

Overcoming the species specificity of the MRSA toxin LukAB with a novel mouse model establishes a role for LukAB in vivo.
Abstract
Many pathogens produce virulence factors that are specific toward their natural host. Clinically relevant methicillin-resistant Staphylococcus aureus (MRSA) isolates are highly adapted to humans and produce an array of human-specific virulence factors. One such factor is LukAB, a recently identified pore-forming toxin that targets human phagocytes by binding to the integrin component CD11b. LukAB exhibits strong tropism toward human, but not murine, CD11b. Here, phylogenetics and biochemical studies lead to the identification of an 11-residue domain required for the specificity of LukAB toward human CD11b, which is sufficient to render murine CD11b compatible with toxin binding. CRISPR-mediated gene editing was used to replace this domain, resulting in a “humanized” mouse. In vivo studies revealed that the humanized mice exhibit enhanced susceptibility to MRSA bloodstream infection, a phenotype mediated by LukAB. Thus, these studies establish LukAB as an important toxin for MRSA bacteremia and describe a new mouse model to study MRSA pathobiology.
INTRODUCTION
Several of the most deadly and common pathogens to mankind display species specificity and can only infect humans or closely related nonhuman primates (1). Other human pathogens have the ability to infect a broader range of species, yet these infections do not faithfully recapitulate human disease. Host tropism can be determined by host restriction factors, incompatible receptors necessary for pathogen invasion or adhesion, variances in interactions with the host immune system, or differences in nutrient availability (1). Coevolution of microbes with their hosts selects for specific host-pathogen interactions necessary for the survival and propagation of the microbe. Concurrently, host immune factors and pathogen-targeted molecules evolve to avoid recognition by pathogens while under selection for executing immunity functions. These interactions can unfold as “molecular arms races” where pathogen and host must continuously adapt for survival. Thus, pathogens may become highly specialized to the species where they thrive.
Staphylococcus aureus is a commensal of approximately 30% of the human population, residing in the nares, skin, and gastrointestinal tract (2). However, when S. aureus moves into deeper tissues, diverse and severe infections can result including skin and soft tissue infections, endocarditis, pneumonia, osteomyelitis, bacteremia, and sepsis, resulting in ~500,000 hospitalizations each year in the United States (3, 4). S. aureus also causes disease in livestock (5) and rodents (6). These animal-associated strains originated from humans but have acquired new traits specific to their animal host and, in some cases, lost the function of genes involved in human pathogenesis (7).
Clinically relevant human S. aureus isolates can cause disease in mice via several routes of infection if administered in high doses. Many studies of S. aureus infection have been performed using murine models; however, these models do not perfectly mimic human diseases (8). This mismatch is perhaps best illustrated by the fact that all S. aureus vaccine clinical trials to date have failed, despite showing efficacy in preclinical murine models. The shortcomings of these models to replicate diseases caused by human-adapted strains can partially be explained by the species specificity of a large array of S. aureus virulence factors (8, 9). One such factor is the bicomponent pore-forming leukotoxin LukAB (9, 10) [also known as LukGH (11)]. LukAB targets and kills human phagocytes by forming pores in target cell membranes (10, 11) following binding to its receptor, CD11b, a component of the αMβ2 integrin (also known as MAC-1 and CR3) (12). Specifically, LukAB binds with high affinity to the I-domain of CD11b (12). LukAB is produced during human infection (13), has been found in more than 99% of sequenced S. aureus isolates (14), and is responsible for killing primary human phagocytes during ex vivo infection with methicillin-sensitive and methicillin-resistant S. aureus isolates (MSSA and MRSA, respectively) (10, 11). Therefore, LukAB is thought to be a key virulence factor involved in disarming the host immune response and a potential drug target. However, LukAB exhibits low activity toward murine cells (12, 15), hampering in vivo studies to investigate the contribution of this toxin to S. aureus pathogenesis. This resistance to LukAB can be explained by the low affinity binding of LukAB to the murine CD11b I-domain, despite 78% amino acid identity to the human I-domain (12).
Here, we set out to dissect the breadth of the LukAB species tropism and exploit this information to generate a susceptible murine model. We examined LukAB binding to CD11b I-domains from a diverse list of mammals and combined this information with evolutionary analysis of the CD11b I-domains of primates and rodents. Our complementary approaches revealed residues displaying signatures of recurrent positive selection, including a residue involved in LukAB binding. Using a series of I-domain chimeras, we identified a region of 11 amino acids within the human I-domain critical for LukAB binding. With this knowledge, we edited the murine genome to encode a humanized LukAB-binding region in the murine CD11b I-domain. The humanized mice exhibit increased susceptibility to infection as well as increased bacterial burdens following bloodstream infection with MRSA in a LukAB-dependent manner. Thus, by combining phylogenetics, biochemistry, and genetic engineering, we have established a role for a human-specific virulence factor in the pathophysiology of S. aureus infection in a new mouse model system.
RESULTS
LukAB binding to the CD11b I-domain is species specific
Previous work demonstrated that LukAB potently targets neutrophils from humans and cynomolgus macaques; however, LukAB targets neutrophils from rabbits with lower affinity, as evidenced by a ~100-fold greater EC50 (median effective concentration) than human cells, and mice with even lower affinity, with an EC50 ~2000-fold greater than human cells (15). These results are consistent with studies showing that LukAB binds with high affinity to recombinant human I-domain, while binding to recombinant murine I-domain is weak or undetectable (12). We expanded on these studies by expressing and purifying recombinant I-domains from different mammals (Fig. 1, A and B) and examining binding of LukAB to the recombinant I-domains (Fig. 1C). As expected, we found that LukAB potently binds in a saturable and specific manner to human I-domain. In contrast, LukAB binds to the murine I-domain weakly and to the rabbit I-domain at intermediate levels. In addition, we found that LukAB binding to horse, rhesus monkey, and pig I-domains is strong and mirrors the binding to human I-domain (Fig. 1C). LukAB binds to rat I-domain in a similar manner as the intermediate binding to rabbit I-domain, while binding to I-domains from Chinese hamster, sheep, and cow is barely detectable (Fig. 1C). I-domains that are bound strongly by LukAB are >80% identical to the human I-domain, while the I-domains with <80% identity to the human I-domain show only weak or intermediate binding (Fig. 1D).
Fig. 1. Binding of LukAB to CD11b I-domain of different species.
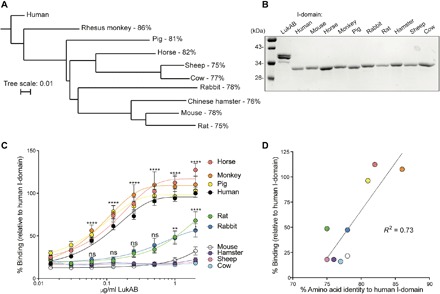
(A) Phylogenetic tree based on amino acid sequences of CD11b I-domains from different species and percent amino acid identities to the human I-domain. (B) Coomassie staining of 1 μg of LukAB and I-domains. (C) Binding of LukAB to recombinant I-domains from the species examined in (A) as measured. Data are normalized to the maximum 450-nm absorbance of LukAB bound to the human I-domain. Data are represented as the average of three independent experiments ± SEM. Statistical significance was determined by two-way ANOVA (****P < 0.0001; **P < 0.01; *P < 0.05; ns, not significant) compared to murine I-domain. (D) Linear regression analysis of % binding (relative to human I-domain) at 1 μg/ml and percent amino acid identity to human I-domain.
CD11b I-domain is subject to positive selection in primates and rodents
CD11b is a receptor for more than 40 diverse ligands and plays critical roles in the immune system, illustrated by the increased susceptibility of CD11b knockout mice to polymicrobial sepsis (16) and a variety of infections (17, 18). CD11b also serves as a target for LukAB and an array of other microbes and microbial factors (12, 19–22). Therefore, CD11b might be under selective pressure to avoid recognition by virulence factors while retaining its key roles in immune functions.
