

New genomic resources for jojoba reveal insights into its phylogeny and the spatial differences of lipid synthesis and storage.
Abstract
Seeds of the desert shrub, jojoba (Simmondsia chinensis), are an abundant, renewable source of liquid wax esters, which are valued additives in cosmetic products and industrial lubricants. Jojoba is relegated to its own taxonomic family, and there is little genetic information available to elucidate its phylogeny. Here, we report the high-quality, 887-Mb genome of jojoba assembled into 26 chromosomes with 23,490 protein-coding genes. The jojoba genome has only the whole-genome triplication (γ) shared among eudicots and no recent duplications. These genomic resources coupled with extensive transcriptome, proteome, and lipidome data helped to define heterogeneous pathways and machinery for lipid synthesis and storage, provided missing evolutionary history information for this taxonomically segregated dioecious plant species, and will support efforts to improve the agronomic properties of jojoba.
INTRODUCTION
Jojoba (Simmondsia chinensis) plants are dioecious desert shrubs that are native to the Sonoran desert and Baja California regions of North America [Fig. 1A and fig. S1, A to D; (1, 2)]. Jojoba is taxonomically classified as the single member of its family Simmondsiaceae (3). Jojoba is widely regarded for its unusual seed oil that consists primarily of liquid wax esters [WEs; Fig. 1A (4–6)]. The seeds of jojoba are one of the world’s only known sustainable sources of liquid WEs and have been used as an eco-friendly replacement for the similar oils that were once harvested from the spermaceti organ of the sperm whale (Physeter macrocephalus), which nearly drove this species to extinction (7, 8). The WEs from jojoba seeds are esters of a monounsaturated long-chain fatty acid (C20-C24) and a fatty alcohol (C20-C24) and can accumulate up to 60% of the seed weight (5, 9, 10). Jojoba seed oils have been demonstrated to have high compatibility with human sebum and promote retention of skin moisture, and because of this, they are highly valued for their use in a wide range of cosmetic products (11–14). The cosmetics industry is currently the largest market for jojoba oil, and consumer demand continues to rise for natural skin care products [e.g., moisturizers, makeup, shampoos, and conditioners (15)]. In addition to its importance to the cosmetics industry, jojoba oils are also widely regarded for their excellent mechanical lubricity properties including stability at high temperatures and pressures (16), antifoaming, antiwear, and antirust properties (17, 18), and oxidative stability (19). Because of the high oil content of its seeds, economic value, and the capacity of the plants to grow in hot, arid climates (35° to 48°C), jojoba has garnered considerable attention for domestication in some of the world’s most unfavorable environments. Currently, jojoba is commercially grown in the United States, Israel, Peru, Argentina, Australia (www.ijec.net), and India (20) on these countries’ most marginal lands.
Fig. 1. Jojoba developing fruit with seeds, Hi-C genome assembly, and genomic features.
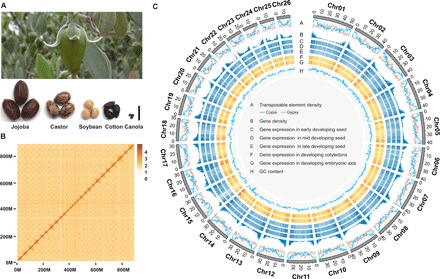
(A) Images of a developing jojoba seed and mature jojoba seeds compared to castor, soybean, cotton, and canola seeds. Scale bar, 1.0 cm. (B) Hi-C assembly of jojoba genome anchored to 26 chromosomes. (C) Mapped features of the jojoba genome including: A, transposable element density; B, gene density; C, gene expression early developing seed; D, mid developing seed; E, late developing seed; F, developing cotyledons; G, developing embryonic axis; H, GC (guanine-cytosine) content. Photo credit: (A) Top: Brenda Singleton, USDA-ARS; bottom: Drew Sturtevant, UNT.
Despite the commercial cultivation of jojoba on marginal lands, crop yields can be reduced markedly by unexpected rainfall, frost, or high temperatures during flowering (2). Over the last decade, there have been considerable efforts to engineer a transgenic row crop with WEs in the seeds including canola (Brassica sp.), camelina (Camelina sativa), crambe (Crambe abyssinica), and Lepidum (Lepidum campestre) (21–24). These efforts were, in part, made possible by the cloning of the jojoba fatty acyl-CoA reductase [ScFAR; (25)] and the wax synthase [ScWS; (26)], which catalyze terminal steps in the synthesis of WEs. However, expression of these jojoba sequences and other allied proteins has resulted in only modest production of WEs in transformed oilseeds (10 to 20%), and nearly all engineered lines were reported to have markedly decreased seed germination rates, especially in seeds with higher WE content (21, 23). As jojoba seeds can accumulate up to 60% seed oil with >95% WE with no observable germination effects, there is likely much to be learned about how jojoba seeds synthesize, package, and mobilize WEs. These challenges and the current lack of genetic resources for jojoba have, in part, motivated our efforts to sequence and annotate the jojoba genome.
RESULTS AND DISCUSSION
Compared to other eukaryotes, plants have considerably complex genomes, which can range in size from 82 Mb [Utricularia gibba, bladderwort; (27)] to 19,600 Mb [Picea abies, Norway Spurce; (28)], contain numerous whole-genome duplications, have a high percentage of transposable elements, and harbor long spanning regions of highly repetitive sequences (29–31). Considering these factors, we used a multifaceted sequencing and assembly approach using a combination of PacBio sequencing reads (107G), Illumina reads (105G), and Hi-C reads (240G) to achieve a high-quality assembly and mapping of the jojoba genome (table S1). Here, we report the 887-Mb jojoba genome assembly anchored to 26 (2n = 26) chromosomes with a contig N50 of 5.2 Mb (Fig. 1, B and C, and Table 1). Using Hi-C assembly mapping, 99.8% of the genomic sequences were assigned unambiguously to discrete chromosome locations (Fig. 1B and table S2), suggesting that the genome assembly is nearly complete (Table 1 and table S3). Genome annotation was completed using 126 Gb of Illumina RNA sequencing (RNA-seq) transcriptome data from 15 plant tissues per seed developmental stages using an integrated pipeline, which included the identification of repetitive elements, noncoding RNAs, and protein-coding genes [fig. S2 and tables S4 to S8; (32)]. The jojoba genome contained 614.7 Mb (69.33%) transposable elements (TEs), where class I (retrotransposons) and class II (DNA transposons) TEs accounted for 62.5 and 6.8% of the genome, respectively (table S4). Long terminal repeats (LTRs) formed the most abundant category of TEs, with LTR/Gypsy and LTR/Copia occupying 21.5 and 21.4% of the jojoba genome, respectively (table S4). Overall activity of LTRs was much lower than that of Spinacia oleracea and Beta vulgaris, which are in the same order, Caryophyllales (fig. S3). Through a combination of ab initio prediction, homology search, and RNA-seq–aided prediction, 23,490 protein-coding genes were annotated in the jojoba genome, with a mean coding length of 1231 base pairs (bp) and an average of six exons per gene (tables S1 and S5). The protein-coding genes were supported by the Illumina RNA-seq reads, and 91.0% of these genes had significant functional annotation matches to the InterPro and Pfam databases (table S6). In addition, we identified sequences for 24,178 noncoding RNAs consisting of ribosomal RNAs, transfer RNAs, microRNAs, and small nuclear RNAs (table S7). Gene region completeness was evaluated by RNA-seq data. On average, more than 95.6% of the RNA-seq reads could be mapped to the jojoba genome assembly (table S8). Further assessments of genome completeness were evaluated with CEGMA (conserved core eukaryotic gene mapping approach) and BUSCO [benchmarking universal single-copy orthologs; (33, 34)], revealing that 98.8% of conserved core eukaryotic genes from CEGMA and 93.5% from BUSCO were captured in our assembly (table S9).
Table 1. Statistics of genome assembly and annotation of S. chinensis.
bp, base pair.
| Genomic feature | S. chinensis | |
| Genome assembly | ||
| PacBio | Total length (bp) | 886,735,837 |
| Total contig number | 994 | |
| Max contig length (bp) | 16,696,083 | |
| Contig N50 (bp) | 5,210,989 | |
| Hi-C | Anchored length (bp) | 885,542,189 |
| Anchored rate | 99.78% | |
| Scaffold N50 (bp) | 38,974,235 | |
| GC content | 37.29% | |
| Genome annotation | ||
| Total repetitive sequences (%) | 582,749,146 (65.73%) | |
| Number of protein-coding genes | 23,490 | |
| Mean transcript length (bp) | 2180.1 | |
| Mean exon length (bp) | 349.6 | |
| Mean intron length (bp) | 1374.0 | |
| Mean exon per mRNA | 6.2 | |
| Mean coding sequence length (bp) | 1230.5 | |
Evolutionary analysis was performed on the assembled jojoba genome (Fig. 2). First, an evolutionary history was reconstructed by comparing the jojoba chromosome assembly to the pre-γ and post-γ ancestral eudicot karyotype (AEK) and three of the evolutionarily least rearranged plant genomes from the Rosid clade (Fig. 2A), Vitis vinifera [grape; (35)], Theobroma cacao [chocolate; (36)], and Prunus persica [peach; (37)]. This evolutionary scenario suggests that jojoba underwent a whole-genome triplication shared among all eudicots and that jojoba diverged from V. vinifera, T. cacao, and P. persica ~100 million years (Ma) ago. These data were supported by analysis of the synonymous substitution rate (Ks) of gene pairs for the shared γ genome triplication of the AEK, V. vinifera, Arabidopsis thaliana, and M. domestica (fig. S4 and table S10), indicating that, except for the γ AEK genome triplication, the jojoba genome has not undergone any additional genome duplications. Genomic regions of jojoba, V. vinifera, and A. thaliana were aligned to compare the syntenic regions of each of these genomes. Jojoba shares a 1:1 or 3:3 synteny with V. vinifera (Fig. 2B, figs. S4 and S5, and tables S5 and S11) and 1:4 relationship with A. thaliana, which has experienced two additional rounds of crucifer genome duplication (Fig. 2B). These observations further support the premise that jojoba has not undergone any additional genome duplications.
Fig. 2. Evolutionary comparison and gene conservation of the jojoba (S. chinensis) genome.
