

Protein modification with ISG15 acts cooperatively with IFIT proteins and preserves glucose homeostasis.
Abstract
Protein modification with ISG15 (ISGylation) represents a major type I IFN–induced antimicrobial system. Common mechanisms of action and species-specific aspects of ISGylation, however, are still ill defined and controversial. We used a multiphasic coxsackievirus B3 (CV) infection model with a first wave resulting in hepatic injury of the liver, followed by a second wave culminating in cardiac damage. This study shows that ISGylation sets nonhematopoietic cells into a resistant state, being indispensable for CV control, which is accomplished by synergistic activity of ISG15 on antiviral IFIT1/3 proteins. Concurrent with altered energy demands, ISG15 also adapts liver metabolism during infection. Shotgun proteomics, in combination with metabolic network modeling, revealed that ISG15 increases the oxidative capacity and promotes gluconeogenesis in liver cells. Cells lacking the activity of the ISG15-specific protease USP18 exhibit increased resistance to clinically relevant CV strains, therefore suggesting that stabilizing ISGylation by inhibiting USP18 could be exploited for CV-associated human pathologies.
INTRODUCTION
Early in an encounter between the pathogen and a cell, receptors located on the cell surface, in the cytosol, or in endosomal compartments engage with nucleic acid or non–nucleic acid pathogen-associated molecular patterns (PAMPs) to mount host responses to combat infection. Specific gene expression patterns and posttranscriptional control mechanisms combat pathogens on multiple levels and stages during disease development. As a first line of defense, cells produce type I interferons (IFNs), which orchestrate a complex defense network in both infected and noninfected cells (1). The ubiquitin family protein IFN-stimulated gene of 15 kDa (ISG15) and its conjugation machinery, comprising the E1-activating enzyme Ube1L, the E2-conjugating enzyme Ube2L6, and the Herc5 E3 ligase (Herc6 in mice), represent a bona fide example of such an IFN-induced, broad-spectrum antimicrobial system (1–4). Infection studies using ISG15- and Ube1L-knockout (ko) mouse models revealed that modification of target proteins with ISG15, a process known as ISGylation, is critical in the defense against pathogens including influenza B, Sindbis viruses, and Listeria monocytogenes (3, 5, 6). The antimicrobial function of ISG15 reported in mice is also present in human ISG15 with proven antiviral activity, for example, against influenza A and B virus, HIV, and human papilloma virus (1). ISGylation is a reversible process, and de-ISGylation is catalyzed by the ISG15-specific protease USP18 (7). In addition to its ISG15-specific protease activity, USP18 acts as a potent negative regulator of IFN signaling (8). Concordantly, complete deletion of USP18 causes an increase in the IFN response, whereas selective inactivation of its protease activity in USP18C61A/C61A (USP18C61A) mice increases the pool of ISGylated proteins without affecting the IFN signaling pathway (9). Notably, enhanced ISGylation in USP18C61A mice increases the antiviral activity against influenza B and vaccinia virus, even in comparison to wild-type controls (9). It has been reported that unconjugated ISG15 in human cells, but not in mouse cells, inhibits proteasomal degradation of USP18, thereby preventing uncontrolled activation of the IFN response (10, 11). Likewise, cells derived from ISG15-deficient patients exhibited reduced levels of USP18, accompanied by increased IFN signaling (10, 11). However, these species-specific differences between ISG15 and USP18 function remain poorly defined.
Furthermore, the underlying mechanisms how ISGylation operates within the context of a whole organism to protect it from infection are largely unknown. Cell culture–based studies from various laboratories provided insights into how ISG15 reduces the pathogenic burden during infection. One mode of action for ISG15-mediated reduction of the viral load involves a direct interference of ISG15 with viral proteins that are required for replication (12, 13). In other cases, ISG15 stimulates antiviral effector pathways of the host (14) or it controls exosome formation, which prevents pathogen spread (15). Moreover, ISG15 modifies endoplasmic reticulum and Golgi proteins to affect the secretion of cytokines (3, 16) and regulates the polarization and function of macrophages (17, 18). Thereby, ISGylation influences multiple important cellular processes needed for pathogen control. Nevertheless, ISG15 is not critical to counteract all kinds of pathogens (1), and it was clearly shown that free ISG15 can exert antimicrobial activity without forming covalent linkages to target substrates (2). Free, intracellular ISG15 can either interact with cellular proteins (19) or, as a secreted protein, bind to lymphocyte function–associated antigen–1 (LFA-1) receptor to stimulate IFN-γ secretion by natural killer (NK) cells (4, 20).
Our previous work documented that ISG15 prevents the manifestation of inflammatory cardiomyopathy (21), a chronic debilitating sequela of experimental coxsackievirus B3 (CV)–induced myocarditis in mice (22). CV disease has substantial medical and socioeconomic impact. During periods of high CV prevalence, particularly newborn infants and young children are at risk of life-threatening hepatitis, myocarditis, encephalomyelitis, and coagulopathy, resulting in multisystem sepsis (23). CV disease can be mimicked in mice, where, similarly to humans, CV triggers a multiphasic systemic disease that affects various organs at different stages with the possibility of viral genome persistence and chronic tissue injury (24). Early upon CV infection, mice show a robust systemic IFN response (25). At this stage, IFN signaling limits viral cytotoxicity and tissue destruction, particularly in the liver, and enhances the chances of survival (26, 27). Although not precisely characterized during CV infection of mice, antiviral responses of the host against CV infection are supported by an IFN-induced remodeling of cellular metabolism (28), with the liver having a central integrating role of systemic IFN input in the context of systemic inflammatory responses (29). The biosynthesis of molecules, needed for an efficient antiviral response against CV, consumes large proportions of cellular adenosine triphosphate (ATP), and in infected cells, IFNs induce processes that activate the uptake and turnover of glucose (28). After the first viremia, when systemic IFN production gradually declines, a second viremia and replication phase of CV emerges in heart tissue, culminating in inflammatory tissue damage and cardiac dysfunction (30). In this state, IFN signaling constrains CV pathology as well (31, 32). Despite the clear physiological relevance of IFN responses, molecular aspects of how ISG15 operates during CV infection remain elusive. Here, we report that protein ISGylation is required in nonhematopoietic cells to set these cells into an antiviral state. ISG15-mediated protection from viral cytotoxicity is accomplished by reprogramming the central liver metabolism, ensuring glucose homeostasis during infection, and an cumulative activity of ISG15 with the expression levels of other required antiviral effectors such as IFIT1/3 proteins. On the basis of these mechanistic aspects, we demonstrate the physiological relevance and therapeutic strategies of ISGylation for CV-associated human pathologies.
RESULTS
Multiphasic inhibition of the CV burden by ISGylation
The ISG15 system is activated by IFNs during the multistage course of CV infection (21, 27). The formation of ISG15 conjugates commenced in liver, pancreas, spleen, and heart tissue 1.5 days (36 hours) after CV infection, and protein ISGylation increased further upon progression of the disease (Fig. 1, A and B, and fig. S1A). In comparison to heart tissue, the protein expression level of free ISG15 dropped in liver tissue on day 6 (Fig. 1, A and B, and fig. S1A). In line with this, we detected differences in both liver and heart tissue concerning the load with infectious viral particles. While we found no infectious virus in the liver on day 6, in heart tissue, virus titers were elevated by this stage (Fig. 1, C and D). To distinguish between ISGylation-dependent functions and those mediated by free ISG15, we investigated the viral burden during CV infection in ISG15−/− and Ube1L−/− mice. Deletion of the ISG15-specific E1 enzyme Ube1L prevents ISGylation, with no relevant effect on the function of free ISG15 (fig. S1B) (33). When we compared wild-type, Ube1L−/−, and ISG15−/− mice, we detected similar viral titers in all organs 1.5 days after CV infection. In contrast, viral titers in the liver, pancreas, and spleen of both ISG15−/− and Ube1L−/− mice were significantly increased in comparison to wild-type controls after 3 days. ISGylation accelerated CV clearance from liver and spleen, as reflected by higher CV titers in ISG15−/− and Ube1L−/− mice compared to wild type after 6 days (Fig. 1C and fig. S1, C and D). Beyond day 3 after infection, we observed no contribution of ISGylation to viral clearance in the pancreas but found increased viral titer in ISG15−/− mice in comparison to wild-type controls and Ube1L−/− mice (fig. S1C). In heart tissue, viral titers increased after 1.5 days and remained at high levels up to day 8. Viral load in the heart from ISG15−/− and Ube1L−/− mice was increased 6 and 8 days after infection, illustrating that ISGylation also suppressed the second CV replication peak, which occurs in heart tissue (Fig. 1D). Together, protein ISGylation blocked virus replication in the liver, spleen, and pancreas most potently after 3 days of infection and in heart tissue after 6 days of infection.
Fig. 1. ISGylation suppresses CV titers at different phases of infection.
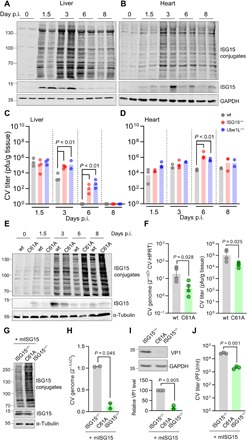
Wild-type (wt), ISG15−/−, and Ube1L−/− mice were infected with 1 × 105 pfu of CV Nancy and sacrificed at the indicated points in time post infection (p.i.). Tissue from (A) liver and (B) heart of wild-type mice was subjected to Western blot analysis using an ISG15-specific antibody. Each lane represents tissue homogenates obtained from a different animal. (C and D) Infectious viral particles were quantified in the respective organs obtained from wild-type, ISG15−/−, and Ube1L−/− mice by plaque assay during CV infection. Each dot represents a different animal; data are summarized as median values. Student’s t tests were conducted. P values of <0.05 are indicated in the graph. (E to J) USP18C61A and wild-type littermate controls were infected with 1 × 105 pfu of CV and sacrificed at the indicated points in time. (E) Heart tissue was homogenized and subjected to Western blot analysis for detection of ISG15. Each lane represents tissue obtained from a different animal, and the shown example for each group and point in time is representative for n = 3 mice. (F) At day 6 after infection, infectious viral particles were determined in heart by TaqMan qPCR and plaque assay. Each dot represents a different animal. Data are summarized as means ± SEM; t tests were performed, and P values of <0.05 are depicted. (G) Cardiomyocytes derived from USP18C61A ISG15−/− and ISG15−/− embryos were transduced with Ad5 vectors encoding murine ISG15 and stimulated with poly(I:C). Cellular lysates were subjected to Western blot analysis. (H to J) ISG15-rescued cardiomyocytes from USP18C61A ISG15−/− and ISG15−/− embryos were infected with CV at an MOI of 0.1 for 24 hours. (H) Total RNA was isolated to determine CV genome copy numbers by TaqMan qPCR; ΔΔCt values obtained from duplicates are shown for two independent experiments. Data are summarized as means ± SEM. (I) Viral load was determined in cellular lysates by Western blot analysis of CV VP1. Densitometric analysis of VP1 and the GAPDH loading control was performed to calculate the mean (± SEM) relative VP1 expression in three independent experiments. (J) Release of infectious viral particles was assessed by plaque assay. One representative out of three independent experiments yielding the same result is depicted. Unpaired t test (plaque assay) and one-sample t tests (VP1 and CV RNA) were performed, and P values are depicted.
Inactivation of ISG15-specific protease USP18 increases resistance to CV infection
To evaluate the physiological relevance and potential therapeutic strategies of ISGylation in the context of CV infection, we analyzed mice selectively lacking the activity of the ISG15-specific protease USP18 (USP18C61A/C61A). CV infection of these USP18C61A mice was characterized by enriched pools of ISGylated proteins, as previously reported (9), and is shown exemplarily for heart tissue (Fig. 1E) and liver (fig. S2A). The first phase of CV replication in USP18C61A was similar to wild-type controls (fig. S2B). In clear contrast, the second phase of viral replication was inhibited once de-ISGylation was blocked, as reflected by suppressed viral titers after 6 days of infection in heart tissue of USP18C61A mice (Fig. 1F). This finding prompted us to investigate the antiviral effect by USP18 protease inactivation in cell culture experiments. Therefore, USP18C61A mice were crossed with ISG15−/− mice, and primary cardiomyocytes were isolated from USP18C61A ISG15−/− embryos. ISGylation was induced in both USP18C61A ISG15−/− and ISG15−/− cells reconstituted with ISG15 by adenoviral transduction. This cell culture model nicely reflected the increased abundance of ISGylated proteins (Fig. 1G) that we observed in the heart of USP18C61A mice (Fig. 1E). Upon infection, hyper-ISGylation, caused by the lack of USP18 protease activity, substantially reduced viral replication. Lower viral genome copy numbers, reduced VP1 expression, and suppressed viral titers were detected in USP18C61A cardiomyocytes, in comparison to USP18 wild-type cardiomyocytes (Fig. 1, H to J). These results support our in vivo data obtained from USP18C61A mice.
