

Summary
Many intracellular bacteria can establish chronic infection and persist in tissues within granulomas comprised of macrophages. Granuloma macrophages exhibit heterogeneous polarization states, or phenotypes, that may be functionally distinct. Here, we elucidate a host-pathogen interaction that controls granuloma macrophage polarization and long-term pathogen persistence during Salmonella Typhimurium (STm) infection. We show that STm persists within splenic granulomas that are densely populated by CD11b+CD11c+Ly6C+ macrophages. STm preferentially persists in granuloma macrophages reprogrammed to an M2 state, in part through the activity of the effector SteE, which contributes to the establishment of persistent infection. We demonstrate that tumor necrosis factor (TNF) signaling limits M2 granuloma macrophage polarization, thereby restricting STm persistence. TNF neutralization shifts granuloma macrophages toward an M2 state, increases bacterial persistence, and these effects are partially dependent on SteE activity. Thus, manipulating granuloma macrophage polarization represents a strategy for intracellular bacteria to overcome host restriction during persistent infection.
Keywords: granuloma, macrophage polarization, Salmonella, SteE, SPI-2, TNF, persistent infection, SarA, alternatively activated
Graphical Abstract
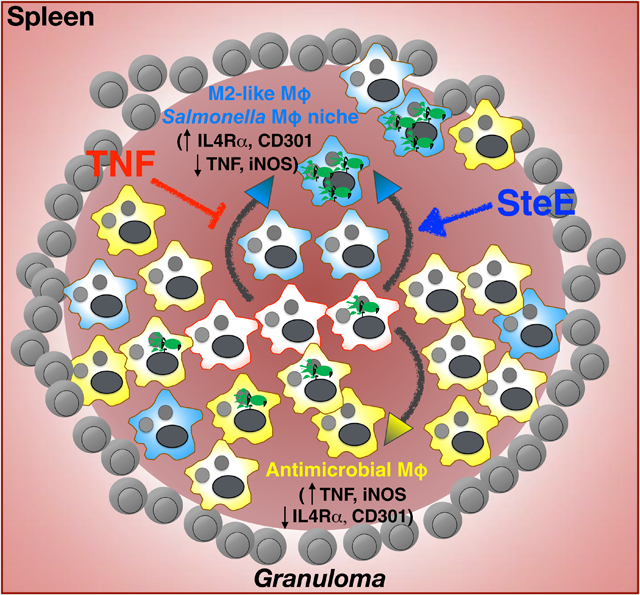
Pham et. al. show that granulomas in Salmonella-infected tissues are comprised of macrophages that exhibit heterogenous polarization states, or phenotypes, and are functionally distinct. They elucidate a pathogen-driven virulence mechanism that controls granuloma macrophage polarization and long-term pathogen persistence during Salmonella infection.
Introduction
Although invasive biopsy is not routinely performed, human histopathological studies suggest macrophage-rich granulomas are a common feature across intracellular bacteria-infected tissues, such as spleens and livers (Florin et al., 2008; Hunt and Bothwell, 1967; Mallory, 1898; Nasrallah and Nassar, 1978). Granuloma formation is thought to be an important immune response to contain infection, but also serves as a crucial mechanism for bacterial persistence (Petersen and Smith, 2013; Ramakrishnan, 2012). Recent studies showed that by residing within macrophages localized to granulomas, Salmonella Typhimurium (STm) can persist in infected tissues despite a robust Th1 response that acts to limit infection (Goldberg et al., 2018). Increasing evidence suggests macrophages of different phenotypes or polarization states exhibit differential functions (Goswami et al., 2017; Porta et al., 2015). During intracellular bacterial infections, infected macrophages typically exhibit an M1-like phenotype and restrict the pathogen through mechanisms such as pyroptosis and production of proinflammatory cytokines, including TNF (Storek and Monack, 2015). Emerging evidence suggests an M2-like phenotype is associated with intracellular bacterial growth and persistence (Eisele et al., 2013; Huang et al., 2018; Kerrinnes et al., 2017; Sahu et al., 2017; Stapels et al., 2018). Computational modeling studies have shown that the ratio of M1 vs. M2 activities within Mycobacterium tuberculosis (Mtb) granulomas can predict controlled infection or disseminated disease (Marino et al., 2015). However, the mechanisms controlling Salmonella granuloma macrophage phenotypes and functions that dictate infection outcome remain to be defined.
TNF signaling is critical for granuloma formation and pathogen restriction during intracellular bacterial infections, but the in vivo mechanisms of TNF are incompletely understood (Dorhoi and Kaufmann, 2014; Matty et al., 2015). Studies exploiting the lack of adaptive immunity in zebrafish embryos to define the role of innate immune cells in mycobacterial granulomas found that disruption of TNF signaling compromised pathogen containment due to increased granuloma macrophage cell death and diminished neutrophil recruitment (Bernut et al., 2016; Clay et al., 2008). The effects of TNF signaling on bacterial granulomas in mammalian hosts during persistent infections, when both innate and adaptive immune cells are operative, have proven to be more complex. Manipulation of TNF signaling in Mtb-infected mice and primates have produced varying impacts on bacterial burdens and immune cells, likely due to differences in the host’s ability to limit the pathogen and control persistent infection (Algood et al., 2004; Allie et al., 2013; Lin et al., 2010; Mohan et al., 2001; Plessner et al., 2007). In acute STm infection in mice, disruption of TNF signaling also impaired pathogen restriction and altered granuloma morphology but the mechanistic basis underpinning these phenotypes is not known (Mastroeni et al., 1995).
Intracellular bacteria utilize a number of mechanisms to compromise host immune responses and establish infection. These pathogens possess macromolecular secretion systems to inject effector proteins into the host cell cytoplasm that modulate immune cellular activities. For example, STm utilizes the SPI-2 T3SS (Salmonella Pathogenicity Island-2 Type-3 Secretion System) effector SteD to promote MHCII ubiquitination and suppress antigen presentation (Bayer-Santos et al., 2016). M. marinum and STm employ ESAT-6 and SseI, respectively, to influence leukocyte migration (McLaughlin et al., 2009; Volkman et al., 2010). More recently, Mtb ESAT-6 and STm SteE have been found to modulate primary macrophage polarization (Refai et al., 2018; Stapels et al., 2018). To establish persistent infection within granulomas, pathogens must overcome immune cellular mechanisms that function to restrict them. Yet little is known about the strategies that intracellular bacteria employ to persist chronically within these microstructures. Here we describe a host pathway that is required for containing STm and a pathogen virulence factor that antagonizes this host pathway by skewing macrophage polarization within granulomas of persistently infected mice. Our findings indicate that granuloma macrophage polarization is a critical intersection of host-pathogen interaction that controls persistent intracellular bacterial infection.
Results
Development and maintenance of CD11b+CD11c+ STm granulomas
The lack of genetically tractable mammalian models of persistent, controlled infection has been a significant limitation in defining the functions and mechanism of bacterial granulomas (Flynn et al., 2015). Our previous work demonstrated that STm-infected 129×1/SvJ mice developed granulomas in systemic tissues such as livers and spleens (Monack et al., 2004). Chronically infected mice recover from pathology such as splenomegaly and limit the pathogen to low levels for long periods of time. Thus, we utilized this tractable mammalian infection model to investigate the role of granulomas during persistent intracellular bacterial infections. We found that in mice orally infected with 108 CFU STm, splenic granulomas developed and morphologically evolved over the course of infection (Figure 1A). At 1 week post-inoculation (p.i.), granulomas contained cells with ample cytoplasm typical of mononuclear phagocytes, with neutrophilic infiltration. At 4 weeks p.i., granulomas became more confluent, with macrophages sometimes having elongated morphologies and infiltration of neutrophils. By 2–3 months p.i., granulomas contained cohesive aggregates of macrophages with peripheral discontinuous rims of lymphocytes and fewer admixed neutrophils. This granuloma development coincided with reduction of tissue bacterial burden to low chronic levels and diminishing CD4+ T cell IFNγ response, which has been shown to be critical for controlling STm infection (Goldberg et al., 2018) (Figure 1B–C, S1A). These data suggest a functional link between granuloma formation and establishment of persistent, controlled infection.
Figure 1: Development and maintenance of CD11b+CD11c+ STm granulomas.
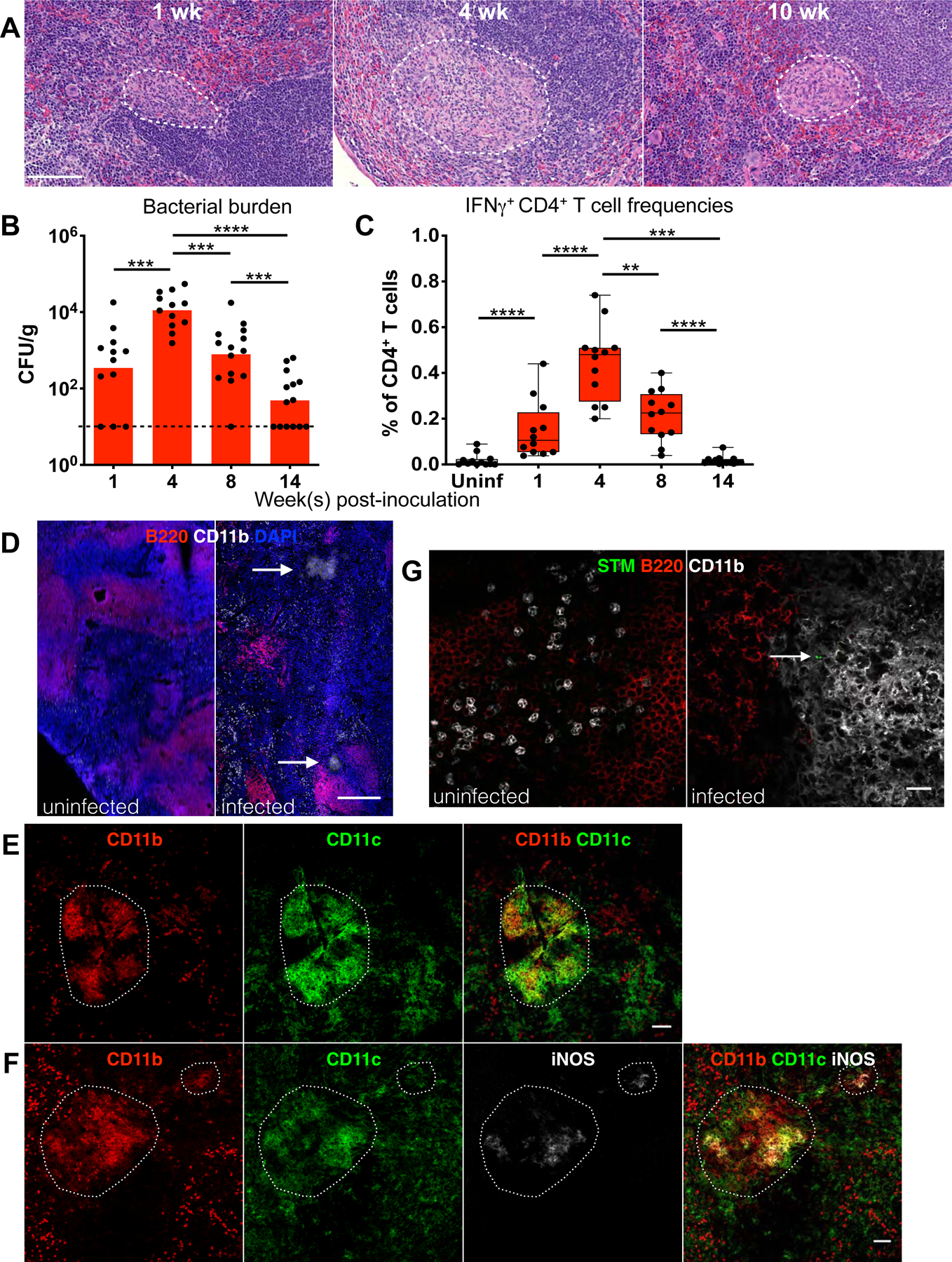
Mice orally infected with S. Typhimurium SL1344 and analyzed at indicated times p.i. (A) Hematoxylin and eosin (H&E) stains of infected spleens. White boundaries demarcate granulomas. Scale bar: 100 μm. (B) Quantitation of splenic bacterial burdens by colony forming units (CFU) using plating assays. Bars: geometric mean. Dots: individual mice. Dotted line: limit of detection. (C) Flow cytometric measurement of IFNγ-producing CD4+ T cells (gated as shown in figure S1) in infected vs uninfected (uninf.) spleens. Box and whisker plots. Horizontal lines indicate minimum, median, and maximum frequencies. Dots: individual mice. (D–E) Confocal microscopy identifying CD11b+CD11c+ granuloma microstructures in STm-infected spleens at 1–3 months p.i. White arrows and boundaries indicate granulomas. Scale bar: 200 μm (D), 50 μm (E). (F) A fraction of granuloma cells is iNOS+. Scale bar: 50 μm. (G) STm bacilli persist within CD11b+CD11c+ splenic granulomas at 4 weeks p.i. Scale bar: 20 μm. White arrow indicates STm bacilli. Significance calculated using a two-tailed Mann-Whitney test. ** p < 0.01, *** p < 0.001, *** p < 0.0001. A-C, data from 3 independent experiments, 3–5 mice per group per experiment. B-C show combined data. D-F, n > 10, multiple sections per mouse from at least 3 independent experiments.