To examine the evolution of CD11b, we performed phylogenetic analyses on sequences collected from 19 anthropoid primate species (table S1) and separately on a sampling of 13 rodent species (table S2). We used complementary maximum likelihood [phylogenetic analysis by maximum likelihood (PAML)] and Bayesian [HyPhy: mixed effects model of evolution (MEME) and fast unbiased Bayesian approximation (FUBAR)] implementations of phylogenetic analysis to calculate ratios of nonsynonymous and synonymous substitution rates (dN/dS) for individual sites within the CD11b coding sequence. Significantly elevated dN/dS values were observed for multiple residues in CD11b (Fig. 2A) in both the primate and rodent clades (table S3). These results indicate that ITGAM, the CD11b coding gene, has undergone recurrent bouts of positive selection reminiscent of many canonical innate immunity factors (23, 24). Sites undergoing positive selection primarily clustered in the CD11b I-domain (Fig. 2A), the domain implicated in binding to endogenous ligands as well as LukAB (25).
Fig. 2. Positive selection on the CD11b I-domain resulting in changes in LukAB binding.
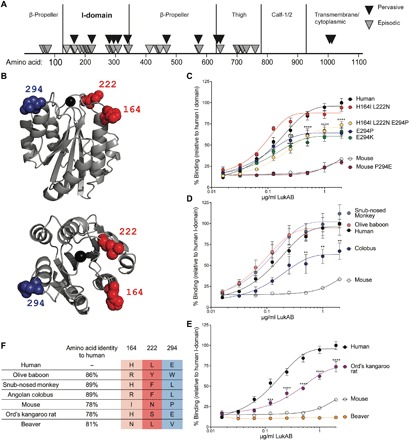
(A) Sites undergoing episodic (MEME and PAML) and pervasive (FUBAR) positive selection in full-length CD11b are indicated by triangles. (B) Structure of the human I-domain [Protein Data Bank (PDB) ID: 1IDO]. Residues displaying signatures of positive selection in three independent algorithms [FUBAR (P > 0.9); PAML (M7 versus M8, P ≤ 0.05); MEME (P ≤ 0.05)] are shown as spheres and labeled. (C to E) Binding of LukAB to I-domain mutants (C) and I-domains from various primates (D) and rodents (E) as measured. Data are normalized to the maximum 450-nm absorbance of LukAB bound to the human I-domain. Data are represented as the average of three independent experiments ± SEM. Statistical significance was determined by two-way ANOVA (****P < 0.0001; ***P < 0.001; **P < 0.01). (F) Table of primates and rodents analyzed in (D) and (E) showing percent amino acid identity to the human I-domain and residues at sites 164, 222, and 294.
We hypothesized that the signatures of positive selection at sites within the CD11b I-domain could be driven by interactions with virulence factors produced by pathogens, including LukAB. In both the anthropoid and rodent clades, amino acid sites 164, 222, and 294 showed shared signatures of pervasive positive selection (Fig. 2B) in three independent analyses (PAML, MEME, and FUBAR). To determine whether amino acid variation at these sites influenced the interaction between CD11b and LukAB, we mutated these residues between the human and murine I-domains (fig. S1A). Mutating the glutamic acid at position 294 (E294) in humans to a proline (murine residue) or a lysine (basic residue) resulted in a significant reduction in LukAB binding compared to the human I-domain (Fig. 2C). However, mutating the proline at position 294 in the mouse I-domain to glutamic acid (P294E) did not result in an increase in LukAB binding, suggesting that additional residues in the human I-domain are required for LukAB binding. Mutating the histidine at site 164 and the leucine at site 222 to the murine residues (H164I L222N) did not affect LukAB binding to the human I-domain. Binding of LukAB to the human I-domain with all three of these residues mutated to the murine residues was similar to E294P and E294K, further supporting that residues 164 and 222 are likely not involved in toxin-receptor interactions but could reflect genetic conflicts arising from other CD11b-pathogen interactions.
Next, the binding of I-domains from additional primates that were included in our analysis and encode divergent amino acids compared to humans at sites 164, 222, and 294 was examined. The I-domain from the Angolan colobus showed a reduction in binding to LukAB, despite high (89%) homology to the human I-domain (Fig. 2, D to F). This reduced binding may be explained by the fact that the I-domain contains a leucine at site 294 (Fig. 2, D to F). In contrast, the golden snub–nosed monkey also has a leucine at this position, but LukAB binds to this I-domain comparably to the human I-domain. Thus, these findings suggest combinatorial contributions in site variation for LukAB binding.
We also examined binding of LukAB to I-domains from additional rodent species. We found that the Ord’s kangaroo rat I-domain showed greatly increased binding compared to the murine I-domain (Fig. 2E). The Ord’s kangaroo rat I-domain contains a glutamic acid at position 294, which is conserved with the human I-domain. The loop surrounding this site is also highly conserved with humans. We hypothesize that this loop is important for LukAB binding to the Ord’s kangaroo rat I-domain, despite this I-domain only having 78% overall amino acid identity to the human I-domain (Fig. 2F).
Together, these data demonstrate that ITGAM has undergone bouts of positive selection at select residues, specifically at the I-domain. We identify site 294 as one such site, which is important for LukAB binding.
Identification of a critical region within the human I-domain involved in LukAB binding
To further demarcate the LukAB-binding interface within the CD11b I-domain, we compared the human and mouse I-domain amino acid sequences (Fig. 3A) and mapped the divergent amino acids on the human I-domain structure (26). We found that the divergent amino acids were mostly surface-exposed (Fig. 3B). We separated the divergent amino acids into seven groups based on location within the I-domain structure (Fig. 3, A and B). Recombinant chimeric I-domains containing murine residues from each of the seven groups in the human I-domain backbone (HMH chimeras) were generated and used for binding studies (fig. S2, A and B). LukAB bound to HMH chimeras 1 to 6 at similar levels as the wild-type (WT) human I-domain (Fig. 3C). In contrast, LukAB bound poorly to HMH chimera 7.
Fig. 3. I-domain chimeras reveal a critical region for LukAB binding.
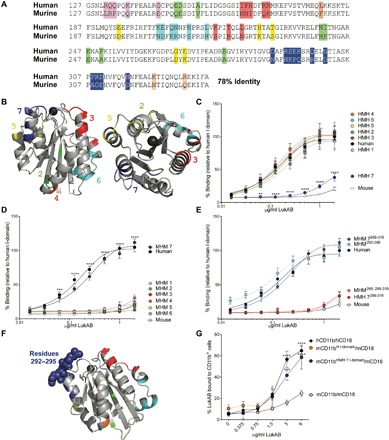
(A) Amino acid alignment of murine and human I-domains. Divergent amino acids are highlighted. Each color represents a different I-domain chimera. (B) Structure of human I-domain (PDB ID: 1IDO). Regions conserved with murine I-domain are shown in gray, and divergent regions are colored as in (A). (C to E) LukAB binding to human, mouse, HMH chimeric (murine residues in human I-domain backbone) (C to E), and MHM chimeric (human residues in murine I-domain backbone) (D and E) I-domains. HMH 7289–316 and MHM 7289–316 refer to HMH 7 and MHM 7 shown in (C and D), respectively (E). Data are normalized to the maximum 450-nm absorbance of LukAB bound to human I-domain. Data are represented as the average of three independent experiments ± SEM. Statistical significance was determined by two-way ANOVA (****P < 0.0001; ***P < 0.001; **P < 0.01) compared to human (C) or murine (D) I-domains. (F) Structure of human I-domain (PDB ID: 1IDO). Regions that are conserved with murine I-domain are shown in gray, and divergent regions are shown following the same color scheme as in (A) and (B). Residues 292 to 295 from chimera 7 are shown as spheres. (G) LukAB binding to HEK293T cells transfected with full-length CD11b and CD18. The percentage of LukAB bound to CD11b + cells was evaluated by flow cytometry. Data are represented as the average of three independent experiments ± SEM. Statistical significance was determined by two-way ANOVA (****P < 0.0001; ***P < 0.001; *P < 0.05) compared to murine CD11b. See also fig. S2.
We also performed the inverse experiment where the same amino acids were mutated to the human residues in the murine I-domain backbone (MHM chimeras). While LukAB bound poorly to MHM chimeras 1 to 6, the toxin bound to MHM 7 with similar levels as the human I-domain (Fig. 2D). Thus, region 7, which contains amino acids 289 to 316, is necessary for LukAB binding to human I-domain and sufficient to render murine I-domain compatible with LukAB binding. Of note, the glutamic acid 294 falls within this region, further supporting the importance of this residue in the LukAB/I-domain interaction.