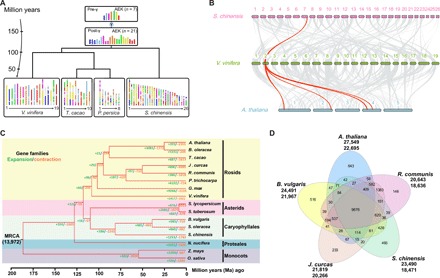
(A) Evolutionary scenario of S. chinensis from the AEK of 7 (pre-γ) and 21 (post-γ) protochromosomes reconstructed from a comparison of the V. vinifera (grape), T. cacao (chocolate), and P. persica (peach) genomes. The modern genomes (bottom) are illustrated with different colors reflecting the seven ancestral chromosomes of AEK origin (top). γ refers to the whole-genome triplication (γ) shared among the eudicots. (B) Macrosynteny between genomic regions of S. chinensis, V. vinifera, and A. thaliana. Macrosynteny patterns between jojoba and grape show that each jojoba region aligns with one region in grape, and each grape region aligns to four syntenic regions in A. thaliana that experienced two additional rounds of crucifer genome duplication. (C) Expansion and contraction of gene families among 16 plant species. The number at the root (13,972) denotes the total number of gene families predicted in the most recent common ancestor (MRCA). A total of 1253 gene families are substantially expanded, and 1783 gene families are contracted in S. chinensis compared with other plant genomes. (D) Shared gene families among S. chinensis, A. thaliana, B. vulgaris, J. curcas, and R. communis. The five species contain 9876 common gene families, and S. chinensis has 493 specific gene families.
Phylogenetic analysis revealed that the jojoba genome is a relatively ancient Astrid genome that has undergone minimal rearrangements, where 1253 gene families are substantially expanded and 1783 families are contracted in the jojoba genome compared with other plant genomes (Fig. 2C and table S12). Of the 23,490 jojoba predicted proteins, 18,471 are clustered into 12,486 families, and of these, 9876 families are shared by five genomes (S. chinensis, A. thaliana, B. vulgaris, Jatropha curcas, and Ricinus communis). In addition, 2034 jojoba proteins are grouped into 493 jojoba-specific gene families (Fig. 2D and table S13). In light of the importance of the unusual WE-containing oils of jojoba seeds, we manually identified and improved the annotation of known genes involved in WE synthesis, glycerolipid synthesis, and lipid droplet (LD) packaging (Figs. 3 and 4).
Fig. 3. Heterogeneous metabolite distributions and gene expression levels in jojoba seeds and tissues.
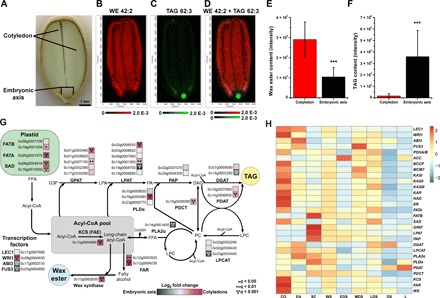
(A) Medial longitude jojoba seed section with labeled seed tissues and structures. Scale bar, 1.0 mm. (B) MALDI-MS image of the major molecular species of WE 42:2 (m/z 655.579, WE 22:1/20:1), which was enriched in the cotyledonary tissues. (C) MALDI-MS image of the major molecular TAG species TAG 62:3 (m/z 1035.872, TAG 20:1/20:1/22:1) highly enriched in the embryonic axis. (D) Composite MALDI-MS image of WE 42:2 (red) and TAG 62:3 (green) demonstrating the differential enrichment of WEs and TAGs in jojoba seeds. In images above, color scale (ion intensity) for each lipid was adjusted to emphasize distribution and does not represent absolute amounts of lipid. Mass tolerance for MALDI-MS images was set to 4 parts per million. (E and F) Total ion signal from each of the cotyledons and embryonic axis was acquired for these tissue areas of the MALDI-MS image. Signal was normalized to the total ion count and then to the number of pixels in the embryonic axis or cotyledons, respectively. The overall WE (E) and TAG (F) content of virtually “dissected” cotyledon and embryonic axis tissues was calculated by summing the intensities of all detected WE and TAG molecular species. Student’s t test was used to calculate significance where *** is P < 0.001. (G) Depiction of metabolic pathways and genes leading to WE and TAG biosynthesis in jojoba. Colored boxes (blue to magenta color scale) adjacent to gene names indicate gene expression bias toward embryonic axis tissues (blue) or toward cotyledon tissues (magenta) and represent a log2 fold change of expression levels (n = 5, *q < 0.05, **q < 0.01, ***q < 0.001). Acyl-CoA, acyl coenzyme A; GPAT, glycerol-3-phosphate acyltransferase; PAP, phosphatidic acid phosphatase. (H) Heat map of gene expression levels of genes involved in glycerolipid and WE synthesis across different jojoba seed tissues and developmental stages. FATB, fatty acyl-ACP thioesterase B; FATA, fatty acyl-ACP thioesterase A; SAD, stearoyl-ACP desaturase; LPAT, lysophosphatidyl acyltransferase; PA phosphatidic acid; DAG, diacylglycerol; FFA, free fatty acid; G3P, glycerol 3 phosphate; LPA, lysophosphatidic acid; DGAT, diacylglycerol acyltransferase; PDAT, phospholipid:diacylglycerol acyltransferase; PDCT, phosphatidylcholine:diacylglycerol cholinephosphotransferase; PLDa, phospholipase D alpha; PLA2a, phospholipase A2 alpha; LPCAT, lysophosphatidylcholine acyltransferase; FAR, fatty acyl-CoA reductase; KCS, ketoacyl-CoA synthase; FAE, fatty acid elongation; PC, phosphatidylcholine; LPC, lysophosphatidylcholine; CO, cotyledons; EA, embryonic axis; SC, seed coat; WS, whole seed; EDS, early developing seed; MDS, mid developing seed; LDS, late developing seed; DS, dry seed; L, leaf. Photo credit: Drew Sturtevant, UNT.
Fig. 4. Gene expression and protein levels of associated with jojoba LDs.
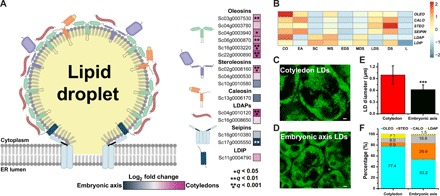
(A) Illustration representing an LD budding from the surface of the endoplasmic reticulum (ER) adorned with various proteins. Colored boxes (blue to magenta scale) adjacent to gene names indicate gene expression bias toward embryonic axis tissues (blue) or toward cotyledon tissues (magenta) and represent a log2 fold change of expression levels (n = 5, *q < 0.05, **q < 0.01, ***q < 0.001). (B) Heat map of gene expression levels of genes involved in LD storage and packaging across different jojoba seed tissues and developmental stages. LDAP, lipid droplet-associated protein; LDIP, LDAP-interacting protein. (C and D) Confocal micrographs of BODIPY-stained LDs from tissues of jojoba (C) cotyledon and (D) embryonic axis tissues. (E) Size comparison of LDs from jojoba cotyledons and embryonic axis tissues. Student’s t test was used to calculate significance where *** is P < 0.001. (F) Bar graph (of 100%) representing an estimation of the percentage of oleosin, steroleosin, caleosin, and LDAP proteins on LDs from jojoba cotyledons and embryonic axis tissues. Proportions of known LD proteins were calculated from normalized MS peptide counts.
Application of tissue-specific RNA-seq and matrix-assisted laser desorption/ionization–mass spectrometry imaging (MALDI-MSI) has previously revealed heterogeneous distributions of lipid metabolites and gene transcripts involved in lipid metabolism across different seed tissues, reflecting an underappreciated spatial regulation to oilseed lipid metabolism (21, 38–45). Here, jojoba seeds were demonstrated to compartmentalize WEs and express genes involved in storage lipid synthesis in a tissue-specific manner. Longitudinal seed sections from mature jojoba seeds were analyzed by MALDI-MS, and these data were searched for major molecular species of WEs and triacylglycerols (TAGs). Unexpectedly, the MALDI-MSI analysis revealed that these two major seed storage lipids were differentially enriched in the seed tissues (Fig. 3, A to D), where WEs were localized primarily to the cotyledonary tissues (WE 42:2 shown in red, Fig. 3, B and D), and TAGs were primarily restricted to the embryonic axis (TAG 62:3 shown in green, Fig. 3, C and D). Total ion counts for both lipid classes were normalized to tissue areas on a per-pixel basis (39, 43, 44), which indicated that cotyledons had a significant (Student’s two-tailed t test; P < 0.001) 4× greater total WE signal intensity than the axis, whereas the embryonic axis had a significant (Student’s two-tailed t test; P < 0.001) 21× higher level in total TAG signal intensity (Fig. 3, E and F). To complement the MALDI-MSI distribution data, total lipids from an intact mature jojoba seed were imaged by magnetic resonance imaging [MRI; (43, 46–48)]. The MRI images revealed that total lipids, presumably WEs, were highly enriched in the cotyledon tissues, whereas the signal from the axis was noticeably less (fig. S6). In addition, the WE and TAG composition of lipid extracts from the cotyledon and embryonic axis tissues of mature jojoba seeds were determined by ultra-performance liquid chromatography (UPLC)–nanoESI (nanoelectrospray ionization)–tandem mass spectrometry (MS/MS) and largely supported the compositions obtained by MALDI-MSI (figs. S6 and S7). Compositions here were consistent with previous findings of jojoba WE and TAG compositions (10, 49).
