S-malate at 1.85 Å resolution (Table S3). This structure is highly similar to the previously determined LmFH-2 structure (PDB entry 5L2R), with a root-mean-square deviation (RMSD) for all of the Cα atoms of 0.14 Å.
As previously described,5 the substrate S-malate is bound in the active site to the unique iron of the [4Fe-4S] cluster via C2-hydroxyl and carboxylate oxygen atoms and interacts with residues from the N- and C-terminal domains (Q134, D135, R173, G216, R421, T467, T468, R471, and K491) and one molecule of water (Figure 2A). Although not directly involved in substrate binding, residue H334 from the other monomer is also considered part of the active site due to its interaction with the proposed catalytic residue D135. The [4Fe-4S] cluster is coordinated to three cysteine residues (C133, C252, and C346), with the fourth iron free to coordinate to the substrate. The active site amino acids are fully conserved in class I FHs.5
Figure 2.

Active sites of LmFH-2 variants reveal similar S-malate-binding modes with LmFH-2-R173A displaying the largest variation. (A) LmFH-2 in a complex with S-malate. The proposed catalytic residues are colored yellow (D135 and H334 from chain B) and light blue (T467, R421, and R471). Residue R173 that is proposed to play a role in the correct positioning of the substrate in the active site is colored magenta. The distance from the T467 OH group to the S-malate C3 is shown as a green dashed line. (B) LmFH-2-H334A in a complex with S-malate. The H334A substitution is colored yellow. (C) LmFH-2-R421A in a complex with S-malate. The R421A substitution is colored light blue. (D) LmFH-2-T467A in a complex with S-malate. The T467A substitution is colored light blue. (E) LmFH-2-R471A in a complex with S-malate. The R471A substitution is colored light blue. (F) LmFH-2-R173A in a complex with S-malate. The R173A substitution is colored magenta. The substrate S-malate is colored green. The [4Fe-4S] cluster is shown as orange (Fe) and yellow (S) spheres. The water molecules are shown as red spheres. The hydrogen bonds are shown as gray dashed lines.
Here we sought to determine how the substitutions of the residues shown above that affect enzyme activity affect the protein structure and S-malate binding. The crystal structures of all variants (LmFH-2-R173A, LmFH-2-H334A, LmFH-2-R421A, LmFH-2-T467A, and LmFH-2-R471A) were determined in a complex with S-malate (Table S4), except for the variant LmFH-2-D135A, which did not crystallize despite considerable effort. The LmFH-2 variant structures are highly similar to each other and also to the structure of wild type LmFH-2 in a complex with S-malate, with an RMSD between 0.1 and 0.4 Å for all of the Cα atoms.
LmFH-2 variant structures show that H334A, R421A, T467A, and R471A substitutions do not change the S-malate-binding mode in comparison with that of wild type LmFH-2 (Figure 2A–E and Figures S1 and S2). In contrast, the crystal structure of the variant LmFH-2-R173A reveals a conformational change in the S-malate-binding mode, where the C3-carboxylate of S-malate has swung back toward residue Q134 (Figure 2F and Figures S1 and S2). In this new conformation, the C3-carboxylate of S-malate has lost its interactions with R421 and now interacts with Q134, T467, and three water molecules (Figure 2F). With R173 replaced with alanine, there is now room for the conserved M538 residue to rearrange, which creates a solvent-exposed pocket with access to the active site (Figure S3). A molecule of the cryoprotectant glycerol is observed in this pocket, where it hydrogen bonds to conserved residues R421 and E539 and a water molecule (Figure S3C). We note that this pocket is distinct from the previously described substrate-access cavity that leads to the active site5 (Figure S3A). Given these structural changes, it is not surprising that the R173A substitution has a drastic effect on enzyme activity. Notably, however, Km values are unchanged (Table 1).