The amino acids in chimera 7 span a loop and part of an α helix proximal to the I-domain’s metal ion–dependent adhesion site (MIDAS). A divalent cation at this site is necessary for the binding of most endogenous CD11b ligands (27), and it is likely that this region is also critical for LukAB binding. Thus, we next tried to further narrow down the LukAB-binding region on the CD11b I-domain. After testing additional chimeras examining smaller groups of residues within chimera 7, we found that a chimera with the murine I-domain backbone where only residues 292 to 295 were humanized (MHM 7292–295) (Fig. 3E) was compatible with LukAB binding in a manner similar to the human I-domain and the full-length MHM 7 (MHM 7289–316). In addition, if we change every other mutated residue in MHM 7 except for residues 292 to 295 to the human sequence in the murine I-domain backbone (MHM 7289, 298–316), LukAB bound poorly and similarly to the murine I-domain.
To further extend the enzyme-linked immunosorbent assay (ELISA) data, we performed surface plasmon resonance (SPR) to measure binding kinetics of LukAB and key chimeras (Table 1). We found that the human I-domain has a dissociation constant (KD) of 39.29 nM, while the mouse I-domain had a KD of more than 6000-fold greater magnitude at 246.6 μM. Our gain-of-binding chimera MHM 7 had a KD of 69.60 nM, a 1.7-fold greater magnitude than the human I-domain. In contrast, the MHM 7292–295 had a KD of 173.98 nm, a 4.4-fold greater magnitude than the human I-domain, suggesting that the other residues outside the 292–295 loop included in the MHM 7 chimera also contribute to toxin binding.
Table 1. SPR data of LukAB binding to recombinant I-domains.
| Protein + LukAB | KD |
| Human | 39.29 ± 8.12 nM |
| Mouse | 246.6 ± 121.9 μM |
| HMH 7 | 78.10 ± 21.9 μM |
| MHM 7 | 69.60 ± 19.51 nM |
| MHM 7292–295 | 173.70 ± 18.9 nM |
Humanization of residues 289 to 316 renders murine CD11b compatible with LukAB binding
The studies described above identified the human residues 289 to 316 as being sufficient to render purified murine I-domain compatible with LukAB binding. We next wanted to determine whether introduction of the human 289 to 316 residues into the full-length, surface-exposed murine CD11b could render the receptor compatible with toxin biding. Human embryonic kidney (HEK) 293T cells were transfected with plasmids to encode for full-length murine CD11b containing the entire human I-domain or the MHM 7 chimeric I-domain along with murine CD18 (fig. S2C). While expression of human CD11b/CD18 rendered cells compatible with LukAB binding, expression of murine CD11b/CD18 did not, validating the whole cell binding assay. We found that LukAB was able to bind to cells producing humanized murine CD11b, containing either the WT human I-domain sequence or the 11 human residues within MHM 7 (Fig. 3G). Therefore, humanizing the LukAB-binding domain within the murine CD11b renders host cells compatible with LukAB binding.
Generation of a humanized CD11b mouse
The notable increase in LukAB binding observed in the MHM 7 chimera prompted us to evaluate whether this region could be humanized in the mouse. Murine Itgam is a large gene, containing 55,852 nucleotides and 30 exons. Luckily, the region identified here as critical for LukAB binding is encoded solely by exon 9. We used CRISPR-Cas9 (28) to edit exon 9 to encode for the human residues 289 to 316 (Fig. 4A and fig. S3A). The “humanized CD11b” mice (hCD11b mice) were generated and found to be normal in appearance, breeding ability, viability, and health.
Fig. 4. Characterization of the hCD11b mouse.
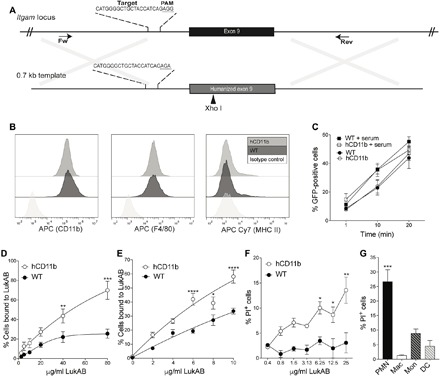
(A) Schematic representation of murine Itgam locus and DNA template used to humanize exon 9. (B to D) iBMDMs from WT and hCD11b mice were stained for CD11b, F4/80, and MHC II and evaluated by flow cytometry (B). (C) WT and hCD11b iBMDMs incubated with GFP-producing S. aureus ± serum and phagocytosis measured as % iBMDMs GFP positive by flow cytometry. Data are represented as the average of three independent experiments performed in duplicate ± SEM. (D and E) WT and hCD11b iBMDMs (D) and peritoneal exudate cells (E) were incubated with biotinylated LukAB, and bound LukAB was quantified by flow cytometry. Data are represented as the average of three independent experiments performed in duplicate ± SEM. Statistical significance was determined by two-way ANOVA (****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05). (F) Peritoneal exudate cells from WT and hCD11b mice were treated with LukAB, and propidium iodide (PI) incorporation was quantified by flow cytometry. Data are represented as the average of three independent experiments performed in duplicate ± SEM. Statistical significance was determined by two-way ANOVA (**P < 0.01 and *P < 0.05). (G) Peritoneal exudate cells treated with LukAB (12.5 μg/ml) were stained, and % PI-positive PMNs (CD11b+ and Ly6G+), macrophages (CD11b+ and F4/80+), monocytes (CD11b+, Ly6C+, and Ly6G−), and DCs (CD11b+, CD11c+, and F4/80−) were quantified by flow cytometry. Data are represented as the average of three independent experiments performed in duplicate ± SEM. Statistical significance was determined by one-way ANOVA (***P < 0.001).
To characterize these mice, we first harvested bone marrow cells from the hCD11b and WT mice and generated immortalized bone marrow–derived macrophages (iBMDMs) (29). iBMDMs from WT and hCD11b mice were found to have identical levels of CD11b, F4/80, and major histocompatibility complex (MHC) class II on their surfaces (Fig. 4B). Moreover, the hCD11b cells are able to phagocytose green fluorescent protein (GFP) S. aureus both with and without serum similar to their WT counterparts (Fig. 4C). Thus, it does not appear that hCD11b mice have grossly altered CD11b-dependent functions or expression levels.
Next, binding of LukAB to iBMDMs from WT and hCD11b mice was measured. Consistent with the HEK293T whole-cell binding studies (Fig. 3G), we observed that LukAB binds to cells from mice producing hCD11b but poorly to cells from WT mice (Fig. 4D). To confirm these data, we measured the interaction between LukAB and murine iBMDMs using whole-cell SPR. The affinity for the iBMDMs expressing hCD11b was 13-fold higher than the iBMDMs expressing WT CD11b, with a KD of 187.7 nM (fig. S3B).
We next performed additional functional studies using freshly isolated primary murine peritoneal exudate cells. We observed improved binding in cells from the hCD11b mouse compared to the WT mice (Fig. 4E). Of note, increased LukAB binding was directly linked with toxin-mediated membrane damage of hCD11b peritoneal exudate cells (Fig. 4F). Peritoneal exudate cells contain a variety of leukocytes that express CD11b, including polymorphonuclear neutrophils (PMNs), macrophages, monocytes, and dendritic cells (DCs). To identify the target(s) of LukAB, we treated primary murine peritoneal exudate cells from the hCD11b mouse with toxin and performed multiparametric flow cytometry analyses. These studies revealed that PMNs are the predominant leukocyte susceptible to LukAB-mediated membrane damage, followed by monocytes and DCs (Fig. 4G).