Protection from CV pathology requires ISGylation in nonhematopoietic cells
Because chemotactic cytokines like CXCL-10 (34), proinflammatory cytokines like tumor necrosis factor–α (TNF-α) (35), interleukin-6 (IL-6) (36), and several other effector functions of innate myeloid cells, such as production of reactive oxygen species (ROS), nitric oxide (NO), or phagocytosis, are critical for CV clearance (37), we investigated whether ISG15 affects these processes. We found increased mRNA expression levels for IL-6, CXCL10, and CCL2 in both Toll-like receptor 3 (TLR3)–activated ISG15−/− bone marrow–derived macrophages (BMMs) and CV-infected ISG15−/− mice in comparison to wild-type controls (fig. S3, A and B). ROS (fig. S3, C and D) and NO (fig. S3E) production, mediated by TLR3, were similar in BMM regardless of whether they were derived from wild-type or ISG15−/− mice. We used three different approaches to investigate the role of ISG15 on the phagocytotic capacity of BMM and found that ISG15 had no effect on phagocytosis (fig. S3F). To gain a closer insight into the biological role of ISG15 in hematopoietic-derived immune cells during CV infection, we generated ISG15−/− bone marrow chimeras. Lethally irradiated ISG15−/− mice were reconstituted with bone marrow cells obtained from either wild-type or ISG15−/− mice (fig. S3G). Analysis of ISG15−/− recipients showed that CV titers in the liver, pancreas, spleen, and heart were identical in mice that were reconstituted with wild-type bone marrow cells (wild-type ➔ ISG15−/− chimeras) or with ISG15−/− bone marrow cells (ISG15−/− ➔ ISG15−/− chimeras) (Fig. 2A). We found that depletion of ISG15 in bone marrow–derived hematopoietic cells had no influence on the production of IL-6 and of CXCL10 and CCL2, as indicated by equal levels of mRNA expression of the respective cytokines in the spleen 3 days after infection (Fig. 2B). Collectively, our data show that ISG15 in immune cells is not required for control of CV infection.
Fig. 2. Protection from CV pathology requires ISGylation in nonhematopoietic cells.
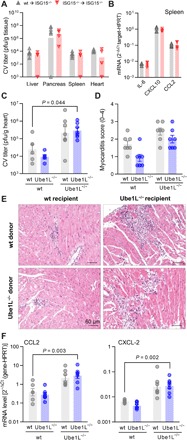
(A and B) ISG15−/− (CD45.2) mice were reconstituted with bone marrow cells from either ISG15−/− (CD45.2) or wild-type (wt, CD45.1) donors before CV infection, and mice were sacrificed 3 days after infection. (A) Infectious viral particles were quantified by plaque assay (wild-type ➔ ISG15−/−, n = 6; ISG15−/− ➔ ISG15−/−, n = 4). Data are summarized as median. (B) Splenic mRNA expression of the indicated cytokines and chemokines was determined by TaqMan qPCR. (C to F) Chimeric wild-type and Ube1L−/− mice were generated upon transfer of wild-type or Ube1L−/− bone marrow cells into lethally irradiated wild-type or Ube1L−/− recipients, respectively. Mice were infected with CV and sacrificed after 8 days (n = 7 in all four groups). (C) Infectious viral particles were quantified in heart tissue by plaque assay. Data are summarized as means ± SEM. (D) Myocarditis was scored microscopically by a blinded pathologist based on cardiac hematoxylin and eosin staining. (E) Representative histopathologic stains of heart tissue of each group are shown. (F) mRNA levels of the indicated genes in heart tissue were determined by TaqMan qPCR. Unequal variance versions of two-way ANOVA were performed, followed by a Sidak-Holm’s multiple comparison test. Data were summarized as means ± SEM if applicable.
On the basis of these findings, we hypothesized that ISG15 mediates protection from CV pathology by protein ISGylation in nonhematopoietic cell types and tissues. To test this hypothesis, we generated Ube1L−/− and wild-type bone marrow chimeras. Whenever lethally irradiated wild-type recipients were reconstituted with bone marrow cells from Ube1L−/− mice (Ube1L−/− ➔ wild-type), we obtained mice where protein ISGylation could be triggered in non–bone marrow–derived somatic cell types and tissues only but not in hematopoietic-derived immune cells. Alternatively, in Ube1L−/− chimeras reconstituted with bone marrow cells from wild-type mice (wild-type ➔ Ube1L−/−), protein ISGylation could be exclusively induced in hematopoietic-derived immune cells. Because of a deletion of the E1-activating enzyme Ube1L, ISGylation was not possible in non–bone marrow–derived cells (fig. S4). Infection studies using this experimental setup and respective controls (wild-type ➔ wild-type and Ube1L−/− ➔ Ube1L−/−) allowed us to distinguish whether ISGylation in non–bone marrow–derived somatic cell types is required for suppression of CV-mediated tissue injury. Upon infection of these chimeras, CV titers were found to be increased in the hearts of Ube1L−/− mice that had been reconstituted with bone marrow cells from Ube1L−/− mice (Ube1L−/− ➔ Ube1L−/−) when compared to wild-type mice reconstituted with bone marrow cells from wild-type mice (wild-type ➔ wild-type) (Fig. 2C). The increased CV load found in Ube1L−/− ➔ Ube1L−/− mice resulted in high-grade inflammation and tissue destruction of the heart (Fig. 2, D and E), as well as increased chemokine expression (Fig. 2F). These results demonstrate the protective role of protein ISGylation to mitigate CV-triggered pathology. As demonstrated by equally high viral titers in Ube1L−/− chimera reconstituted with wild-type bone marrow cells (wild-type➔ Ube1L−/−), the aggravated phenotype detected in Ube1L−/−➔ Ube1L−/− mice was not improved once immune cells were equipped with an operative ISGylation machinery (Fig. 2, C to F). This argues against a biological role of protein ISGylation in bone marrow–derived immune cells during CV infection. Vice versa, in comparison to wild-type ➔ wild-type mice, knockdown of the E1-activating enzyme Ube1L specifically in bone marrow cells (Ube1L−/− ➔ wild-type) had no effect on CV titers or on inflammatory injury (Fig. 2, C to F). Together, our data demonstrate that ISGylation in non–bone marrow–derived somatic cell types is essential to prevent viral cytotoxicity and inflammatory tissue damage during CV infection.
ISGylation increases expression levels of IFIT1/3 proteins
To identify molecular mechanisms involved in how protein ISGylation suppresses the viral titer in cells targeted by viral infection, we performed protein profiling on infected liver tissue using mass spectrometry (MS)–based proteomics. In line with published data (32, 38), our proteomics analysis confirmed that, in addition to ISG15, IFN-induced proteins with tetratricopeptide repeats (IFIT) 1 and 3 are among the most intensively up-regulated proteins during CV infection (Fig. 3A). Because different ISGs often act cooperatively (39) and IFIT proteins are required for control of CV infection (32), our analysis focused, in particular, on these antiviral effectors. Notably, an MS–based search for ISG15 substrates in HeLa cells identified IFIT family proteins as targets of ISG15 (13, 40). On the basis of this, we tested ISGylation of IFIT proteins and performed pull-down experiments using cellular lysates from IFN-β–treated FLAG-ISG15-HeLa cells. Higher molecular weight versions of IFIT1 and IFIT3 proteins were detected within the pool of enriched ISGylated proteins (Fig. 3B), which appeared to be ISGylation of IFIT1 and IFIT3. To be sure, we transfected FLAG-tagged IFIT1/3 into HeLa cells along with ISG15 and plasmids encoding the E1/E2/E3 proteins. Pull down of both IFIT1 and IFIT3 revealed higher molecular weight versions of both IFIT proteins, thereby confirming ISGylation of IFIT1 and IFIT3 (Fig. 3C).
Fig. 3. ISGylation increases expression levels of IFIT1/3 proteins.
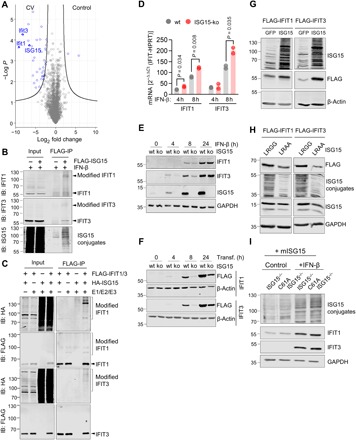
(A) Liver samples obtained from wild-type (wt) mice (n = 3) before and 3 days after infection were subjected to LC-MS/MS analysis. In the depicted volcano plots, each protein identified at baseline and after CV infection in the proteome screen of liver tissue is represented by a dot. The x axis specifies log2 fold changes and the y axis specifies −log10 of P values obtained from t test. (B) 3×FLAG-6×His-ISG15-HeLa cells and controls were stimulated with IFN-β for 24 hours, and cellular lysates were subjected to FLAG immunoprecipitation (IP). Total cell lysates (input) and FLAG-IP enriched proteins were immunoblotted using antibodies for ISG15, IFIT3, and IFIT1. Data are representative of three independent experiments. (C) HeLa cells transfected with a four-plasmid combination (HA-ISG15, Ube1L, Ube2L6, and Herc5) and with either FLAG-tagged IFIT1 or IFIT3 were subjected to pull-down experiments using anti-FLAG agarose beads. Total cell lysates and FLAG-immunoprecipitated proteins were subjected to immunoblotting using antibodies for FLAG and HA. Data are representative of three independent experiments. (D and E) ISG15-ko HeLa cells generated by CRISPR-Cas9–mediated gene editing and respective control cells were stimulated with IFN-β (100 U/ml). IFIT1/IFIT3 transcripts and IFIT1/IFIT3 protein expression levels were determined by (D) TaqMan qPCR and (E) Western blot analysis. One representative of three independently performed experiments is shown. Unpaired t tests were conducted. (F) ISG15-ko HeLa cells and controls were transfected with plasmids encoding either FLAG-tagged IFIT1 or IFIT3, and IFIT expression was determined by Western blot analysis. Date are representative of three independent experiments. (G) ISG15-ko HeLa cells were transfected with either GFP or ISG15 cDNA together with IFIT1 or IFIT3 expression plasmids, respectively. Subsequently, cells were stimulated with IFN-β for 24 hours, and IFIT protein expression levels were determined by Western blot analysis. Data are representative of two independent experiments. (H) Following the experimental approach outlined in (G), ISG15-ko cells were transfected with either ISG15 or the unconjugatable ISG15-LRAA mutant as well as with IFIT1/3 cDNA. IFIT protein expression was determined after 24 hours by Western blot analysis. Data are representative for three independent experiments. (I) Primary cardiomyocytes were generated from USP18C61A ISG15−/− and ISG15−/− mice, and ISGylation was induced by transduction of Ad5 vectors encoding mISG15. Cells were left unstimulated or stimulated with IFN-β (100 U/ml) to induce the expression of endogenous IFIT1 and IFIT3.
To explore the proposed additive activity of ISG15 and IFIT proteins, we investigated how ISG15 alters IFIT1/3 protein expression, if at all. We used ISG15-ko HeLa cells generated by CRISPR-Cas9–mediated gene editing. Stimulation of the IFNAR (interferon-a/b receptor) induced higher IFIT1/3 transcript levels in ISG15-ko HeLa cells compared to wild-type controls (Fig. 3D). In clear contrast, the protein expression levels of IFIT1 and IFIT3 were increased in wild-type cells, which did not reflect increased transcript levels found in ISG15-ko cells (Fig. 3E). Using FLAG-tagged IFIT1/3 complementary DNA (cDNA) constructs in the same cell culture model, we obtained similar results, confirming profoundly increased IFIT1/3 protein expression in wild-type cells in comparison to ISG15-ko HeLa cells (Fig. 3F). Moreover, small interfering RNA (siRNA)–mediated knockdown of ISG15 in HeLa cells resulted in decreased IFN-induced IFIT1/3 expression in comparison to control cells (fig. S5). To explore whether reduced protein expression of IFIT1/3 in ISG15−/− cells can be restored by ISG15, we investigated IFIT protein expression in ISG15-ko cells, with and without the restoration of ISG15 by exogenous ISG15 expression. Again, induction of ISG15/ISGylation resulted in increased IFIT1 and IFIT3 protein expression levels (Fig. 3G), thereby proving ISG15-mediated increased expression of IFIT1/3 proteins at the posttranscriptional level.