Overall, our kinetic and structural results indicate that residues H334, R421, T467, and R471 play a role in catalysis because their substitution to alanine does not change the mode of substrate binding but does substantially decrease kcat (Figure 2 and Table 1). The H334A variant is the most active of the variants studied. We expected the loss of the histidine side chain to affect the reactivity of D135 because these two residues interact with each other in the wild type enzyme, and thus, we expected the substitution to substantially impair enzyme activity. Our LmFH-2-H334A structure suggests that negative effects on catalysis by loss of the H334 side chain might be mitigated by a structural rearrangement caused by the amino acid substitution. In particular, the structure shows that in the absence of H334, residue D135 slightly changes conformation and now interacts with R173 (Figure 2B and Figure S2A). Thus, R173 might fulfill the role of activating D135 in the absence of H334.
Structures of LmFH-2 Variants Bound to Succinate and Fumarate Reveal Monodentate Binding to the Unique Iron of the [4Fe-4S] Cluster
We have tried unsuccessfully to determine a structure of wild type LmFH-2 with the substrate fumarate. However, the LmFH-2 variant R173A does crystallize with fumarate, providing the first structure of an LmFH enzyme with fumarate bound at 3.3 Å resolution (Table S4). Here, we have also obtained a structure of wild type LmFH-2 with the close fumarate analogue, succinate, at 2.35 Å resolution (Table S3). The structures of LmFH-2 with succinate and LmFH-2-R173A with fumarate are highly similar to each other and also to the structure of LmFH-2 with S-malate described above, with an RMSD between 0.17 and 0.4 Å for all of the Cα atoms.
Despite the similarity of succinate to fumarate (Scheme 2), we show here that succinate is a poor inhibitor of LmFH-2, with an IC50 of 34.2 ± 1.2 mM against S-malate and 33.7 ± 1.9 mM against fumarate (Figure 3E). The crystal structure of LmFH-2 in a complex with succinate (Figure 3A) reveals that succinate is bound in the active site to the unique iron of the [4Fe-4S] cluster via a carboxylate oxygen atom similar to the S-malate-binding mode (Figure 2A). A water molecule coordinates the [4Fe-4S] cluster, occupying a position similar to that of the C2-hydroxyl oxygen atom of S-malate (Figures 2A and 3A and Figure S4). Succinate interacts with the same residues that are involved in binding S-malate, except that D135 and G216 interact with the bound water molecule rather than with succinate (Figure 3A).
Scheme 2. Structures of Fumarate, S-Malate, Succinate, and Malonate.
Figure 3.

Structures of LmFH-2 variants bound to succinate or fumarate reveal coordination of the ligand to the unique iron of the [4Fe-4S] cluster. (A) Wild type LmFH-2 in a complex with succinate (cyan). The distances from the T467 OH group to the succinate C3 (3.2 Å) and the water (coordinated to the [4Fe-4S] cluster) to the succinate C2 (2.9 Å) are shown as green dashed lines. (B) LmFH-2-R173A in a complex with fumarate (light orange). R173A is colored magenta. The distance from the T467 OH group to the fumarate C3 (3.3 Å) is shown as a green dashed line. (C) Superposition of LmFH-2 with succinate (cyan) and LmFH-2-R173A with fumarate (light orange). (D) The left panel shows a 2Fo – Fc electron density map contoured at 1.5 RMSD (blue mesh) for succinate (cyan), the [4Fe-4S] cluster, and a water molecule. The right panel shows a 2Fo – Fc electron density map contoured at 1.5 RMSD (blue mesh) for fumarate (light orange) and the [4Fe-4S] cluster. The [4Fe-4S] cluster is shown as orange (Fe) and yellow (S) spheres. The water molecule is shown as red spheres. (E) Dose–response curve of the inhibition of LmFH-2 by succinate against S-malate (left) and fumarate (right). Error bars represent three independent measurements.
The crystal structure of fumarate-bound LmFH-2-R173A shows that fumarate binds the catalytic [4Fe-4S] cluster via a carboxylate oxygen atom similar to the succinate-binding mode (Figure 3B–D). However, due to the R173A substitution, the molecule of fumarate is shifted closer to the position formerly occupied by R173 (Figure 3C). This structure suggests that residue R173 is key for the proper positioning of fumarate in the active site, although not essential for fumarate’s binding affinity. A water molecule was not observed bound to the [4Fe-4S] cluster in the structure of the LmFH-2-R173A–fumarate complex (Figure 3D), likely due to the modest resolution.