Humanizing CD11b renders mice susceptible to S. aureus infection
We next determined whether the hCD11b mice are more susceptible to S. aureus intravenous infection. We used the MRSA strain LAC (Los Angeles County), a representative of the pulsed-field gel electrophoresis type USA300, the most common cause of community-acquired MRSA infections, as well as an emerging cause of hospital-associated MRSA infections in the United States (30). Age-matched WT and hCD11b mice were infected retro-orbitally with 3 × 106 colony-forming units (CFUs) of USA300, and bacterial burdens in the organs were examined 3 days after infection. In this experimental setting, only ~30% of the WT mice exhibit detectable bacterial burdens in the liver. In contrast, the hCD11b mice had detectable bacterial burdens in 86% of the mice, a phenotype dependent on LukAB (Fig. 5A). Thus, humanization of CD11b reduced the barrier of infection exhibited by WT mice. By examining bacterial burden in organs, we noticed over 1-log increase in CFU in the livers of hCD11b mice compared to WT mice (Fig. 5B). Of note, there was no statistically significant difference in bacterial burdens in the spleen, kidney, heart, and lungs and no difference in weight loss or survival (fig. S4, A and B). The increased CFU phenotype observed in the hCD11b mice is also dependent on LukAB, as hCD11b mice infected with ΔlukAB USA300 phenocopied WT mice infected with WT USA300 (Fig. 5B).
Fig. 5. The hCD11b mouse is susceptible to MRSA infection.
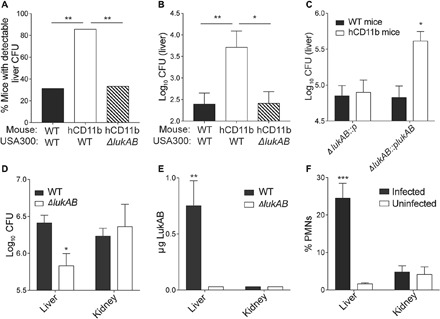
(A and B) WT and hCD11b mice were infected intravenously with ~3 × 106 CFU of WT USA300 strain LAC or an isogenic ΔlukAB LAC strain. Three days after infection, % of mice with detectable CFUs in the liver was determined (A). Statistical significance was determined by chi-square test (**P < 0.01). (B) CFUs in the livers were also quantified. Data are represented as the average of three independent experiments with 15 total mice per group. Statistical significance was determined by one-way ANOVA (**P < 0.01 and *P < 0.05). (C) WT and hCD11b mice were infected intravenously with 1 × 107 ΔlukAB::p LAC or ΔlukAB::plukAB LAC. One day after infection, CFUs in the livers were quantified. Data are represented as the average of two independent experiments with 10 total mice per group. Statistical significance was determined by one-way ANOVA (*P < 0.05). (D and E) hCD11b mice were infected intravenously with 1 × 108 CFU of WT LAC or an isogenic ΔlukAB LAC strain. (D) CFUs in the livers and kidneys 1 day after infection. (E) Tissue homogenates from (D) were also used to quantify LukAB. Data are represented as the average of three independent experiments with 15 total mice per group. Statistical significance was determined by one-way ANOVA (**P < 0.01). See also fig. S4. (F) WT mice were infected with S. aureus intravenously. One day after infection, the % Ly6G+ CD11b+ PMNs out of the total CD45% leukocytes were quantified. Data are represented as the average of two independent experiments with six total mice per group. Statistical significance was determined by one-way ANOVA (***P < 0.001).
We next performed complementation studies by infecting mice with 1 × 107 CFUs of ΔlukAB USA300 or an isogenic strain where lukAB was expressed on a plasmid. Organs were harvested 1 day after infection to minimize loss of the complementing plasmid. These studies further demonstrated an increase in liver CFU in the hCD11b mice in a LukAB-dependent manner (Fig. 5C). Thus, humanizing CD11b lowers the resistance of mice to bloodstream infection with MRSA, in a manner dependent on LukAB.
LukAB exhibits tissue-specific tropism in vivo
We hypothesized that the observed liver-specific increase in bacterial burden may be explained by differential regulation/production of LukAB in vivo. To tackle this, we developed an ELISA to quantify the amount of LukAB produced in vivo. We examined LukAB levels in the livers and the kidneys 1 day after infection. Despite comparable bacterial burdens at this time point (Fig. 5D), we were able to detect LukAB in the liver, but not the kidney, of infected mice (Fig. 5E and fig. S4, C and D). Therefore, LukAB seems to be produced at higher levels in the liver, one of the first organs infected upon bloodstream infections (31) compared to other organs, providing a potential explanation for the liver-specific LukAB-mediated increase in bacterial burden.
A tissue-specific phenotype could also be explained by the recruitment and/or local quantities of susceptible cells. As PMNs are the predominant hCD11b cell type susceptible to LukAB (Fig. 4G), we quantified the PMNs in murine livers and kidneys 1 day after infection and in uninfected controls. These studies revealed that the proportion of PMNs were enhanced more than 10-fold in infected livers 1 day after infection compared with uninfected livers, while PMNs in the kidneys remained low at this time point (Fig. 5F and fig. S4). Together, these data suggest that quantities of both LukAB and the cells that LukAB targets are enhanced in the liver at early time points after infection, providing an explanation for the tissue-specific LukAB-mediated increase in bacterial burden.
DISCUSSION
Here, we defined the molecular determinants of the species specificity exhibited by the LukAB toxin and used this information to establish an improved mouse model of S. aureus infection. To gain insight into how LukAB binds to the human CD11b I-domain but not the murine I-domain, we first took a broad evolutionary genetic approach focusing on the substantial variation observed among primates and rodents. We found that several sites within CD11b, particularly within the I-domain, showed signatures of pervasive positive selection. This suggests that these sites have been under strong and repeated selective pressure favoring substitutions that decrease recognition by pathogens. Because these signals were inferred over ~40 million years of primate divergence, and a similar evolutionary interval in our sampling of rodents, pinpointing outcomes of selection by specific ancient pathogens over that long period of time is challenging. However, recurrent sites of selection in the I-domain are consistent with repeated interactions with pathogens encoding LukAB-like functions. These repeated bouts of selection can leave modern species either more resistant or sensitive to contemporary infections owing to evolutionary consequential interactions from the past. Of these sites in the I-domain, we identified one residue, glutamic acid at site 294 (E294), that plays a role in LukAB binding. By examining the residues at this site in rodents, we identified the Ord’s kangaroo rat I-domain to be compatible with LukAB binding despite its overall low homology with the human I-domain. We hypothesize that this is due to the glutamic acid at site 294 and perhaps the amino acid sequences of the surrounding loop, which are highly overlapping with humans. However, E294 is not solely responsible for LukAB binding. Using a series of I-domain chimeras, we found a region containing 11 divergent amino acids that is necessary for binding. Replacing this region with the human sequence results in high-affinity LukAB binding to mouse CD11b.
As LukAB binds poorly to murine CD11b, the contribution of this toxin to S. aureus infection in current murine models is underestimated. To address this deficiency, we generated a mouse containing a murine CD11b receptor that was engineered to harbor the 11 human amino acids critical for LukAB binding to human CD11b I-domain. The hCD11b mice appear to have undisturbed CD11b functions, likely due to the small size of the region we mutated and the fact that this domain has not been implicated in the binding to other ligands of CD11b. Collectively, our data demonstrate that these mice are more susceptible to MRSA infections. Infection of WT mice with WT USA300 phenocopied infection of hCD11b mice with ΔlukAB USA300, which highlights that the increased susceptibility described here is mediated by LukAB and that the hCD11b mice are not inherently more susceptible to S. aureus infection by strains lacking LukAB.
We found that LukAB is produced at high levels in the liver compared to the kidney, despite similar bacterial burdens 1 day after intravenous infection. In addition, PMNs, the predominant phagocyte susceptible to LukAB in the hCD11b mouse, markedly increase in infected livers 1 day after infection while remaining low in infected kidneys. These findings are complementary to studies that have shown that abscess formation in the kidneys, which is accompanied by massive PMN infiltration, is first observed ~3 days after intravenous S. aureus infection despite high bacterial burdens (32). The livers, in contrast, have been shown to play a critical role early in intravenous S. aureus infection. Following infection, the bacteria are rapidly sequestered and controlled by Kupffer cells in the liver, forming a reservoir of bacteria (31). Ultimately, either the bacteria will escape and disseminate to the other organs or the infection will be contained. Together, this can explain why we observe LukAB-dependent phenotypes in the liver but not in other organs like the kidneys under the condition examined. CD11b on PMNs has been shown to be up-regulated in the presence of S. aureus (33), while LukAB has been shown to be up-regulated in the presence of PMNs (34). Thus, the host responds to the presence of bacteria by recruiting PMNs with high levels of CD11b, yet the bacteria exploit this by producing LukAB to injure these cells.