We found that persistent STm granulomas are densely populated by CD11b+CD11c+ myeloid cells and positive for inducible nitric oxide synthase (iNOS), which mediates macrophage production of reactive nitrogen metabolites to restrict intracellular bacteria (Bekker et al., 2001) (Figure 1D–F). Within granulomas, there is a small number of admixed cells positive for Gr-1, a marker that stains mature neutrophils more intensely than other myeloid cells (Kim et al., 2017) (Figure S1B). T and B cells typically localize to the surrounding areas (Figure S1B). To determine if these CD11b+CD11c+ granulomas are a site of STm persistence in infected spleens (Goldberg et al., 2018), we stained for bacilli using a Salmonella-specific antibody and found rod-shaped STm within these microstructures (Figure 1G).
CD11b+CD11c+Ly6C+ macrophages populate STm granulomas
Classically, granulomas are thought to be enriched with macrophages (Pagan and Ramakrishnan, 2018). Thus, we hypothesized that the CD11b+CD11c+ cells within STm granulomas are a distinct macrophage population in the infected tissues and sought to functionally characterize them. Using flow cytometry, we found two major cell populations in the infected spleens that are CD11b+CD11c+: one positive for the macrophage/monocyte marker Ly6C and the other Ly6C negative (Figure 2A). The CD11b+CD11c+Ly6C+ population exhibited surface markers consistent with activated macrophages: CD8α−, MHCII high, CD64 high, F4/80+, CCR2 intermediate, and CD115 intermediate/high (Figure 2B). In contrast, the CD11b+CD11c+Ly6C− population displayed markers suggestive of splenic CD11b+ dendritic cells that had been described previously (Cohn and Delamarre, 2014). To test if the presence of the CD11b+CD11c+Ly6C+ activated macrophages in the infected spleens is induced by STm infection, we compared this population from uninfected and infected mice. In uninfected mice, ~0.02% (SD ± 0.007) of total splenocytes were comprised of CD11b+CD11c+Ly6C+ cells that had low F4/80, MHCII, and CD64 expression. STm infection increased the number of CD11b+CD11c+Ly6C+ cells to as much as 0.92% (± 0.52%) of total splenocytes and these cells had an activated macrophage phenotype as indicated by high levels of MHCII and CD64 (Figure 2C). Using confocal microscopy, we found that the CD11b+CD11c+ myeloid cells within STm granulomas are also Ly6C and CD64 positive (Figure 2D). Hereafter, this cell population—CD11b+, CD11c+, Ly6C- intermediate, F4/80+, CD64-high, MHCII-high—is referred to as granuloma macrophages; whereas, we refer to the CD11b+CD11c+Ly6C− population as CD11b+CD11c+ DC (dendritic cells) (see Figure S2A for gating scheme). Quantitation of granuloma macrophages in infected spleens over a 3-month infection showed this population increased as much as 200-fold by 4 weeks p.i., compared to uninfected spleens, before contracting and persisting (Figure 2E, S2B). Notably, this cell population expands markedly more than neutrophils and classical monocytes (Figure S2C–D), both of which have been found to have critical roles in the host immune response in persistent STm infection (Gopinath et al., 2013; Tam et al., 2014).
Figure 2: CD11b+CD11c+Ly6C+ macrophages populate STm granulomas.
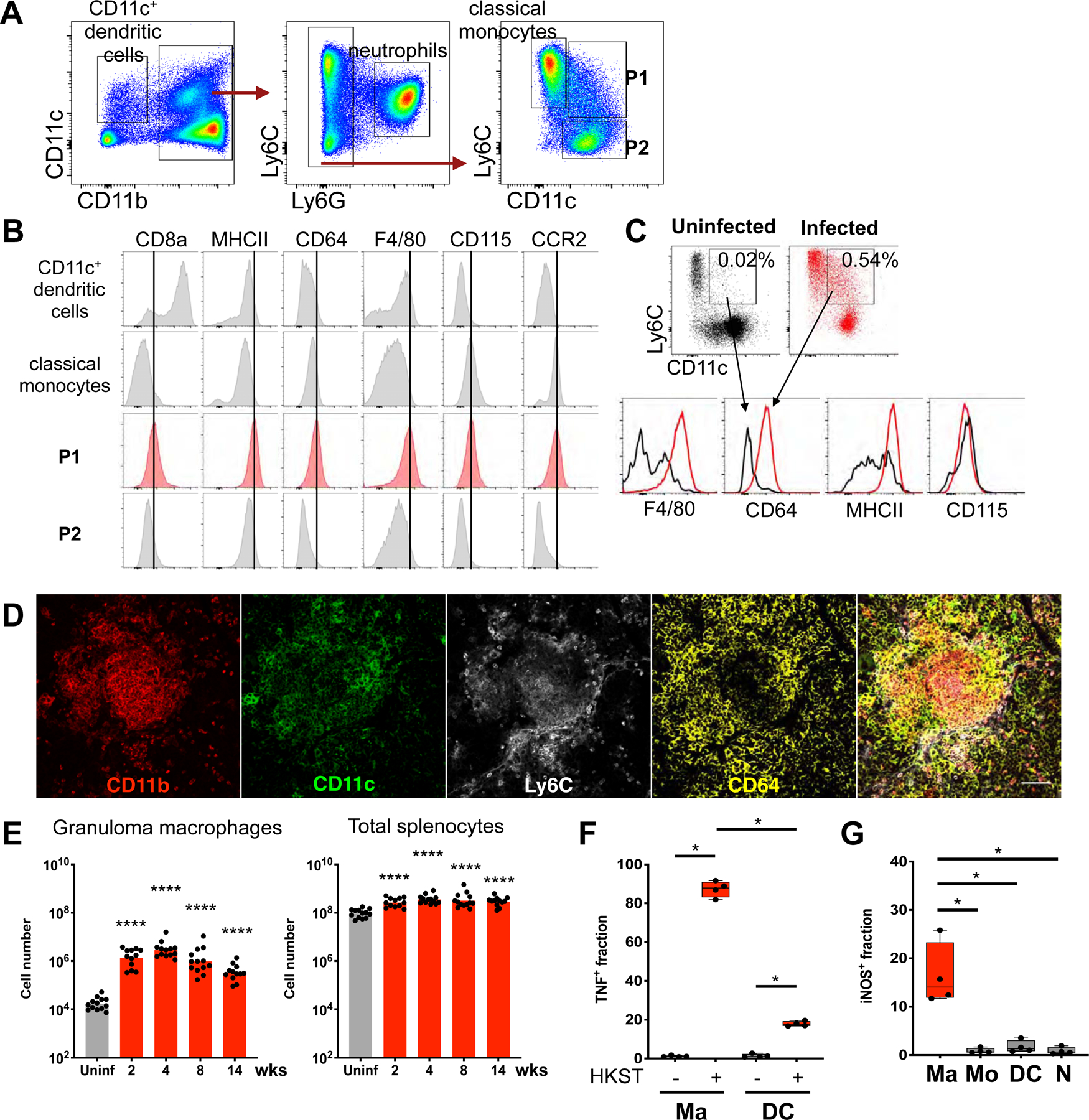
(A) Flow cytometric analysis of splenic CD11b+CD11c+ granuloma cells at 4 weeks p.i. Splenocytes gated for live, singlet, CD19−CD3−NK1.1− population, then further gated as shown. (B) Histograms illustrating expression of phenotypic and functional markers of myeloid cell populations as indicated in (A). (C) Splenocytes gated as in (A). Dot plots and histograms showing frequencies and phenotype of CD11b+CD11c+Ly6C+ cells in uninfected vs. infected spleens. Percentages indicate frequencies of gated cells among total splenocytes. (D) Visualization of STm granuloma macrophage phenotype by confocal microscopy. Scale bar: 50 μm (E) Quantitation of splenic granuloma macrophages (gated as shown in figure S2A) and total splenocytes at indicated times p.i. Bars: geometric means. Dots: individual mice. Significance derived from comparing infected animals at each time point to uninfected animals. (F) Intracellular TNF staining of splenocytes from infected animals at 4 weeks p.i. after stimulation ex vivo with medium or heat-killed S. Typhimurium (HKST). (G) Quantitation of endogenous iNOS+ splenocytes from infected animals. Ma: granuloma macrophages; N: neutrophils; Mo: classical monocytes; DC: CD11b+CD11c+ DC as shown in figure S2A. Significance calculated using a two-tailed Mann-Whitney test. * p < 0.05, **** p < 0.0001. A-C and E-H, data from at least 3 independent experiments 4–5 mice per groups per experiment. E-F show combined data. D, n > 5, multiple sections per mouse.
To further interrogate the importance of splenic granuloma macrophages, we characterized their functional features. TNF and iNOS productions are hallmarks of macrophage antimicrobial response during intracellular bacterial infections (Bekker et al., 2001; Dorhoi and Kaufmann, 2014). Furthermore, TNF signaling is critical for bacterial restriction and protective immunity during STm infection (Mastroeni et al., 1995; Tite et al., 1991). Thus, we examined granuloma macrophages for their capacity to produce TNF and iNOS. Splenocytes from infected spleens were stimulated with heat-killed STm (HKST) in the presence of brefeldin A, a protein transport inhibitor, for 3 hours ex vivo, followed by intracellular TNF staining. We found that most granuloma macrophages robustly produced TNF (Figure 2F). In contrast, the fraction of CD11b+CD11c+ DC producing TNF was significantly lower. In addition, flow cytometric measurement of endogenous iNOS showed that the frequencies of iNOS+ granuloma macrophages were typically 10 to 50-fold higher than other major myeloid cell populations in the infected spleens (Figure 2G).