In consideration of the heterogeneous lipid metabolite distributions observed in jojoba cotyledons and embryonic axis tissues, transcriptome analysis was performed on these two tissues from seeds collected during the peak of oil accumulation [~75 days after pollination (DAP); Fig. 3, G and H, and tables S14 and S15], from whole developing seeds from early, mid, late developmental time points, and from mature, desiccated seeds (Fig. 3H and table S16). From the jojoba genome annotation, we identified genes known to be involved in acyl-lipid metabolism (Fig. 3, G and H) and the previously described jojoba fatty acyl-CoA elongase 1 [ScFAE1; (50)], ScFAR (25), and ScWS (26) and used them for expression analysis. In Fig. 3G, the acyl-lipid metabolic pathway is diagrammed with the corresponding genes from the jojoba genome, and a magenta-to-blue colored box indicates the differential transcript abundances (log2 fold scale) between the cotyledons (magenta) and embryonic axis (blue). Genes known to be involved in WE synthesis including the ScFAE1, ScFAR, and ScWS were all highly expressed and significantly biased toward the cotyledonary tissues (Fig. 3G and table S14). In addition, nearly all genes involved in fatty acid synthesis and export from the plastid were similarly significantly highly expressed and biased toward the cotyledons (Fig. 3G and tables S14 and S17). The expression pattern of these genes likely reflects the cotyledons’ capacity to serve as the major seed storage organs for WEs. By contrast, the jojoba acyl-CoA:diacylglycerol O-acyltransferase 1 (ScDGAT1), which is a terminal enzyme in TAG synthesis, was significantly expressed and biased toward the embryonic axis tissues. The differential expression of the genes encoding the jojoba ScWS and ScDGAT1 offers a molecular explanation for the observed WE and TAG distributions, respectively. In general, the genes associated with storage lipid synthesis increased concomitant with seed development and seed maturation, as would be anticipated (Fig. 3, G and H).
Because different lipids and pathways predominated in different seed parts, we examined the cellular LD packaging machinery for potential differences, both in terms of transcripts for major LD proteins and in terms of proteins on isolated LDs (Fig. 4, A to F). Differential transcript abundances were noted for transcripts for several major LD proteins including oleosins (51, 52), steroleosins (53), caleosins (54) and LD-associated proteins [LDAPs; (55–58)]. Several oleosins and one LDAP especially seemed to be significant differentially expressed in cotyledonary tissues (Fig. 4, A and B), and this appeared to be reflected in the relative abundances of these proteins on the isolated LDs from cotyledons (Fig. 4F and tables S18 to S21). In the endoplasm reticulum membrane, SEIPIN proteins are involved in the biogenesis of LDs and are not LD surface proteins per se (59, 60); however, one isoform appeared to be significantly differentially expressed in the embryonic axis. In addition to differences in LD gene expression profiles, the morphology of LDs was also different between the cotyledons and embryonic axis, where LDs in cotyledons were significantly larger on average, ~1.0 μm in diameter (61), than those in the embryonic axis, ~0.65 μm in diameter (Fig. 4, C to E). Since the LDs of these two tissues contain different types of hydrophobic molecules in their cores (WEs and TAGs), differences in LD surface protein composition may be required for the proper packaging of WE-containing LDs versus TAG-containing LDs and also may influence LD size. The prevalence of LDAP1 (Fig. 4, A, B, and F), in particular, may have functional significance, since this isoform is not substantially expressed in other oilseeds during TAG accumulation (57, 58). LDAPs are homologous to small rubber particle proteins, which localize to the surface of and stabilize rubber particles, compartments specific for the packaging of the hydrophobic polyisoprenoids [reviewed in (62)]. Perhaps jojoba LDAP1 is part of the process specifically required for the proper packaging of WEs.
Overall, our results provide a reference-quality genome for jojoba that facilitates the placement of this minimally studied plant species into evolutionary context. In addition, these genomic resources enabled insights into a previously unrecognized heterogeneity of neutral lipid synthesis and storage in jojoba seeds. These resources will be valuable to others interested in the utilization of this species as an oilseed crop and will also fill important knowledge gaps for this taxonomically isolated dioecious species and its life history and adaptations.
MATERIALS AND METHODS
Plant material
S. chinensis (accession no. PARL 940) developing seeds were collected from plants grown under rainfed conditions at the U.S. Department of Agriculture, U.S. Arid Land Agricultural Research Center, Maricopa AZ (latitude, 33.077514; longitude −111.974276; elevation, 375 m). Developing seeds were collected from wind-pollenated plants. Developing seeds for the RNA-seq developmental time series were collected in 2018 and staged developmentally by their seed weight, with average seed weights of 0.2 g (early), 0.45 g (mid), and 0.7 g (late). Seeds were frozen in liquid nitrogen immediately after harvesting and stored at −80°C. Developing seeds for the tissue-specific RNA-seq experiments were collected in 2017, and the average seed weight of these tissues was ~0.45 g and were at the mid to mid-late oil accumulation seed stage. Seeds were vacuum-infiltrated with RNAlater solution immediately after harvesting and removal from the capsule and stored at −20°C. Mature seeds from these plants were harvested after seeds were desiccated and used here for additional analyses.
Vegetative plant tissues used for gene annotation were collected from 1-year-old jojoba plants grown at the University of North Texas greenhouses at 30°C under long-day (~16 hours) conditions, with day length supplemented by Na-vapor lamps. Tissues were immediately frozen in liquid nitrogen after harvest and stored at −80°C.
Jojoba seed germination and plant growth
Mature jojoba seeds were sterilized in 50% bleach solution for 1 hour at room temperature, and then, seeds were washed/imbibed for 16 hours under running water. Seeds were germinated on wet vermiculite in the dark. Vermiculite moisture content was checked every 2 days and adjusted accordingly. After germination, seedlings were removed from the dark and transplanted to soil. Seed germination and seedling growth conditions were 100 μmol m−2 s−1 16-hour light/8-hour dark at 28°C.
DNA sequencing library construction and sequencing
High-quality genomic DNA was extracted from leaves of 3-month-old plant using the DNAsecure Plant Kit (DP320, www.tiangen.com). DNA samples and tissues from a single plant were sent to Novogene (www.novogene.com) for genome sequencing and Hi-C assembly, respectively.
Libraries for SMRT PacBio genome sequencing were constructed according to the released protocol from PacBio. Approximately 20 μg of high-quality genomic DNA was sheared to approximately 20 kb and evaluated by an Agilent 2100 Bioanalyzer. After shearing, DNA samples were subjected to damage and end repair, blunt-end adaptor ligation, and size selection. The final libraries were sequenced by the PacBio Sequel platform (Pacific Biosciences).
Libraries for Illumina sequencing were generated using the Truseq Nano DNA HT Sample preparation Kit (Illumina, USA) according to the manufacturer’s protocol. The DNA sample was fragmented to generate 350 bp in size by sonication. Resulting DNA fragments were end-polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing with further polymerase chain reaction (PCR) amplification. PCR products were purified by the AMPure XP system and quantified by real-time PCR. The Illumina libraries were sequenced by the Illumina HiSeq X Ten platform.
For Hi-C libraries, jojoba leaves from a single plant were fixed with 1% formaldehyde solution in MS buffer and were used for the preparation of two independent libraries. Subsequently, the DNA libraries were subjected to nuclei extraction, nuclei permeabilization, chromatin digestion (Hind III), and proximity ligation treatments. The constructed libraries were sequenced on the Illumina HiSeq X Ten platform.
Estimation of the genome size and heterozygosity
The genome size was estimated by K-mer frequency analysis. The distribution of K-mer depends on the characteristic of the genome and follows a Poisson’s distribution. Before assembly, the K-mer distribution of 100G Illumina short reads was generated using Jellyfish [v2.1.3; (63)], and we obtained an estimated haploid genome size of 1003.02 Mb with a 0.76% heterozygous rate.
Hi-C–based genome assembly
The S. chinensis genome was de novo assembled using FALCON (https://github.com/PacificBiosciences/FALCON) based on PacBio long reads. Contigs were polished using raw PacBio long reads and corrected using Illumina short reads with Pilon [v1.22; (64)]. The raw assembled genome consisted of 994 contigs with a N50 length of 5.21 Mb. On the basis of Hi-C chromatin interactions information, using 3d-dna [v180419; -r 0 -I 5000; (65)] software and manual correction, scaffolds were assembled into 26 chromosomes. As a result, we obtained a high-quality reference genome of 887 Mb with a N50 length of 38.97 Mb and anchored 99.78% scaffolds into chromosome scale.
Genome assessment
CEGMA (34) pipeline analysis was used to validate the genome completeness, and 93% completeness was obtained of Core Eukaryotic Genes database. In addition, the BUSCO (v3.0.1) (33) database was also used to assess the genome assembly.
Repeat annotation
Transposable elements in the genome assembly were identified both at the DNA and protein levels. RepeatModeler (www.repeatmasker.org/RepeatModeler/) software was used to develop a de novo transposable element library. RepeatMasker (www.repeatmasker.org) was applied for identifying transposable element from Repbase (66) and de novo library. At the protein level, RepeatProteinMask (www.repeatmasker.org) was used to conduct WU-BLASTX searches against the transposable element protein database. Overlapping transposable elements belonging to the same type of repeats were combined together.
Gene prediction
Gene annotation of S. chinensis genome was conducted by combining de novo prediction, homology information, and RNA-seq data. First, Augustus [v3.2.1; (67)], GlimmerHMM [v3.0.4; (68)], SNAP (http://korflab.ucdavis.edu/software.html), Geneid [v1.4.4; (69)], and Genscan [v1.0; (70)] were used on the repeat masked genome with trained parameters. Then, the nonredundant proteins from six sequenced species, Amaranthus hypochondriacus, A. thaliana, Oryza sativa, Dianthus caryophyllus, B. vulgaris, and Fagopyrum tataricum were mapped onto the S. chinensis genome by using TBLASTN [v2.7.1+; (71)] with an E-value cutoff of 1 × 10−5.
For each BLAST hit, Genewise [v2.4.1; (72)] was used to predict the exact gene structure in the corresponding genomic regions. Furthermore, RNA-seq data were mapped to the genome using TopHat [v2.1.1; (73)], and Cufflinks [v2.2.1; (74)] was used to assemble transcripts to gene models. Last, all predictions were combined with EVidenceModeler [EVM v1.1.1; (75)] to get a nonredundant gene set, and PASA [v2.3.0; (76)] was used to correct the predicted result and annotate alternatively spliced isoforms to finalize the gene set.