Here, we exclusively characterize a bloodstream infection model. However, S. aureus is responsible for a variety of diverse diseases, and the role of LukAB needs to be characterized in other models (e.g., skin infection, pneumonia, osteomyelitis, indwelling device–associated infections, and more). In the future, it will be interesting to determine whether this liver phenotype is dependent on the site of infection.
LukAB is one of many S. aureus virulence factors that exhibit human tropism. We report here a road map on how combining evolutionary studies with biochemistry and genetic engineering can facilitate in vivo studies of species-specific host-pathogen interactions. By humanizing or knocking-in human-specific targets, it should be feasible to generate improved mouse models that more closely mimic human infections. These models would also facilitate studies to evaluate the effects of anti–S. aureus therapies designed against previously untested virulence factors, increasing our ability to identify appropriate targets for the generation of much needed anti–S. aureus agents.
MATERIALS AND METHODS
Ethics statement
All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of New York University and were performed according to guidelines from the National Institutes of Health (NIH), the Animal Welfare Act, and the U.S. Federal Law.
Bacterial strains
Escherichia coli DH5a was used in cloning procedures. E. coli T7 LysY/LacQ was used for human FLAG-tagged CD11b I-domain expression. All E. coli strains were grown in Luria-Bertani (LB) broth.
S. aureus strains used in the study are listed in table S4. For recombinant protein expression and infection studies, S. aureus strains were streaked to single colonies on tryptic soy agar (TSA) plates. Single colonies were inoculated in tryptic soy broth (TSB) for overnight culture and then subsequently subcultured 1:100 for 3 hours in TSB.
Generation of I-domain expression plasmids and purification of recombinant I-domains
The I-domains from human ITGAM (NM_001145808.1) and murine Itgam (NM_001082960.1), along with a 5′ Nde I site, 3′ 6-glycine linker, 3′ 3×FLAG tag, 3′ Xho I site, and the desired mutations (table S6), were produced as gBlocks Gene Fragments from Integrated DNA Technologies (IDT) and cloned into pET15b using standard cloning methods. Plasmids were transformed into E. coli T7 LysY/LacQ and purified as described previously (12). Briefly, strains were grown at 37°C, 180 rpm in LB broth supplemented with ampicillin (100 μg/ml) to an OD600 (optical density at 600 nm) of 0.5 and then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 hours. Bacteria were lysed with xTracter buffer (Clontech) supplemented with protease inhibitor (Roche), lysozyme (1 mg/ml), and Benzonase nuclease (Sigma) at 3 U/ml of culture. Lysates were incubated with nickel–nitrilotriacetic acid (NTA) resin (Qiagen), washed, and his-tagged. I-domains were eluted with 500 mM imidazole. Purified proteins were dialyzed in 1× tris-buffered saline (TBS) + 10% glycerol at 4°C overnight and then stored at −80°C.
Purification of LukAB
LukAB with a 5′ 6×-histidine tag was copurified from S. aureus (table S4), as previously described (34). Briefly, strains were grown in TSB with chloramphenicol (10 μg/ml) for 5 hours at 37°C, 180 rpm, to an OD600 of approximately 1.5 (which represents 1 × 109 CFU/ml). The bacteria were then pelleted, and the supernatant was collected and filtered. NTA resin (Qiagen) was incubated with culture supernatant, washed, and eluted with 500 mM imidazole. The protein was dialyzed in 1× TBS plus 10% glycerol at 4°C overnight and then stored at −80°C.
ELISA to measure binding of LukAB to I-domains
Recombinant I-domains (10 μg/ml) were coated onto the wells of an Immulon 2HB 96-well plate in phosphate-buffered saline (PBS) overnight at 4°C. Wells were blocked with blotto buffer (2% dry fat-free milk, 0.9% NaCl, 0.05% Tween 20, PBS), washed, and incubated with the indicated concentration of LukAB in blotto buffer for 30 min at room temperature with shaking conditions. Wells were washed and incubated with anti-LukA rabbit polyclonal antibody at a 1:3000 dilution for 1 hour at room temperature with shaking. Next, wells were washed and incubated with anti-rabbit horseradish peroxidase (HRP) at a 1:2500 dilution for 1 hour at room temperature with shaking. Wells were washed and incubated with 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate for 5 min. The reaction was terminated with 100 μl of sulfuric acid, and absorbance at 450 nm was measured.
Evolutionary analysis
Full-length complementary DNA sequences of CD11b were retrieved from the National Center for Biotechnology Information (NCBI) database using BLAST searches. All DNA sequences were aligned using MUSCLE with default settings. Sequences were manually trimmed to eliminate all insertions, deletions, and stop codons. The evolutionary analysis was performed using the trimmed alignment file as well as a constrained species tree that represented the most currently accepted evolutionary relationships of anthropoids and rodents.
The anthropoid and rodent clades were individually assessed for signatures of positive selection. The trimmed alignment files and phylogenetic trees were used as input files for three complementary software packages: PAML (v4.8, M7 versus M8), as well as the MEME and FUBAR algorithms available through the HyPhy package. Sites that were identified to have signatures of both episodic and pervasive diversifying selection across all three algorithms were considered robust candidates for further functional characterization.
Antibodies
The following antibodies were used: anti-LukA rabbit polyclonal antibody (10), goat anti-rabbit immunoglobulin G HRP (SC-2004), allophycocyanin (APC) anti-mouse/human CD11b, clone M1/70 (BioLegend, 101212); APC anti-mouse F4/80, clone BM8 (BioLegend, 123115); APC-Cy7 anti-mouse I-A/I-E, clone M5/114.15.2 (BioLegend, 107627); phycoerythrin (PE)–Cy7 anti-mouse/human CD11b, clone M1/70 (BioLegend, 101215); fluorescein isothiocyanate (FITC) anti-mouse Ly6G, clone 1A8 (BD Biosciences, 551460); APC anti-mouse F4/80, clone BM8 (BioLegend, 123116); peridinin chlorophyll protein (PerCP) Cy5.5 anti-mouse Ly6C, HK1.4 (BioLegend, 128011); BV421 anti-mouse CD11c (BioLegend, 117329); V500 anti-mouse/human CD11b, clone M1/70 (BD, 562128); PE-Cy7 anti-mouse CD45, clone I3/2.3 (BioLegend, 147704); PE anti-mouse Ly6G, clone 1A8 (BD, 551461); anti-LukA rabbit polyclonal sera (35); and anti-LukAB monoclonal antibody.
The anti-LukAB monoclonal antibody was custom-made at Envigo Inc. according to their approved standard operating procedures for mouse monoclonal hybridoma generation. Briefly, recombinant LukAB (rLukAB) was emulsified with Freud’s complete adjuvant for the primary immunization, followed by one boost of rLukAB emulsified with Freud’s incomplete adjuvant and one to two additional boosts of rLukAB emulsified with TiterMax adjuvant. The immunized mouse was sacrificed, and splenocytes were harvested and fused with NS01 myeloma cells to generate hybridoma. The monoclonal hybridoma cell lines were selected by ELISA.
SPR analysis of LukAB binding to CD11b I-domains
SPR was run using the Biacore T200 system (GE) as described previously with the following modifications (12). Briefly, recombinant I-domain (mouse, human, and mutants) was immobilized onto flow cell 2 to 4 of a series S sensor chip CM5 (GE) using an NHS capture kit, and flow cell 1 was run as a blank immobilization. LukAB was performed across two lots of five concentration ranges 0.008 to 5 μg/ml and 0.16 to 100 μg/ml using single-cycle kinetics. The best range for the interaction was performed three times. The running buffer for all SPR experiments was 1× PBS at pH 6.8, and all data were double reference–subtracted.