Taken together, our S-malate-, fumarate-, and succinate-bound structures indicate that both substrates and inhibitor bind similarly in the active site.
Malonate Binds to the FH Active Site but Not to the Fe–S Cluster
We have previously shown that malonate is a weak inhibitor of LmFH-2, with IC50 values of 9.8 ± 0.3 mM against S-malate and 5.6 ± 0.3 mM against fumarate.5 We also showed previously by crystallography that in the presence of both S-malate and malonate, S-malate binds in the active site and malonate binds out of the active site, both at the dimer interface and on the top face of the protein.5
To investigate if malonate can also occupy the substrate-binding site, we crystallized wild type LmFH-2 only in the presence of malonate. The crystal structure of LmFH-2 with malonate was determined at 1.95 Å resolution (Table S3) and reveals that the inhibitor binds in the active site, interacting with Q134, R173, R421, T467, T468, and R471, many of which are important catalytic residues (see Table 1), and two water molecules (Figure 4A). The observation that malonate binds to the active site and interacts with catalytic residues suggests that its mode of inhibition is not due to its binding to the previously identified remote sites on the protein5 but rather to active site interactions.
Figure 4.

Malonate binds to the active site of LmFH-2 but not to the Fe–S cluster. (A) LmFH-2 in a complex with malonate (salmon). This structure contains an inactive [3Fe-4S] cluster. (B) Snapshot of the Fe–S cluster destruction by oxidation. LmFH-2 with malonate was exposed to molecular oxygen in the anaerobic chamber, which oxidizes the [4Fe-4S]2+ cluster to [3Fe-4S]+ and releases Fe (orange sphere; labeled in white). The 2Fo – Fc electron density map (blue mesh) is contoured at 1.5 RMSD for malonate (salmon), [3Fe-4S] cluster, and Fe. (C) EPR spectrum of oxygen-exposed LmFH-2 showing a signal characteristic of a [3Fe-4S]+ cluster with a g value of 2.02. (D) LmFH-2-T467A in a complex with malonate (salmon). This structure contains an active [4Fe-4S] cluster. The T467A substitution is colored light blue. (E) The 2Fo – Fc electron density map contoured at 1.5 RMSD (blue mesh) for malonate (salmon), [4Fe-4S] cluster, water molecule, and T467A substitution (light blue). (F) Superposition of LmFH-2-T467A in a complex with malonate (salmon) and LmFH-2 in a complex with succinate (cyan). The [3Fe-4S] cluster and [4Fe-4S] cluster are shown as orange (Fe) and yellow (S) spheres. The water molecules are shown as red spheres. The hydrogen bonds are shown as gray dashed lines.
Notably, this structure contains a [3Fe-4S] cluster instead of the catalytic [4Fe-4S] cluster, presumably due to a temporary oxygen contamination of the anaerobic chamber (∼180 ppm) resulting in cluster oxidation and the conversion of the catalytic [4Fe-4S]2+ cluster to a [3Fe-4S]+ cluster with the release of iron (Figure 4B). Interestingly, the loss of an iron ion from the cluster is accompanied by the appearance of a nearby electron density peak that is consistent with the size, shape, and scattering of an iron ion. An iron ion at half-occupancy refines well into the electron density, yielding a B-factor that is the same as the average B-factor for the protein molecule [27 Å2 (see Table S3)] and no positive or negative difference density. This putative iron ion-binding site is found in the substrate-access cavity adjacent to conserved residues N219, T468, and R471 (Figure 4B).
In agreement with our structure, the EPR spectrum of LmFH-2 under aerobic conditions shows a signal characteristic of a [3Fe-4S]+ cluster with a g value of 2.0225 (Figure 4C). Thus, the inactivity of LmFHs under aerobic conditions9 is likely due to the presence of the inactive [3Fe-4S] cluster that lacks the catalytic iron atom required for substrate binding. We found no evidence of further cluster degradation to a [2Fe-2S] state.