Our finding that ISG15 increases protein expression levels for IFIT1/3, despite reciprocal effects on IFIT transcription, prompted us to question whether the stabilizing effect of ISG15 on IFIT1/3 protein expression involves ISGylation. To verify a role for ISGylation relating to IFIT expression levels, we transfected ISG15-ko HeLa cells with either hISG15-LRGG (wild-type) or an unconjugatable hISG15-LRAA mutant. IFIT protein expression was induced in these cells, and we found higher IFIT1 and IFIT3 protein expression levels in cells with an operative ISGylation machinery compared to cells expressing hISG15-LRAA mutants (Fig. 3H), confirming that ISGylation increases IFIT1 and IFIT3 protein expression. Further supportive evidence for a specific contribution of ISGylation to IFIT1/3 protein expression comes from cell culture experiments using primary cardiomyocytes from USP18C61A ISG15−/− mice. Both USP18C61A ISG15−/− and ISG15−/− cells were resupplied with ISG15 activity by adenoviral transduction to trigger ISGylation with detection of higher ISGylation levels in cells from USP18C61A ISG15−/− mice. Upon induction of IFIT1/3 expression by IFN, the expression levels of IFIT1/3 were elevated in ISG15-reconstituted USP18C61A ISG15−/− cells compared with ISG15−/− cells (Fig. 3I), thereby verifying the stabilizing role of ISGylation on IFIT1/3 proteins. Together, our findings prove that the ISG15 system controls protein expression levels for IFIT1/3 at posttranscriptional level through its capacity to form covalent protein conjugates.
ISG15 reprograms central liver metabolism during CV infection
Viral infection with the subsequent activation of host defense pathways alters the cellular demands for ATP, requiring remodeling of central metabolic processes (29, 41). In addition, we found that HeLa cells infected with CV increased their glucose consumption, showing, together with a higher release of lactate, elevated glycolysis rates in infected cells. Compared to control cells, glucose consumption and release of lactate were significantly increased in infected ISG15-ko HeLa cells (Fig. 4A). IFN treatment, which inhibits CV replication in cell culture studies (32), decreased glucose consumption back to control levels in both wild-type and ISG15-ko cells (Fig. 4A). In the context of a whole organism, in addition to elevated glucose uptake by infected cells, alternative mechanisms such as an impaired function of the exocrine pancreas (42) may substantiate the requirement for metabolic reprogramming after CV infection. We found a significant decline of blood glucose and triglyceride concentrations after CV infection in both wild-type and ISG15−/− mice (Fig. 4B). Free fatty acids (FFAs), reflecting mobilized sources of stored triglycerides, also declined (Fig. 4B). We quantified glucose and its branched storage form in the liver and found a significant reduction of both glucose and glycogen after CV infection (Fig. 4, C and D), providing further evidence for infection-triggered hypoglycemia. Together, these findings indicate that, after CV infection, mice acquire a hypoglycemic state with an increased energy demand accompanied with a malnutrition phase and lower glucose store in the liver.
Fig. 4. ISG15 influences the metabolic state in healthy mice and during CV infection.
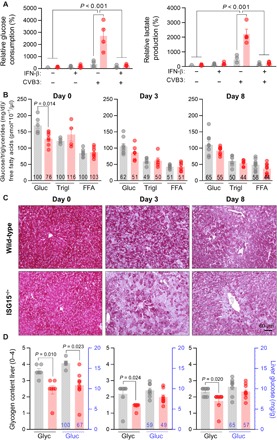
(A) Unstimulated or IFN-β pretreated, ISG15-ko HeLa, and control cells were infected with CV (MOI 1.0). After 32 hours, the glucose and lactate concentrations were determined in cell culture supernatants. Values normalized to cell counts per well are presented as increase relative to the untreated wild-type control. The data of four independent experiments have been summarized as means ± SEM. A two-way ANOVA test was performed followed by a Sidak’s multiple comparison test. (B to D) Wild-type (wt) and ISG15−/− mice sacrificed before (wild-type, n = 7; ISG15−/−, n = 7), 3 days (wild-type, n = 8; ISG15−/−, n = 6), and 8 days (wild-type, n = 9; ISG15−/−, n = 8) after CV infection. (B) Blood glucose and serum triglycerides/FFAs at baseline and 3 and 8 days after CV infection in wild-type and ISG15−/− mice summarized as the means ± SEM. Numbers in bar graphs are the percentage of the metabolite compared to the mean value in naive wild-type mice. Multiple t tests with Holm-Sidak multiple comparison correction were performed. (C) Glycogen content visualized in liver tissue section with periodic acid Schiff reaction. Representative images are shown. (D) Glycogen content scored semiquantitatively from 0 to 4 based on histological analysis and glucose concentration assessed in liver tissue homogenates. Bars are means ± SEM. Multiple t tests with Holm-Sidak multiple comparison correction were performed; P values are depicted.
A comparison of glucose and glycogen in blood and liver of ISG15−/− mice and wild-type controls revealed a hypoglycemic state in ISG15−/− mice already before infection (Fig. 4B), reflected by both a reduced blood glucose level and a lower glycogen and glucose content in the liver (Fig. 4, C and D). Early after CV infection, blood and liver glucose declined to lower levels in ISG15−/− mice with a substantially reduced glycogen content in liver tissue compared to wild-type mice, demonstrating a role of ISG15 for glucose homeostasis in CV infection. To obtain more insights into the function of ISG15 on central liver metabolism during infection, we investigated how CV infection affects the protein abundance of key metabolic enzymes in liver tissue from ISG15−/− mice and wild-type controls. Quantitative proteomic data were obtained from shotgun liquid chromatography–tandem MS (LC-MS/MS) analysis of liver samples derived from wild-type and ISG15−/− mice at baseline as well as 3 and 8 days after infection. Using a label-free quantitative setup, 3982 proteins were identified by LC-MS/MS analysis, of which 1094 proteins were found to be significantly up- or down-regulated. Cluster analysis revealed six distinct protein profiles in both wild-type and ISG15−/− mice during CV infection (Fig. 5A). A direct comparison of the different protein clusters revealed up to 73% of the identified proteins to be regulated differentially in wild-type and ISG15−/− mice (fig. S6A). Gene Ontology (GO) enrichment analysis of these clusters uncovered a reduction of proteins involved specifically in FA oxidation and oxidative phosphorylation (OXPHOS) in the absence of ISG15 at an early stage of infection. In wild-type mice, these alterations of protein levels were only observed in later stages of infection (Fig. 5B). Enzymes of carbohydrate metabolism involving key enzymes of glycolysis, such as fructose-1,6-bisphosphate-aldolase, pyruvate kinase, or lactate dehydrogenase, were up-regulated in wild-type mice at this advanced state of infection (Fig. 5B).
Fig. 5. ISG15 reprograms the central liver metabolism during CV infection.
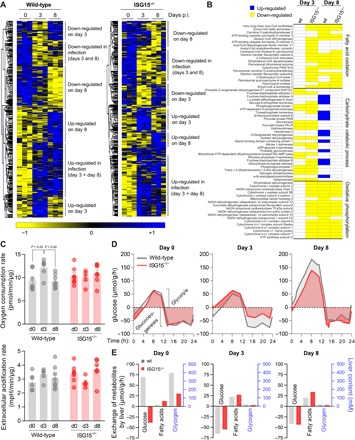
(A) Hepatic tissue obtained from wild-type (wt) and ISG15−/− mice (n = 3) during early (day 3) and late (day 8) state of CV infection was subjected to LC-MS/MS analysis. Heatmaps summarizing all differentially regulated hepatic proteins during infection for both strains are depicted. The relative abundance of each protein is color-coded based on the z score normalized log2-transformed LFQ intensities. Blue color indicates proteins of high abundance, and yellow color indicates proteins of low abundance as compared to row means. A hierarchical clustering resolved six distinct clusters, with annotation shown on the right. (B) Heatmap-based clusters were subjected to Gene Ontology (GO) analysis, and proteins involved in selected enriched metabolic GO terms with catabolic ATP-generating function (FA oxidation, carbohydrate catabolic process, and OXPHOS) are depicted at an early and late state of CV infection, applying the same color code as used in (A) (blue, up-regulation; yellow, down-regulation). If the GO term of interest was not found within a dataset, individual proteins were not plotted. (C) At the indicated time points of infection, liver biopsies were obtained from wild-type and ISG15−/− mice. The basal oxygen consumption (top) and extracellular acidification (bottom) rates were monitored using a Seahorse Biosciences extracellular flux analyzer. Values were normalized to protein content in the biopsies. Data of at least six mice per group were summarized as means ± SEM. A one-way ANOVA was performed followed by a Tukey’s multiple comparison test. (D and E) Liver proteome data together with HEPATOKIN1, a model of central liver metabolism (43), were used to assess the metabolic alterations in liver tissue of wild-type and ISG15−/− mice during viral infection. Metabolic models for the different conditions were constructed by scaling the maximal activity for each enzyme using the LFQ intensities for each protein obtained from MaxQuant analysis at the respective point in time. (D) For a standard 24-hour profile metabolite plasma profile, diurnal glucose exchange fluxes were simulated in wild-type and ISG15−/− mice at each time point of viral infection. Negative exchange fluxes indicate net release from the liver to the plasma (gluconeogenesis), while positive values indicate hepatic glucose uptake (glycolysis). (E) For each condition, experimentally determined blood glucose levels as depicted in Fig. 4B were used as model input to calculate realistic exchange fluxes and glycogen levels.
As a next step, we investigated whether these altered protein abundances of metabolic enzymes or proteins regulating OXPHOS and carbohydrate metabolism in ISG15−/− mice influence mitochondrial metabolism of liver tissue during CV infection. We used the Seahorse metabolic assay platform to monitor both the oxygen consumption and extracellular acidification rate as parameters for mitochondrial metabolism in liver biopsies obtained from ISG15−/− mice and wild-type controls 3 and 8 days after CV infection. Three days after CV infection, liver tissue of wild-type mice had a higher oxygen consumption and extracellular acidification rate compared to uninfected controls; however, both parameters remained at baseline levels in ISG15−/− mice at this stage (Fig. 5C). After 8 days, oxygen consumption and extracellular acidification rate reached baseline levels in wild-type mice, with no alteration found in ISG15−/− mice (Fig. 5C). Together, these functional data show that ISG15 activates oxidative mitochondrial metabolism in liver tissue early after CV infection.
Although alterations in enzyme abundances as detected by LC-MS/MS analysis and functional investigation of liver tissue by Seahorse technology deliver substantial information on specific metabolic reactions, the biomedical challenge is to integrate these data into a modeling platform that enables in silico prediction of organ metabolism. To determine how ISG15 affects liver metabolism during CV infection, we integrated various effectors of metabolism by computing proteome data for metabolic enzymes during CV infection using HEPATOKIN1, a multiscale kinetic model of central liver metabolism (43). First, we assumed a 24-hour standard metabolite profile to compute the mean diurnal metabolic effort of different metabolic pathways after infection under standardized conditions. Our simulations show that the metabolic alterations progress during the stages of CV infection. In line with the aforementioned findings in the GO enrichment analysis, FA oxidation increases specifically in wild-type mice during the early phase of CV infection. FA synthesis rates were reduced during infection, yielding similar values in naive controls and 3 days after infection in both wild-type and ISG15−/− mice, respectively (fig. S6B). Glucose production in wild-type controls maintained at baseline levels early upon infection, whereas these processes were reduced in ISG15−/− mice, revealing a lower capacity for hepatic glucose production in the absence of ISG15 (Fig. 5D and fig. S6B). After 8 days, the glycolytic capacity of liver tissue increased in both wild-type and ISG15−/− mice. This adaptation of carbohydrate metabolism was more pronounced in wild-type mice (Fig. 5D and fig. S6B). Overall, these data demonstrate that ISG15 influences the central liver metabolism at the different stages of CV infection by increasing the capacity of liver tissue for endogenous glucose production during the early stage and by equipping the liver with an enzymatic machinery needed for efficient glycolysis at later stages of CV infection.
We used the metabolic model to investigate how ISG15 affects exchangeable metabolites in liver tissue before and during infection for strain- and disease-specific stage conditions. Instead of standardized conditions, we used experimentally determined blood glucose levels as the data input into the model to determine the metabolic response of the liver in both wild-type and ISG15−/− mice in terms of glucose and FA exchange flux as well as hepatic glycogen content (Fig. 4C). Under baseline conditions, we found that the healthy liver of wild-type control mice is a net consumer of glucose. At 170 mg/ml, which is the mean nonfasting blood glucose detected in this group (Fig. 4B), the liver consumes glucose with a computed rate of 69 μmol g−1 hour−1 (Fig. 5E and fig. S6C). In contrast, naïve ISG15−/− mice are not net glucose consumer but instead produce glucose at a rate of 38 μmol g−1 hour−1 under baseline conditions. A 50% reduction of the glycogen content in liver tissue of ISG15−/− mice calculated by metabolic modeling (Fig. 5E) corresponds to the lower glycogen store shown by histologic examination of liver tissue from hypoglycemic ISG15−/− mice (Fig. 4, C and D). Early after CV infection (day 3), when blood glucose levels drop significantly (Fig. 4B), the liver responds with a pronounced shift toward glucose production, particularly in wild-type mice. The computed glucose production rate by liver tissue was 66 μmol g−1 hour−1 in wild-type mice and 55 μmol g−1 hour−1 in ISG15−/− mice (Fig. 5E and fig. S6C). Eight days after infection, when blood glucose levels remain hypoglycemic but gradually shift back to normal (110 mg/ml in wild-type mice and 93 mg/ml in ISG15−/− mice; Fig. 4B), the rates of gluconeogenesis decrease slightly (42 μmol g−1 hour−1 in wild-type mice and 44 μmol g−1 hour−1 in ISG15−/− mice). The computed glycogen content of liver tissue was reduced during CV infection, corresponding to experimental findings 3 and 8 days after infection. Carbohydrate metabolism in liver tissue is closely linked to FA metabolism. During CV infection, the catabolic phase of acute viral infection is accompanied by reduced levels of FFA in mouse serum (Fig. 4B), reflected by the increase in uptake of FA by liver tissue (Fig. 5E). Compared to wild-type mice, ISG15−/− mice had a higher uptake of FA by liver tissue during CV infection (Fig. 5E). In conclusion, ISG15 ensures efficient storage of glucose in liver tissue of healthy mice and reprograms liver metabolism toward greater glucose production early after CV infection.