Phenotypic and functional heterogeneity of STm granuloma macrophages
Macrophages mediate critical antimicrobial immune responses to control intracellular bacterial pathogens. Yet intracellular bacteria, including STm, can exploit macrophages as a cellular niche for persistence. We hypothesized that granuloma macrophages have differential phenotypes, or polarization states, that are functionally distinct to control persistent infection within the granuloma microenvironment. To interrogate this, we stained splenic STm granulomas for the alternatively activated, M2 macrophage marker CD301. We previously found that CD301 expression on CD11b+F4/80+ mononuclear phagocytes was associated with a metabolic state that facilitated STm persistence (Eisele et al., 2013). Imaging by confocal microscopy revealed that a small fraction of granuloma macrophages was CD301+ and frequently encircled the granuloma periphery (Figure 3A). Using flow cytometry, we determined that approximately 13.5% (± 4.3%) of granuloma macrophages were also CD301+ by 4 weeks p.i. (Figure 3B–C, S3A). Functionally, a significantly lower fraction of CD301+ granuloma macrophages produced TNF upon stimulation with HKST, compared to CD301− cells (Figure 3D). Furthermore, the fraction of CD301− granuloma macrophages that expressed iNOS was typically 2 to 5-fold higher than CD301+ cells (Figure 3E). Thus, among granuloma macrophages, CD301+ cells are less likely to exhibit antimicrobial functions.
Figure 3: Phenotypic and functional heterogeneity of STm granuloma macrophages.
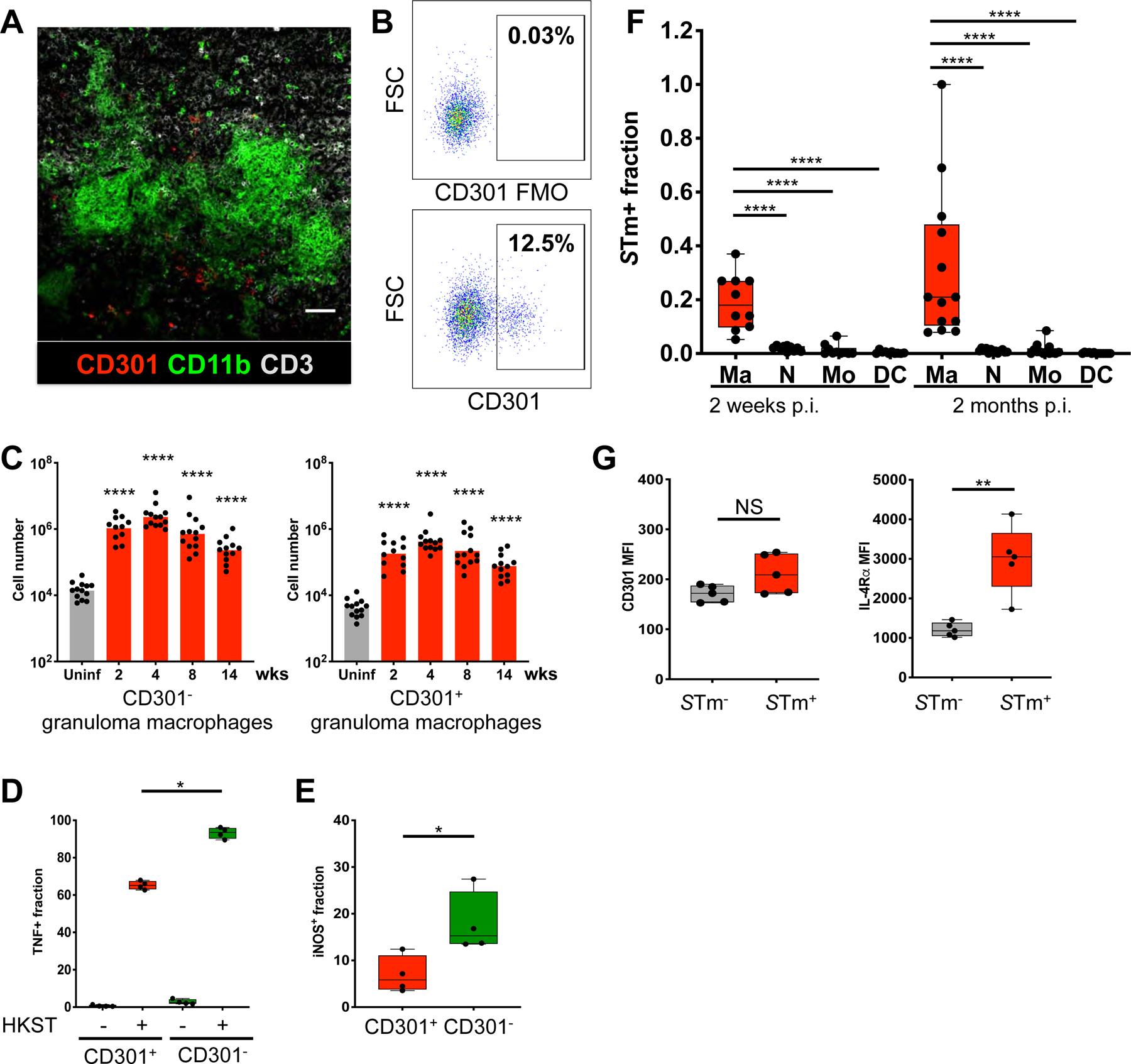
(A) Confocal microscopy showing cells expressing the alternative macrophage marker CD301 within STm granulomas. (B) Granuloma macrophages gated as in figure S2A then examined for CD301 expression as shown. Percentages indicate frequencies of gated cells among granuloma macrophages. FMO: fluorescence-minus-one control. Samples from 4 week-infected mice shown. (C) Quantitation of splenic CD301+ and CD301− granuloma macrophages at indicated times p.i. Significance derived from comparing infected to uninfected animals. (D) TNF response to HKST ex vivo of CD301− and CD301+ granuloma macrophages from infected animals. (E) Quantitation of iNOS+ cells among CD301− and CD301+ granuloma macrophages from infected animals by flow cytometry. (D-E) Samples from 4 week-infected mice shown. (F) STm+ fractions among splenic myeloid cell populations from orally infected mice in early and chronic, persistent infections. Ma: granuloma macrophages; N: neutrophils; Mo: classical monocytes; DC: CD11b+CD11c+ DC as shown in figure S2A. (G) Mean fluorescence intensity (MFI) of CD301 and IL-4Rα on granuloma macrophages from mice infected with STm intraperitoneally (i.p.) at 2 weeks post-infection. Significance calculated using a two-tailed Mann-Whitney test. * p < 0.05, ** p < 0.01, **** p <0.0001. A, n > 10, multiple sections per mouse. B-G, data from at least 3 independent experiments, 4–6 mice per group per experiment. C and F show combined data.
Next, we investigated if granuloma macrophages have differential potential to harbor STm. Mice were infected with 108 CFU STm orally or 103 CFU intraperitoneally (i.p.), which yielded a higher splenic CFU in the early stage of infection. Splenocytes analyzed by flow cytometry were first gated for live, singlet, CD19−CD3−NK1.1− population, followed by gating for granuloma macrophages and other major myeloid cell populations as shown in figure S2A. Gated myeloid cell populations were then plotted for intracellular STm staining using an anti-STm antibody (S3B-C). We found that the fraction of granuloma macrophages harboring intracellular STm was markedly higher than other myeloid populations (Figure 3F). By 8 weeks p.i. the STm+ macrophage fraction was 20 to 30-fold higher than that of neutrophils and classical monocytes, similar to our prior confocal microscopy analysis in which more than 80% of the bacteria in chronically infected mesenteric lymph nodes were localized within cells expressing the macrophage marker MOMA-2 (Monack et al., 2004). Although a recent report found a splenic CD11b+CD11c+MHCII+iNOS+Ly6C− macrophage population that harbors STm bacilli, those cells have variable Ly6C expression and only account for a small fraction of STm+ CD64+ cells in the infected spleens (Goldberg et al., 2018).
IL-4Rα expression is a canonical functional signature of M2-like macrophages. Functional and transcriptional analyses suggest the IL-4 signaling capacity of bone-marrow derived macrophages (BMDMs) and CD11b+F4/80+ mononuclear phagocytes is highly associated with bacterial intracellular growth and persistence (Eisele et al., 2013; Saliba et al., 2016; Stapels et al., 2018). To determine if granuloma macrophage CD301 and IL-4Rα levels are associated with a higher potential to harbor STm, we compared their levels on granuloma macrophages with or without intracellular bacilli. We found that granuloma macrophages containing intracellular STm expressed higher IL-4Rα levels but not CD301 (Figure 3G). Thus, within the splenic granuloma macrophage population, cells with high IL-4Rα expression preferentially serve as a cellular niche for STm, whereas cells with high CD301 expression provide a more permissive cytokine and intracellular metabolic environment for pathogen persistence.
TNF limits M2 granuloma macrophage polarization to restrict STm
Our findings that granuloma macrophages with M2-like features are less likely to exhibit antimicrobial functions and preferentially harbor intracellular STm suggest that regulation of granuloma macrophage polarization may directly affect bacterial persistence. TNF signaling has been implicated in restricting intracellular bacteria and regulating granuloma formation through a number of mechanisms such as modulating macrophage and T cell death, neutrophil recruitment, and nitric oxide production (Bernut et al., 2016; Dorhoi and Kaufmann, 2014; Matty et al., 2015). Recently, it was found that TNF signaling suppressed the emergence of M2 tumor-associated macrophages (Kratochvill et al., 2015). This led us to investigate whether a TNF-dependent mechanism may restrict STm during persistent infection by limiting M2 polarization of granuloma macrophages. Mice infected for 2–3 months were injected i.p. with either 500 μg TNF neutralizing antibody (Ab) or control Ab on day 0 and day 4, followed by analysis on day 8. At this dose, the anti-TNF Ab has been shown to effectively neutralize TNF activity in systemic tissues of Mtb infected mice (Plessner et al., 2007). We measured IL-4Rα levels of granuloma macrophages and quantitated the number of CD301+ cells within this population to determine the impact of TNF neutralization on polarization. We found that disruption of TNF signaling led to increased IL-4Rα expression of splenic granuloma macrophages but not CD11b+CD11c+ dendritic cells (Figure 4A). In addition, the CD301+ granuloma macrophage population expanded by approximately 7-fold in the spleens of mice treated with anti-TNF Ab compared to controls (Figure 4B–C). Concurrent with the increased M2 granuloma macrophage polarization, TNF-neutralized mice had 1–1.5 log more STm in the spleens and livers (Figure 4D–E).
Figure 4: TNF limits M2 granuloma macrophage polarization to restrict STm persistence.