Generation of iBMDMs
Twelve-week-old mice were euthanized with CO2, sprayed with 70% ethanol, and mounted on a Styrofoam block. Murine bone marrow cells were collected as follows. Skin and muscle from the hind legs were removed, and the femurs were cut off above the hip with sharp sterile scissors. Additional muscle was removed, and the femur and tibia were separated and placed in cold RPMI/10% fetal bovine serum (FBS) in a six-well plate on ice. Epiphyses of the bones were removed, and the bone marrow was flushed out with 10 ml of RPMI/10% FBS. Bone marrow was then passed through a cell strainer and rinsed with the rest of the medium, and cells were collected by centrifugation at 1500 rpm for 5 min. To remove red blood cells (RBCs), 2 ml of ACK (Ammonium-Chloride-Potassium) Lysing Buffer was added to the freshly isolated bone marrow cells and incubated for 2 min at room temperature. Lysis buffer was then neutralized by adding 10 ml of RPMI medium, and the cells were centrifuged at 1500 rpm for 5 min. Cells were then resuspended in 10 ml of RPMI/10% FBS and counted.
Bone marrow cells (1 × 106) were plated in an untreated 10-cm dish in Dulbecco’s modified Eagle’s medium (DMEM) with 30% L-929 supernatants (ATCC CCL-1). Five milliliters of additional medium was added on day 3 of differentiation. On day 7 of macrophage differentiation, J2 fibroblast (Kagan laboratory) (29) supernatant was collected and filtered with a 0.45-μm filter. BMDMs were grown in 50% J2 conditioned supernatant and 50% L-929 supernatants for 7 days, with one new batch of mixed J2 and L-929 supernatants added on day 3. Transduced BMDMs were then cultured in complete DMEM plus 30% L-929 supernatant until 90% confluent. Twenty percent of cells were then passaged into medium containing 25% L-929 supernatant. Following this trend, L-929 supernatant concentration in complete DMEM was decreased by 5% during each passage until the BMDMs grew normally in DMEM in the absence of L-929 supernatant.
Cell culture
HEK293T cells and iBMDMs were maintained at 37°C with 5% (v/v) CO2 in DMEM, supplemented with 10% (v/v) FBS and penicillin (100 U ml−1) and streptomycin (0.1 mg ml−1) unless stated otherwise.
iBMDM staining
iBMDMs were grown in DMEM + 10% FBS + 1× penicillin/streptomycin. Cells (2 × 105) were plated in V-bottom 96-well plates, washed two times with PBS, and stained with 25 μl of APC anti-mouse/human CD11b, APC anti-mouse F4/80, or APC-Cy7 anti-mouse I-A/I-E at a 1:300 dilution along with Fc block (BioLegend, 101320) at a 1:300 dilution in PBS. Cells were stained for 30 min on ice, washed two times with PBS, and fixed in 2% paraformaldehyde (PFA), 2% heat-inactivated FBS, and 0.5% sodium azide in PBS. Flow cytometry data were acquired using a CytoFLEX flow cytometer (Beckman Coulter), and data were analyzed using FlowJo software (TreeStar Inc.).
Phagocytosis assay
Overnight cultures AH-LAC USA300-∆lukAB hlg::tet lukED::kan lukSF::spec-pOS1sGFP of were subcultured 1:100 in TSB + chloramphenicol (10 μg/ml) and grown to midlogarithmic phase. Bacteria were washed and resuspended in medium and diluted to a concentration of 5 × 107 CFU/ml. Confluent iBMDMs were washed and resuspended in medium at 5 × 105 cells/ml. Twenty-five microliters of 5 × 107 CFU/ml bacteria and 50 μl of medium or 2% murine serum were added to wells of a 96-well U-bottom plate and preincubated for 1 min at 37°C under shaking conditions. Twenty-five microliters of 5 × 105 iBMDMs per ml were added and incubated at 37°C under shaking conditions. The reaction was stopped at the indicated times by placing the samples on ice and adding 100 μl of fluorescence-activated cell sorting (FACS) fixing buffer [PBS + 2% FBS + 2% PFA + 0.05% (w/v) sodium azide]. Flow cytometry data were acquired using a CytoFLEX flow cytometer (Beckman Coulter), and data were analyzed using FlowJo software (TreeStar Inc.). Phagocytosis was determined as % GFP-positive cells.
Plasmids for protein expression in mammalian cells
Plasmids used in this study are listed in table S5. pCMV6-Entry mouse CD11b H-I-domain and pCMV6-Entry mouse CD11b HMH 7 I-domain were cloned by FastCloning (36) using the oligonucleotides in table S6 and confirmed by sequencing.
HEK293T cell transfection
HEK293T cells were transfected with calcium phosphate with the plasmids listed in table S6. In brief, 10 μg/ml of each plasmid and 75 μl of 2.5 M CaCl2 were added to deionized water to a final volume of 750 μl. Seven hundred fifty microliters of 2× HEPES buffered saline (HBS) [50 mM HEPES (pH 7.05), 280 mM NaCl, and 1.5 mM Na2PO4] was added dropwise while aerating the mixture. Solution was incubated for 5 min at room temperature before the dropwise addition to a 10-cm plate with 30 to 40% confluent HEK293T cells. Six hours after transfection, the medium was changed to prewarmed DMEM +10% FBS. Experiments were performed 14 hours after transfection.
Biotinylation of LukAB
Proteins were biotinylated using EZ-Link NHS-PEG4-Biotin, No-Weigh Format (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, proteins were purified as described above and dialyzed overnight in 1× PBS. EZ-Link NHS-PEG4-Biotin reagent was added to the protein at a 1:20 molar ratio and incubated for 30 min at room temperature. Unreacted biotin reagent was removed by dialyzing overnight in 1× PBS. Labeled protein was then filter-sterilized, aliquoted, and stored at −80°C.
Cell binding assays
Transfected HEK293T cells (1 × 105), iBMDMs, or peritoneal exudate cells were added to each well of a V-bottom plate, washed with PBS, and incubated with 50 μl of biotinylated LukAB in PBS at the indicated concentrations for 10 min on ice. iBMDMs were preincubated with Fc block (1:100) at 37°C for 1 hour before LukAB treatment. Cells were washed two times with PBS and stained with PerCP Cy5.5 streptavidin (1:300) (BioLegend, 405214), APC CD11b (1:300) (BioLegend, 101212), and Fixable Viability Dye eFluor 450 (1:1500) (eBioscience, 65-0863-18) in PBS for 40 min on ice. Cells were washed two times with PBS and resuspended in FACS fixing buffer [PBS + 2% FBS + 2% PFA + 0.05% (w/v) sodium azide]. Flow cytometry data were acquired using a CytoFLEX flow cytometer (Beckman Coulter), and data were analyzed using FlowJo software (TreeStar Inc.).
SPR analysis of LukAB binding to murine iBMDMs
Whole murine iBMDMs expressing WT or hCD11b were fixed using 4% formaldehyde and washed three times with PBS and resuspended at 106 cells/ml. Cells were immobilized onto a Series S C1 sensor chip using the C1 wizard methodology on the Biacore T200 control system using similar methodology to that described by Mubaiwa et al. (37). Cells were flowed at 5 μl/min for 900 s to load the chip to saturation. Flow cell 1 was blank-immobilized to allow for double reference subtraction. LukAB was flowed over the immobilized cells at 8 nM to 5 μM.
Peritoneal exudate cell membrane damage assay
Eight- to 9-week-old WT or hCD11b C57BL/6 mice were injected intraperitoneally with 1 × 108 CFU of heat-killed S. aureus strain Newman 24 and 48 hours before harvest. Mice were euthanized with CO2, and the peritoneal cavity was injected with RPMI + 0.1% human serum albumin (HSA) + 10 mM HEPES and massaged. Cells were collected and washed. Cells (150,000 ) were incubated with the indicated concentration of LukAB for 3 hours at 37°C with 5% (v/v) CO2. Cells were washed with PBS and stained with PE-Cy7 CD11b (BioLegend, 101215), FITC Ly6G (BD, 557396), APC F4/80 (BioLegend, 123116), PerCP Cy5.5 Ly6C (BioLegend, 128011), BV 421 CD11c (BioLegend, 117329), and Fc block at 1:200 dilution in PBS for 20 min on ice. Cells were washed twice and resuspended in PBS + 5% FBS + propidium iodide (PI) (2 μg/ml) (G-Biosciences). Flow cytometry data were immediately acquired using a CytoFLEX flow cytometer (Beckman Coulter), and data were analyzed using FlowJo software (TreeStar Inc.).