Identification of the antiviral capacity of human ISG15 for CV infection
Our finding that inactivation of the ISG15-specific protease USP18 renders cells more resistant to CV infection (Fig. 1, E to J) strongly suggests that stabilizing ISGylation by USP18 inhibition could be exploited therapeutically. Therefore, we asked whether the antiviral activity of ISGylation observed in the mouse also applies to the human system. First, CV-permissive HeLa cells, in which ISG15 expression was shut down by CRISPR-Cas9 gene editing, were used to explore the role of human ISG15 during CV infection. Viral replication, as monitored by the detection of both the CV structural protein VP1 and infectious viral titer, was increased in ISG15-ko cells compared to control cells (Fig. 6A). Correspondingly, a transient knockdown of ISG15 achieved by siRNA technology resulted in higher VP1 production in infected cells (Fig. 6B). In a reverse approach, HeLa cells with stable ISG15 overexpression (3) were infected with CV, resulting in reduced VP1 protein expression levels in comparison to control cells (Fig. 6C). In a complementary approach, human ISG15 was transduced into primary cardiomyocytes obtained from ISG15−/− mice, and together with ISG15−/− controls, these human ISG15-expressing cells were infected with CV. Cells expressing human ISG15 showed a significantly decreased production of VP1 and a lower virus titer (Fig. 6D). Last, we asked whether inactivation of the ISG15-specific protease USP18, achieved by the C61A mutation (9), might be used to increase the antiviral activity exerted by human ISG15. Therefore, ISGylation was restored in cardiomyocytes obtained from USP18C61A ISG15−/− or ISG15−/− mice by adenoviral vector transduction of human ISG15, resulting in hyper-ISGylation in USP18C61A ISG15−/− cells. After CV infection, as reflected by both profoundly reduced VP1 production and lower viral titer in USP18C61A ISG15−/− cardiomyocytes with reconstituted hISG15 expression, enhanced linkage of human ISG15 to target proteins conferred increased antiviral activity (Fig. 6E). Together, these results show that there is no particular human/mouse species barrier in the role of ISGylation counteracting CV infection.
Fig. 6. Human ISG15 suppresses CV replication.
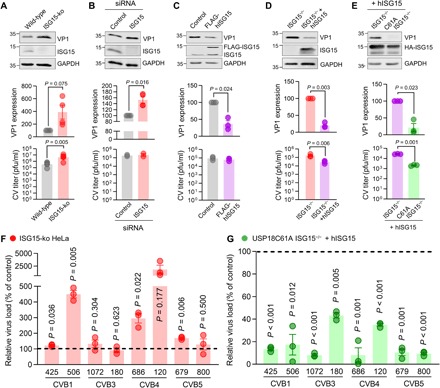
(A) ISG15 expression was deleted in HeLa cells using CRISPR-Cas9 gene editing as described in Fig. 3. ISG15-ko cells and wild-type cells were infected with CV (MOI 0.1) for 16 hours. Expression of CV VP1 was determined by Western blot analysis in four independent experiments, and the obtained signal normalized to GAPDH was compared to wild-type samples. Infectious virus particles were quantified in five independent experiments by plaque assay. Data are summarized as means ± SEM. (B) HeLa cells were transfected with siRNA targeting human ISG15 or a nontargeting control siRNA. Cells were subsequently infected with CV (MOI 0.01) for 16 hours. VP1 protein expression was determined and normalized to the control sample in four independent experiments. Plaque assays were performed in two independent experiments. Data are summarized as described in (A). (C) HeLa cells stably expressing FLAG-tagged human ISG15 and respective control cells were infected with CV (MOI 0.1) for 16 hours. VP1 expression and virus titer were determined as described in (A) in three independent experiments. (D) Primary embryonic cardiomyocytes obtained from ISG15−/− mice were transduced with Ad5 vectors expressing human ISG15 (hISG15) or control for 48 hours at MOI 25 before CV infection (MOI 0.1) for 24 hours. VP1 levels were determined by Western blot analysis, and infectious viral particles were quantified by plaque assay in three independent experiments. (E) Cardiomyocytes derived from USP18C61A ISG15−/− and ISG15−/− embryos were transduced with Ad5 vectors encoding hISG15 and infected with CV in three independent experiments for detection of the viral load by Western blot analysis of VP1 as well as plaque assay. One-sample t tests were performed for all summarized VP1 data. Unpaired t tests were conducted for all plaque assay data. (F and G) CVB isolates were obtained from patients presenting with neurological symptoms that may have been of infectious origin. (F) ISG15-ko HeLa cells and control cells were infected with the indicated CV serotypes (MOI 0.1), and infectious virus particles were determined after 16 hours by plaque assay. The relative increase of the viral titer in ISG15-ko HeLa cells as compared to control cells is depicted for a representative experiment. Three independent experiments demonstrated similar results. (G) The ISG15 system was induced in USP18C61A ISG15−/− and ISG15−/− cardiomyocytes by transduction of Ad5 vectors encoding hISG15. Cells were infected with CV serotypes as follows: CVB1 425, MOI 1 (2 days); CVB1 506, MOI 1 (1 day); CVB3 1072, MOI 10 (2 days); CVB3 180, MOI 10 (1 day); CVB4 686, MOI 10 (2 days); CVB4 120, MOI1 (2 days); CVB5 679, MOI 1 (1 days); CVB5 800, MOI 10 (1 day). Infectious viral particles were quantified by plaque assay. Relative reduction of the viral load in USP18C61A ISG15−/− cells as compared to ISG15−/− cardiomyocytes with restored ISG15 expression is depicted for a representative of at least three independent experiments. Data are means ± SEM; one-sample t tests were performed, and P values are depicted.
Targeting ISGylation as an antiviral approach for clinical CVB isolates
Our results demonstrated that ISGylation is an effective antiviral effector operative on a laboratory CV strain. Because a laboratory viral strain might differ from clinically relevant viruses, we tested the latter for their sensitivity to ISG15. Two isolates of the CV serotypes CVB1, CVB2, CVB3, and CVB5 were obtained from patients undergoing clinical investigation due to suspected meningitis or encephalitis. To investigate whether ISG15 influences the replication of these CVB serotypes, after virus propagation in cell culture, we infected ISG15-ko HeLa cells and wild-type controls with the clinical strains. This approach enabled us to study the effect mediated by ISG15 expression under baseline conditions, because ISG15 is induced neither by CV infection nor by IFN before infection, as this would completely block the replication of CV (27). After two replication cycles, viral titers were determined for each serotype by plaque assay. As depicted in Fig. 6F, in comparison to wild-type cells, we found an increased viral load in ISG15-ko cells for one of the CVB5 and both of the CVB1 and CVB4 isolates, confirming an antiviral activity of human ISG15 for these clinical CV strains. This finding prompted us to evaluate whether a therapeutic strategy of enhancing ISGylation might be used for the clinical CVB serotypes. Therefore, we made use of our cell culture system, isolating USP18C61A ISG15−/− and ISG15−/− cardiomyocytes and restoring ISGylation by adenovirus vector–mediated transduction of human ISG15, as outlined above (Fig. 6E). After triggering hyper-ISGylation in cells from USP18C61A ISG15−/− and ISGylation in cells from ISG15−/− mice, cells were infected with the CVB1, CVB2, CVB3, and CVB5 serotypes, and viral titers were determined by plaque assay. Hyper-ISGylation originating from USP18 protease inactivation significantly reduced the viral load for all CV serotypes, with some strains being more vulnerable to this approach than others, yielding an overall reduction of viral titers to 18.1 ± 4.8% in comparison to control cells (Fig. 6G). In conclusion, enhancing ISGylation inhibited the replication of all clinical CV isolates tested.
DISCUSSION
The findings of our study are briefly summarized in Fig. 7. Our data indicate that ISG15 inhibits viral cytotoxicity during CV infection by suppressing the viral burden both at an early stage in liver, pancreas, and spleen as well as at later stages in heart tissue, thereby mitigating viral pathology and attenuating inflammatory tissue damage. Our results demonstrate that protein ISGylation is required in nonhematopoietic cells to promote cellular resistance to CV infection. Both ablation of the ISG15 gene and deficiency of the E1 enzyme Ube1L increased viral titers equally at the different stages of CV infection. This finding emphasizes the physiological requirement of ISGylation in vivo, an important aspect regarding the increasing number of reports on various immunomodulatory functions of free ISG15 and their protective role in infectious disease (1). ISG15, in its unconjugated form, activates IFN-γ production by NK cells (4, 20). However, IFN-γ secretion by NK cells is not affected in Ube1L−/− mice, which express free ISG15 but are ISGylation incompetent (21). Apart from an advanced stage of pancreatic injury (day 6), Ube1L−/− mice exhibited the same CV pathology as ISG15−/− mice. The finding that viral titers are higher in ISG15−/− mice in comparison to both wild-type and Ube1L−/− mice in the pancreas at day 6 might be indicative of a contribution of free ISG15 to combating CV infection. On the other hand, viral cytotoxicity attacks the pancreas as early as between day 1 and day 2 after infection (25), and Ube1L−/− mice showed similarly increased viral titers as ISG15−/− mice 3 days after infection. Therefore, we conclude that free ISG15-triggered IFN-γ release by NK cells is of minor biological relevance in counteracting CV-triggered pathology. Moreover, ISG15 influences other functions of innate immune cells, such as phagocytosis and cytokine production by macrophages (17, 18). Together with their progenitors, these immune cells profoundly aggravate CV-triggered inflammatory pathology (37). Most data on the role of ISG15 in macrophages during viral infection are derived from in vitro infection studies with vaccinia virus, where ISG15-triggered reprogramming of macrophages combats the infection (17, 18). Other reports provide evidence that protein ISGylation exacerbates inflammatory responses (3, 16). In accordance with studies using vaccinia virus (18), here, we demonstrate that ISGylation suppresses cytokine production during CV infection. In our hands, ISG15 had no effect on phagocytosis, ROS, and NO production by macrophages. Moreover, the absence of ISG15 did not affect manifestation of experimental autoimmune myocarditis (AM) (fig. S8), which is also affected profoundly by chemotactic cytokines produced by monocytes/macrophages (44). Correspondingly, the various bone marrow chimera experiments shown in this study unequivocally attributed the biological activity of ISG15 to ISGylation in non–bone marrow–derived cells and tissues. Similar to influenza B virus (5), protein ISGylation during CV infection sets nonhematopoietic cells into an antiviral state, thereby reducing CV-triggered cytotoxicity and consequently inhibiting the production of proinflammatory mediators. This differentiates the function of ISG15 for CV-induced pathology from other IFN-dependent pathogens. There are other examples, such as influenza A and Sindbis viruses, where ISGylation is protective, but this phenotype is seen regardless of pathogen titer (45). During infection with chikungunya virus, however, ISG15 protects the host from virus-triggered injury of lung tissue, independent of its protein modifier function (2).
Fig. 7. Protein modification with ISG15 acts cooperatively with IFIT proteins and preserves glucose homeostasis.
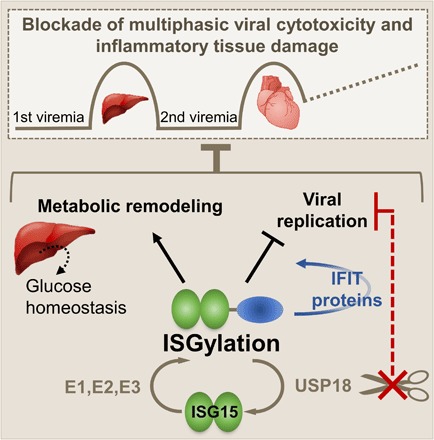
CV infection is a bona fide example for multiphasic state infectious disease with primary injury of liver and pancreas followed by a second viremia culminating in cardiac damage and chronic tissue damage. Early upon infection, IFNs trigger the ubiquitin-like modifier ISG15, which, in a three-step enzymatic cascade, forms covalent linkages with proteins in both infected and noninfected cells. In non–bone marrow–derived somatic cells and tissues, ISGylation inhibits viral replication, and this involves augmented protein expression levels of antiviral effectors such as IFIT1 and IFIT3. ISG15 ensures efficient storage of glucose in liver tissue of healthy mice and reprograms liver metabolism toward improved glucose production early after CV infection. Cells lacking activity of the ISG15-specific protease USP18 show a marked increased resistance against CV infection, thus providing a rationale that USP18 inhibition could be a novel host-directed approach countered to CV-associated human pathology.