We have also obtained a malonate-bound LmFH-2 structure that shows an intact [4Fe-4S] cluster, allowing us to evaluate whether malonate coordinates the cluster when the unique Fe is present. This 1.95 Å resolution crystal structure of an LmFH-2-T467A variant, determined in the presence of S-malate, malonate, and succinate (Table S4), displays malonate bound in the active site (Figure 4D,E) in a fashion identical to that of LmFH-2–malonate complex (Figure 4A), but with the [4Fe-4S] cluster intact. With the caveat that this structure is of the LmFH-2 variant, it supports a binding mode for malonate that does not involve the [4Fe-4S] cluster.
Interestingly, we find a water molecule bound to the unique iron and interacting with D135 in the structure of the malonate-bound LmFH-2-T467A variant (Figure 4D,E), similar to what was observed in the structure of LmFH-2 with succinate (Figure 4F). We believe that this water-binding site is catalytically relevant, representing the position from which water reacts with fumarate to generate S-malate. The appearance of water in the absence of fumarate indicates that substrate-binding events are independent. The most surprising finding from this structure of LmFH-2-T467A is that malonate was observed in the active site when this enzyme variant was incubated with both S-malate and succinate in addition to the malonate. As mentioned above, with wild type protein, S-malate is preferentially bound to the active site over malonate when both are present in the crystallization buffer.
C-Terminal Domain Mobility May Regulate Active Site Access
In the LmFH-2 structure with S-malate, a second molecule of S-malate was found at the entrance of the cavity that leads to the active site, located between the N- and C-terminal domains. The C-terminal domain, whose fold was described previously as a “swivel domain”, has been proposed to regulate access to the active site through its mobility.5 To investigate whether the C-terminal domain changes positions in the absence of substrates and inhibitors, we determined the crystal structure of substrate- and inhibitor-free LmFH-2 (LmFH-2-holo).
The LmFH-2-holo structure was determined at 3.2 Å resolution (Table S3) and contains four copies of the functional homodimeric enzyme in the asymmetric unit. The superposition of Cα atoms between LmFH-2 with S-malate and each LmFH-2-holo dimer shows major structural differences, with maximum RMSDs between 2.91 and 4.39 Å (Figure 5C and Figure S5). Excitingly, these structural differences are observed mainly in the C-terminal domain, whereas the N-terminal domain remains stationary (Figure 5). This structure reveals four different conformations of the C-terminal domain showing a swiveling movement that opens and closes the cavity to the active site. Thus, C-terminal domain flexibility may regulate the entry of the substrate into the active site.
Figure 5.

Conformationally flexible LmFH-2 C-terminal domain. (A) Two views of the superposition of LmFH-2-holo dimers (chains A and B, C and D, E and F, and G and H) showing the C-terminal domain mobility. The N-terminal domain is colored light blue, and the C-terminal domain of each dimer is colored light pink (chains A and B), hot pink (chains C and D), purple (chains E and F), and violet (chains G and H). The [4Fe-4S] cluster is shown as orange (Fe) and yellow (S) spheres. (B) Another view of the LmFH-2-holo C-terminal domains showing its swiveling movement. (C) Table of RMSD values between LmFH-2 with the S-malate dimer and each LmFH-2-holo dimer. The Cα RMSD values were calculated using the ColorByRMSD PyMol script.
Discussion
Class I FHs are central metabolic enzymes that contain an oxygen-sensitive [4Fe-4S] cluster required for the reversible dehydration/hydration of S-malate to fumarate. Recently, we determined the crystal structures of mitochondrial and cytosolic isoforms of class I FHs in L. major, a parasite responsible for the neglected tropical disease leishmaniasis.5,14 These structures revealed that mitochondrial LmFH-1 and cytosolic LmFH-2 have very similar homodimeric structures with each monomer composed of two domains, an N-terminal domain and a C-terminal domain, arranged around the catalytic [4Fe-4S] cluster.5,14 A co-crystal structure of LmFH-2 revealed substrate S-malate bound to the unique iron of the [4Fe-4S] cluster as well as conserved active site residues that play a key role in catalysis.5 In this study, we present mutagenesis, biochemical, and structural characterization of LmFH-2 and propose a catalytic mechanism for class I FHs.