Functional annotation of protein-coding genes was evaluated by BLASTP [v2.7.1+; (71)] with an E-value cutoff of 1 × 10−5 using two integrated protein sequence databases—SwissProt and TrEMBL (77). Protein domains were annotated by searching InterPro and Pfam databases, using InterProScan [v5.28; (78)] and Hmmer [v3.1b2; (79)], respectively. Gene ontology (GO) terms for each gene were obtained from the corresponding InterPro or Pfam entry. The pathways, in which the gene might be involved, were assigned by BLAST against the Kyoto Encyclopedia of Genes and Genomes (KEGG) (80) database.
Genome evolution analysis
Paralogous pairs of S. chinensis, V. vinifera, A. thaliana, and M. domestica proteins were identified using all-versus-all search in BLAST and used to identify syntenic blocks [MCScan v1.1; (81)]. Synonymous substitutions (Ks) were calculated from the syntenic blocks using KaKs_calculator (82) using the Nei and Gojobori’s (NG) method (83). The same method was used to identify the collinear blocks between S. chinensis, V. vinifera, and A. thaliana. Both LTRharvest [v1.5.9; -minlenltr 100 -maxlenltr 3000 -motif TGCA -motifmis 1 -mintsd 4 -maxtsd 20; (84)] and LTR_FINDER [v1.07; -D 15000 -d 1000 -L 7000 -l 100 -p 20; (85)] were used to de novo detect full-length LTR retrotransposons in jojoba, sugar beet, and spinach genomes. LTR_retriever [v20170512; (86)] was used to integrate the results of LTRharvest and LTR_FINDER and calculate LTR insertion time using the formula T = K/2r, where r was set to 7.0 × 10−9 substitutions per site per year according to Ossowski et al. (87).
Gene family analysis
Five sequenced plant genomes (S. chinensis, A. thaliana, B. vulgaris, J. curcas, and R. communis) were selected for gene family analysis. Proteins from these genomes were aligned to each other using BLASTP. OrthoMCL [v2.0.9; (88)] was used to cluster proteins and identify paralogs and orthologs.
Phylogenetic reconstruction and gene family expansion/contraction
A total of 501 single-copy orthologous genes were identified using OrthoMCL for S. chinensis and 15 other flowering plants (A. thaliana, B. vulgaris, Brassica oleracea, Glycine max, Populus trichocarpa, J. curcas, O. sativa, R. communis, Solanum tuberosum, Solanum lycopersicum, T. cacao, Nelumbo nucifera, V. vinifera, Zea mays, and S. oleracea). Amino acid sequences encoded by single-copy genes from these 16 species were aligned using MUSCLE [v3.8.31; (89)], and RaxML [v8.2.12; (90)] was used to construct a phylogenetic tree based on multiple sequence alignments. Last, the iTOL (91) tools were used to visualize the phylogenetic tree. To estimate divergence time of all the 16 species in the phylogenetic tree, we used the MCMCTree program in the PAML package [v4.8a; (92)], and A. thaliana–B. oleracea split time (mean, 25 Ma ago) and monocot-dicot split time (mean, 150 Ma ago) were chosen. The MCMCTree runs 505,000 iterations to calculate divergence time. CAFÉ [v3.1; (93)] was used to calculate the expansion and contraction of gene family numbers based on the phylogenetic tree and gene family statistics.
Total RNA extraction and RNA-seq library preparation
Developing seeds were removed from RNAlater, and the seed coat, cotyledons, and embryonic axis tissues were hand-dissected. Tissues of the whole developing seeds, cotyledons, embryonic axis, and seed coats were pulverized in liquid nitrogen into a fine powder. Tissues of three seeds were combined per replicate, with five replicates per tissue (n = 5 per tissue). For the total RNA extraction, 100 mg of tissue (whole developing seeds, seed coat, and cotyledons) per replicate was used. Because of the small size of the embryonic axis tissues, only 25 mg of tissue was used per replicate.
Total RNA was isolated using a combination of hot borate buffer and the Qiagen RNeasy Plant Mini Kit (94). Assessment of total RNA quality and quantity was accomplished using a Qubit analyzer and an Agilent 2100 Bioanalyzer.
A sequencing library was prepared from the total RNA for 2 × 150 bp sequencing (n = 5 per tissue, n = 1 for mature seed) using an Illumina NextSeq. The sequencing reactions were prepared at the University of North Texas (UNT) BioDiscovery Institute Genomics Core Facility from 1.0 μg of total RNA using the Illumina TruSeq Stranded mRNA prep kit with minor modifications. The Frag-Prime enzymatic digestion step was reduced from 8 to 4 min. After the adaptor ligation of the sequencing library, the average fragment length was approximately 420 bp (2 × 150 bp library), measured by an Agilent 4200 TapeStation. The 2× 150 paired-end sequencing reads were performed using an Illumina NextSeq instrument with a high-output cassette (120 Gb).
Read preparation for gene expression analysis
For gene expression analysis, the 2 × 150 bp raw reads from Illumina RNA-seq were trimmed to remove any remaining adaptor sequences using Trimmomatic [v0.32; (95)]. Read quality after trimming was assessed using FastQC toolkit.
Processed reads were used for gene expression analyses using STAR using the default parameters. Quantification of differential gene expression analysis was completed using DESeq2 [v3.7; (96, 97)] software using the default settings. The significance of differential gene expression was calculated as q values (*q < 0.05, **q < 0.01, ***q < 0.001), which are more stringent than P values, in that they also account for the false discovery rate rather than the false-positive rate (98). Principal components analysis and gene expression analysis plots were generated using the DESeq2 package pcaExplorer (96, 97). Venn diagrams were assembled using an online Venn diagram builder from http://bioinformatics.psb.ugent.be/webtools/Venn/.
Seed embedding and cryosectioning
A 10% (w/v) porcine gelatin solution was prepared and equilibrated in a 40°C water bath with shaking for 2 hours. Mature desiccated jojoba seeds were embedded in the prepared gelatin solution, frozen at −80°C overnight, and then transferred to −20°C for 48 hours before sectioning. Embedded seeds were cryosectioned at a tissue thickness of 30 μm using a cryo-microtome (Leica Microsystems). Cryosections were collected using Cryo-Tape (Leica Microsystems) and then taped to glass slides. Slides containing the thaw-mounted sections were lyophilized for 6 hours and then stored in a desiccator until processed for MALDI-MSI. All MALDI-MSI occurred within 36 hours of cryosectioning. Bright-field images were taken for all sections used for MSI.
Matrix application and MALDI-MS imaging analysis
For MALDI-MSI, the matrix 2,5-dihydroxybenzoic acid (DHB) was used for analysis of WEs and TAGs. DHB was applied by sublimation using an adapted method developed by Hankin et al. (99). MALDI-MSI data were collected on a hybrid MALDI-LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). The instrument was equipped with a N2 laser with a spot size of ~40 μm. MALDI-MSI data acquisition conditions were as follows: Laser energy was 12 uJ per pulse, a raster step size of 40 μm, and 10 laser shots per raster step with one sweepshot; data were acquired using the Orbitrap mass analyzer with a resolution of 60,000, and data were collected with a mass/charge ratio (m/z) scan range of 500 to 1200. Raw mass spectra were processed into MALDI-MS images using ImageQuest software (Thermo Fisher Scientific). Images were generated by searching the exact mass of WE or TAG molecular species using a 4–parts per million mass tolerance. Processed images were normalized to the total ion count for each pixel and then presented on a red (WE) or green (TAG) scale, where the brightest color represents the highest intensity. After processing, the background of the images was removed, and the images were placed on a black background. The virtual dissection intensities were achieved by selecting the area of each cotyledon and embryonic axis tissues with open source software Metabolite Imager (100), averaging the intensities for all WE and TAG molecular species across all cotyledon or embryonic axis tissues, and lastly normalizing the intensities to the number of pixels per tissue type (32,650 cotyledon pixels and 431 embryonic axis pixels). This method is described in more detail elsewhere (43, 44).
Nuclear MRI of jojoba seeds
The measurement of the lipid distribution of the jojoba seed was measured on a Bruker Advance II AMX spectrometer (Bruker BioSpin, Rheinstetten, Germany) equipped with a 660 mT/m imaging gradient system. The seed was placed inside an inhouse-built birdcage coil (inner diameter, 17 mm), and the lipid-selective custom spin echo sequence was adjusted with the following parameters: repetition time, 850 ms; echo time, 7.8 ms; number of averages, 2; matrix size, 140 × 100 × 90; isotropic resolution, 100 μm; RF (radio-frequency) pulse bandwidth, 2000 Hz; and pulse shape, hermite. The data were further analyzed in MATLAB (MathWorks, Natick, MA, USA) with inhouse written algorithms and were exported to AMIRA (Mercury Computer Systems, Chelmsford, USA) for visualization.
Lipid extractions and thin-layer chromatography
Tissues from the cotyledons and embryonic axes of jojoba seeds were dissected by hand using a scalpel. Dissected tissues were homogenized in hot (70°C) isopropanol (IPA) with glass beads using a Beadbeater (BioSpec Mini-Beadbeater-16). Total lipids were extracted using hot (70°C) IPA with 0.01% butylated hydroxytoluene and incubated at 70°C for 30 min to inactivate endogenous phospholipases (101). After cooling to room temperature, chloroform (CHCl3) and water (H2O) were added to the extracts at a ratio of 2:1:0.45 (IPA:CHCl3:H2O; v/v/v). Lipids were extracted overnight at 4°C. Total lipids were partitioned with additional chloroform and further purified by washing with 1.0 M KCl three times. To examine the neutral lipid content of cotyledon and axis tissues, total lipids were separated using thin-layer chromatography (TLC). Aliquots containing 80 μg of total lipids were spotted on TLC Silica gel 60 plates (Merck) and resolved using 80:20:1 (hexane:diethyl ether:acetic acid; v/v/v). Neutral lipids from extracts were compared to a TLC Neutral standard mix 18-5C and the WE standard, heptadecanyl stearate (Nu-Chek Prep), for identification. Three TLC plates (per tissue) containing nine spots of 80 μg of total lipids were prepared for the embryonic axis and cotyledon tissues. One lane was cut from the TLC plate and stained with iodine vapors to determine the Rf (retention factor) values of the spots. Spots corresponding to WEs and TAGs were scraped, re-extracted (2× with hexane), and dried under nitrogen. WE extracts were suspended in methanol:chloroform (2:1, v/v) containing 5 mM ammonium acetate, whereas TAGs were suspended in tetrahydrofuran:methanol:water (4:4:1, v/v/v).