Second, we demonstrate a mechanistic aspect of how ISGylation might enhance cellular resistance to CV infection. The E3 ligase Herc5 is associated with polyribosomes; as a result, newly synthesized proteins are accessible substrates for ISGylation in a cotranslational manner (12). Late in the infection cycle, viral proteins make up the main bulk of de novo synthesized proteins. Our previous in vitro study demonstrated that ISG15 combats the degradation of the host translation initiation factor eIF4G by the viral 2A protease, and this was partially attributed to ISGylation of this protease (21), revealing one aspect of how ISG15 might protect infected cells from CV-triggered cytotoxicity. IFN release after viral infection orchestrates the synthesis of antiviral ISGs in both infected and noninfected cells, and ISGylation of these host proteins may be an alternate mechanism for ISG15 to disrupt viral replication upon infection (1). During CV infection, it is plausible that other ISGs, such as the IFIT proteins, are modified by ISG15 (14, 46), and we verified that both IFIT1 and IFIT3 are targets of ISG15 conjugation. As mediators of antiviral immunity, IFIT proteins sequester viral RNA and block viral protein translation, resulting in suppression of viral titers (47). Here, we demonstrate that ISG15 strongly increases cellular protein expression levels of IFIT1 and IFIT3 by protein ISGylation, which is in line with a cumulative activity among the ISGs for improved pathogen control (39). Regarding the function of protein ISGylation, the increase caused by IFIT1/3 protein expression may contribute to enhanced cellular resistance against CV infection. Recent data from the Whitton group demonstrate that the IFIT locus is required to limit CVB3 replication (32). IFITs play a role in protecting many tissues against CVB3 infection, with an IFN-induced phase of IFIT activity specific for the heart. RNA-activated protein kinase (PKR) is another example where ISGylation affects the activity of ISGs and prepares as yet uninfected cells to better resist infection (14). Further supportive evidence for cooperative activity of ISG15 with other ISGs comes from the transcription factor IRF3, where ISGylation prevents ubiquitylation of IRF3 and thus stabilizes the protein (48). Because both IFIT1 and IFIT3 proteins are substrates of ubiquitylation (49), it is conceivable that a modification of IFIT1/3 proteins with ISG15, as shown here, might impair proteasomal processing of degradation-prone IFIT proteins. Alternatively, because protein ISGylation is known to reduce the overall turnover of ubiquitylated proteins by the proteasome, these processes might also influence IFIT protein expression levels (16, 48).
Enforced activity of the cellular translation machinery in infected and IFN-activated cells is a highly energy demanding process that requires an adaption of the host metabolism for the swift production of ATP (50). IFN-regulated glycolytic metabolism is important for the acute induction of an antiviral response during infection with CV (28). Here, we report increased glucose consumption rates by CV-infected ISG15-ko cells, demonstrating that ISG15, most likely by attenuating CV protein synthesis, influences the magnitude of glucose uptake. At the same time, in mice, ISG15 ensures a normoglycemic state before infection, resulting in fully loaded hepatic glycogen stores, allowing rapid adaptation to hypoglycemia. Hypoglycemia in naïve ISG15−/− mice alters liver carbohydrate metabolism, leading to low hepatic glycogen stores and a shift toward glucose production. Currently, it is not well understood how, under baseline conditions, ISG15 controls blood glucose. Among many factors, the efficient uptake of nutrients by the intestine is one variable, promoting storage of surplus metabolites. We found that the plasma triglyceride level, being an indicator of external supply of nutrients, was not affected by ISG15, suggesting that at least the uptake of lipids is not altered in ISG15−/− mice. In noninfected cells, we also investigated the mitochondrial oxidative metabolism and consumption of glucose and found no influence of ISG15 on the demand or on the production of ATP (fig. S9). Therefore, future studies focusing on the role of ISG15, for example, in the endocrine pancreas, are required to dissect the function of ISG15 for glucose homeostasis under baseline conditions.
After infection with CV, when the energy expenditure increases and hepatic glycogen stores are reduced, a rapid remodeling of central liver metabolism becomes important, ensuring the efficient supply with metabolites for the whole organism. The increased energy demand of infected and cytokine-activated cells (41) is countered to some degree by the diminished uptake of nutrients by CV-infected mice (42), resulting in a catabolic state with decreased plasma levels of glucose, triglycerides, and FFAs. At this stage, experimental analysis of mitochondrial oxidative metabolism and shotgun proteomics from liver tissue, in combination with metabolic network modeling, revealed that ISG15 profoundly influences the central liver metabolism toward enforced production of glucose. Our data demonstrate that ISG15 reprograms carbohydrate metabolism in the liver to favor glucose production after the initial viremia. Early after infection, when blood glucose drops profoundly and gluconeogenesis is essential, kinetic modeling of liver metabolism showed a reduced capacity of liver tissue from ISG15−/− mice compared to wild-type mice to produce glucose. The ISG15-deficient liver tissue was less able to compensate for the profound hypoglycemia mice developed early after infection. These metabolic perturbations in ISG15−/− mice can be considered as stressor in the battle against CV. Glucose production by the liver is an ATP-consuming anabolic pathway, rendering glucose production sensitive to ATP depletion. Corresponding to a previous in vitro finding regarding the ISG15-dependent increase of the oxygen consumption rate in IFN-stimulated macrophages (17), our Seahorse measurements of oxidative metabolism in infected liver samples, together with proteomics data and results from metabolic modeling, demonstrate that ISG15 shifts OXPHOS toward higher oxidative capacity in the liver at an early state of infection. This metabolic remodeling improves the energetic state of liver tissue during infection (41), required for ATP-consuming glucose production. In addition to the role of ISG15 for adaptation of OXPHOS, ISG15 reduces the energy demands of hepatocytes for other cellular processes by lowering both cytokine responses and viral protein synthesis in CV-infected liver tissue. All these factors explain why hepatocytes with an intact ISG15 system at an early stage of virus-triggered hypoglycemia maintain their physiological capacity to promote glucose production in response to hypoglycemia by liver tissue. After the second viremia, when the catabolic phase is maintained in both wild-type and ISG15−/− mice, there is only a marginal increase of blood glucose and hepatic glycogen. Persisting hypoglycemia at this stage requires a compensation by the liver with higher production of glucose. Nevertheless, metabolic modeling under standardized conditions at this stage revealed, together with preserved gluconeogenesis, an increased glycolytic capacity of liver tissue. In keeping with a higher protein abundance of key glycolytic enzymes in the presence of ISG15, the metabolic shift toward higher glycolytic capacity in the liver is supported by ISG15. Therefore, ISG15 promotes an adaptation of organ-specific metabolism toward lower availability of metabolic resources, as reflected by lowered levels of plasma metabolites in infected mice at this stage, presumably by the enhanced activity of adenosine monophosphate (AMP)–activated kinases (28).
Precise demarcation of ISG15 function in antiviral and metabolic rewiring tremendously improved our understanding of the ISG15 system for combating CV infection. Despite this impressive antiviral activity for CV, ISG15 is not effective for all viruses (51). A constant battle of immune evasion mechanisms by the pathogen and corresponding host immune response can result in the evolution of resistance by viruses (52, 53). Supportive evidence for the pathophysiological relevance of ISGylation for CV control, however, comes from experiments in which—similar to influenza B and vaccinia virus (9)—inactivation of the ISG15-specific protease USP18 enhanced the antiviral capacity of the ISG15 system for clinically relevant CV serotypes. The deconjugating activity of USP18 is selective for ISG15 (7). In clear contrast to the physical requirement of the USP18 protein for blocking IFNAR signaling (8), inhibition of the protease activity of USP18 maintains IFNAR signaling at basal levels (9). Together with the antiviral function that we demonstrate here for human ISG15 during CV infection, our data support the notion that USP18 protease inhibition might be exploited as a host-directed antiviral approach to combat CV pathology. Our findings provide a rationale for therapeutic application of the ISG15 system for patients with severe CV pathology. Targeting host factors, as a means of antiviral therapy, is attractive, because the antiviral activity mediated by ISGylation involves many potent antiviral effector pathways (1) such as IFIT proteins and multiple CV serotypes are affected. While minimizing the potential evolution of resistance by the virus as observed for many direct antivirals (54), stabilizing ISGylation by USP18 inhibitors represents a favorable approach to counter CV pathology in man.
METHODS
Mice
C57BL/6J mice were obtained from in-house breeding pairs. ISG15−/−, Ube1L−/−, and USP18C61A/C61A (USP18C61A) transgenic mice (all C57BL/6) were originally described elsewhere (9, 33, 51). USP18C61A and wild-type littermate controls originated from the offspring of USP18C61A/wt × USP18C61A/wt mice. ISG15−/− USP18C61A mice were generated by successive cross breeding of ISG15−/− and USP18C61A/C61A mice. All mice were kept at the animal facilities of the Charité University Medical Center. If not indicated otherwise, 4- to 6-week-old male mice received a single intraperitoneal injection of 1 × 105 plaque-forming units (pfu) of CV (Nancy strain) (22). Eight days after infection, heart tissue was isolated, fixed in HistoFix [1× phosphate-buffered saline (PBS), 4% ROTI Histofix] overnight, and embedded in paraffin. To visualize cardiac infiltration, cross sections were stained with hematoxylin and eosin and judged by a blinded pathologist as described elsewhere (21). For induction of AM, ISG15−/− mice originally on a C57BL/6 background were backcrossed to an A/J background for seven to eight generations. To induce AM, mice were subcutaneously immunized with a solution of 150 μg of murine cardiac TnI-peptide (Peptide Specialty Laboratories, Heidelberg, Germany) diluted in complete Freund’s adjuvant, which was supplemented with Mycobacterium tuberculosis H37Ra (5 mg/ml) (Sigma-Aldrich, St Louis, MO, USA). Injections were repeated after 7 and 14 days. All experiments investigating AM were conducted in Heidelberg. Acid fuchsin orange G (Afog) staining was performed to detect collagen deposition. Five sections of each heart were inspected in a double-blinded fashion by two independent investigators under light microscopy to determine respective inflammation and fibrosis scores as described elsewhere (55). This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the German animal welfare act, which is based on the directive of the European parliament and of the council on the protection of animals used for scientific purposes and follows the Institutional Animal Care and Use Committee guidelines. This study conforms to the Berlin and Baden-Württemberg state guidelines for animal welfare. The protocol was approved by the local authorities for animal welfare in Berlin or Karlsruhe (permit numbers: G0279/11, G0272/14, H0076/08, and G161/14). All efforts were made to minimize suffering.
Bone marrow chimeras
Six- to 10-week-old female congenic mice with CD45.1 (wild-type) versus CD45.2 alleles (Ube1L−/− and ISG15−/− mice) were irradiated with 10 Gy using a Gammacell 40 exactor (MDS Nordion). Mice received 1 × 107 bone marrow cells from donor mice upon intravenous injection into the tail vein. After 3 to 4 weeks, blood samples were collected by facial vein puncture for enumeration of CD45.1- and CD45.2-expressing white blood cells. Following red blood cell lysis with 10 mM KHCO3, 155 mM NH4Cl, and 0.1 mM EDTA, Fc receptor blocking reagent (1:50; Miltenyi) was added for 20 min at 4°C. Cells were stained with CD45.1 (A20; BD Biosciences) and CD45.2 (104, BD Biosciences) antibodies and acquired on a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed using FlowJo v10.0 software (Tree Star). To obtain ISG15 chimeric mice, bone marrow cells from either ISG15−/− (CD45.2) or wild-type (CD45.1) donors were injected into ISG15−/− recipient mice, and after 4 weeks, mice were infected with 1 × 105 pfu of CV. Ube1L−/− chimera were generated by transfer of wild-type (CD45.1) or Ube1L−/− (CD45.2) bone marrow cells into wild-type (CD45.1) and Ube1L−/− (CD45.2) recipient mice 4 weeks before CV infection (1 × 104 pfu per mouse).