Enzymatic kinetics of LmFH isoforms reveal a preference for fumarate as a substrate as indicated by a 7–10-fold higher catalytic efficiency. A comparison between LmFH activity in aerobic9 and anaerobic (this study) conditions shows the inactivation of both enzymes by oxygen. Our LmFH-2 structures with the inhibitor malonate and EPR analysis confirmed that oxygen inactivation is due to the oxidation of the active [4Fe-4S] cluster to the inactive [3Fe-4S] cluster, which loses the iron atom required to bind the substrate. Together, these results indicate that the [4Fe-4S] cluster is essential for LmFHs activity, as observed for other members of the class I FHs.25,26
Class I FHs are proposed to catalyze the stereospecific reversible dehydration/hydration of S-malate to fumarate by an acid–base catalytic mechanism, with the [4Fe-4S] cluster acting as a Lewis acid25,27 (Figure 6). To catalyze the dehydration of S-malate to fumarate, the first step is thought to be deprotonation at C3 of S-malate. The second step is protonation at the C2-hydroxyl group of S-malate for anti-elimination as water and subsequent formation of fumarate.
Figure 6.

Proposed catalytic mechanism for class I FHs. In the dehydration of S-malate to fumarate, first, a proton is abstracted from S-malate C3 by T467 that is activated by either of the two arginine residues (R421 and R471), which accept the proton. In the second step, D135 donates a proton to the S-malate C2-hydroxyl group for elimination as water and subsequent formation of fumarate. The negative charge of D135 is stabilized by H334. In the hydration of fumarate to S-malate, D135 abstracts a proton from the water molecule bound to the [4Fe-4S] cluster to form a hydroxyl group for addition to C2 of fumarate, and then T467 and either of its partners R421 and R471 donate a proton to C3 of fumarate for subsequent formation of S-malate. The [4Fe-4S] cluster acts as a Lewis acid to activate the hydroxyl group from S-malate for elimination or water for addition.
The crystal structure of LmFH-2 in a complex with S-malate tells us about the substrate-bound state for the forward reaction, and the complex with succinate informs on the product-bound state because succinate is a mimic for fumarate. These structures reveal that T467 is the closest residue to C3 of S-malate, with a distance from the T467 hydroxyl group to the substrate C3 of 3.2 Å (Figures 2A and 3A). In addition, this T467 hydroxyl group is close to residues R421 and R471 (2.5–3.0 Å), either of which could act as a proton acceptor from C3 of S-malate via the T467 side chain (Figure 6). Although the involvement of arginine residues in acid/base chemistry is unusual given its high pKa, there are a number of examples in the literature for arginine acting in this role (reviewed in ref (28)). Furthermore, we would expect the pKa of the C3–H bond to be decreased, and thus deprotonation facilitated, by the interaction of S-malate with the cluster. For the second step of the forward reaction, the protonation at the C2-hydroxyl group of S-malate, D135 is the closest residue. Importantly, D135 is found to interact with H334 in the active site. We expect that D135 and H334 serve as a catalytic dyad, acting as a catalytic acid to protonate the C2-hydroxyl group of S-malate, facilitating its loss as water (Figure 6). The [4Fe-4S] cluster serves to position the hydroxyl group next to D135 and may also facilitate its release as water.
In the reverse direction of the reaction, the hydration of fumarate to S-malate, the first step is deprotonation of the active site water to form a hydroxyl group for anti-addition to C2 of fumarate, followed by protonation at the fumarate C3 and subsequent formation of S-malate (Figure 6). Using our succinate-bound wild type LmFH-2 structure (Figure 3A) and fumarate-bound LmFH-2-R173A structure (Figure 3B) as models for the fumarate-bound wild type state of LmFH-2, we predict that fumarate will bind to the unique iron of the cluster in the wild type enzyme using one oxygen atom, leaving an open coordination site on that iron for a water molecule. Coordination of this water molecule to the cluster positions the water molecule perfectly for interaction with fumarate C2 at a distance of approximately 2.9 Å. The water molecule is also in the proximity of the D135-H334 dyad (2.5 Å), which can act as a catalytic base to activate the water for addition to the C2 position of the substrate. Finally, as described above, the residue closest to C3 of the substrate is T467, which could provide a proton via R421 or R471 (Figure 6).