NanoESI-MS/MS analysis of wax esters and UPLC-nanoESI-MS/MS analysis of TAGs
WE extracts from Jojoba cotyledons and embryonic axis tissues (n = 3 per tissue) were analyzed on an Applied Biosystems 4000 triple-quadrupole mass spectrometer (ABSciex) equipped with a direct inject nanoESI chip ion source (TriVersa NanoMate, Advion BioSciences). Before analysis, 5 nmol of internal standard, heptadecanoyl heptadecanoate was added to extracts. Samples (10 μl) of each WE extract were analyzed using the following ionization parameters: positive ionization mode, voltage of 1.5 kV, back pressure of 0.5 psi, source temperature of 40°C, and curtain gas set to 10 (arbitrary units). Using multiple reaction monitoring (MRM), 785 WE molecular species were monitored. For the MRM precursor ion, the Q1 mass analyzer was set to the ammonium adduct of the WE, and the Q3 mass analyzer was set for the [RCO2H2]+ or [RCO+]+ ion fragment. Each WE molecular species was measured using 150-ms dwell time with 6 cycles (121.6 s/cycle), and total analysis time per sample was 12.1 min. Data were collected using Analysist software (v 1.5.1), and signal intensities were analyzed using LipidView software (ABSciex). WE signals were selected using a 0.5–atomic mass unit window, a minimal signal-to-noise ratio of 1.0, and a maximum intensity of 0%. Data were normalized to type I 13C isotope correction and were corrected by using a WE calibration response factor (10).
UPLC-nanoESI-MS/MS analysis was used to determine the TAG composition of jojoba cotyledon, embryonic axis, and whole seed tissues. TAGs from lipid extracts, prepared as described above, were separated by UPLC (ACQUITY UPLC, Waters) equipped with an AQUITY UPLC HSS T3 column (100 mm by 1 mm, 1.8 μm; Waters). UPLC instrument settings were as follows: A total of 2 μl was injected using a needle overfill mode with a flow rate of 0.1 ml min −1, temperature was set to 35°C, solvent B was tetrahydrofuran/methanol/20 mM ammonium acetate (6:3:1, v/v/v) with 0.1% (v/v) acetic acid, and solvent A was methanol/20 mM ammonium acetate (3:7 v/v) containing 0.1% (v/v) acetic acid. TAG species were separated with the following linear binary gradient: 90% solvent B held for 2 min, linear increase to 100% solvent B for 2 min, and 100% solvent B held for 4 min and re-equilibration to start conditions in 4 min (102). Separated TAGs were analyzed on an Applied Biosystems 6500 QTRAP triple-quadrupole mass spectrometer (ABSciex) equipped with a direct inject nanoESI chip ion source (TriVersa NanoMate, Advion BioSciences). Ionization parameters were as follows: positive ionization mode, voltage of 1.3 kV, source temperature of 40°C, and curtain gas set to 20 (arbitrary units). MRM transitions were measured with a dwell time of 5 ms per transition and used to detect TAG molecular species and acyl composition by the fatty acid–associated neutral loss from [M + NH4]+ molecular ions. Analyst and LipidView software packages were used to analyze the TAG composition, and identifications were made using a custom TAG fragment database specific for the acyl composition of jojoba.
Staining and confocal imaging of LDs in seed sections
Hand sections of developing (65 to 80 days after flowering) jojoba cotyledons and embryonic axis seed tissues were prepared and then fixed in a 4% paraformaldehyde 50 mM Pipes NaOH (pH 7.2) solution overnight. Sections were washed two times (15 min per wash) with 50 mM Pipes NaOH (pH 7.2) buffer and then stained with BODIPY 493/503 [2 μg/ml in 50 mM Pipes NaOH (pH 7.2)] for 45 min under house vacuum. The stained sections were washed two times (15 min per wash under house vacuum) with 50 mM Pipes NaOH (pH 7.2 buffer) and one time in ultrapure water (18.2 megohms) for 10 min under house vacuum. Stained sections were placed on slides with a coverslip and sealed with clear nail polish. Sections were immediately imaged by confocal microscopy. A Zeiss LSM710 confocal laser scanning microscope was used to image BODIPY-stained LDs with the following settings: 63× objective, image resolution of 1024 × 1024, pinhole size of 62.2 Airy Units (AU), and master gain set between 650 and 750. The fluorophore BODIPY 493/503 was excited by a 488-nm laser, and emission signal was collected between 500 and 540 nm. Images were processed using Zeiss Zen imaging software (v8.1).
For the jojoba seed sections, the LD diameter of LDs of the cotyledon and embryonic axis tissues were measured using the ImageJ software package (103). LDs were measured only if the entire circumference of the LD could be distinguished. The diameters of 600 and 400 LDs were measured for three separate seed sections of cotyledon and embryonic axis tissues, respectively.
LD flotation and protein isolation
LDs were isolated from ~80-DAP developing jojoba seed cotyledon and embryonic axis tissues by flotation centrifugation using a protocol modified by Chapman and Trelease (104). Two solutions were prepared, solution A, which was 100 mM potassium phosphate (pH 7.5), 400 mM sucrose, 1 mM EDTA, and 10 mM KCl, and solution B, which was 100 mM potassium phosphate (pH 7.5), 600 mM sucrose, 1 mM EDTA, and 10 mM KCl, and stored on wet ice until/during use. Cotyledon and embryonic axis tissues were each finely chopped in solution B (2:1 solution:tissue, v/w) in a glass petri dish on ice using a razor blade. Resulting homogenates were filtered through Miracloth dampened with solution B into a high-speed centrifuge tube. Tubes were centrifuged at 500g at 4°C for 5 min to pellet large tissue debris. The supernatant, or crude homogenate, was transferred to a new tube, and an aliquot was reserved for protein precipitation. Solution A was carefully layered on top of the crude homogenate and centrifuged in a Sorvall HB-6 swinging bucket rotor at 10,500g at 4°C for 60 min. After centrifugation, the fat pad was removed and placed into a tube with 2.0 ml of buffer B and placed on wet ice. The supernatant, containing the cytosol and microsomes, was placed in a separate tube on wet ice, and the pellet, enriched in plastids and mitochondria, was suspended in 1 ml of ice-cold tris-HCl [100 mM (pH 7.5)] and reserved on wet ice. Solution A was carefully layered on top of the fat pad resuspended in solution B and centrifuged again at 10,500g at 4°C for 60 min. After centrifugation, the fat pad at the top of the tube was carefully removed and suspended in 2.0 ml of solution B, and then, 4.0 ml of buffer A was carefully layered on top. This process was repeated once more to achieve purified LDs. An aliquot (2 μl) of purified LDs in solution B was examined by confocal microscopy, whereas the remaining LD fraction was suspended in 1 ml of ice cold tris-HCl [100 mM (pH 7.5)].
The 10,500g supernatant fraction was centrifuged at 100,000g at 4°C for 60 min (fixed angle rotor, Sorvall Discovery 90) to obtain a microsomal pellet and cytosolic fraction. The supernatant, containing the cytosol fraction, was removed and kept on wet ice, and the pellet, containing the microsomes, was suspended in 1 ml of ice-cold tris-HCl [100 mM (pH 7.5)]. The isolated fractions of the crude homogenate, mitochondria/plastids, purified LDs, cytosol, and microsomes were each combined with four volumes of −20°C acetone and placed at −20°C overnight for protein precipitation. Proteins were pelleted 13,000g at 4°C for 15 min, after which the acetone was removed and the pellet was briefly dried. The protein pellets were suspended in 4× Laemmli buffer (Bio-Rad) and heated at 70°C for 15 min. Proteins were loaded on a 4 to 12% bis-tris gel and run at 160 V for ~15 min. The gel was fixed in a solution of 50:40:10 (water:ethanol:acetic acid; v/v/v), stained with QC Colloidal Coomassie Blue Stain (Bio-Rad) for 1 hour, and then destained in ultrapure water (18.2 megohms). For visualizing the proteins in each fraction, the samples were electrophoresed until the dye front was just at the bottom of the gel. For samples to be analyzed for proteomics, the proteins were concentrated by electrophoresis in the stacking gel and continued until the protein samples had just entered the separating gel. Bands in lanes corresponding to each fraction were excised from the gel and stored in a 5% acetic acid solution until prepared for proteomic analysis.
The aliquots of purified LDs were stained with BODIPY 493/503 (0.4 μg/ml) and imaged using a Zeiss confocal scanning microscope. A Zeiss LSM710 confocal laser scanning microscope was used to image BODIPY-stained LDs with the following settings: 63× objective, image resolution of 1024 × 1024, pinhole size of 62.2 AU, and master gain set between 650 and 750. The fluorophore BODIPY 493/503 was excited by a 488-nm laser, and emission signal was collected between 500 and 540 nm. Images were processed using Zeiss Zen imaging software (v8.1).
Proteomic analysis
Proteomics analysis was performed at the Michigan State University Proteomics Facility. Protein gel bands were dehydrated using 100% acetonitrile and incubated with 10 mM dithiothreitol in 100 mM ammonium bicarbonate (pH 8) at 56°C for 45 min, dehydrated again, and incubated in the dark with 50 mM iodoacetamide in 100 mM ammonium bicarbonate for 20 min. Dehydrated gel bands were then washed with ammonium bicarbonate and dehydrated again. Sequencing grade modified trypsin was prepared to 0.01 μg/μl in 50 mM ammonium bicarbonate, and ~50 μl of this was added to each gel band so that the gel was completely submerged. Bands were then incubated at 37°C overnight.
Peptides were extracted from the gel into a solution of 60% acetonitrile/1% trichloroacetic acid by water bath sonication and vacuum-dried to ~2 μl. Extracted peptides were then resuspended in 2% acetonitrile/0.1% trifluoroacetic acid to 20 μl. From this, 5 μl was automatically injected by a Thermo Fisher (www.thermofisher.com) EASYnLC 1000 onto a Thermo Acclaim PepMap 0.1 mm × 20 mm C18 peptide trap and washed for ~5 min. Bound peptides were eluted onto a Thermo Acclaim PepMap RSLC 0.075 mm × 500 mm C18 column over 95 min with a gradient of 5% B to 28% B in 84 min, ramping to 90% B at 85 min, and held at 90% B for the duration of the run at a constant flow rate of 0.3 μl/min (buffer A = 99.9% water/0.1% formic acid, buffer B = 99.9% acetonitrile/0.1% formic acid). The column was maintained at 50°C using an integrated column heater (PRSO-V1, Sonation GmbH, Biberach, Germany).