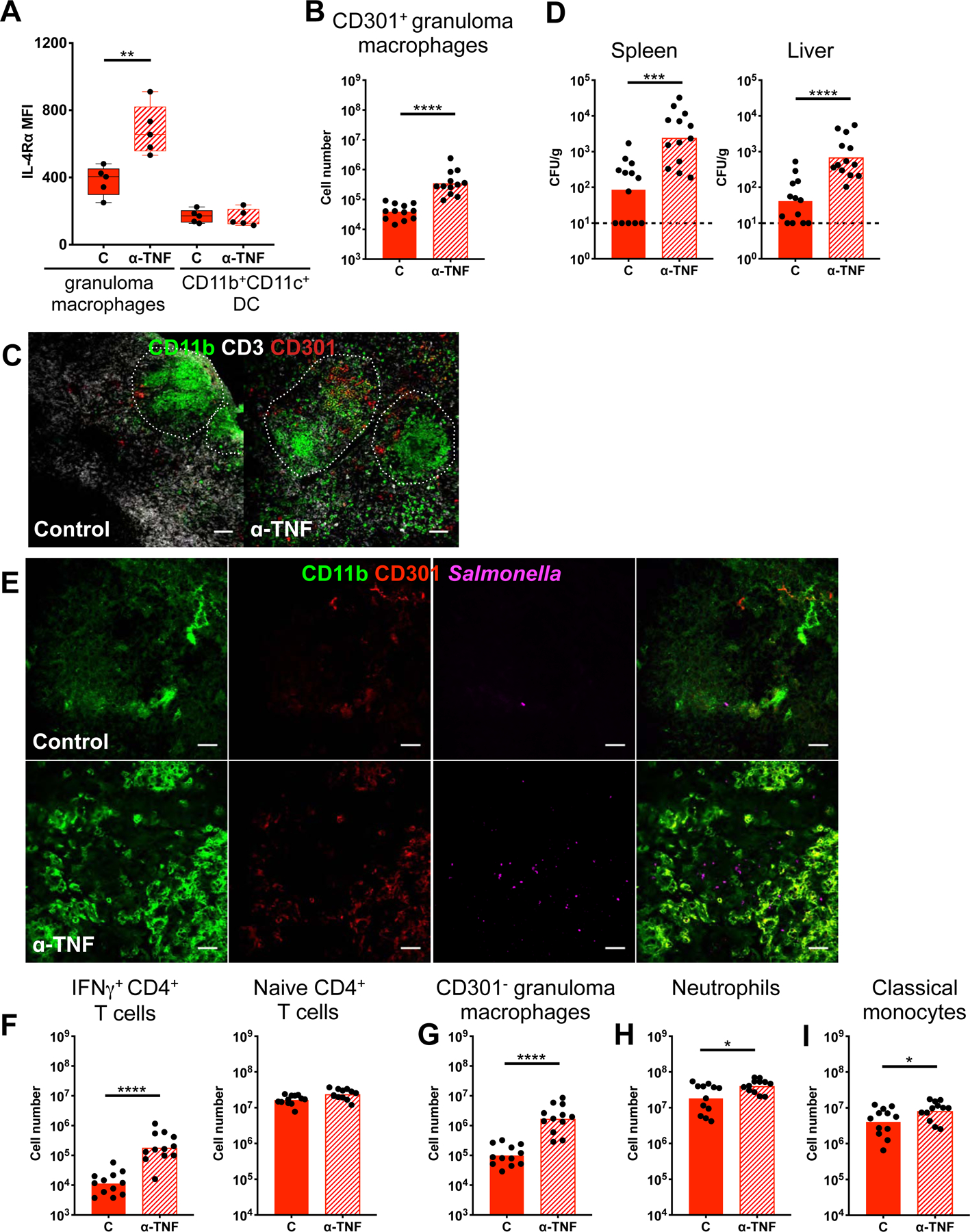
Mice orally infected with STm for 3 months and treated with either anti-TNF Ab or control Ab on day 0 and 4, followed by analysis on day 8. (A) IL-4Rα levels on granuloma macrophages and CD11b+CD11c+ DC. (B) Quantitation of splenic CD301+ granuloma macrophages. (C) Confocal microscopy showing expansion of CD301+ cells among granuloma macrophages with TNF neutralization. Two month-infected mice shown. Scale bar: 50 μm. (D) Bacterial burdens in infected spleens and livers quantitated by CFU. Dotted line: limit of detection. (E) Confocal microscopy showing STm dynamics within granulomas. Scale bar: 20 μm (F) Quantitation of naïve and IFNγ+ CD4+ T cells. Gating for these cells shown in figure S1. (G-I) Quantitation of splenic CD301− granuloma macrophages, neutrophils, and classical monocytes. Significance calculated using a two-tailed Mann-Whitney test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p <0.0001. A, B, D, F-I, data from 3 independent experiments 3–5 mice per group per experiment. B, D, F-I show combined data, C and E, n > 5 per group, multiple sections per mouse.
In addition, TNF neutralization resulted in 30 times more IFNγ+ CD4+ T cells (Figure 4F), indicating that diminished T-helper IFNγ response was unlikely to be the driving factor underpinning the loss of pathogen restriction. The number of naïve T cells were similar in the spleens of neutralized and control mice (Figure 4F). Furthermore, there was a marked increase in the number of CD301− granuloma macrophages, which are more likely to produce TNF and iNOS, as well as a modest increase of neutrophils and classical monocytes from TNF inactivation (Figure 4G–I). Taken together these data demonstrate that TNF signaling limits M2 granuloma macrophage polarization to restrict STm during persistent infection. Disruption of this critical signal leads to increased tissue bacterial burden and antigen availability, which triggers exacerbated inflammatory innate and adaptive cellular responses.
STm SteE drives M2 granuloma macrophage polarization and promotes pathogen persistence
The SPI-2 T3SS effector SteE (Stm 2585, also known as SarA) has been shown to be important for STm persistence in systemic tissues and to mediate M2 reprogramming in primary macrophages (Jaslow et al., 2018; Lawley et al., 2006; Stapels et al., 2018). However, the impact of SteE on host immune responses and STm persistence in vivo is not well understood. We tested if STm establishes systemic persistence by modulating M2 granuloma macrophage polarization in a SteE-dependent manner. First, we determined the impact of SteE on pSTAT3, a transcriptional regulator of M2 polarization, and IL-4Rα levels in BMDMs from the 129×1/SvJ mice. Consistent with recent reports (Jaslow et al., 2018; Stapels et al., 2018), 129×1/SvJ BMDMs infected with ΔsteE mutant showed diminished M2 polarization, as indicated by the reduced pSTAT3 and IL-4Rα levels compared to wild-type (WT) STm-infected cells at 20 hours of infection (Figure S4A–C).
Next, we infected mice with either WT or ΔsteE STm via i.p. injection to bypass potential effects of SteE deletion on bacterial gut invasion and examined its effect on STm systemic persistence. The ΔsteE STm bacterial levels in the spleens and livers were approximately one log lower than WT STm at 4 weeks p.i. (Figure 5A–B). Mice infected with ΔsteE STm exhibited less severe splenomegaly compared to those infected with WT STm, suggesting that the lack of SteE augmented the ability of the host to limit STm and minimize immunopathology (Figure 5C, S4D). Previous reports showed that ΔsteE did not exhibit a growth defect in vitro or within macrophages (Figueira et al., 2013; Jaslow et al., 2018; Stapels et al., 2018). By affecting cellular IL-10 production and reprogramming the intra-macrophage metabolic and immune environment, SteE activity is thought to promote bacterial persistence. Since IL-10 is a negative regulator of Th1 T cell response (Couper et al., 2008), we determined if enhanced Th1 T cell response contributed to the attenuated persistence of ΔsteE STm in vivo. Quantitation of splenic IFNγ+ CD4+ T cells showed a 5-fold decrease in these cells in ΔsteE STm-infected compared to WT STm-infected mice (Figure 5D), suggesting that the reduced ΔsteE STm levels resulted in lower antigen loads and dampened T cellular response. There was no difference in the number of naïve T cells (Figure 5D). In addition, we found that the numbers of neutrophils, classical monocytes, and CD301− granuloma macrophages, which produce high levels of TNF and iNOS, were reduced in ΔsteE STm-infected spleens (Figure 5E–G, S4E). Furthermore, granuloma macrophages from ΔsteE and WT STm-infected spleens produced TNF similarly in response to HKST (Figure S4F). Collectively, these findings demonstrate that the attenuated persistence of the ΔsteE STm mutant was not due to increased proinflammatory immune cells in infected tissues.
Figure 5: STm effector SteE drives M2 granuloma macrophage polarization and pathogen persistence.
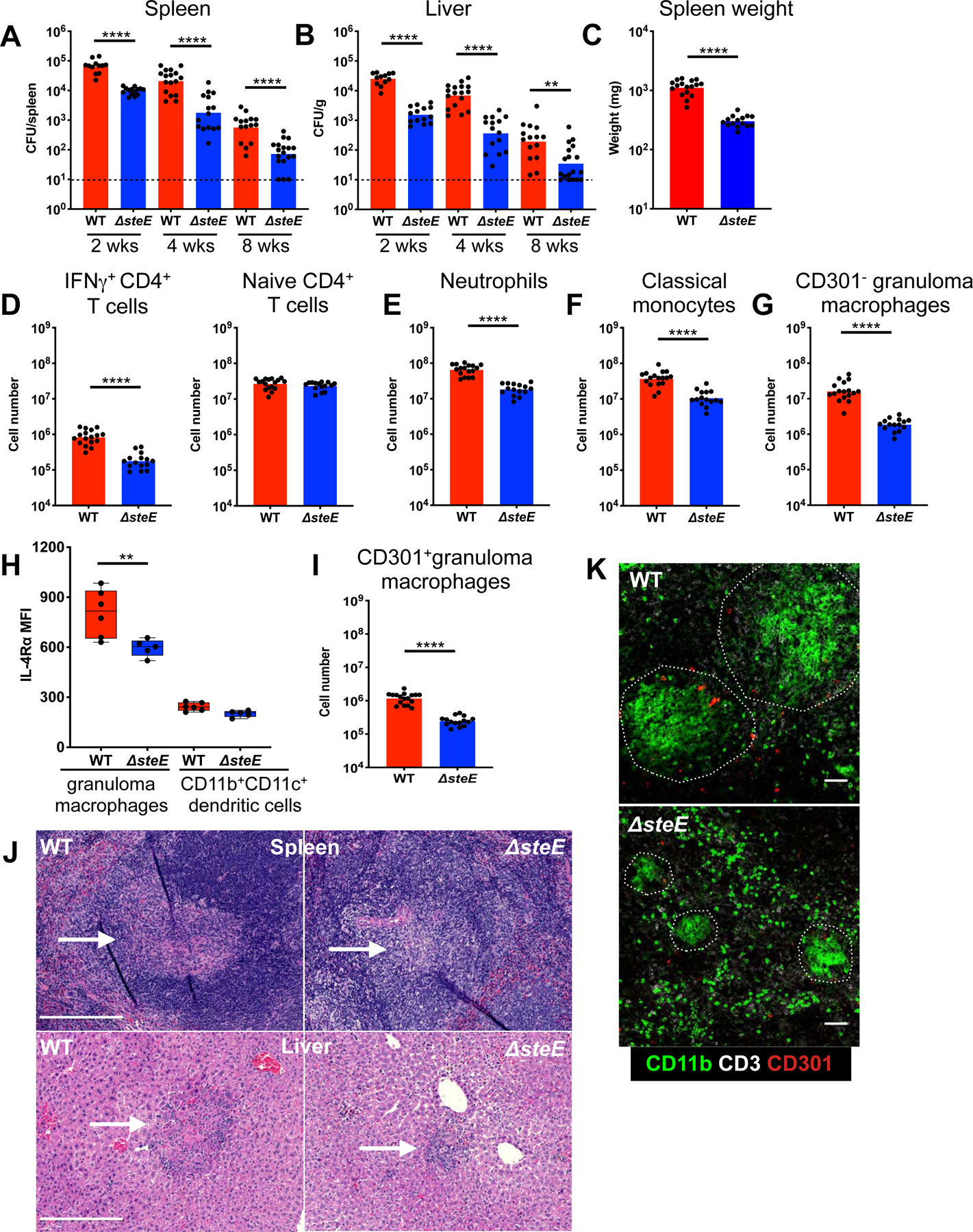
(A-B) Mice infected with either WT or ΔsteE STm i.p. and tissue CFU quantitated at indicated time points p.i. Line: limit of detection. (C-K) Mice analyzed at 4 weeks p.i. (C) Spleen weights of infected mice. (D) Quantitation of naïve and IFNγ+ CD4+ T cells in infected spleens. Gating for these cells shown in figure S1. (E-G) Quantitation of splenic CD301− granuloma macrophages, neutrophils, and classical monocytes. (H) IL-4Rα levels of splenic granuloma macrophages and CD11b+CD11c+ dendritic cells. (I) Quantitation of CD301+ splenic granuloma macrophages. (J) H&E stains showing granulomas in the infected spleens and livers. White arrows indicate granulomas. Scale bar: 200 μm. (K) Staining of granulomas by confocal microscopy. Scale bar: 50 μm. Significance calculated using a two-tailed Mann-Whitney test. ** p < 0.01, **** p <0.0001. A-K, data from at least 3 independent experiments, 3–6 mice per group per experiment. A–G, I show combined data.