Notably, residue substitutions, LmFH-2-T467A, LmFH-2-R421A, LmFH-2-R471A, LmFH-2-D135A, and LmFH-2-H334A, drastically affect catalytic activity (Figure 1E) but do not significantly affect the S-malate-binding mode (Figure 2). These findings are consistent with the proposed catalytic roles put forth in Figure 6, in which a triad of T467, R421, and R471 and a dyad of D135 and H334 function as catalytic acids or bases, depending on the direction of the reaction. Remarkably, our structures show that the water molecule, D135, and [4Fe-4S] cluster are on the opposite side of the substrate compared to the T467 residue, which is consistent with an anti-addition/elimination of water in class I FHs.27 The class II FH is proposed to catalyze the same reactions without iron and using S318 and a dyad of H188 and E331 (E. coli fumarase C numbering) as acid–base catalytic amino acids.29,30
Our structure of the LmFH-2 variant T467A also indicates that T467 is important for substrate specificity. Whereas other LmFH-2 variants studied preferentially bind substrate S-malate over malonate, LmFH-2-T467A appears to prefer malonate as indicated by the crystal structure. Interestingly, malonate does not coordinate the cluster in contrast to the inhibitors succinate (this work) and 2-thiomalate.14 These observations indicate that inhibitors can bind to LmFH in multiple ways to afford inactivation, albeit some of the inhibition measured is quite modest.
Finally, the first structure of a substrate-free and inhibitor-free class I FH has provided insight into how substrates or inhibitors access the active site and how products leave. The holo LmFH-2 structure reveals C-terminal domain conformational changes that appear to open a path to the active site that lies between the C-terminal and N-terminal domains. A comparison between ligand-free and ligand-bound structures shows that a ligand in the active site limits the conformational flexibility of the C-terminal domain. We suspect that it is advantageous to have the [4Fe-4S] cluster tucked away between domains for protection but that this protection necessitates an access channel. Movement of the C-terminal domain appears to provide that channel.
Conclusions
In summary, this work provides insights into the mechanism of action of class I FHs that may consist of movement of the C-terminal domain to open the active site cavity allowing substrate entrance, followed by the C-terminal domain closing to perform the reaction and then opening to release the product. Our results show that class I FHs require an active [4Fe-4S] cluster and two sets of acid–base residues, located on opposite sides from one another, to catalyze the anti-addition/elimination of water. The differences between class I parasitic FH and class II human FH catalytic mechanisms provide valuable knowledge for structure-based drug design aiming to fight parasitic neglected tropical diseases.
Acknowledgments
The authors thank Professor Antonio J. Costa-Filho from the University of São Paulo, Ribeirão Preto, São Paulo, Brazil, for assistance with the collection of the EPR spectrum of LmFH-2.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.9b00923.
Primers used for site-directed mutagenesis of LmFH-2 (Table S1), tacsimate composition and its variations to obtain structures with particular ligands bound (Table S2), data collection and refinement statistics of LmFH-2 (Table S3), data collection and refinement statistics of LmFH-2 variants (Table S4), numbers of residues visualized in each LmFH-2 structure (Table S5), electron density maps of LmFH-2 and variants with S-malate (Figure S1), stereoviews of LmFH-2 and variants in a complex with S-malate (Figure S2), LmFH-2-R173A structure revealing a solvent-exposed pocket that is not observed in wild type LmFH-2 (Figure S3), stereoview of the superposition of LmFH-2 active sites in a complex with S-malate and succinate (Figure S4), and LmFH-2-holo dimers colored by RMSD (Figure S5) (PDF)
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank as entries 6UNZ, 6UO0, 6UOI, 6UOJ, 6UP9, 6UPM, 6UPO, 6UQ8, 6UQ9, 6UQB, 6UQL, 6UQM, and 6UQN. LmFH-1, UniProtKB entry Q4QAU9. LmFH-2, UniProtKB entry E9AE57.