Eluted peptides were sprayed into a Thermo Fisher Q-Exactive mass spectrometer (www.thermofisher.com) using a FlexSpray spray ion source. Survey scans were taken in the Orbitrap (70000 resolution, determined at m/z 200), and the top 10 ions in each survey scan were then subjected to automatic higher-energy collision-induced dissociation with fragment spectra acquired at a resolution of 17,500. The resulting MS/MS spectra were converted to peak lists using Mascot Distiller, v2.7 (www.matrixscience.com) and searched against a database of jojoba protein sequences acquired from the de novo transcriptome assembly appended with common laboratory contaminants [downloaded from www.thegpm.org, cRAP (common Repository of Adventitious Proteins) project] using the Mascot searching algorithm, v 2.6.0. The Mascot output was then analyzed using Scaffold, v4.8.7 (www.proteomesoftware.com) to probabilistically validate protein identifications. Assignments validated using the Scaffold 1% false discovery rate confidence filter were considered true.
Protein counts taken from Scaffold were used to make relative quantifications of the proteins enriched in each protein fraction collected from the cotyledon and embryonic axis seed tissues. The LD protein fraction often contains protein contaminants from other cellular fractions. Thus, protein levels of the other fractions are needed as controls to eliminate contaminant proteins. Four criteria were used to develop a candidate list of proteins that were highly enriched in the LD protein fraction. Proteins were counted as an LD protein candidate if (i) peptide counts were the highest in the LD fraction, (ii) peptide counts for the protein in the LD fraction were two times greater than mitochondria/plastid fraction, (iii) peptide counts for the candidate LD protein in the cytosolic fraction must be less than 10, and (iv) the peptide counts in the LD fraction must be greater than 5.
Supplementary Material
Acknowledgments
Genome sequencing and assembly were performed by Novogene Corporation. Sequencing and preliminary analysis of transcriptome data were performed by the UNT BioDiscovery Institute Genomics Core Facility, and the advice of T. Kim is gratefully acknowledged. The jojoba genotype for these studies is available as accession no. PARL 940 from the National Arid Land Plant Genetic Resource Unit (NALPGRU), USDA-ARS, Parlier, CA, USA, curated by C. Heinitz. Funding: This work was funded by The National Key Research and Development Program of China (2016YFD0101000) and the Fundamental Research Funds for the Central Universities (2662015PY090 and 2662017PY043). D.S. received a UNT Dissertation Fellowship. Jojoba transcriptomes, MSI, and LD packaging studies were supported by the U.S. Department of Energy, Office of Science, BES–Physical Biosciences Program (DE-SC0016536). Lipidomic studies were supported by the Deutsche Forschungsgemeinschaft (DFG; INST 186/1167-1). Author contributions: K.D.C., L.G., and L.-L.C. designed this study. D.S. and S.L. performed the experiments. Z.-W.Z., Y.S., S.W., J.-M.S., J.Z., Z.-Q.Y., Q.-Y.Y., D.J.B., A.E.C., X.W., and R.K.A. led or assisted in bioinformatics and genetics analysis. C.H. and I.F. contributed to the lipidomic analysis. G.F.V. provided support for MSI experiments. J.M.D. and B.S. identified and collected plant material and conducted seed staging for experiments. L.B. and E.M. performed the whole seed nuclear magnetic resonance analyses. D.S., S.L., Z.-W.Z., Y.S., K.D.C., L.-L.C., and L.G. drafted the manuscript with input from other authors. All authors read and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The jojoba genome assembly and meta-data were deposited in the genome warehouse in Beijing Institute of Genomics (BIG) data center, BIG under accession no. GWHAASQ00000000 that is publicly accessible at http://bigd.big.ac.cn/gwh. RNA-seq transcriptomic data were deposited to NCBI GEO repository and can be accessed using accession no. GSE130603.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay3240/DC1
Fig. S1. Jojoba plants, flowers, and seed.
Fig. S2. The integrated annotation pipeline for the S. chinensis genome.
Fig. S3. Distribution of LTR insertion time of S. chinensis, S. oleracea, and B. vulgaris.
Fig. S4. Density distributions of the Ks values for homologous genes.
Fig. S5. Homologous gene dot plots.
Fig. S6. MRI-based quantitative imaging of lipid distributions in an intact mature seed of jojoba.
Fig. S7. NanoESI-MS/MS quantification of WE molecular species composition and content in jojoba cotyledon and embryonic axis tissues.
Fig. S8. UPLC-nanoESI-MS/MS quantification of TAG molecular composition in jojoba cotyledon and embryonic axis tissues.
Fig. S9. Transcriptome analysis of jojoba cotyledon and embryonic axis tissues.
Fig. S10. GO enrichment analysis of differentially expressed genes between jojoba cotyledon and embryonic axis tissues.
Table S1. Raw sequencing data.
Table S2. Anchored chromosome lengths of the S. chinensis genome.
Table S3. The statistics of Illumina reads mapped to the assembled genome.
Table S4. Statistics of transposable elements within the S. chinensis genome.
Table S5. Characterization of genes in the S. chinensis genome.
Table S6. Gene functional annotation statistics.
Table S7. Statistics of annotated noncoding RNAs.
Table S8. Evaluation of assembly completeness with RNA-seq data.
Table S9. Evaluation of assembly completeness with respect to genespace using CEGMA and BUSCO.
Table S10. Summary of the peaks in Ks distribution of S. chinensis paralogs and orthologs.
Table S11. The syntenic blocks detected by MCScanX.
Table S12. Orthology of protein-coding genes among 16 plants.
Table S13. Statistics of OrthoMCL analysis.
Table S14. Differential expression analysis between cotyledon and embryonic axis tissues.
Table S15. GO enrichment analysis of differentially expressed genes between cotyledon and embryonic axis tissues.
Table S16. Gene expression in different stages of a developing seed.
Table S17. Gene expression in different tissues of a developing seed.
Table S18. Filtered list of MS peptide counts from known and candidate cotyledon LD proteins.
Table S19. Filtered list of MS peptide counts from known and candidate embryonic axis LD proteins.
Table S20. Normalized peptide counts from different cellular fractions of jojoba developing cotyledon tissues.
Table S21. Normalized peptide counts from different cellular fractions of jojoba developing embryonic axis tissues.
REFERENCES AND NOTES
- 1.Gentry H. S., The natural history of jojoba (Simmondsia chinensis) and its cultural aspects. Econ. Bot. 12, 261–295 (1958). [Google Scholar]
- 2.National Research Council, Jojoba: New Crop for Arid Lands, New Raw Material for Industry (National Academies Press, 1985), vol. 53. [Google Scholar]
- 3.Angiosperm Phylogeny Group , An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20 (2016). [Google Scholar]
- 4.Greene R. A., Foster E. O., The liquid wax of seeds of Simmondsia californica. Bot. Gaz. 94, 826–828 (1933). [Google Scholar]
- 5.Miwa T. K., Jojoba oil wax esters and derived fatty acids and alcohols: Gas chromatographic analyses. J. Am. Oil Chem. Soc. 48, 259–264 (1971). [Google Scholar]
- 6.Ohlrogge J. B., Pollard M. R., Stumpf P. K., Studies on biosynthesis of waxes by developing jojoba seed tissue. Lipids 13, 203–210 (1978). [Google Scholar]
- 7.Gisser H., Messina J., Chasan D., Jojoba oil as a sperm oil substitute. Wear 34, 53–63 (1975). [Google Scholar]
- 8.Spencer G. F., Tallent W. H., Sperm whale oil analysis by gas chromatography and mass spectrometry. J. Am. Oil Chem. Soc. 50, 202–206 (1973). [Google Scholar]
- 9.Busson-Breysse J., Farines M., Soulier J., Jojoba wax: Its esters and some of its minor components. J. Am. Oil Chem. Soc. 71, 999–1002 (1994). [Google Scholar]
- 10.Iven T., Herrfurth C., Hornung E., Heilmann M., Hofvander P., Stymne S., Zhu L.-H., Feussner I., Wax ester profiling of seed oil by nano-electrospray ionization tandem mass spectrometry. Plant Methods 9, 24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aburjai T., Natsheh F. M., Plants used in cosmetics. Phytother. Res. 17, 987–1000 (2003). [DOI] [PubMed] [Google Scholar]
- 12.M. Katoh, M. Taguchi, T. Kunimoto, in Proceedings Seventh international Conference on Jojoba and Its Uses, A. R. Baldwin, Ed. (American Oil Chemists’ Society, 1988), pp. 318–342. [Google Scholar]
- 13.Meyer J., Marshall B., Gacula M. Jr., Rheins L., Evaluation of additive effects of hydrolyzed jojoba (Simmondsia chinensis) esters and glycerol: A preliminary study. J. Cosmet. Dermatol. 7, 268–274 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Ranzato E., Martinotti S., Burlando B., Wound healing properties of jojoba liquid wax: An in vitro study. J. Ethnopharmacol. 134, 443–449 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Jojoba Oil Market Size, Share & Trends Analysis Report By Application (Pharmaceutical, Cosmetics & Personal Care, Industrial), By Region (North America, Asia Pacific, Europe, MEA, Central & South America), And Segment Forecasts, 2019–2025; www.grandviewresearch.com/industry-analysis/jojoba-oil-market.