We then investigated whether the reduced capacity of the SteE-deficient strain to persist in systemic tissues is due to a diminished ability to promote M2 polarization of granuloma macrophages. We found that the granuloma macrophages from mice infected with ΔsteE STm for 4 weeks expressed significantly less IL-4Rα, compared to those from WT STm-infected animals (Figure 5H). In addition, the CD301+ granuloma macrophage population in the mutant-infected spleen was 6 times smaller than WT STm-infected spleens (Figure 5I, S4G). SteE complementation fully restored the granuloma macrophage polarization and persistence defects of the ΔsteE mutant (Figure S5A–B). To gain further insight into the role of SteE in persistence, we examined granuloma formation. Granulomas in the livers and spleens of ΔsteE STm-infected mice were markedly smaller and morphologically less well-defined compared to those from WT STm-infected animals (Figure 5J–K, S5C). Taken together, our data indicate that SteE drives M2 granuloma macrophage polarization and contributes to persistent systemic STm infection, suggesting that SteE acts to overcome host restriction.
SteE activity antagonizes TNF-mediated restriction of M2 granuloma macrophage polarization and STm persistence
To directly test if SteE opposes TNF-mediated restriction, we determined the impact of SteE activity on granuloma macrophage polarization and pathogen persistence when TNF signaling is abrogated. TNF levels are upregulated in STm-infected lymphoid tissues at days 7–14 p.i. (Eisele et al., 2013), during the period of time when nascent granulomas develop (Figure 1A). Thus, we neutralized TNF in mice that had been infected with either ΔsteE or WT STm for 10 days. When TNF was neutralized for 8 days (Figure 4), a large proportion of WT STm-infected animals treated with anti-TNF Ab became moribund (Figure S6A), likely due to overwhelming bacterial burden (Mastroeni et al., 1995). Therefore, mice were infected for 10 days and then injected with either anti-TNF or control Ab, followed by analysis on day 4 post-treatment. We found that when TNF was neutralized, granuloma macrophages in both WT and ΔsteE-infected animals became more M2-like, as indicated by their higher IL-4Rα levels and increased numbers of CD301+ but not CD301− granuloma macrophages (Figure 6A–C). However, M2 polarization was markedly diminished in neutralized, ΔsteE STm-infected spleens, compared to neutralized, WT STm-infected spleens (Figure 6A–B, striped red vs. striped blue columns). In addition, TNF neutralization led to increased pathogen burdens in both WT and ΔsteE STm-infected spleens and livers (Figure 6D, S6B). However, the bacterial levels were reduced by more than 1 log in neutralized ΔsteE STm-infected mice, compared to neutralized WT STm-infected animals (Figure 6D, striped red vs. striped blue columns). These results suggest that when TNF-mediated suppression of M2 state is abolished, unopposed SteE activity promotes granuloma macrophage M2 polarization and pathogen persistence to a greater extent in WT STm-infected animals, compared to those infected with ΔsteE STm.
Figure 6: SteE antagonizes TNF-mediated restriction of STm.
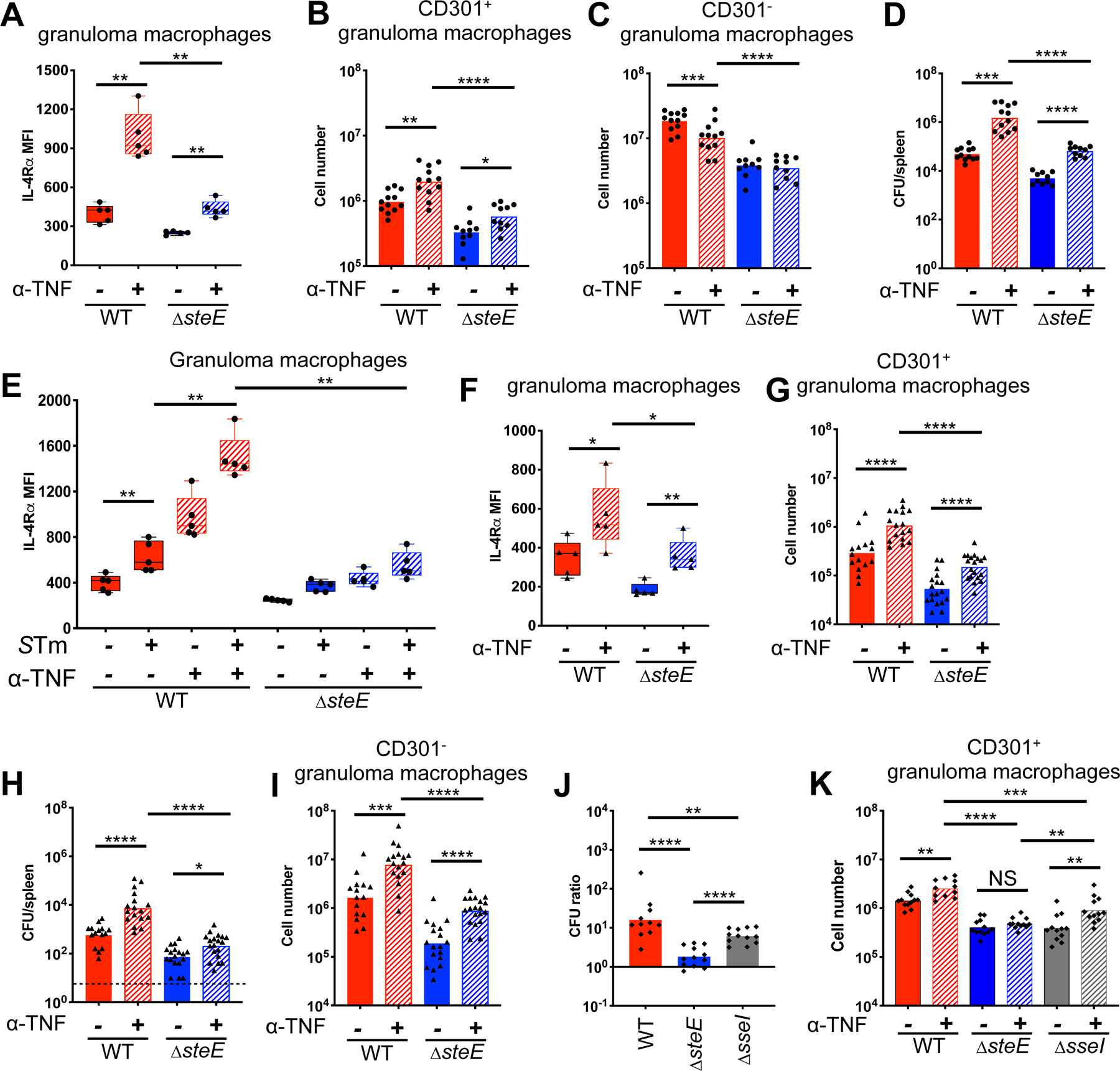
Red: WT STm-infected mice; blue: ΔsteE STm-infected mice; gray: ΔsseI STm-infected mice. (A-E) Mice infected with either WT or ΔsteE STm i.p. for 10 days before treatment with either anti-TNF or control Ab on day 0, followed by analysis on day 4. (A) IL-4Rα levels on splenic granuloma macrophages. (B-C) Quantitation of CD301+ and CD301− granuloma macrophages in infected spleens. (D) Quantitation of splenic bacterial burdens by CFU. (E) IL-4Rα levels on uninfected, bystander vs. infected granuloma macrophages from infected animals using anti-Salmonella Ab to stain for intracellular STm. (F-I) Mice infected with either WT or ΔsteE STm i.p. for 8 weeks, then treated with either anti-TNF or control Ab on day 0 and day 4, followed by analysis on day 8. (F) IL-4Rα levels on splenic granuloma macrophages. (G) Quantitation of CD301+ granuloma macrophages in infected spleens. (H) CFU quantitation of splenic bacterial burdens. (I) Quantitation of CD301− granuloma macrophages. (J-K) Mice infected with either WT, ΔsteE, ΔsseI STm i.p. for 4 weeks before treatment with either anti-TNF or control Ab on day 0, followed by analysis on day 4. (J) Splenic CFU ratio of individual anti-TNF treated mice to the mean CFU of the control Ab-treated mice that were infected with the same STm strain. (K) Quantitation of CD301+ granuloma macrophages in infected spleens. Significance calculated using a two-tailed Mann-Whitney test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p <0.0001. A-E, data from 2 independent experiments, 5–7 mice per group per experiment; F-I, 4 independent experiments, 3–5 mice per group per experiment. J-K, 2 independent experiments, 5–6 mice per group. B-D, G-K show combined data.
To determine the impact of intracellular bacilli on SteE-mediated counteraction of TNF effects, we compared IL-4Rα expression of uninfected, bystander vs. infected granuloma macrophages within infected spleens. As expected, the IL-4Rα level on WT STm-infected splenic granuloma macrophages was higher than uninfected cells and was further enhanced when TNF was neutralized (Figure 6E). In contrast, the IL-4Rα level on ΔsteE STm-infected splenic granuloma macrophages was marginally increased upon TNF neutralization (Figure 6E, striped red vs. striped blue columns). Neutrophils expressed much lower IL-4Rα levels compared to granuloma macrophages, and the level was not modulated by intracellular STm (data not shown).
To test whether SteE activity antagonizes TNF effects to maintain STm persistence within granulomas during persistent infection, mice were infected with either ΔsteE or WT STm for 2 months and then injected with anti-TNF or control Ab on day 0 and day 4, followed by analysis on day 8. TNF neutralization led to increased M2 polarization of splenic granuloma macrophages, as indicated by increased IL-4Rα levels and CD301+ fraction among these cells (Figure 6F–G). However, M2 polarization was diminished in neutralized, ΔsteE STm-infected mice, compared to neutralized, WT STm-infected animals (Figure 6F–G). Furthermore, the tissue bacterial burdens in neutralized, ΔsteE STm-infected spleens and livers remained 1.5 to 2-log lower than in neutralized, WT STm-infected tissues (Figure 6H and S6C, striped red vs. striped blue columns). The diminished persistence of ΔsteE STm was not due to increased CD301− granuloma macrophages, which produce higher levels of antimicrobials such as TNF and iNOS (Figure 6I). Collectively, our results suggest that intracellular STm drives M2 granuloma macrophage polarization via a SteE-dependent mechanism to overcome TNF-mediated suppression of its preferred cellular niche.