This work was supported by Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) Grant 2014/22246-4 (P.R.F.) and the National Institute of General Medical Sciences of the National Institutes of Health via Grant R35 GM126982 (C.L.D.). C.L.D. is a Howard Hughes Medical Institute Investigator. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences of the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on the 24-ID-C beamline is funded by an NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
The authors declare no competing financial interest.
Supplementary Material
References
- Chen B. S.; Otten L. G.; Hanefeld U. (2015) Stereochemistry of enzymatic water addition to C = C bonds. Biotechnol. Adv. 33, 526–546. 10.1016/j.biotechadv.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Coustou V.; Besteiro S.; Riviere L.; Biran M.; Biteau N.; Franconi J. M.; Boshart M.; Baltz T.; Bringaud F. (2005) A mitochondrial NADH-dependent fumarate reductase involved in the production of succinate excreted by procyclic Trypanosoma brucei. J. Biol. Chem. 280, 16559–16570. 10.1074/jbc.M500343200. [DOI] [PubMed] [Google Scholar]
- Yogev O.; Yogev O.; Singer E.; Shaulian E.; Goldberg M.; Fox T. D.; Pines O. (2010) Fumarase: A Mitochondrial Metabolic Enzyme and a Cytosolic/Nuclear Component of the DNA Damage Response. PLoS Biol. 8, e1000328 10.1371/journal.pbio.1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustou V.; Biran M.; Besteiro S.; Riviere L.; Baltz T.; Franconi J. M.; Bringaud F. (2006) Fumarate is an essential intermediary metabolite produced by the procyclic Trypanosoma brucei. J. Biol. Chem. 281, 26832–26846. 10.1074/jbc.M601377200. [DOI] [PubMed] [Google Scholar]
- Feliciano P. R.; Drennan C. L.; Nonato M. C. (2016) Crystal structure of an Fe-S cluster-containing fumarate hydratase enzyme from Leishmania major reveals a unique protein fold. Proc. Natl. Acad. Sci. U. S. A. 113, 9804–9809. 10.1073/pnas.1605031113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver T. M.; Levitt D. G.; Donnelly M. I.; Stevens P. P. W.; Banaszak L. J. (1995) Themultisubunitactive-site of fumarase C from Escherichiacoli. Nat. Struct. Biol. 2, 654–662. 10.1038/nsb0895-654. [DOI] [PubMed] [Google Scholar]
- van Vugt-Lussenburg B. M. A.; van der Weel L.; Hagen W. R.; Hagedoorn P. L. (2009) Identification of two 4Fe-4S cluster-containing hydro-lyases from Pyrococcus furiosus. Microbiology 155, 3015–3020. 10.1099/mic.0.030320-0. [DOI] [PubMed] [Google Scholar]
- van Vugt-Lussenburg B. M. A.; van der Weel L.; Hagen W. R.; Hagedoorn P.-L. (2013) Biochemical Similarities and Differences between the Catalytic 4Fe-4S Cluster Containing Fumarases FumA and FumB from Escherichiacoli. PLoS One 8, e55549 10.1371/journal.pone.0055549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano P. R.; Gupta S.; Dyszy F.; Dias-Baruffi M.; Costa-Filho A. J.; Michels P. A. M.; Nonato M. C. (2012) Fumarate hydratase isoforms of Leishmaniamajor: Subcellular localization, structural and kinetic properties. Int. J. Biol. Macromol. 51, 25–31. 10.1016/j.ijbiomac.2012.04.025. [DOI] [PubMed] [Google Scholar]
- Jayaraman V.; Suryavanshi A.; Kalale P.; Kunala J.; Balaram H. (2018) Biochemical characterization and essentiality of Plasmodium fumarate hydratase. J. Biol. Chem. 293, 5878–5894. 10.1074/jbc.M117.816298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Padua R. A.P.; Kia A. M.; Costa-Filho A. J.; Wilkinson S. R.; Nonato M. C. (2017) Characterisation of the fumarate hydratase repertoire in Trypanosoma cruzi. Int. J. Biol. Macromol. 102, 42–51. 10.1016/j.ijbiomac.2017.03.099. [DOI] [PubMed] [Google Scholar]