Cell culture
Primary cardiomyocytes were isolated and cultured from embryonic mouse hearts at embryonic day 14, yielding >90% troponin I–positive cells as described elsewhere (56). Adenoviral vectors were applied to cardiomyocytes at a multiplicity of infection (MOI) of 25 for 48 hours to induce ISG15 expression. HeLa cells stably expressing 3×FLAG-6×His-ISG15 (3) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and puromycin (2 μg/ml). Cells were infected with CV (Nancy strain) at the indicated MOI for 1 hour at 37°C. Clinical isolates of CVB serotypes 1, 2, 3, and 5 were isolated from patients with suspected meningitis or encephalitis and passaged once on HEp-2 or rhabdomyosarcoma RD cells. Viral titers were determined on HeLa cell monolayer by plaque assay. Aliquots were stored at −80°C before use. For siRNA studies, HeLa cells (American Type Culture Collection) were grown to approximately 70% confluence and medium was changed to Opti-MEM before transfection. Cells were transfected with 25 nM human ISG15 siRNA (L-004235-03; Dharmacon) or a nontargeting pool (D0018101005; Dharmacon) using Lipofectamine RNAiMAX transfection reagent (Invitrogen). IFIT expression was induced upon transfection of pcDNA3.1-3×FLAG-IFIT1 (Addgene) or pcDNA3.1-3×FLAG-IFIT3 (Addgene) plasmids using Lipofectamine 2000 (Thermo Fisher Scientific). BMMs were generated using L929 cell conditioned medium (LCCM) as a source of granulocyte/macrophage colony-stimulating factor following procedures described recently (25). In brief, bone marrow was passed through 40-μm cell strainers, centrifuged, resuspended with RPMI 1640, and plated in 60-mm dishes. After 24 hours, nonadherent cells were collected and replated in 10-mm dishes with RPMI 1640 supplemented with 30% LCCM. After 4 days, fresh BMM growth medium was added. After days 7 to 9, cells were plated in RPMI 1640 supplemented with 15% LCCM and used for experiments. Flow cytometric analysis of F4/80 and CD11b coexpression revealed more than 90% macrophage purity. The ISG15 system was induced by IFN-β (100 U/ml; Roche) or polyinosinic:polycytidylic acid (poly(I:C)) (50 μg/ml; InvivoGen) for 12 to 24 hours.
ISG15 gene editing using CRISPR-Cas9 technology
Single-guide RNAs (sgRNAs) targeting ISG15 were designed using CRISPOR (CRISPOR.tefor.net): 5′-CACCGGAACTCATCTTTGCCAGTACAGG-3′ and 5′-AAACGTACTGGCAAAGATGAGTTCC-3′. The single-strand gRNAs were duplexed and ligated into the Bsm BI–digested and dephosphorylated lentiCRISPRv2 vector. After validation of correct sequences, the sgRNA-containing plasmid was transfected into human embryonic kidney (Hek) 293 T cells together with lentiviral packaging plasmids psPAX2 (Addgene) and pMD2.G (Addgene) using polyethylenimine as transfection reagent. The green fluorescent protein (GFP) carrying lentiviral plasmid pLJM1-EGFP (Addgene) was transfected together with the two packaging plasmids to obtain GFP lentiviruses that served as a transduction control. Sterile-filtered cell culture supernatant was used to transduce HeLa cells 72 hours after transfection. Medium was changed 4 to 6 hours after transduction, followed by selection of successfully transduced cells (72 to 96 hours after transduction) using puromycin treatment at a concentration of 1.5 μg/ml. Single-clone selection took place 24 to 48 hours after puromycin treatment by dilution of the positive cells. ISG15 deficiency was tested by IFN-β stimulation and subsequent Western blotting for ISG15. Control cells were generated by transduction of lentiviral vectors containing the Cas9 sequence without gRNAs.
Generation of adenoviral vectors
The adenoviral shuttle plasmid containing hemagglutinin (HA)–tagged human ISG15 (pZs2-HA-hISG15) was cloned using the following primers: 5′-GAATTCATGTATCCTTATGACGTGCCTGACTATGCCAGCCTGGGAGGACCTATGGGCTGGGACCTGACGGTGAA-3′ and 5′-ACCGGATCCTTAGCCTCCCCGCAGGCGCAGATTCATGAACACGGTGCTCA-3′. The recombinant, replication-deficient adenovector Ad5-HA-hISG15 as well as the Ad5-control vector were cloned by in vitro ligation of the 5′ long arm of Xba I–digested ΔE1ΔE3 adenovirus mutant RR5. Following transfection of the constructs into Hek293 cells, recombinant vector preparation was performed as described elsewhere (57). Concentration of the obtained vector preparation was determined by standard plaque assay on Hek293 cells.
Phagocytosis assay
BMMs were plated in 24-well plates at a density of 3 × 105 cells per well. After 15 min of synchronization on ice, cells were loaded with target particles for phagocytosis and placed again on ice to allow equal attachment of the particles to the cell surface. The following phagocytosis particles were used: pHrodo Red Escherichia coli Bio Particles (Life Technologies, according to the manufacturer’s instructions), polystyrene carboxylate-modified latex beads with yellow-green fluorescence (1 × 107 beads per well; Sigma-Aldrich), and PKH67-labeled, heat-inactivated Streptococcus pneumoniae (D38Δcps, MOI 25). After incubation at 37°C for the indicated length of time, cells were rinsed with ice-cold PBS to stop phagocytosis. Fluorescence intensity was analyzed by flow cytometry.
Determination of ROS and NO
Generation of ROS in BMMs was analyzed by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; 10 μM) staining performed in triplicate. After 1 hour of incubation at 37°C, cells were rinsed twice with PBS and fluorescence was analyzed by flow cytometry. The capacity of BMMs to produce NO was determined by Griess assay. Cell culture supernatants of 1 × 105 per 96-well BMMs were incubated with Griess reagent (Molecular Probes). Nitrite concentrations were calculated using a reference curve.
RNA isolation and quantitative real-time polymerase chain reaction
RNA was isolated using the TRIzol (Ambion) method according to the manufacturer’s instructions. The remaining DNA was removed by digestion with deoxyribonuclease I (Invitrogen) at 37°C for 15 min followed by enzyme deactivation at 65°C for 10 min. RNA (250 to 1000 ng) was reverse-transcribed by MLV Reverse Transcriptase (Promega) in combination with random hexamer primers (Roche). TaqMan polymerase chain reaction (PCR) was performed using primers and probes of TaqMan gene expression assays (Life Technologies, Germany) as well as the following combinations of primers and probes: human HPRT, 5′-AGTCTGGCTTATATCCAACACTTCG-3′ (forward), 5′-GACTTTGCTTTCGGTCAGG-3′ (reverse), and 5′-TTTCACCAGCAAGCTTGCGACCTTGA-3′ (probe); murine HPRT, 5′-ATCATTATGCCGAGGATTTGGAA-3′ (forward), 5′-TTGAGCACACAGAGGGCCA-3′ (reverse), and 5′FAM-TGGACAGGACTGAAAGACTTGCTCGAGATG-3′TAMRA (probe); and CV, 5′-CCCTGAATGCGGCTAATCC-3′ (forward), 5′-ATTGTCACCATAAGCAGCCA-3′ (reverse), and 5′-FAM-TGCAGCGGAACCG-MGB3′ (probe). A StepOnePlus real-time PCR system was used for quantitative PCR (qPCR). TaqMan assays for HPRT served as endogenous controls and were used to calculate relative expression using the ΔCt or ΔΔCt method.
Quantification of infectious viral particles
Plaque assays were performed on subconfluent monolayers of green monkey kidney cells incubated with serial 10-fold dilutions of cell culture supernatant or supernatant from homogenized mouse tissue. After incubation at 37°C for 1 hour, supernatants were removed and monolayers were overlaid with DMEM containing 1 mM pyruvate, 2.5% fetal calf serum, 3.75% NaHCO3, and 0.6% agar. Two days later, cells were fixed with 75% methanol/25% acetic acid and stained with 0.25% crystal violet solution to count virus plaques. In some experiments, virus titers were determined by median tissue culture infectious dose (TCID50) assay on green monkey kidney cells.
Immunoprecipitation
After IFN-β treatment [IFN-β (100 U/ml) for 24 hours], FLAG-His ISG15 cells (pBabe-FLAG-His ISG15) and respective controls (pBabe plasmid empty vector) (3) were lysed in 20 mM Hepes, 8 mM EDTA, 2 mM EGTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 5 mM N-ethylmaleimide (NEM), 1% Triton X-100, and cOmplete Protease Inhibitor Cocktail (Roche). Total cell lysate was (2 mg) was incubated with 50 μl of anti-FLAG M2 affinity gel (Sigma-Aldrich) for 3 hours at 4°C on a rotation wheel. After washing with lysis buffer, beads were incubated in Laemmli buffer for 10 min at room temperature and samples were subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Alternatively, HeLa cells were transfected with pcDNA3.1-hUbe1L, pcDNA3.1-UbcM8, pTriEx2-hHERC5, and pcDNA3.1-HA-hISG15. FLAG-tagged IFIT1 or IFIT3 constructs were transfected a day later. After 24 hours, cells were lysed with Hepes buffer containing 1% CHAPS, 8 mM EDTA, 2 mM EGTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 5 mM NEM, and cOmplete Protease Inhibitor Cocktail (Roche), and 2 mg of cell lysate was used for FLAG immunoprecipitation. Beads were washed with lysis buffer, high-salt buffer (200 mM Hepes, 300 mM NaCl, and 0.1 mM EDTA), and washing buffer (200 mM Hepes and 0.1 mM EDTA) before elution with Laemmli buffer.
Western blot analysis
For SDS-PAGE, cells or tissues were lysed using 8 M urea buffer containing 1% Triton X-100, 0.1% SDS, 20 mM Hepes, 8 mM EDTA, 2 mM EGTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 1 mM tris (2-Carboxyethyl)phosphine (TCEP), and cOmplete Protease Inhibitor Cocktail (Roche, Germany). Western blot analysis was performed according to standard protocols. The following primary antibodies were used: β-actin (C4; Merck), FLAG (M2; Sigma-Aldrich), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (FL-335; Santa Cruz Biotechnology or 6C5; Invitrogen), ISG15 (lab stocks), human IFIT1 (polyclonal; Abcam), human IFIT3 (polyclonal; Abcam), murine IFIT1 (polyclonal; Abcam), murine IFIT3 (polyclonal; Antibodies Online), α-tubulin (GT114; GeneTex), and VP1 (31A2; Mediagnost). Secondary IRD680CW- or IRDye800CW-labeled antibodies were detected by an Odyssey CLx infrared imaging system (LI-COR Biosciences). Densitometric analysis was performed using Image Studio (LI-COR Biosciences), and values were normalized to internal loading controls.
Proteomics sample preparation and LC-MS/MS analysis
Male wild-type and ISG15−/− mice were infected with 1 × 105 pfu of CVB3 and sacrificed at day 3 or 8 after infection. Mice that were treated with DMEM served as control. Liver samples (three mice per group, 18 samples in total) were flushed with PBS before they were collected and frozen in liquid nitrogen. Tissue was homogenized in 20 mM Hepes (pH 8.0) containing 8 M Urea before samples were sonicated by three pulses of 10 s at an amplitude of 20% and centrifuged for 15 min at 16,000 relative centrifugal force (rcf) at room temperature. Proteins were first reduced by addition of 5 μl of dithiothreitol to a final concentration of 5 mM and incubated for 30 min at 55°C and then alkylated by addition of chloroacetamide to a concentration of 10 mM and incubation for 15 min at room temperature. The samples were diluted to a urea concentration of 4 M, and 1 mg of protein was digested with lysyl endopeptidase (1:250, w/w; Wako) for 4 hours at 37°C. After further dilution to 2 M urea, proteins were digested with trypsin (1:200, w/w; Promega) overnight at 37°C. The resulting peptide mixture was acidified by addition of 1% trifluoroacetic acid (TFA), and insoluble components were removed by centrifugation at 7000 rcf for 15 min at room temperature. Peptides were purified on C18 Sep-Pak columns (Waters), lyophilized, and stored at −80°C before LC-MS/MS analysis. Peptides were redissolved in loading solvent A [0.1% TFA in water/ACN (98:2, v/v)], and the peptide concentration was determined on a Lunatic instrument (Unchained Labs) to inject 3 μg of peptides for LC-MS/MS analysis on an Ultimate 3000 RSLCnano system in-line connected to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific). Trapping was performed at 10 μl/min for 4 min in loading solvent A on a 20-mm trapping column [100 μm internal diameter (I.D.), 5-μm beads; C18 Reprosil-HD, Dr. Maisch, Germany] before the samples were separated on a 400-mm analytical column packed in the needle (75 μm I.D., 1.9-μm beads; C18 Reprosil-HD, Dr. Maisch). Before packing of the column, the fused silica capillary had been equipped with a laser-pulled electrospray tip using the P-2000 Laser Based Micropipette Puller (Sutter Instruments). Peptides were eluted from the analytical column by a nonlinear gradient from 2 to 56% solvent B [0.1% FA in water/acetonitrile (2:8, v/v)] over 145 min at a constant flow rate of 250 nl/min, followed by a 5-min increase to 97% solvent B and an additional 10 min wash in solvent B. The column was then reequilibrated with 98% solvent A (0.1% FA in water) for 20 min. The column temperature was kept constant at 50°C in a column oven (Sonation COControl). The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the 16 most abundant ion peaks per MS spectrum. Full-scan MS spectra [375 to 1500 mass/charge ratio (m/z)] were acquired at a resolution of 60,000 in the Orbitrap analyzer after accumulation to a target value of 3 × 106. The 16 most intense ions above a threshold value of 1.3 × 104 were isolated for fragmentation at a normalized collision energy of 28% after filling the trap at a target value of 1 × 105 for maximum 80 ms. MS/MS spectra (200 to 2000 m/z) were acquired at a resolution of 15,000 in an Orbitrap analyzer.