- 16.Miwa T., Rothfus J. A., Dimitroff E., Extreme—Pressure lubricant tests on jojoba and sperm whale oils. J. Am. Oil Chem. Soc. 56, 765–770 (1979). [Google Scholar]
- 17.El Kinawy O., Comparison between jojoba oil and other vegetable oils as a substitute to petroleum. Energy Source 26, 639–645 (2004). [Google Scholar]
- 18.Bisht R. P. S., Sivasankaran G. A., Bhatia V. K., Additive properties of jojoba oil for lubricating oil formulations. Wear 161, 193–197 (1993). [Google Scholar]
- 19.Kampf A., Grinberg S., Galun A., Oxidative stability of jojoba wax. J. Am. Oil Chem. Soc. 63, 246–248 (1986). [Google Scholar]
- 20.Arya D., Khan S., A review of Simmondsia chinensis (Jojoba) "the desert gold": A multipurpose oil seed crop for industrial uses. J. Pharm. Sci. Res. 8, 381–389 (2016). [Google Scholar]
- 21.Ivarson E., Iven T., Sturtevant D., Ahlman A., Cai Y., Chapman K., Feussner I., Zhu L.-H., Production of wax esters in the wild oil species Lepidium campestre. Ind. Crops Prod. 108, 535–542 (2017). [Google Scholar]
- 22.Iven T., Hornung E., Heilmann M., Feussner I., Synthesis of oleyl oleate wax esters in Arabidopsis thaliana and Camelina sativa seed oil. Plant Biotechnol. J. 14, 252–259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L.-H., Krens F., Smith M. A., Li X., Qi W., van Loo E. N., Iven T., Feussner I., Nazarenus T. J., Huai D., Taylor D. C., Zhou X.-R., Green A. G., Shockey J., Klasson K. T., Mullen R. T., Huang B., Dyer J. M., Cahoon E. B., Dedicated industrial oilseed crops as metabolic engineering platforms for sustainable industrial feedstock production. Sci. Rep. 6, 22181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Lopez N., Broughton R., Usher S., Salas J. J., Haslam R. P., Napier J. A., Beaudoin F., Tailoring the composition of novel wax esters in the seeds of transgenic Camelina sativa through systematic metabolic engineering. Plant Biotechnol. J. 15, 837–849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metz J. G., Pollard M. R., Anderson L., Hayes T. R., Lassner M. W., Purification of a jojoba embryo fatty acyl-coenzyme A reductase and expression of its cDNA in high erucic acid rapeseed. Plant Physiol. 122, 635–644 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lardizabal K. D., Metz J. G., Sakamoto T., Hutton W. C., Pollard M. R., Lassner M. W., Purification of a jojoba embryo wax synthase, cloning of its cDNA, and production of high levels of wax in seeds of transgenic Arabidopsis. Plant Physiol. 122, 645–655 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibarra-Laclette E., Lyons E., Hernández-Guzmán G., Pérez-Torres C. A., Carretero-Paulet L., Chang T.-H., Lan T., Welch A. J., Juárez M. J. A., Simpson J., Fernández-Cortés A., Arteaga-Vázquez M., Góngora-Castillo E., Acevedo-Hernández G., Schuster S. C., Himmelbauer H., Minoche A. E., Xu S., Lynch M., Oropeza-Aburto A., Cervantes-Pérez S. A., de Jesús Ortega-Estrada M., Cervantes-Luevano J. I., Michael T. P., Mockler T., Bryant D., Herrera-Estrella A., Albert V. A., Herrera-Estrella L., Architecture and evolution of a minute plant genome. Nature 498, 94–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nystedt B., Street N. R., Wetterbom A., Zuccolo A., Lin Y.-C., Scofield D. G., Vezzi F., Delhomme N., Giacomello S., Alexeyenko A., Vicedomini R., Sahlin K., Sherwood E., Elfstrand M., Gramzow L., Holmberg K., Hällman J., Keech O., Klasson L., Koriabine M., Kucukoglu M., Käller M., Luthman J., Lysholm F., Niittylä T., Olson Å., Rilakovic N., Ritland C., Rosselló J. A., Sena J., Svensson T., Talavera-López C., Theißen G., Tuominen H., Vanneste K., Wu Z.-Q., Zhang B., Zerbe P., Arvestad L., Bhalerao R., Bohlmann J., Bousquet J., Garcia Gil R., Hvidsten T. R., de Jong P., MacKay J., Morgante M., Ritland K., Sundberg B., Lee Thompson S., Van de Peer Y., Andersson B., Nilsson O., Ingvarsson P. K., Lundeberg J., Jansson S., The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Hamilton J. P., Robin Buell C., Advances in plant genome sequencing. Plant J. 70, 177–190 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Schatz M. C., Witkowski J., McCombie W. R., Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 13, 243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michael T. P., VanBuren R., Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 24, 71–81 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Yuan Y., Jin X., Liu J., Zhao X., Zhou J., Wang X., Wang D., Lai C., Xu W., Huang J., Zha L., Liu D., Ma X., Wang L., Zhou M., Jiang Z., Meng H., Peng H., Liang Y., Li R., Jiang C., Zhao Y., Nan T., Jin Y., Zhan Z., Yang J., Jiang W., Huang L., The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9, 1615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Parra G., Bradnam K., Korf I., CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Jaillon O., Aury J.-M., Noel B., Policriti A., Clepet C., Casagrande A., Choisne N., Aubourg S., Vitulo N., Jubin C., Vezzi A., Legeai F., Hugueney P., Dasilva C., Horner D., Mica E., Jublot D., Poulain J., Bruyère C., Billault A., Segurens B., Gouyvenoux M., Ugarte E., Cattonaro F., Anthouard V., Vico V., Fabbro C. D., Alaux M., Gaspero G. D., Dumas V., Felice N., Paillard S., Juman I., Moroldo M., Scalabrin S., Canaguier A., Clainche I. L., Malacrida G., Durand E., Pesole G., Laucou V., Chatelet P., Merdinoglu D., Delledonne M., Pezzotti M., Lecharny A., Scarpelli C., Artiguenave F., Pè M. E., Valle G., Morgante M., Caboche M., Adam-Blondon A.-F., Weissenbach J., Quétier F., Wincker P.; French-Italian Public Consortium for Grapevine Genome Characterization , The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Argout X., Salse J., Aury J.-M., Guiltinan M. J., Droc G., Gouzy J., Allegre M., Chaparro C., Legavre T., Maximova S. N., Abrouk M., Murat F., Fouet O., Poulain J., Ruiz M., Roguet Y., Rodier-Goud M., Barbosa-Neto J. F., Sabot F., Kudrna D., Ammiraju J. S. S., Schuster S. C., Carlson J. E., Sallet E., Schiex T., Dievart A., Kramer M., Gelley L., Shi Z., Bérard A., Viot C., Boccara M., Risterucci A. M., Guignon V., Sabau X., Axtell M. J., Ma Z., Zhang Y., Brown S., Bourge M., Golser W., Song X., Clement D., Rivallan R., Tahi M., Akaza J. M., Pitollat B., Gramacho K., D'Hont A., Brunel D., Infante D., Kebe I., Costet P., Wing R., McCombie W. R., Guiderdoni E., Quetier F., Panaud O., Wincker P., Bocs S., Lanaud C., The genome of Theobroma cacao. Nat. Genet. 43, 101–108 (2011). [DOI] [PubMed] [Google Scholar]
- 37.International Peach Genome Initiative, Verde I., Abbott A. G., Scalabrin S., Jung S., Shu S., Marroni F., Zhebentyayeva T., Dettori M. T., Grimwood J., Cattonaro F., Zuccolo A., Rossini L., Jenkins J., Vendramin E., Meisel L. A., Decroocq V., Sosinski B., Prochnik S., Mitros T., Policriti A., Cipriani G., Dondini L., Ficklin S., Goodstein D. M., Xuan P., Fabbro C. D., Aramini V., Copetti D., Gonzalez S., Horner D. S., Falchi R., Lucas S., Mica E., Maldonado J., Lazzari B., Bielenberg D., Pirona R., Miculan M., Barakat A., Testolin R., Stella A., Tartarini S., Tonutti P., Arús P., Orellana A., Wells C., Main D., Vizzotto G., Silva H., Salamini F., Schmutz J., Morgante M., Rokhsar D. S., The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Lu S., Sturtevant D., Aziz M., Jin C., Li Q., Chapman K. D., Guo L., Spatial analysis of lipid metabolites and expressed genes reveals tissue-specific heterogeneity of lipid metabolism in high- and low-oil Brassica napus L. seeds. Plant J. 94, 915–932 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Sturtevant D., Romsdahl T. B., Yu X.-H., Burks D. J., Azad R. K., Shanklin J., Chapman K. D., Tissue-specific differences in metabolites and transcripts contribute to the heterogeneity of ricinoleic acid accumulation in Ricinus communis L. (castor) seeds. Metabolomics 15, 6 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Sturtevant D., Horn P., Kennedy C., Hinze L., Percy R., Chapman K., Lipid metabolites in seeds of diverse Gossypium accessions: Molecular identification of a high oleic mutant allele. Planta 245, 595–610 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Sturtevant D., Lee Y.-J., Chapman K. D., Matrix assisted laser desorption/ionization-mass spectrometry imaging (MALDI-MSI) for direct visualization of plant metabolites in situ. Curr. Opin. Biotechnol. 37, 53–60 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Horn P. J., Sturtevant D., Chapman K. D., Modified oleic cottonseeds show altered content, composition and tissue-specific distribution of triacylglycerol molecular species. Biochimie 96, 28–36 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Woodfield H. K., Sturtevant D., Borisjuk L., Munz E., Guschina I. A., Chapman K., Harwood J. L., Spatial and temporal mapping of key lipid species in Brassica napus seeds. Plant Physiol. 173, 1998–2009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sturtevant D., Dueñas M. E., Lee Y.-J., Chapman K. D., Three-dimensional visualization of membrane phospholipid distributions in Arabidopsis thaliana seeds: A spatial perspective of molecular heterogeneity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 268–281 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Marmon S., Sturtevant D., Herrfurth C., Chapman K., Stymne S., Feussner I., Two acyltransferases contribute differently to linolenic acid levels in seed oil. Plant Physiol. 173, 2081–2095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munz E., Rolletschek H., Oeltze-Jafra S., Fuchs J., Guendel A., Neuberger T., Ortleb S., Jakob P. M., Borisjuk L., A functional imaging study of germinating oilseed rape seed. New Phytol. 216, 1181–1190 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Horn P. J., Silva J. E., Anderson D., Fuchs J., Borisjuk L., Nazarenus T. J., Shulaev V., Cahoon E. B., Chapman K. D., Imaging heterogeneity of membrane and storage lipids in transgenic Camelina sativa seeds with altered fatty acid profiles. Plant J. 76, 138–150 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Horn P. J., Korte A. R., Neogi P. B., Love E., Fuchs J., Strupat K., Borisjuk L., Shulaev V., Lee Y.-J., Chapman K. D., Spatial mapping of lipids at cellular resolution in embryos of cotton. Plant Cell 24, 622–636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Boven M., Holser R. A., Cokelaere M., Decuypere E., Govaerts C., Lemey J., Characterization of triglycerides isolated from jojoba oil. J. Am. Oil Chem. Soc. 77, 1325–1329 (2000). [Google Scholar]