- Ajalla Aleixo M. A.; Rangel V. L.; Rustiguel J. K.; de Padua R. A. P.; Nonato M. C. (2019) Structural, biochemical and biophysical characterization of recombinant human fumarate hydratase. FEBS J. 286, 1925–1940. 10.1111/febs.14782. [DOI] [PubMed] [Google Scholar]
- Cordeiro A. T.; Feliciano P. R.; Pinheiro M. P.; Nonato M. C. (2012) Crystal structure of dihydroorotate dehydrogenase from Leishmania major. Biochimie 94, 1739–1748. 10.1016/j.biochi.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Feliciano P. R.; Drennan C. L.; Nonato M. C. (2019) Crystal Structures of Fumarate Hydratases from Leishmania major in a Complex with Inhibitor 2-Thiomalate. ACS Chem. Biol. 14, 266–275. 10.1021/acschembio.8b00972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papworth C.; Bauer J. C.; Braman J.; Wright D. A. (1996) Site-Directed Mutagenesis in One day with >80% Efficiency. Strategies 9, 3. 10.1080/08924562.1996.11000299. [DOI] [Google Scholar]
- Nakamaru-Ogiso E.; Yano T.; Ohnishi T.; Yagi T. (2002) Characterization of the iron-sulfur cluster coordinated by a cysteine cluster motif (CXXCXXXCX27C) in the Nqo3 subunit in the proton-translocating NADH-quinoneoxidoreductase (NDH-1) of Thermusthermophilus HB-8. J. Biol. Chem. 277, 1680–1688. 10.1074/jbc.M108796200. [DOI] [PubMed] [Google Scholar]
- McPherson A.; Cudney B. (2006) Searching for silver bullets: An alternative strategy for crystallizing macromolecules. J. Struct. Biol. 156, 387–406. 10.1016/j.jsb.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Luft J. R.; DeTitta G. T. (1999) A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 55, 988–993. 10.1107/S0907444999002085. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 2.2.3.
- Morin A.; Eisenbraun B.; Key J.; Sanschagrin P. C.; Timony M. A.; Ottaviano M.; Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint D. H.; Emptage M. H.; Guest J. R. (1992) Fumarase A from Escherichia coli: purification and characterization as an iron-sulfur cluster containing enzyme. Biochemistry 31, 10331–10337. 10.1021/bi00157a022. [DOI] [PubMed] [Google Scholar]
- Van Kuijk B. L. M.; Van Loo N. D.; Arendsen A. F.; Hagen W. R.; Stams A. J. M. (1996) Purification and characterization of fumarase from the syntrophic propionate-oxidizing bacterium strain MPOB. Arch. Microbiol. 165, 126–131. 10.1007/s002030050307. [DOI] [PubMed] [Google Scholar]
- Flint D. H. (1994) Initial kinetic and mechanistic characterization of Escherichia coli fumarase A. Arch. Biochem. Biophys. 311, 509–516. 10.1006/abbi.1994.1269. [DOI] [PubMed] [Google Scholar]
- Guillén Schlippe Y. V.; Hedstrom L. (2005) A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch. Biochem. Biophys. 433, 266–278. 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Weaver T.; Banaszak L. (1996) Crystallographic studies of the catalytic and a second site in fumarase C from Escherichiacoli. Biochemistry 35, 13955–13965. 10.1021/bi9614702. [DOI] [PubMed] [Google Scholar]
- Mechaly A. E.; Haouz A.; Miras I.; Barilone N.; Weber P.; Shepard W.; Alzari P. M.; Bellinzoni M. (2012) Conformational changes upon ligand binding in the essential class II fumarase Rv1098c from Mycobacterium tuberculosis. FEBS Lett. 586, 1606–1611. 10.1016/j.febslet.2012.04.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.