Generation of hCD11b mouse
C57BL/6J fertilized zygotes were collected from superovulated C57BL/6J mice. Microinjection was performed with 45 μl of filtered injection mix consisting of guide RNA (50 ng/μl) [Alt-R CRISPR-Cas9 crRNA (IDT, guide sequence: CATGGGGCTGCTACCATCAG)] + Alt-R CRISPR-Cas9 tracrRNA (IDT, 1072533), GeneArt Platinum Cas9 nuclease (80 ng/μl; Invitrogen), and template DNA (10 ng/ml) (table S6) in tris-EDTA buffer. Guide RNA and DNA template were designed on Benchling.com. An Xho I restriction site was engineered into the DNA template to facilitate genotyping.
Injected zygotes were implanted into pseudopregnant CD-1 (Charles River Laboratory) females, and the resulting 15 pups were screened for alleles encoding the expected modifications in Itgam exon9. Genomic DNA samples were amplified (table S6), and the 379–base pair (bp) polymerase chain reaction (PCR) product was purified and cut with Xho I. Samples from 7 of 15 animals were cleaved by Xho I, and PCR products from these animals were sequenced. Two founders with the expected modifications were backcrossed to C57BL/6J mice for four generations. G4 animals with the expected modifications were then intercrossed to produce homozygous breeder pairs from two independent lines for colony enrichment and downstream studies.
Mice were bred in-house and maintained under specific pathogen–free conditions and used age-matched and sex-matched at 8 to 9 weeks of age. Mice from experiments using hCD11b and WT C57BL/6J mice were siblings generated from hCD11b het × het crosses. Mice were randomly mixed within their genotypes and sex and then assigned to groups.
Murine infections with S. aureus
Eight- to 9-week-old WT or hCD11b homozygote C57BL/6 mice were infected retro-orbitally with approximately 3 × 106, 1 × 107, or 1 × 108 CFU of S. aureus. Initial experiments were performed with hCD11b homozygote mice derived from G4 het × het crosses. WT littermates were used as controls. Subsequent experiments were performed with hCD11b homozygote mice derived from G4 homo × homo crosses. S. aureus strains used in this study were derived from USA300 strain LAC and are detailed in table S4. One or 3 days after infection, mice were euthanized with CO2. The livers and kidneys were removed, homogenized in sterile PBS, serially diluted, and plated on TSA for CFU counts. Two to three independent experiments were performed per strain.
ELISA to detect LukAB levels in vivo
Mouse anti-LukA (2 μg/ml) monoclonal antibody was coated onto the wells of an Immulon 2HB 96-well plate in carbonate-bicarbonate buffer overnight at 4°C. Wells were blocked with blotto buffer (2% dry fat-free milk, 0.9% NaCl, 0.05% Tween 20, PBS), washed, and incubated with infected organs digested in 1× radioimmunoprecipitation assay buffer (Abcam) + 1× Halt Protease Inhibitor Cocktail (Thermo Scientific) for 1 hour at room temperature with shaking conditions. Wells were washed and incubated with anti-LukA rabbit sera (35) at a 1:500 dilution for 1 hour at room temperature with shaking. Next, wells were washed and incubated with anti-rabbit HRP at a 1:2500 dilution for 1 hour at room temperature with shaking. Wells were washed and incubated with 100 μl of TMB substrate for 5 min. The reaction was terminated with 100 μl of sulfuric acid, and absorbance at 450 nm was measured. LukAB (μg/organ) was determined by comparison to a standard curve determined by measuring LukAB in organs with known quantities of LukAB added.
Isolating leukocytes from murine livers and kidneys
Eight- to 9-week-old C57BL/6 mice were injected retro-orbitally with PBS or approximately 1 × 107 CFU of S. aureus. One day after infection, mice were euthanized. The livers and kidneys were removed, homogenized in RPMI, and digested in 200 U of type VIII collagenase (Sigma-Aldrich, C2139) and 0.2 mg of DNase I (Sigma-Aldrich, DN25). Homogenate was strained, washed, and loaded onto a 40/80% Percoll gradient. The gradient was centrifuged, and the leukocyte layer was removed. RBCs were lysed with 1× BD Pharm Lyse and washed. Cells were stained with V500 CD11b (BD, 562128), PE-Cy7 CD45 (BioLegend, 147704), PE Ly6G (BD, 551461), APC F4/80 (BioLegend, 123116), and Fc block at 1:200 dilution in PBS for 20 min on ice, washed twice, and resuspended in FACS fixing buffer [PBS + 2% FBS + 2% PFA + 0.05% (w/v) sodium azide]. Flow cytometry data were acquired using a CytoFLEX flow cytometer (Beckman Coulter), and data were analyzed using FlowJo software (TreeStar Inc.).
Graphical and statistical analyses
Statistical details (“n” numbers, tests used, and definition of the error bars) are described in the figure legends. Analyses of flow cytometric data were performed using FlowJo. Statistical significance was determined using Prism 7.0 b, with two-way analysis of variance (ANOVA) with Tukey’s post hoc test for multiple comparisons, chi-square test, or unpaired one- or two-tailed Student’s t test with SEM as indicated.
Supplementary Material
Acknowledgments
We thank current and former members of the Torres laboratory for fruitful discussions. We thank J. Kagan for providing reagents to immortalize BMDMs and J. Muller and M. Dustin for providing CRISPR reagents. We also thank the NYU Langone Health’s Rodent Genetic Engineering Laboratory for help making the hCD11b mouse. Funding: This work was supported by NIH National Institute of General Medical Sciences (NIGMS) award numbers GM007308 (to K.M.B.) and GM114514 (to N.C.E.), an MSTP Vilcek Scholar Award (to K.M.B.), a Damon Runyon Cancer Research Foundation Postdoctoral Fellowship (to A.N.M.), NIH National Institute of Allergy and Infectious Diseases (NIAID) award numbers T32AI007180 (to K.T.) and R01AI099394 and HHSN272201400019C ( to V.J.T.), National Health and Medical Research Council (NHMRC) Program grant number 1071659 (to M.P.J), and NHMRC Principal Research fellowship APP1138466 (to M.P.J.). Work in S.B.K. laboratory was supported by NIH National Heart, Lung, and Blood Institute award number R01HL125816, the Judith and Stewart Colton Center for Autoimmunity Pilot grant, The Irma T. Hirschl Trust award, and a grant from the Drs. Martin and Dorothy Spatz Foundation. Flow cytometry technologies were provided by NYU Langone’s Cytometry and Cell Sorting Laboratory, which is supported, in part, by grant P30CA016087 from the NIH National Cancer Institute. N.C.E. and V.J.T. are Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Diseases. Author contributions: K.M.B., S.B.K., N.C.E., and V.J.T. conceived and designed experiments. A.N.M. and N.C.E. performed evolutionary analyses. C.J.D. and M.P.J. performed SPR experiments. K.T. generated the anti-LukAB monoclonal antibody. N.V. bred mice. K.M.B., S.Y.K., and S.B.K. generated the hCD11b mice. K.M.B. and K.A.L. performed the flow cytometry phagocyte experiments. K.M.B. performed all the remaining experiments. K.M.B. and V.J.T. wrote the manuscript with input from coauthors. Competing interests: V.J.T. is an inventor on patent applications related to this work filed by New York University (no. WO/2011/140337, filed on 5 May 2010, and no. WO/2013/165613, filed on 2 May 2012). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the lead contact. Reagents, including bacterial strains and humanized mice, can be provided pending scientific review, a completed material transfer agreement, and a licensing fee. Requests for reagents should be submitted to V.J.T.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaax7515/DC1
Fig. S1. Purification of CD11b I-domains.
Fig. S2. Purification and transfection of CD11b I-domain chimeras.
Fig. S3. Characterization of the hCD11b mouse.
Fig. S4. LukAB exhibits liver-specific tropism in vivo.
Table S1. Signatures of positive selection on primate ITGAM.
Table S2. Signatures of positive selection on rodent ITGAM.
Table S3. Primate and rodent sequences analyzed for selection on ITGAM.
Table S4. S. aureus strains used in this study.
Table S5. Plasmids used in this study.