Data processing and GO term enrichment analysis
Data analysis was performed with MaxQuant (version 1.6.3.4) using the Andromeda search engine with default search settings including a false discovery rate (FDR) set at 1% on both the peptide and protein level. The recorded spectral data files were searched together against all mouse proteins in the UniProt/Swiss-Prot database [database release version of January 2019 containing 17,006 murine protein sequences (taxonomy ID 9606), downloaded from www.uniprot.org] supplemented with all precursor and mature proteins encoded in the CVB3 genome in the UniProt/Swiss-Prot database [database release version of January 2019 containing 17 CVB3 protein sequences (taxonomy ID 103903), downloaded from www.uniprot.org]. The mass tolerance for precursor and fragment was set to 4.5 and 20 parts per million, respectively, during main search. Enzyme specificity was set as C-terminal to arginine and lysine (trypsin), also allowing cleavage at arginine/lysine-proline bonds with a maximum of two missed cleavages. Carbamidomethylation of cysteine residues was set as a fixed modification, and variable modifications were set to oxidation of methionine (to sulfoxides) and acetylation of protein N termini. Matching between runs was enabled with an alignment time window of 20 min and a matching time window of 1 min. Only proteins with at least one unique peptide were retained, leading to the identification of 3982 mouse proteins in all 18 samples. Further data analysis was performed with the Perseus software (version 1.6.1.1) after loading the proteinGroups table from MaxQuant. Hits identified in the reverse database and as potential contaminants were removed, and protein label-free quantification (LFQ) intensities for each sample were log2-transformed. Replicate samples were grouped, proteins with less than three valid values in at least one group were removed, and missing values were imputed from a normal distribution around the detection limit to compile a list of 2655 quantified proteins (table S1A). To reveal significantly regulated proteins, two t tests (FDR = 0.05, S0 = 1) were performed to compare protein intensities between days 0 and 3 after infection in wild-type mice (table S1B). To identify proteins that were significantly regulated across all samples, groups were defined based on genotypes (wild-type versus ISG15−/−) and time after infection (day 0 versus day 3 versus day 8), and a two-way analysis of variance (ANOVA) test was performed to compare the protein intensities in the infection group with the genotype group. For each protein, this test calculated a P value for infection, a P value for genotype, and a P value for the interaction between both groups. This test revealed 1094 significantly regulated proteins with P ≤ 0.05 in at least one of the three conditions. The intensities of these proteins are shown in the heatmap in Fig. 4A after z scoring and nonsupervised hierarchical clustering. To further dissect the regulation between wild-type and ISG15−/− mice, the ANOVA-regulated proteins were clustered in two separate heatmaps and are reported in table S1C. The first heatmap displays the protein intensities of wild-type mice at days 0, 3, and 8 after infection (Fig. 4A, left), while the second heatmap shows the protein intensities of ISG15−/− mice at days 0, 3, and 8 after infection (Fig. 4A, right). In both heatmaps, six clusters with the same regulation pattern were distinguished. To visualize the overlap between each cluster in both heatmaps for wild-type versus ISG15−/− mice, Venn diagrams were created using MATLAB (R2018a) (Fig. 4B). Last, GO enrichment analysis was performed on four protein lists derived from both heatmaps. The first list contains the proteins up-regulated at day 3 after infection, and the second list contains the proteins up-regulated at day 8 after infection. Proteins up-regulated at both days were included into both lists. Likewise, two lists per genotype were generated for proteins that were down-regulated on days 3 and 8 after infection. GO term enrichment analysis was performed using the GO consortium bioinformatics resources (58), and proteins associated with selected GO terms were listed in Figs. 4C and 6C using MATLAB (R2018a).
Modeling of central liver metabolism
We used HEPATOKIN1, a model of central liver metabolism (43), to assess the metabolic alterations during viral infection. We constructed strain- and condition-dependent metabolic models by scaling the maximal activity for each enzyme and transporter in the model according to proteomic data. Therefore, we used the LFQ intensities obtained from MaxQuant analysis according to , where is the maximal enzyme activity for E in the considered condition, E* is the LFQ intensity in the considered condition, Eref is the LFQ intensity in the reference state, and is the maximal enzyme activity in the reference state. The reference values used were taken from the generic model for a healthy liver (43). The strain- and infection-dependent metabolic response of the liver was calculated according to varying plasma metabolite profiles and hormones including glucose, lactate, pyruvate, glycerol, FFAs, acetoacetate, β-hydroxybutyrate, oxygen, ammonia, glutamine, glutamate, serine, alanine, insulin, and glucagon. We simulated either (i) the metabolic output of the liver over a standardized diurnal cycle defined in (43) (Fig. 5D and fig. S6B) or (ii) the glucose exchange flux of the liver for the physiological range of plasma glucose concentrations between 3 mM (54 mg/dl) and 10 mM (180 mg/dl) (Fig. 5E and fig. S6C). For (i), we evaluated the metabolic state of the liver at any time point of the 24-hour cycle in response to the time-dependent variations of the metabolite and hormone concentrations in the blood plasma profile, followed by averaging of glucose and lactate exchange fluxes as well as the FA uptake rates over 24 hours (fig. S6B). For (ii), we systematically varied the plasma glucose concentration in the physiological range while, at the same time, varying the FA plasma concentrations and the plasma concentrations of insulin and glucagon using both the glucose-FFA and hormone-glucose transfer function defined in (43). These calculations are plausible because the pancreas, in response to altered concentrations of plasma glucose, secretes insulin and glucagon. Both hormones also control the plasma concentration of FAs, mainly by altered lipolysis of triglycerides in adipose tissue. For details, see (43).
Measurement of metabolites
The levels of glucose and lactate in cell culture supernatants of CV-infected HeLa cells were determined using an ABL800 FLEX blood gas analyzer. Cell culture supernatants were collected 32 hours after infection, and cell debris was removed by centrifugation for 5 min at 500 rcf before duplicate samples were applied to the blood gas analyzer. Baseline concentrations in medium were subtracted, and values were normalized to cell number per well. To monitor metabolite concentrations in vivo, wild-type and ISG15−/− mice were sacrificed at days 0, 3, and 8 after infection. Blood samples were taken, and the glucose concentration was determined using an Accu-Check glucometer (Roche). Serum FFA concentrations were measured using the colorimetric Free Fatty Acid Kit (Abcam) according to the manufacturer’s instructions. The concentration of triglycerides in sera of infected mice was analyzed by the Institute for Veterinary Diagnostics (Berlin). To determine the hepatic glucose content, tissue samples were homogenized in PBS using a FastPrep 120 cell disrupter (Thermo Savant), followed by three freeze-thaw cycles. Cell debris was removed by centrifugation for 10 min at 13,000 rcf. Supernatants were collected and applied to the ABL800 FLEX blood gas analyzer. Paraffin-embedded tissue sections were used to visualize hepatic glycogen. Samples were first oxidized in 0.5% periodic acid solution, followed by incubation with Schiff’s reagent for 15 min as well as counterstaining with Mayer’s hematoxylin.
Seahorse metabolic assessment
Analysis of mitochondrial oxidative metabolism was conducted using the XFe 96well Extracellular Flux Analyzer (Agilent). Mitochondrial function was assessed by evaluating basal oxygen consumption and extracellular acidification rate in DMEM-based assay medium with 2 mM glutamine and 10 mM glucose from liver samples (1-mm punches) (59). All data were normalized to total protein content per well as determined by the Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Outliers were excluded after analysis using Grubbs’ test in GraphPad Prism 8.
Statistics/data availability
Statistical analysis of the data was performed in GraphPad Prism v7.00 for Windows (GraphPad Software, La Jolla, USA). All data are plotted as individual points. If not indicated otherwise, data summaries are given as means ± SEM. Unpaired t tests were used for two-group comparisons. If samples had unequal variances (determined by an F test), an unpaired t test with the Welch correction was used. If values were normalized to an internal control, one-sample t tests were applied. For multiple group comparison, unequal variance versions of ANOVA (one-way or two-way ANOVA) were performed followed by multiple comparison test. The significance threshold for all tests was set at the 0.05 level. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (60) partner repository with the dataset identifier PXD013259.
Supplementary Material
Acknowledgments
We acknowledge A. Lindner, K. Voss, R. Öttl, M.-C. Gaerz, and X. Wang for excellent technical assistance. C. C. Goetzke and S. Prokop supported bone marrow chimera studies. Ube1L−/−mice were a donation of D.-E. Zhang, and CD45.1 mice were provided by G. Willimsky from an in-house stock. PKH67-labeled, heat-inactivated S. pneumoniae were provided by F. Kirschner. K. Renko (kostja.renko@charite.de) is acknowledged for the generation of the tissue restraints for the XFe96 metabolic analyzer. S. Diedrich is acknowledged for providing access to clinical CV isolates. We acknowledge N. Althof and S. Pinkert for methodological advice. Funding: This project was funded by the German Research Foundation: BE 6335/5-1 and 6335/6-1 to A.B. and KN 590/7-1. The Federal Ministry for Education and Research supported this project with individual grants to K.-P.K. and A.B. (Infect-ERA BacVIRISG15). A.B. was supported by the Hengstberger research fellowship and received support from the Foundation for Experimental Biomedicine (Zürich, Switzerland). M.K., C.B., and M.Z. were supported by International Max Planck Research School for Infectious Diseases and Immunology (IMPRS-IDI), Berlin. F.I. was supported by Infect-ERA BacVIRISG15 and Odysseus grant G0F8616N from the Research Foundation Flanders (FWO). This work was supported by the DFG (KN490/7-1) and the Federal Ministry for Education and Research (ERAinfect, BacVirISG15) to K.-P.K. N.B. was supported by the German Systems Biology Programs “LiSyM” (31L0057) sponsored by the Federal Ministry of Education and Research. Author contributions: Conceptualization: A.B., K.-P.K., M.K., and A.P.; supervision: A.B., K.-P.K., F.I., and J.S.; methodology: A.P., M.K., N.B., F.I., F.T., M.Z., W.P., H.F., M.V., L.R., E.K.W., M.F., and Z.K.; data acquisition: M.K., C.B., A.P., K.K., F.T., M.V., Z.K., E.K.W., and M.F.; data analysis: M.K., C.B., N.B., A.P., F.T., M.V., J.E., L.R., K.K., and E.K.W.; data visualization: A.B., M.K., C.B., J.E., A.P., and K.K.; funding acquisition: A.B., K.-P.K., F.I., and J.S.; writing ± original draft: M.K., K.-P.K., and A.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Materials. Additional data related to this paper may be requested from the authors. Clinical CV isolates can be provided by the Robert-Koch-Institut, Berlin, Germany (RKI) pending a completed material transfer agreement. Requests for these viruses should be submitted to the legal representative of the RKI.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay1109/DC1
Fig. S1. CV-induced ISG15/ISGylation and its impact on virus load.
Fig. S2. Influence of USP18 inactivation in the early phase of CV infection.
Fig. S3. Intact function of innate myeloid cells in ISG15−/− mice during CV infection.
Fig. S4. Generation of Ube1L−/− bone marrow chimeric mice.
Fig. S5. Impact of ISG15 on IFIT1 and IFIT3 protein expression.
Fig. S6. Hepatic glucose flux in dependence of blood glucose.
Fig. S7. Cell culture models to investigate the antiviral activity of human ISG15.
Fig. S8. Influence of ISG15 on experimental troponin I–induced AM.
Fig. S9. Analysis of mitochondrial respiration in wild-type and ISG15−/− hepatocytes.
Table S1. MaxQuant data liver tissue.