- 50.Lassner M. W., Lardizabal K., Metz J. G., A jojoba beta-Ketoacyl-CoA synthase cDNA complements the canola fatty acid elongation mutation in transgenic plants. Plant Cell 8, 281–292 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.A. H. C. Huang, R. Qu, Y. K. Lai, C. Ratnayake, K. L. Chan, G. W. Kuroki, K. C. Oo, Y. Z. Cao, Structure, synthesis and degradation of oil bodies in maize, in Compartmentation of Plant Metabolism in Non-Photosynthetic Tissues, M. J. Emes, Ed. (Cambridge Univ. Press, 1991), vol. 42, pp. 43. [Google Scholar]
- 52.Huang A. H. C., Oil bodies and oleosins in seeds. Annu. Rev. Plant Biol. 43, 177–200 (1992). [Google Scholar]
- 53.Lin L.-J., Tai S. S. K., Peng C.-C., Tzen J. T. C., Steroleosin, a sterol-binding dehydrogenase in seed oil bodies. Plant Physiol. 128, 1200–1211 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Næsted H., Frandsen G. I., Jauh G.-Y., Hernandez-Pinzon I., Nielsen H. B., Murphy D. J., Rogers J. C., Mundy J., Caleosins: Ca 2+−binding proteins associated with lipid bodies. Plant Mol. Biol. 44, 463–476 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Pyc M., Cai Y., Greer M. S., Yurchenko O., Chapman K. D., Dyer J. M., Mullen R. T., Turning over a new leaf in lipid droplet biology. Trends Plant Sci. 22, 596–609 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Horn P. J., James C. N., Gidda S. K., Kilaru A., Dyer J. M., Mullen R. T., Ohlrogge J. B., Chapman K. D., Identification of a new class of lipid droplet-associated proteins in plants. Plant Physiol. 162, 1926–1936 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gidda S. K., Watt S., Collins-Silva J., Kilaru A., Arondel V., Yurchenko O., Horn P. J., James C. N., Shintani D., Ohlrogge J. B., Chapman K. D., Mullen R. T., Dyer J. M., Lipid droplet-associated proteins (LDAPs) are involved in the compartmentalization of lipophilic compounds in plant cells. Plant Signal. Behav. 8, e27141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gidda S. K., Park S., Pyc M., Yurchenko O., Cai Y., Wu P., Andrews D. W., Chapman K. D., Dyer J. M., Mullen R. T., Lipid droplet-associated proteins (LDAPs) are required for the dynamic regulation of neutral lipid compartmentation in plant cells. Plant Physiol. 170, 2052–2071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai Y., Goodman J. M., Pyc M., Mullen R. T., Dyer J. M., Chapman K. D., Arabidopsis SEIPIN proteins modulate triacylglycerol accumulation and influence lipid droplet proliferation. Plant Cell 27, 2616–2636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szymanski K. M., Binns D., Bartz R., Grishin N. V., Li W.-P., Agarwal A. K., Garg A., Anderson R. G. W., Goodman J. M., The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. U.S.A. 104, 20890–20895 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moreau R. A., Huang A. H., Gluconeogenesis from storage wax in the cotyledons of jojoba seedlings. Plant Physiol. 60, 329–333 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berthelot K., Lecomte S., Estevez Y., Peruch F., Hevea brasiliensis REF (Hev b 1) and SRPP (Hev b 3): An overview on rubber particle proteins. Biochimie 106, 1–9 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Marçais G., Kingsford C., A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C. A., Zeng Q., Wortman J., Young S. K., Earl A. M., Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dudchenko O., Batra S. S., Omer A. D., Nyquist S. K., Hoeger M., Durand N. C., Shamim M. S., Machol I., Lander E. S., Aiden A. P., Aiden E. L., De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurka J., Kapitonov V. V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J., Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Stanke M., Waack S., Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Majoros W. H., Pertea M., Salzberg S. L., TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Blanco E., Parra G., Guigó R., Using geneid to identify genes. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.3 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Burge C. B., Karlin S., Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 8, 346–354 (1998). [DOI] [PubMed] [Google Scholar]
- 71.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 72.Birney E., Clamp M., Durbin R., GeneWise and Genomewise. Genome Res. 14, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., Pachter L., Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., White O., Buell C. R., Wortman J. R., Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haas B. J., Delcher A. L., Mount S. M., Wortman J. R., Smith R. K. Jr., Hannick L. I., Maiti R., Ronning C. M., Rusch D. B., Town C. D., Salzberg S. L., White O., Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bairoch A., Apweiler R., The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zdobnov E. M., Apweiler R., InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Mistry J., Finn R. D., Eddy S. R., Bateman A., Punta M., Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang H., Bowers J. E., Wang X., Ming R., Alam M., Paterson A. H., Synteny and collinearity in plant genomes. Science 320, 486–488 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Wang D., Zhang Y., Zhang Z., Zhu J., Yu J., KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nei M., Gojobori T., Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986). [DOI] [PubMed] [Google Scholar]
- 84.Ellinghaus D., Kurtz S., Willhoeft U., LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu Z., Wang H., LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ou S., Jiang N., LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ossowski S., Schneeberger K., Lucas-Lledó J. I., Warthmann N., Clark R. M., Shaw R. G., Weigel D., Lynch M., The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327, 92–94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li L., Stoeckert C. J. Jr., Roos D. S., OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stamatakis A., RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Letunic I., Bork P., Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang Z., PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997). [DOI] [PubMed] [Google Scholar]
- 93.De Bie T., Cristianini N., Demuth J. P., Hahn M. W., CAFE: A computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Wu Y., Llewellyn D. J., Dennis E. S., A quick and easy method for isolating good-quality RNA from cotton (Gossypium hirsutum L.) tissues. Plant Mol. Biol. Rep. 20, 213–218 (2002). [Google Scholar]
- 95.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.S. Anders, W. Huber, Differential Expression of RNA-Seq Data at the Gene Level–The DESeq Package [European Molecular Biology Laboratory (EMBL), 2012].
- 97.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Storey J. D., Tibshirani R., Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hankin J. A., Barkley R. M., Murphy R. C., Sublimation as a method of matrix application for mass spectrometric imaging. J. Am. Soc. Mass Spectrom. 18, 1646–1652 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Horn P. J., Chapman K. D., Metabolite Imager: Customized spatial analysis of metabolite distributions in mass spectrometry imaging. Metabolomics 10, 337–348 (2014). [Google Scholar]
- 101.Chapman K. D., Moore T. S. Jr., N-acylphosphatidylethanolamine synthesis in plants: Occurrence, molecular composition, and phospholipid origin. Arch. Biochem. Biophys. 301, 21–33 (1993). [DOI] [PubMed] [Google Scholar]
- 102.Tarazona P., Feussner K., Feussner I., An enhanced plant lipidomics method based on multiplexed liquid chromatography-mass spectrometry reveals additional insights into cold- and drought-induced membrane remodeling. Plant J. 84, 621–633 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chapman K. D., Trelease R. N., Acquisition of membrane lipids by differentiating glyoxysomes: Role of lipid bodies. J. Cell Biol. 115, 995–1007 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaay3240/DC1
Fig. S1. Jojoba plants, flowers, and seed.
Fig. S2. The integrated annotation pipeline for the S. chinensis genome.
Fig. S3. Distribution of LTR insertion time of S. chinensis, S. oleracea, and B. vulgaris.
Fig. S4. Density distributions of the Ks values for homologous genes.
Fig. S5. Homologous gene dot plots.
Fig. S6. MRI-based quantitative imaging of lipid distributions in an intact mature seed of jojoba.
Fig. S7. NanoESI-MS/MS quantification of WE molecular species composition and content in jojoba cotyledon and embryonic axis tissues.
Fig. S8. UPLC-nanoESI-MS/MS quantification of TAG molecular composition in jojoba cotyledon and embryonic axis tissues.
Fig. S9. Transcriptome analysis of jojoba cotyledon and embryonic axis tissues.
Fig. S10. GO enrichment analysis of differentially expressed genes between jojoba cotyledon and embryonic axis tissues.
Table S1. Raw sequencing data.
Table S2. Anchored chromosome lengths of the S. chinensis genome.
Table S3. The statistics of Illumina reads mapped to the assembled genome.
Table S4. Statistics of transposable elements within the S. chinensis genome.
Table S5. Characterization of genes in the S. chinensis genome.
Table S6. Gene functional annotation statistics.
Table S7. Statistics of annotated noncoding RNAs.
Table S8. Evaluation of assembly completeness with RNA-seq data.
Table S9. Evaluation of assembly completeness with respect to genespace using CEGMA and BUSCO.
Table S10. Summary of the peaks in Ks distribution of S. chinensis paralogs and orthologs.
Table S11. The syntenic blocks detected by MCScanX.
Table S12. Orthology of protein-coding genes among 16 plants.
Table S13. Statistics of OrthoMCL analysis.
Table S14. Differential expression analysis between cotyledon and embryonic axis tissues.
Table S15. GO enrichment analysis of differentially expressed genes between cotyledon and embryonic axis tissues.
Table S16. Gene expression in different stages of a developing seed.
Table S17. Gene expression in different tissues of a developing seed.
Table S18. Filtered list of MS peptide counts from known and candidate cotyledon LD proteins.
Table S19. Filtered list of MS peptide counts from known and candidate embryonic axis LD proteins.
Table S20. Normalized peptide counts from different cellular fractions of jojoba developing cotyledon tissues.
Table S21. Normalized peptide counts from different cellular fractions of jojoba developing embryonic axis tissues.