Table S6. Oligonucleotides and gene sequences used in this study.
REFERENCES AND NOTES
- 1.Douam F., Gaska J. M., Winer B. Y., Ding Q., von Schaewen M., Ploss A., Genetic dissection of the host tropism of human-tropic pathogens. Annu. Rev. Genet. 49, 21–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Belkum A., Verkaik N. J., de Vogel C. P., Boelens H. A., Verveer J., Nouwen J. L., Verbrugh H. A., Wertheim H. F. L., Reclassification of Staphylococcus aureus nasal carriage types. J. Infect. Dis. 199, 1820–1826 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Klein E., Smith D. L., Laxminarayan R., Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg. Infect. Dis. 13, 1840–1846 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lowy F. D., Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald J. R., Livestock-associated Staphylococcus aureus: Origin, evolution and public health threat. Trends Microbiol. 20, 192–198 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Holtfreter S., Radcliff F. J., Grumann D., Read H., Johnson S., Monecke S., Ritchie S., Clow F., Goerke C., Bröker B. M., Fraser J. D., Wiles S., Characterization of a mouse-adapted Staphylococcus aureus strain. PLOS ONE 8, e71142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzgerald J. R., Holden M. T. G., Genomics of natural populations of Staphylococcus aureus. Annu. Rev. Microbiol. 70, 459–478 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Salgado-Pabón W., Schlievert P. M., Models matter: The search for an effective Staphylococcus aureus vaccine. Nat. Rev. Microbiol. 12, 585–591 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Spaan A. N., van Strijp J. A. G., Torres V. J., Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 15, 435–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumont A. L., Nygaard T. K., Watkins R. L., Smith A., Kozhaya L., Kreiswirth B. N., Shopsin B., Unutmaz D., Voyich J. M., Torres V. J., Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 79, 814–825 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ventura C. L., Malachowa N., Hammer C. H., Nardone G. A., Robinson M. A., Kobayashi S. D., DeLeo F. R., Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLOS ONE 5, e11634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuMont A. L., Yoong P., Day C. J., Alonzo F. III, McDonald W. H., Jennings M. P., Torres V. J., Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. U.S.A. 110, 10794–10799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen I. P., Dumont A. L., James D. B. A., Yoong P., Saville B. R., Soper N., Torres V. J., Creech C. B., Children with invasive Staphylococcus aureus disease exhibit a potently neutralizing antibody response to the cytotoxin LukAB. Infect. Immun. 82, 1234–1242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Copin R., Shopsin B., Torres V. J., After the deluge: Mining Staphylococcus aureus genomic data for clinical associations and host–pathogen interactions. Curr. Opin. Microbiol. 41, 43–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malachowa N., Kobayashi S. D., Braughton K. R., Whitney A. R., Parnell M. J., Gardner D. J., Deleo F. R., Staphylococcus aureus leukotoxin GH promotes inflammation. J. Infect. Dis. 206, 1185–1193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J.-R., Han X., Soriano S. G., Yuki K., The role of macrophage 1 antigen in polymicrobial sepsis. Shock 42, 532–539 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Prince J. E., Brayton C. F., Fossett M. C., Durand J. A., Kaplan S. L., Smith C. W., Ballantyne C. M., The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with streptococcus pneumoniae. J. Immunol. 166, 7362–7369 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Pilione M. R., Agosto L. M., Kennett M. J., Harvill E. T., CD11b is required for the resolution of inflammation induced by Bordetella bronchiseptica respiratory infection. Cell Microbiol. 8, 758–768 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Osicka R., Osickova A., Hasan S., Bumba L., Cerny J., Sebo P., Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 4, e10766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jennings M. P., Jen F. E.-C., Roddam L. F., Apicella M. A., Edwards J. L., Neisseria gonorrhoeae pilin glycan contributes to CR3 activation during challenge of primary cervical epithelial cells. Cell. Microbiol. 13, 885–896 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliva C. R., Swiecki M. K., Griguer C. E., Lisanby M. W., Bullard D. C., Turnbough C. L. Jr., Kearney J. F., The integrin Mac-1 (CR3) mediates internalization and directs Bacillus anthracis spores into professional phagocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 1261–1266 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orrskog S., Rounioja S., Spadafina T., Gallotta M., Norman M., Hentrich K., Fälker S., Ygberg-Eriksson S., Hasenberg M., Johansson B., Uotila L. M., Gahmberg C. G., Barocchi M., Gunzer M., Normark S., Henriques-Normark B., Pilus adhesin RrgA interacts with complement receptor 3, thereby affecting macrophage function and systemic pneumococcal disease. MBio 4, e00535-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deschamps M., Laval G., Fagny M., Itan Y., Abel L., Casanova J.-L., Patin E., Quintana-Murci L., Genomic signatures of selective pressures and introgression from archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 98, 5–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb A. E., Gerek Z. N., Morgan C. C., Walsh T. A., Loscher C. E., Edwards S. V., O’Connell M. J., Adaptive evolution as a predictor of species-specific innate immune response. Mol. Biol. Evol. 32, 1717–1729 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickeson S. K., Santoro S. A., Ligand recognition by the I domain-containing integrins. Cell. Mol. Life Sci. 54, 556–566 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J. O., Rieu P., Arnaout M. A., Liddington R., Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 80, 631–638 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Diamond M. S., Garcia-Aguilar J., Bickford J. K., Corbi A. L., Springer T. A., The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 120, 1031–1043 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., Jaenisch R., One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evavold C. L., Ruan J., Tan Y., Xia S., Wu H., Kagan J. C., The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrel M., Perencevich E. N., David M. Z., USA300 methicillin-resistant Staphylococcus aureus, United States, 2000–2013. Emerg. Infect. Dis. 21, 1973–1980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Surewaard B. G. J., Deniset J. F., Zemp F. J., Amrein M., Otto M., Conly J., Omri A., Yates R. M., Kubes P., Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 213, 1141–1151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng A. G., Kim H. K., Burts M. L., Krausz T., Schneewind O., Missiakas D. M., Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23, 3393–3404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janesch P., Rouha H., Weber S., Malafa S., Gross K., Maierhofer B., Badarau A., Visram Z. C., Stulik L., Nagy E., Selective sensitization of human neutrophils to LukGH mediated cytotoxicity by Staphylococcus aureus and IL-8. J. Infect. 74, 473–483 (2017). [DOI] [PubMed] [Google Scholar]
- 34.DuMont A. L., Yoong P., Surewaard B. G., Benson M. A., Nijland R., van Strijp J. A., Torres V. J., Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect. Immun. 81, 1830–1841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melehani J. H., James D. B. A., DuMont A. L., Torres V. J., Duncan J. A., Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLOS Pathog. 11, e1004970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C., Wen A., Shen B., Lu J., Huang Y., Chang Y., FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biol. 11, 92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mubaiwa T. D., Hartley-Tassell L. E., Semchenko E. A., Jen F. E. C., Srikhanta Y. N., Day C. J., Jennings M. P., Seib K. L., The glycointeractome of serogroup B Neisseria meningitidis strain MC58. Sci. Rep. 7, 5693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duthie E. S., Lorenz L. L., Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6, 95–107 (1952). [DOI] [PubMed] [Google Scholar]
- 39.Berends E. T. M., Zheng X., Zwack E. E., Menager M. M., Cammer M., Shopsin B., Torres V. J., Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB toxin. MBio 10, e01918-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller L. G., Perdreau-Remington F., Rieg G., Mehdi S., Perlroth J., Bayer A. S., Tang A. W., Phung T. O., Spellberg B., Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N. Engl. J. Med. 352, 1445–1453 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaax7515/DC1
Fig. S1. Purification of CD11b I-domains.
Fig. S2. Purification and transfection of CD11b I-domain chimeras.
Fig. S3. Characterization of the hCD11b mouse.
Fig. S4. LukAB exhibits liver-specific tropism in vivo.
Table S1. Signatures of positive selection on primate ITGAM.
Table S2. Signatures of positive selection on rodent ITGAM.
Table S3. Primate and rodent sequences analyzed for selection on ITGAM.
Table S4. S. aureus strains used in this study.
Table S5. Plasmids used in this study.
Table S6. Oligonucleotides and gene sequences used in this study.