REFERENCES AND NOTES
- 1.Perng Y.-C., Lenschow D. J., ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 16, 423–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Werneke S. W., Schilte C., Rohatgi A., Monte K. J., Michault A., Arenzana-Seisdedos F., Vanlandingham D. L., Higgs S., Fontanet A., Albert M. L., Lenschow D. J., ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLOS Pathog. 7, e1002322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radoshevich L., Impens F., Ribet D., Quereda J. J., Tham T. N., Nahori M.-A., Bierne H., Dussurget O., Pizarro-Cerdá J., Knobeloch K.-P., Cossart P., ISG15 counteracts Listeria monocytogenes infection. eLife 4, e06848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogunovic D., Byun M., Durfee L. A., Abhyankar A., Sanal O., Mansouri D., Salem S., Radovanovic I., Grant A. V., Adimi P., Mansouri N., Okada S., Bryant V. L., Kong X.-F., Kreins A., Velez M. M., Boisson B., Khalilzadeh S., Ozcelik U., Darazam I. A., Schoggins J. W., Rice C. M., Al-Muhsen S., Behr M., Vogt G., Puel A., Bustamante J., Gros P., Huibregtse J. M., Abel L., Boisson-Dupuis S., Casanova J.-L., Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai C., Struckhoff J. J., Schneider J., Martinez-Sobrido L., Wolff T., García-Sastre A., Zhang D.-E., Lenschow D. J., Mice lacking the ISG15 E1 enzyme UbE1L demonstrate increased susceptibility to both mouse-adapted and non-mouse-adapted influenza B virus infection. J. Virol. 83, 1147–1151 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giannakopoulos N. V., Arutyunova E., Lai C., Lenschow D. J., Haas A. L., Virgin H. W., ISG15 Arg151 and the ISG15-conjugating enzyme UbE1L are important for innate immune control of Sindbis virus. J. Virol. 83, 1602–1610 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basters A., Geurink P. P., Röcker A., Witting K. F., Tadayon R., Hess S., Semrau M. S., Storici P., Ovaa H., Knobeloch K.-P., Fritz G., Structural basis of the specificity of USP18 toward ISG15. Nat. Struct. Mol. Biol. 24, 270–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malakhova O. A., Kim K. I., Luo J.-K., Zou W., Kumar K. G. S., Fuchs S. Y., Shuai K., Zhang D.-E., UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 25, 2358–2367 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ketscher L., Hannß R., Morales D. J., Basters A., Guerra S., Goldmann T., Hausmann A., Prinz M., Naumann R., Pekosz A., Utermöhlen O., Lenschow D. J., Knobeloch K.-P., Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc. Natl. Acad. Sci. U.S.A. 112, 1577–1582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speer S. D., Li Z., Buta S., Payelle-Brogard B., Qian L., Vigant F., Rubino E., Gardner T. J., Wedeking T., Hermann M., Duehr J., Sanal O., Tezcan I., Mansouri N., Tabarsi P., Mansouri D., Francois-Newton V., Daussy C. F., Rodriguez M. R., Lenschow D. J., Freiberg A. N., Tortorella D., Piehler J., Lee B., García-Sastre A., Pellegrini S., Bogunovic D., ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 7, 11496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Bogunovic D., Payelle-Brogard B., Francois-Newton V., Speer S. D., Yuan C., Volpi S., Li Z., Sanal O., Mansouri D., Tezcan I., Rice G. I., Chen C., Mansouri N., Mahdaviani S. A., Itan Y., Boisson B., Okada S., Zeng L., Wang X., Jiang H., Liu W., Han T., Liu D., Ma T., Wang B., Liu M., Liu J.-Y., Wang Q. K., Yalnizoglu D., Radoshevich L., Uzé G., Gros P., Rozenberg F., Zhang S.-Y., Jouanguy E., Bustamante J., García-Sastre A., Abel L., Lebon P., Notarangelo L. D., Crow Y. J., Boisson-Dupuis S., Casanova J.-L., Pellegrini S., Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durfee L. A., Lyon N., Seo K., Huibregtse J. M., The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 38, 722–732 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao C., Hsiang T.-Y., Kuo R.-L., Krug R. M., ISG15 conjugation system targets the viral NS1 protein in influenza A virus–infected cells. Proc. Natl. Acad. Sci. U.S.A. 107, 2253–2258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okumura F., Okumura A. J., Uematsu K., Hatakeyama S., Zhang D.-E., Kamura T., Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 288, 2839–2847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villarroya-Beltri C., Baixauli F., Mittelbrunn M., Fernández-Delgado I., Torralba D., Moreno-Gonzalo O., Baldanta S., Enrich C., Guerra S., Sánchez-Madrid F., ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat. Commun. 7, 13588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan J. B., Miyauchi-Ishida S., Arimoto K.-i., Liu D., Yan M., Liu C.-W., Győrffy B., Zhang D.-E., Type I IFN induces protein ISGylation to enhance cytokine expression and augments colonic inflammation. Proc. Natl. Acad. Sci. U.S.A. 112, 14313–14318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldanta S., Fernández-Escobar M., Acín-Perez R., Albert M., Camafeita E., Jorge I., Vázquez J., Enríquez J. A., Guerra S., ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLOS Pathog. 13, e1006651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yángüez E., García-Culebras A., Frau A., Llompart C., Knobeloch K.-P., Gutierrez-Erlandsson S., García-Sastre A., Esteban M., Nieto A., Guerra S., ISG15 regulates peritoneal macrophages functionality against viral infection. PLOS Pathog. 9, e1003632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okumura A., Pitha P. M., Harty R. N., ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. U.S.A. 105, 3974–3979 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swaim C. D., Scott A. F., Canadeo L. A., Huibregtse J. M., Extracellular ISG15 signals cytokine secretion through the LFA-1 integrin receptor. Mol. Cell 68, 581–590.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahnefeld A., Klingel K., Schuermann A., Diny N. L., Althof N., Lindner A., Bleienheuft P., Savvatis K., Respondek D., Opitz E., Ketscher L., Sauter M., Seifert U., Tschöpe C., Poller W., Knobeloch K.-P., Voigt A., Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation 130, 1589–1600 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Klingel K., Hohenadl C., Canu A., Albrecht M., Seemann M., Mall G., Kandolf R., Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. U.S.A. 89, 314–318 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abzug M. J., Prognosis for neonates with enterovirus hepatitis and coagulopathy. Pediatr. Infect. Dis. J. 20, 758–763 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Whitton J. L., Cornell C. T., Feuer R., Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3, 765–776 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Althof N., Goetzke C. C., Kespohl M., Voss K., Heuser A., Pinkert S., Kaya Z., Klingel K., Beling A., The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 10, 200–218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wessely R., Klingel K., Knowlton K. U., Kandolf R., Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: Implications for mortality and early viral replication. Circulation 103, 756–761 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Koestner W., Spanier J., Klause T., Tegtmeyer P.-K., Becker J., Herder V., Borst K., Todt D., Lienenklaus S., Gerhauser I., Detje C. N., Geffers R., Langereis M. A., Vondran F. W. R., Yuan Q., van Kuppeveld F. J. M., Ott M., Staeheli P., Steinmann E., Baumgärtner W., Wacker F., Kalinke U., Interferon-beta expression and type I interferon receptor signaling of hepatocytes prevent hepatic necrosis and virus dissemination in Coxsackievirus B3-infected mice. PLOS Pathog. 14, e1007235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burke J. D., Platanias L. C., Fish E. N., Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J. Virol. 88, 3485–3495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritsch S. D., Weichhart T., Effects of interferons and viruses on metabolism. Front. Immunol. 7, 630 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beling A., Kespohl M., Proteasomal protein degradation: Adaptation of cellular proteolysis with impact on virus—And cytokine-mediated damage of heart tissue during myocarditis. Front. Immunol. 9, 2620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Althof N., Harkins S., Kemball C. C., Flynn C. T., Alirezaei M., Whitton J. L., In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J. Virol. 88, 5087–5099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura T., Flynn C. T., Alirezaei M., Sen G. C., Whitton J. L., Biphasic and cardiomyocyte-specific IFIT activity protects cardiomyocytes from enteroviral infection. PLOS Pathog. 15, e1007674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim K. I., Yan M., Malakhova O., Luo J.-K., Shen M.-F., Zou W., de la Torre J. C., Zhang D.-E., Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling. Mol. Cell. Biol. 26, 472–479 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinzierl A. O., Szalay G., Wolburg H., Sauter M., Rammensee H.-G., Kandolf R., Stevanović S., Klingel K., Effective chemokine secretion by dendritic cells and expansion of cross-presenting CD4−/CD8+ dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis. J. Virol. 82, 8149–8160 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane J. R., Neumann D. A., Lafond-Walker A., Herskowitz A., Rose N. R., Interleukin 1 or tumor necrosis factor can promote Coxsackie B3-induced myocarditis in resistant B10.A mice. J. Exp. Med. 175, 1123–1129 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanda T., McManus J. E., Nagai R., Imai S., Suzuki T., Yang D., McManus B. M., Kobayashi I., Modification of viral myocarditis in mice by interleukin-6. Circ. Res. 78, 848–856 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Meyer I. S., Goetzke C. C., Kespohl M., Sauter M., Heuser A., Eckstein V., Vornlocher H.-P., Anderson D. G., Haas J., Meder B., Katus H. A., Klingel K., Beling A., Leuschner F., Silencing the CSF-1 axis using nanoparticle encapsulated siRNA mitigates viral and autoimmune myocarditis. Front. Immunol. 9, 2303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szalay G., Meiners S., Voigt A., Lauber J., Spieth C., Speer N., Sauter M., Kuckelkorn U., Zell A., Klingel K., Stangl K., Kandolf R., Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes. Am. J. Pathol. 168, 1542–1552 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., Rice C. M., A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong J. J., Pung Y. F., Sze N. S.-K., Chin K.-C., HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. U.S.A. 103, 10735–10740 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu D., Sanin D. E., Everts B., Chen Q., Qiu J., Buck M. D., Patterson A., Smith A. M., Chang C.-H., Liu Z., Artyomov M. N., Pearce E. L., Cella M., Pearce E. J., Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44, 1325–1336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kallewaard N. L., Zhang L., Chen J.-W., Guttenberg M., Sanchez M. D., Bergelson J. M., Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 6, 91–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berndt N., Bulik S., Wallach I., Wünsch T., König M., Stockmann M., Meierhofer D., Holzhütter H.-G., HEPATOKIN1 is a biochemistry-based model of liver metabolism for applications in medicine and pharmacology. Nat. Commun. 9, 2386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Göser S., Ottl R., Brodner A., Dengler T. J., Torzewski J., Egashira K., Rose N. R., Katus H. A., Kaya Z., Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation 112, 3400–3407 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Morales D. J., Monte K., Sun L., Struckhoff J. J., Agapov E., Holtzman M. J., Stappenbeck T. S., Lenschow D. J., Novel mode of ISG15-mediated protection against influenza A virus and Sendai virus in mice. J. Virol. 89, 337–349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao C., Denison C., Huibregtse J. M., Gygi S., Krug R. M., Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 10200–10205 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fensterl V., Sen G. C., Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 89, 2462–2468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi H.-X., Yang K., Liu X., Liu X.-Y., Wei B., Shan Y.-F., Zhu L.-H., Wang C., Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol.Cell Biol. 30, 2424–2436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akimov V., Barrio-Hernandez I., Hansen S. V. F., Hallenborg P., Pedersen A.-K., Bekker-Jensen D. B., Puglia M., Christensen S. D. K., Vanselow J. T., Nielsen M. M., Kratchmarova I., Kelstrup C. D., Olsen J. V., Blagoev B., UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25, 631–640 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Wu B., Li J., Ni H., Zhuang X., Qi Z., Chen Q., Wen Z., Shi H., Luo X., Jin B., TLR4 activation promotes the progression of experimental autoimmune myocarditis to dilated cardiomyopathy by inducing mitochondrial dynamic imbalance. Oxid. Med. Cell. Longev. 2018, 3181278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osiak A., Utermöhlen O., Niendorf S., Horak I., Knobeloch K.-P., ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus. Mol. Cell. Biol. 25, 6338–6345 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerra S., Cáceres A., Knobeloch K.-P., Horak I., Esteban M., Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLOS Pathog. 4, e1000096 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao C., Sridharan H., Chen R., Baker D. P., Wang S., Krug R. M., Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat. Commun. 7, 12754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thibaut H. J., De Palma A. M., Neyts J., Combating enterovirus replication: State-of-the-art on antiviral research. Biochem. Pharmacol. 83, 185–192 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Bangert A., Andrassy M., Müller A.-M., Bockstahler M., Fischer A., Volz C. H., Leib C., Göser S., Korkmaz-Icöz S., Zittrich S., Jungmann A., Lasitschka F., Pfitzer G., Müller O. J., Katus H. A., Kaya Z., Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. U.S.A. 113, E155–E164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Opitz E., Koch A., Klingel K., Schmidt F., Prokop S., Rahnefeld A., Sauter M., Heppner F. L., Völker U., Kandolf R., Kuckelkorn U., Stangl K., Krüger E., Kloetzel P. M., Voigt A., Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLOS Pathog. 7, e1002233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marienfeld U., Haack A., Thalheimer P., Schneider-Rasp S., Brackmann H. H., Poller W., ‘Autoreplication’ of the vector genome in recombinant adenoviral vectors with different E1 region deletions and transgenes. Gene Ther. 6, 1101–1113 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neville K. E., Bosse T. L., Klekos M., Mills J. F., Weicksel S. E., Waters J. S., Tipping M., A novel ex vivo method for measuring whole brain metabolism in model systems. J. Neurosci. Methods 296, 32–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay1109/DC1
Fig. S1. CV-induced ISG15/ISGylation and its impact on virus load.
Fig. S2. Influence of USP18 inactivation in the early phase of CV infection.
Fig. S3. Intact function of innate myeloid cells in ISG15−/− mice during CV infection.
Fig. S4. Generation of Ube1L−/− bone marrow chimeric mice.
Fig. S5. Impact of ISG15 on IFIT1 and IFIT3 protein expression.
Fig. S6. Hepatic glucose flux in dependence of blood glucose.
Fig. S7. Cell culture models to investigate the antiviral activity of human ISG15.
Fig. S8. Influence of ISG15 on experimental troponin I–induced AM.
Fig. S9. Analysis of mitochondrial respiration in wild-type and ISG15−/− hepatocytes.
Table S1. MaxQuant data liver tissue.