Macrophage polarization and heterogeneity are influenced not only by cell-intrinsic programming, but also microenvironmental signaling (Gordon and Pluddemann, 2019). To determine if the diminished M2 granuloma macrophage polarization and pathogen levels in neutralized, ΔsteE STm-infected animals was specifically due to the loss of SteE-dependent macrophage reprogramming and/or a change in the immune milieu associated with reduced pathogen persistence, we compared infections with ΔsteE STm and with STm lacking the SPI2 T3SS effector SseI. Our previous studies demonstrated that SseI promotes STm long-term persistence by inhibiting macrophage and dendritic cell migration to compromise host immune responses (McLaughlin et al., 2009). To determine whether SseI impacts macrophage polarization, BMDMs were infected with ΔsseI STm and IL-4Rα levels were measured. The levels of IL-4Rα were increased to the same level in WT and ΔsseI STm-infected BMDMs, but not with ΔsteE STm macrophages (Figure S6D). Similar to our previous results (McLaughlin et al., 2009), the levels of ΔsseI STm in the spleens of 129×1/SvJ mice at 4 weeks p.i. were significantly lower than WT STm and were the same as the levels of the SteE-deficient strain (Figure S6E). We next determined the impact of TNF neutralization on pathogen persistence and M2 granuloma macrophage polarization in mice infected with ΔsseI STm. Mice were infected for 4 weeks and then injected with either anti-TNF or control Ab, followed by analysis on day 4 post-treatment. We expressed the ratio of splenic CFUs from mice injected with TNF neutralizing antibody to the splenic CFU mean from mice injected with the control antibody. TNF neutralization led to a 10- and 7-fold increase in the splenic levels of WT- and ΔsseI STm, respectively, and a 2-fold increase in the splenic levels of ΔsteE STm (Figure 6J and S6E). Granuloma macrophage IL- 4Rα levels increased similarly between neutralized ΔsteE STm- and ΔsseI STm-infected mice, although to a lesser extent than neutralized WT STm-infected animals (Figure S6F). In addition, we quantitated the levels of CD301+ splenic granuloma macrophages upon neutralization. Although the levels of this cell population were higher in WT STm-infected mice, the expansion of these cells in TNF-neutralized ΔsseI STm-infected mice was significantly greater than the expansion in neutralized ΔsteE STm-infected mice (Figure 6K). Thus, when TNF-mediated restriction is abrogated, SteE activity provides ΔsseI STm with a greater ability to shift granuloma macrophages toward a CD301+ state than ΔsteE STm, but remains insufficient to polarize this cellular compartment toward an IL-4Rαhi state or promote similar levels of granuloma macrophage M2 polarization as WT STm. Taken together, these findings suggest that a SteE-mediated mechanism partially opposes TNF-mediated restriction by driving granuloma macrophage polarization.
Discussion
Our finding that STm granulomas are comprised of CD11b+CD11+ macrophages suggests these microstructures are analogous to the CD11c+ granulomas in Mtb- infected non-human primates (NHP) described previously (Mattila et al., 2013). As seen with Mtb granulomas, many macrophages within STm granulomas express iNOS (Figure 1 and 2) (Goldberg et al., 2018; Mattila et al., 2013). STm and Mtb granulomas similarly contain a mixture of M1-like and M2-like macrophages with distinct functional characteristics and regional distributions (Figure 3)(Mattila et al., 2013). Macrophages can arise from cells of embryonic origin or from bloodborne monocyte progenitors that are recruited to tissues during homeostatic and inflammatory states (Gordon and Pluddemann, 2019). The phenotype of splenic STm granuloma macrophages—CD11b+, CD11c+, Ly6C intermediate, CD115+, F4/80+, CD64 high, and MCHII high—suggests that they may arise from a monocyte lineage. Future studies elucidating the ontogeny and the developmental relationship between granuloma macrophages and the antimicrobial monocytes that have been described in recent years will provide further insights into granuloma development (Xiong and Pamer, 2015).
The juxtaposition of M1 and M2 macrophages within the granuloma microenvironment provides a structural basis for how granulomas may be both protective to the host and beneficial to intracellular bacterial pathogens (Pagan and Ramakrishnan, 2018). Although we have utilized the M2 markers CD301 and IL-4Rα to define and track STm granuloma macrophage subsets for mechanistic interrogations, a spectrum of activities of multiple macrophage determinants that are shaped by macrophage pre-existing programming, local environmental cues, and pathogen factors likely determine whether an individual macrophage plays a predominantly antimicrobial role or serves as a cellular niche for pathogen persistence. Our results suggest a model for granuloma macrophage regulation in which localized TNF signaling within the granuloma microenvironment restrains M2 polarization to limit the STm cellular niche, thereby restricting the pathogen. On the other hand, STm SteE activity polarizes granuloma macrophages toward an M2 state, enabling the pathogen to overcome host suppression of its long-term cellular niche and to establish persistent infection. When the balance between these competing drivers of granuloma macrophage polarization is disrupted, such as when TNF is neutralized, SteE-driven M2 polarization is unopposed, leading to expansion of the STm cellular niche and increased pathogen persistence. Higher bacterial burden increases the antigen loads in infected tissues and triggers enhanced host cellular immune responses, including increased IFNγ+ CD4+ T cells, neutrophils, monocytes, and CD301− granuloma macrophages—which produce more TNF and iNOS—to regain control of the infection. Conversely, when pathogen mechanisms for modulating M2 granuloma macrophage polarization are compromised, such as SteE deficiency, TNF-mediated restraint of the pathogen cellular niche becomes dominant, leading to more rapid pathogen eradication.
TNF is a pleotropic cytokine that may exert multiple, non-mutually exclusive functions during intracellular bacterial infections. Dissection of TNF-mediated mechanisms regulating mycobacterial granulomas has yielded varying effects of this pathway on pathogen control and host cellular immune responses, depending on the host-pathogen system and the stage of infection (Dorhoi and Kaufmann, 2014; Matty et al., 2015). In our present study, we found that TNF neutralization in mice infected with STm for 2–3 months, a chronic stage in which the pathogen is kept at a low level, led to more than a 10-fold increase in splenic granuloma macrophages, antigen-experienced CD4+ T cells, and to a lesser extent, neutrophils (Figure 4). These cellular immune changes were linked to 1–1.5 log STm expansion in systemic tissues, suggesting that granuloma macrophage and T cell death, as well as impaired neutrophil recruitment were not the primary factors underlying the loss of pathogen control during the persistent stage of infection. These findings are consistent with TNF neutralization in chronic Mtb infection in NHP (Lin et al., 2010). However, they vary from findings in zebrafish demonstrating that disruption of TNF signaling in early mycobacterial granulomas led to increased granuloma macrophage cell death, impaired neutrophil recruitment, and subsequent compromised pathogen restriction (Bernut et al., 2016; Clay et al., 2008). Other studies have found that a loss of TNF signal leads to increased T-cell death in Mtb infected mice, an infection model in which tissue bacterial burdens are high as the host fails to limit the pathogen (Plessner et al., 2007). Collectively, these findings suggest that loss of TNF signaling may have differential impacts due to the levels of tissue pathogen and extent of inflammation. Indeed, in our study, we find that TNF neutralization at 2 weeks p.i. leads to a 2-fold decrease in total splenic granuloma macrophages, in contrast to our findings for TNF neutralization during chronic stage of infection (Figure 4 and 6). Regardless of the infection stage in which we disrupted TNF signaling, STm tissue expansion was linked to a marked increase in M2 granuloma macrophage polarization, indicating that limiting M2 granuloma macrophages is an important mechanism by which TNF functions to restrict pathogen persistence throughout all stages of infection. Thus, our data revealed a mechanism of multifaceted TNF signaling during intracellular bacterial infections that had not been previously appreciated. During the course of infection, TNF-mediated restraint of M2 polarization likely operates in concert with other TNF functions to limit the pathogen.
While SteE had been found to be an important factor for long-term STm infection in systemic tissues, the in vivo mechanism of SteE remained largely unexplored (Jaslow et al., 2018; Lawley et al., 2006). In in vitro broth culture, ΔsteE mutant exhibits similar fitness to the WT strain (Jaslow et al., 2018). However, by modulating host cytokine response and reprogramming the STm intracellular metabolic and immune environment to an M2 state, SteE activity is coupled to STm persistence within its cellular niche and host tissue (Jaslow et al., 2018; Stapels et al., 2018). Extending from prior reports, our results indicate that SteE promotes the establishment of persistent infection in systemic tissues by driving M2 granuloma macrophage polarization to provide STm with a favorable long-term cellular niche. Consequentially, SteE activity also antagonizes a TNF-mediated mechanism that functions to limit M2 granuloma macrophages, thereby restricting STm during persistent infection. Our finding that ΔsseI STm, which possesses a functional steE gene, polarizes granuloma macrophages and antagonizes TNF-mediated restriction to a greater extent than ΔsteE mutant but not as much as WT STm suggests SteE activity is necessary but not sufficient for the pathogen to overcome TNF-mediated effects. During infection in vivo, macrophage polarization is likely impacted not only by the pathogen but also multiple host-regulated factors, including microenvironmental signals and developmental pre-programming. For example, the ontogeny of tissue macrophages was recently shown to shape their polarization, metabolic state, and susceptibility to intracellular bacterial infection (Huang et al., 2018). Furthermore, there are several factors that could temper SteE’s effect on granuloma macrophages in a ΔsseI STm-infected spleen. Firstly, SseI, which modulates macrophage migration, may be required to position granuloma macrophages in the optimal microenvironment to maximize pathogen-driven M2 polarization. Secondly, the low levels of ΔsseI STm in the tissue, which is due to a defect in the ability of the mutant bacteria to compromise host immune responses (McLaughlin et al., 2009), may result in a lower overall level of SteE-mediated polarization of the granuloma macrophage compartment, compared to a WT STm-infected spleen. Finally, changes in the immune milieu when there are decreased bacterial burdens may affect availability of the cellular target of SteE, rendering SteE less effective.
By modulating STAT3 activity, SteE manipulates a master transcriptional regulator that promotes the alternative M2 phenotype (Figure 4S and 5)(Jaslow et al., 2018). Effectively, SteE activity opposes TNF-mediated suppression of M2 macrophage reprogramming. Our finding that ΔsteE mutant-infected granuloma macrophages expressed substantially less IL-4Rα than those infected with WT bacteria when TNF was neutralized provides evidence that during persistent STm infection, these opposing regulations operate at the level of granuloma macrophages to dictate infection outcome (Figure 6). SteE immunoprecipitates with STAT3 (Jaslow et al., 2018). How SteE activity enhances pSTAT3 levels is a fascinating subject for futures studies, including molecular structural analysis to examine the impact of SteE binding to STAT3. Our data indicate that modulating granuloma polarization is an effective pathogenesis strategy for intracellular bacteria to establish persistent infection. Whether other intracellular bacteria secrete effectors to promote M2 granuloma macrophage polarization to persist in host tissues is not known. SteE coding sequence is absent from Salmonella Typhi, which is well known to cause persistent systemic infections in humans (Jennings et al., 2017). However, given the multitude of regulatory checkpoints in macrophage polarization, it is conceivable that different intracellular bacteria might exploit unique host pathways to achieve similar macrophage polarization effects. Indeed, a recent in vitro study showed Mtb utilizes ESAT-6 to modulate macrophage polarization, suggesting that diverse intracellular bacteria may employ evolutionarily convergent strategies to exploit granuloma macrophages (Refai et al., 2018).
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Denise M. Monack (dmonack@stanford.edu)
Experimental Model and Subject Details
Ethics Statement
Experiments involving animals were performed in accordance with NIH guidelines, the Animal Welfare Act, and US federal law. All animal experiments were approved by the Stanford University Administrative Panel on Laboratory Animal Care (APLAC) and overseen by the Institutional Animal Care and Use Committee (IACUC) under Protocol ID 12826. Animals were housed in a centralized research animal facility accredited by the Association of Assessment and Accreditation of Laboratory Animal Care (AAALAC) International.
Mouse Strains and Husbandry
129X1/SvJ mice were obtained from Jackson Laboratories or an in-house 129X1/SvJ colony. Male and female mice (7–16 weeks old) were housed under specific pathogen-free conditions in filter-top cages that were changed bi-monthly by veterinary or research personnel. Sterile water and food were provided ad libitum. Mice were given at least one week to acclimate to the Stanford Animal Biohazard Research Facility prior to experimentation.
Bacterial Strains and Growth Conditions
Salmonella enterica serovar Typhimurium strain SL1344 was utilized in this study. SL1344 ΔsteE and SL1344 ΔsteE + psteE were generated as described previously (Stapels et al., 2018). For all mouse infections, S. Typhimurium strains were maintained aerobically on LB agar supplemented with 200 g/mL streptomycin, ± 40 g/mL kanamycin, ± 100 g/mL carbenicillin, and grown aerobically to stationary phase overnight at 37 °C with broth aeration. Bacterial c ultures were spun down and washed with sterile phosphate-buffered saline (PBS) before suspension in PBS for infection.
Methods Details
Mouse Infections and TNF neutralization
Mice were allocated to control and experimental groups randomly, sample sizes were chosen based on previous experience to obtain reproducible results and the investigators were not blinded. For oral infections, food was removed 16 hours prior to inoculation with 108 CFU S. Typhimurium in 100 μL PBS by oral gavage. For intraperitoneal infections, mice were injected with 103 CFU in 200 μL PBS. For TNF neutralization, infected mice were injected i.p. with either 500 g anti-TNF monoclonal Ab, clone MP6-XT22 (Biolegend), or isotype control Ab in sterile PBS in 400 L total volume every 4 days until analysis. Mice were euthanized at the indicated time points post-inoculation by CO2 asphyxiation followed by cervical dislocation as the secondary method of euthanasia. Organs were collected, weighted, and either homogenized in PBS for CFU enumeration, used to make single cell suspension for flow cytometric analysis, or prepared or histopathological examinations.
Flow Cytometry
Spleens from mice were minced with surgical blades No. 22 and incubated in digestion buffer (HBSS+Ca2++Mg2+ + 50 g/mL DNase (Roche) + 25 g/mL Liberase TL (Sigma)) at 37 °C for 25 min, mixing at 200 rpm. EDTA was ad ded at a final concentration of 5 mM to halt digestion. Single cell suspensions were passed through a 70-μm filter and washed with R5 buffer (RPMI containing 5% FBS and 10 mM Hepes). Red blood cells were lysed with ACK Lysis Buffer (Fisher Scientific) for 3 min at room temperature, washed, and resuspended in R5 buffer until they were stained for flow cytometry.
For myeloid intracellular TNF staining, heat-killed S. Typhimurium (HKST) was prepared from stationary-phase broth cultures grown aerobically overnight followed by heat treatment for 30 min at 56 °C. Digested splenocytes were washed and resuspended in RPMI containing 10% FBS, 10mM Hepes, and 50 ug/mL gentamicin. A total of 9 × 106 splenocytes in 2-mL volume was added into each well of 6-well plates and allowed to equilibrate for 30 min at 37 °C. HKST at MOI 10:1 a nd brefeldin A to final concentration of 3 g/mL were then added to stimulate splenocytes. After 3 hours of stimulation at 37 °C, splenocytes were harvested and stained.
Single-cell suspensions were incubated in Fc Block (TruStain fcX anti-mouse CD16/32, Biolegend) for at 15 min on ice and washed with PBS. Cells were stained on ice for 30 min in PBS with a cocktail of Live/Dead Fixable Blue Viability Dye (Invitrogen) and antibodies for surface antigens (list of antibodies used provided in the Key Resources table). Cells were washed with FACS buffer (PBS containing 2% FBS and 2 mM EDTA), followed by fixation for 15 min with Cytofix/Cytoperm solution (BD Biosciences). Cells were washed twice with Perm/Wash buffer (BD Biosciences) and stained for intracellular Salmonella, TNF, iNOS, and IFNγ. After washing, cells were resuspended in FACS buffer and analyzed on a LSRFortessa or LSRII cytometer (Becton Dickinson). Data were acquired with DIVA software (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Salmonella chicken antibody | Aves Lab | Custom-made |
| Anti-Salmonella CSA-1 | KPL | Cat# 02-91-99 |
| B220 Alexa Fluor® 488 | Biolegend | Cat# 103225 RRID:AB_389308 |
| B220 Alexa 594 | Biolegend | Cat# 103254 RRID:AB_2563229 |
| CCR2 PE | R&D Systems | Cat# FAB5538P RRID:AB_10718414 |
| CD11b Alexa Fluor® 647 | Biolegend | Cat# 101218 RRID:AB_389327 |
| CD11b Biotin | Biolegend | Cat# 101204 RRID:AB_312787 |
| CD11b Brilliant Violet 785™ | Biolegend | Cat# 101243 RRID:AB_2561373 |
| CD11b FITC | Biolegend | Cat# 101206 RRID:AB_312789 |
| CD11c Alexa Fluor® 700 | eBioscience | Cat# 56-0114-80 RRID:AB_493993 |
| CD11c APC | BD Biosciences | Cat# 550261 RRID:AB_398460 |
| CD11c eFluor® 450 | eBioscience | Cat# 48-0114-82 RRID:AB_1548654 |
| CD11c FITC | eBioscience | Cat# 11-0114-82 RRID:AB_464940 |
| CD11c PE-Cy7 | BD Pharmingen | Cat# 558079 RRID:AB_647251 |
| CD115 Brilliant Violet 605™ | Biolegend | Cat# 135517 RRID:AB_2562760 |
| CD19 APC eFluor® 780 | eBioscience | Cat# 47-0193-82 RRID:AB_10853189 |
| CD19 BUV 395 | BD Biosciences | Cat# 563557 RRID:AB_2722495 |
| CD3 FITC | Biolegend | Cat# 100203 RRID:AB_312660 |
| CD3ε APC eFluor® 780 | eBioscience | Cat# 47-0031-82 RRID:AB_11149861 |
| CD3ε eFluor® 450 | eBioscience | Cat# 48-0032-82 RRID:AB_1272193 |
| CD301 Alexa Fluor® 488 | Bio-Rad | Cat# MCA2392A488 RRID:AB_872011 |
| CD301 Alexa Fluor 647 | Bio-Rad | Cat# MCA2392A647 RRID:AB_872012 |
| CD301 PE | Biolegend | Cat# 145704 RRID:AB_2561961 |
| CD4 PerCP/Cy5.5 | Biolegend | Cat# 100434 RRID:AB_893324 |
| CD44 Brilliant Violet 785™ | Biolegend | Cat# 103041 RRID:AB_11218802 |
| CD44 FITC | Biolegend | Cat# 103006 RRID:AB_312957 |
| CD49b APC eFluor® 780 | eBioscience | Cat# 47-5971-82 RRID:AB_11218895 |
| CD64 PE | Biolegend | Cat# 139304 RRID:AB_10612740 |
| CD64 PerCP/Cy5.5 | Biolegend | Cat# 139307 RRID:AB_2561962 |
| CD8α Alexa Fluor® 700 | eBioscience | Cat# 56-0081-82 RRID:AB_494005 |
| CD8α FITC | Biolegend | Cat# 100706 RRID:AB_312745 |
| F4/80 Brilliant Violet 650™ | Biolegend | Cat# 123149 RRID:AB_2564589 |
| F4/80 PE | eBioscience | Cat# 12-4801-82 RRID:AB_465923 |
| Goat anti-chicken Alexa Fluor® 488 | ThermoFisher | Cat# A11039 RRID:AB_2534096 |
| Goat anti-chicken Alexa Fluor® 594 | ThermoFisher | Cat# A11042 RRID:AB_2534099 |
| Gr-1 FITC | eBioscience | Cat# 11-5931-81 RRID:AB_465313 |
| IFNγ APC | Biolegend | Cat# 505810 RRID:AB_315404 |
| IL-10 APC | eBioscience | Cat# 17-7101-81 RRID:AB_469501 |
| IL-18Rα PE | eBioscience | Cat# 12-5183-82 RRID:AB_2572617 |
| Ly-6C APC | eBioscience | Cat# 17-5932-82 RRID:AB_1724153 |
| Ly-6C Biotin | Biolegend | Cat# 128003 RRID:AB_1236552 |
| Ly-6C PerCP/Cy5.5 | eBioscience | Cat# 45-5932-82 RRID:AB_2723343 |
| IL-4Rα Biotin | Biolegend | Cat# 552508 RRID:AB_394406 |
| IL-4Rα PE-Cy7 | Biolegend | Cat# 144806 RRID:AB_2565599 |
| iNOS Alexa Fluor® 488 | eBioscience | Cat# 53-5920-82 RRID:AB_2574423 |
| iNOS APC | eBioscience | Cat# 17-5920-80 RRID:AB_2573243 |
| iNOS unconjugated | ThermoFisher | Cat#
PA3–030A RRID:AB_2152737 |
| Ly-6C Pacific Blue™ | Biolegend | Cat# 128014 RRID:AB_1732079 |
| Ly-6G BUV 395 | BD Biosciences | Cat# 563978 RRID:AB_2716852 |
| Ly-6G PE | Biolegend | Cat# 127608 RRID:AB_1186099 |
| MHC class II Alexa Fluor® 700 | eBioscience | Cat# 56-5321-82 RRID:AB_494009 |
| STAT1 (pY701) Alexa Fluor® 488 | BD Biosciences | Cat# 612696 |
| STAT1 (pY701) Alexa Fluor® 647 | BD Biosciences | Cat# 612697 |
| STAT3 (pY705) Alexa Fluor® 488 | BD Biosciences | Cat# 557814 RRID:AB_647098 |
| STAT3 (pY705) Alexa Fluor® 647 | BD Biosciences | Cat# 557815 RRID:AB_647144 |
| TCRαβ APC eFluor® 780 | eBioscience | Cat# 47-5961-82 RRID:AB_1272173 |
| TNF Brilliant Violet 510™ | Biolegend | Cat# 506339 RRID:AB_2563127 |
| Ultra-LEAF™ Purified Rat IgG1, κ Isotype | Biolegend | Cat# 400458 |
| Ultra-LEAF™ Purified anti-mouse TNF | Biolegend | Cat# 506348 RRID:AB_2616672 |
| Bacterial and Virus Strains | ||
| Salmonella enterica serovar Typhimurium, strain SL1344, StrepR | Monack lab strain collection and Stapels et. al., 2018 | n/a |
| steE mutant Salmonella (SL1344), StrepR/KanR | this study | n/a |
| sseI mutant Salmonella (SL1344), StrepR/KanR | Monack lab strain collection, McLaughlin et. al. 2009 | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ACK lysis buffer | Lonza | Cat# BW10–548E |
| BD cytofix/cytoperm solution kit | BD Biosciences | Cat# 554714 |
| DNase I | Sigma | Cat# 10104159001 |
| Liberase TL | Sigma | Cat# 05401020001 |
| Live/Dead Fixable Viability Dye | Invitrogen | Cat# L-34962 |
| OCT | Fisher | Cat# 23-730-571 |
| TruStain fcX | BioLegend | Cat# 101320 |
| Experimental Models: Organisms/Strains | ||
| 129X1/SvJ mice | Jackson Laboratory and in house colony | Stock #: 000691 |
| Software | ||
| FlowJo | FlowJo LLC | https://www.flowjo.com |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| NDP.view 2 | Hamamatsu Inc. | https://www.hamamatsu.com/us/en/product/type/U12388-01/index.html |
| Prism | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
Immunofluorescence Microsopy
A full list of antibodies used for immunohistochemistry can be found in the Key Resources table. Spleens were harvested, frozen in OCT compound (Fisher Scientific), and frozen sections 8 m in thickness were placed on SuperFrost Plus cryosection slides (Fisher Scientific). Sections were fixed in ice-cold acetone at −20 °C for 10 minutes and then allowed to dry. A boundary was drawn around tissue sections using a pap pen (Fisher Scientific). Sections were washed with PBS and then blocked with staining buffer (PBS with 3% bovine serum albumin, ± 1% saponin, 5 % normal mouse serum) for 30 min at room temperature. After blocking, sections were stained with the primary anti-Salmonella antibody in staining buffer for 4 hours, or with primary antibodies against surface antigens for 1.5 hours, at room temperature. Sections were washed and then stained for 2 hours at room temperature with fluorescent conjugated secondary antibodies. Slides were washed in PBS and then mounted using ProLong Diamond (Life Technologies). Images were acquired on a Zeiss LSM 700 or 880 confocal microscope with the ZEN 2010 software (Zeiss) and processed using FIJI software.
Histology
Tissues were fixed in 10% neutral buffered formalin. After fixation, tissues were routinely processed for paraffin embedding and stained with hematoxylin and eosin. Histopathological analyses were performed by a veterinarian pathologist (J.G. V-M). Images were collected using a Hamamatsu Nanozoomer scanner and processed with NDP.view2 software.
Quantification and Statistical Analysis
Sample sizes were chosen based on previous experience to obtain reproducible results. On rare occasions, an infected mouse within a cohort developed signs of meningitis or severe infection characterized by ruffling, ataxia, inability to attain water and food, and hunching. Such animals were humanely euthanized and excluded from analyses. Data were consistently reproduced in at least 3 independent experiments (except for figures 6A–E, 6J–K, S5A–B, S6B, S6E–F, for which 2 independent experiments were performed), with a minimum of 3 mice analyzed per group in each experiment, as indicated in the legends. All statistics were calculated in GraphPad Prism v8.0 software using two-tailed Mann-Whitney tests and are noted in figure legends. P values are expressed as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Data and Code Availability
This study did not generate datasets or code
Supplementary Material
Highlights.
M2-like granuloma macrophages are more permissive for Salmonella persistence
TNF signaling limits M2-like granuloma macrophages, thereby restricting Salmonella
Salmonella SteE promotes M2 phenotype to overcome TNF-mediated restriction
Acknowledgements
The authors thank the late Dr. Stanley Falkow, Dr. Manuel Amieva, and members of the Monack and Amieva laboratories for valuable discussions. Research reported in this publication was supported by grant R01-AI116059 from the NIAID (DM), the Pediatric Infectious Diseases Society Fellowship Award funded by Stanley A. Plotkin Sanofi Pasteur (TP), grant K08-AI143796 from the NIAID (TP), and the Maternal and Child Health Research Institute (TP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
All authors declare no competing interests.
REFERENCES
- Algood HM, Lin PL, Yankura D, Jones A, Chan J, and Flynn JL (2004). TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J Immunol 172, 6846–6857. [DOI] [PubMed] [Google Scholar]
- Allie N, Grivennikov SI, Keeton R, Hsu NJ, Bourigault ML, Court N, Fremond C, Yeremeev V, Shebzukhov Y, Ryffel B, et al. (2013). Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci Rep 3, 1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer-Santos E, Durkin CH, Rigano LA, Kupz A, Alix E, Cerny O, Jennings E, Liu M, Ryan AS, Lapaque N, et al. (2016). The Salmonella Effector SteD Mediates MARCH8-Dependent Ubiquitination of MHC II Molecules and Inhibits T Cell Activation. Cell Host Microbe 20, 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker LG, Freeman S, Murray PJ, Ryffel B, and Kaplan G (2001). TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 166, 6728–6734. [DOI] [PubMed] [Google Scholar]
- Bernut A, Nguyen-Chi M, Halloum I, Herrmann JL, Lutfalla G, and Kremer L (2016). Mycobacterium abscessus-Induced Granuloma Formation Is Strictly Dependent on TNF Signaling and Neutrophil Trafficking. PLoS Pathog 12, e1005986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Volkman HE, and Ramakrishnan L (2008). Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L, and Delamarre L (2014). Dendritic cell-targeted vaccines. Front Immunol 5, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couper KN, Blount DG, and Riley EM (2008). IL-10: the master regulator of immunity to infection. J Immunol 180, 5771–5777. [DOI] [PubMed] [Google Scholar]
- Dorhoi A, and Kaufmann SH (2014). Tumor necrosis factor alpha in mycobacterial infection. Semin Immunol 26, 203–209. [DOI] [PubMed] [Google Scholar]
- Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, Mukundan L, Chawla A, and Monack DM (2013). Salmonella require the fatty acid regulator PPARdelta for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 14, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueira R, Watson KG, Holden DW, and Helaine S (2013). Identification of salmonella pathogenicity island-2 type III secretion system effectors involved in intramacrophage replication of S. enterica serovar typhimurium: implications for rational vaccine design. MBio 4, e00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin TA, Zaoutis TE, and Zaoutis LB (2008). Beyond cat scratch disease: widening spectrum of Bartonella henselae infection. Pediatrics 121, e1413–1425. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Gideon HP, Mattila JT, and Lin PL (2015). Immunology studies in non-human primate models of tuberculosis. Immunol Rev 264, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MF, Roeske EK, Ward LN, Pengo T, Dileepan T, Kotov DI, and Jenkins MK (2018). Salmonella Persist in Activated Macrophages in T Cell-Sparse Granulomas but Are Contained by Surrounding CXCR3 Ligand-Positioned Th1 Cells. Immunity 49, 1090–1102 e1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath S, Hotson A, Johns J, Nolan G, and Monack D (2013). The systemic immune state of super-shedder mice is characterized by a unique neutrophil-dependent blunting of TH1 responses. PLoS Pathog 9, e1003408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, and Pluddemann A (2019). The Mononuclear Phagocytic System. Generation of Diversity. Front Immunol 10, 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, and Baral R (2017). Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol 316, 1–10. [DOI] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AC, and Bothwell PW (1967). Histological findings in human brucellosis. J Clin Pathol 20, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaslow SL, Gibbs KD, Fricke WF, Wang L, Pittman KJ, Mammel MK, Thaden JT, Fowler VG Jr., Hammer GE, Elfenbein JR, et al. (2018). Salmonella Activation of STAT3 Signaling by SarA Effector Promotes Intracellular Replication and Production of IL-10. Cell Rep 23, 3525–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings E, Thurston TLM, and Holden DW (2017). Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms And Physiological Consequences. Cell Host Microbe 22, 217–231. [DOI] [PubMed] [Google Scholar]
- Kerrinnes T, Winter MG, Young BM, Diaz-Ochoa VE, Winter SE, and Tsolis RM (2017). Utilization of host polyamines in alternatively activated macrophages promotes chronic infection by Brucella abortus. Infect Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Yang D, Kim M, Kim SY, Kim D, and Kang SJ (2017). A late-lineage murine neutrophil precursor population exhibits dynamic changes during demand-adapted granulopoiesis. Sci Rep 7, 39804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, McEvoy J, Roussel MF, Dyer MA, Qualls JE, et al. (2015). TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep 12, 1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, and Monack DM (2006). Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog 2, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, Grieser H, Chiosea I, Voitenek NN, Capuano SV, et al. (2010). Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum 62, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory FB (1898). A Histological Study of Typhoid Fever. J Exp Med 3, 611–638. [PMC free article] [PubMed] [Google Scholar]
- Marino S, Cilfone NA, Mattila JT, Linderman JJ, Flynn JL, and Kirschner DE (2015). Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect Immun 83, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni P, Skepper JN, and Hormaeche CE (1995). Effect of anti-tumor necrosis factor alpha antibodies on histopathology of primary Salmonella infections. Infect Immun 63, 3674–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, Via LE, Barry CE 3rd, Klein E, Kirschner DE, et al. (2013). Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol 191, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matty MA, Roca FJ, Cronan MR, and Tobin DM (2015). Adventures within the speckled band: heterogeneity, angiogenesis, and balanced inflammation in the tuberculous granuloma. Immunol Rev 264, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin LM, Govoni GR, Gerke C, Gopinath S, Peng K, Laidlaw G, Chien YH, Jeong HW, Li Z, Brown MD, et al. (2009). The Salmonella SPI2 effector SseI mediates long-term systemic infection by modulating host cell migration. PLoS Pathog 5, e1000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, and Chan J (2001). Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun 69, 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Bouley DM, and Falkow S (2004). Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J Exp Med 199, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah SM, and Nassar VH (1978). Enteric fever: a clinicopathologic study of 104 cases. Am J Gastroenterol 69, 63–69. [PubMed] [Google Scholar]
- Pagan AJ, and Ramakrishnan L (2018). The Formation and Function of Granulomas. Annu Rev Immunol 36, 639–665. [DOI] [PubMed] [Google Scholar]
- Petersen HJ, and Smith AM (2013). The role of the innate immune system in granulomatous disorders. Front Immunol 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessner HL, Lin PL, Kohno T, Louie JS, Kirschner D, Chan J, and Flynn JL (2007). Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J Infect Dis 195, 1643–1650. [DOI] [PubMed] [Google Scholar]
- Porta C, Riboldi E, Ippolito A, and Sica A (2015). Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol 27, 237–248. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan L (2012). Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 12, 352–366. [DOI] [PubMed] [Google Scholar]
- Refai A, Gritli S, Barbouche MR, and Essafi M (2018). Mycobacterium tuberculosis Virulent Factor ESAT-6 Drives Macrophage Differentiation Toward the Proinflammatory M1 Phenotype and Subsequently Switches It to the Anti-inflammatory M2 Phenotype. Front Cell Infect Microbiol 8, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu SK, Kumar M, Chakraborty S, Banerjee SK, Kumar R, Gupta P, Jana K, Gupta UD, Ghosh Z, Kundu M, et al. (2017). MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPbeta regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog 13, e1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba AE, Li L, Westermann AJ, Appenzeller S, Stapels DA, Schulte LN, Helaine S, and Vogel J (2016). Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat Microbiol 2, 16206. [DOI] [PubMed] [Google Scholar]
- Stapels DAC, Hill PWS, Westermann AJ, Fisher RA, Thurston TL, Saliba AE, Blommestein I, Vogel J, and Helaine S (2018). Salmonella persisters undermine host immune defenses during antibiotic treatment. Science 362, 1156–1160. [DOI] [PubMed] [Google Scholar]
- Storek KM, and Monack DM (2015). Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev 265, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam JW, Kullas AL, Mena P, Bliska JB, and van der Velden AW (2014). CD11b+ Ly6Chi Ly6G-immature myeloid cells recruited in response to Salmonella enterica serovar Typhimurium infection exhibit protective and immunosuppressive properties. Infect Immun 82, 2606–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tite JP, Dougan G, and Chatfield SN (1991). The involvement of tumor necrosis factor in immunity to Salmonella infection. J Immunol 147, 3161–3164. [PubMed] [Google Scholar]
- Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, and Ramakrishnan L (2010). Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, and Pamer EG (2015). Monocytes and infection: modulator, messenger and effector. Immunobiology 220, 210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code