

A massively parallel array of monoclonal antibodies enables rapid antibody development and target discovery across species.
Abstract
Antibodies are essential for elucidating gene function. However, affordable technology for proteome-scale antibody generation does not exist. To address this, we developed Proteome Epitope Tag Antibody Library (PETAL) and its array. PETAL consists of 62,208 monoclonal antibodies (mAbs) against 15,199 peptides from diverse proteomes. PETAL harbors binders for a great multitude of proteins in nature due to antibody multispecificity, an intrinsic antibody feature. Distinctive combinations of 10,000 to 20,000 mAbs were found to target specific proteomes by array screening. Phenotype-specific mAb-protein pairs were found for maize and zebrafish samples. Immunofluorescence and flow cytometry mAbs for membrane proteins and chromatin immunoprecipitation–sequencing mAbs for transcription factors were identified from respective proteome-binding PETAL mAbs. Differential screening of cell surface proteomes of tumor and normal tissues identified internalizing tumor antigens for antibody-drug conjugates. By finding high-affinity mAbs at a fraction of current time and cost, PETAL enables proteome-scale antibody generation and target discovery.
INTRODUCTION
Facilitated by the ever-growing capability of current DNA-sequencing technologies, more than 1300 genomes of animals and 496 genomes of plants and many others have already been sequenced, representing millions of genes, and the number will increase faster from projects such as G10K, i5k, and so on (1). To understand the roles of these genes, the functions of the gene-coded proteins need to be explored, and antibodies, especially renewable monoclonal antibodies (mAbs) generated at a proteome scale, are urgently needed. mAbs produced by hybridoma technologies for human proteins have long been recognized as the most direct tools for diagnostic and therapeutic target discovery (2). Classic therapeutic targets sialyl Lewis Y, prostate-specific membrane antigen, and, more recently, a previously unidentified target for multiple myeloma were found by mAbs for cell surface proteins (3–5).
Despite the power of mAbs and mAb-based discovery, large-scale generation of mAbs remains difficult since traditional hybridoma development is time consuming (4 to 6 months starting from antigens), expensive ($3000 to $8000 per antigen), and challenging to scale. Further, mAb generation by immunization typically requires a milligram of purified antigens, a substantial challenge for many proteins, especially for membrane proteins of primary research interest. Even for human proteins, most of the 6000 membrane proteins have not been directly explored as diagnostic or therapeutic targets due to a lack of high-quality antibodies for applications such as flow cytometry [fluorescence-activated cell sorting (FACS)] and immunofluorescence (IF) (6, 7).
The Human Protein Atlas (HPA) offers an alternative approach for proteome-scale antibody development. HPA has generated more than 25,000 affinity-purified polyclonal antibodies against >17,000 human proteins covering more than 80% of the human proteome (8, 9). However, it is impractical to replicate the success of HPA on the majority of other species with a need for proteome-scale antibodies due to the great human and capital resources required for such a project. Furthermore, polyclonal HPA antibodies are not renewable, making reproduction of these antibodies with consistent quality difficult. Thus, proteome-scale antibody generation has remained elusive for most sequenced genomes.
Over the past few decades, numerous attempts have been made to address high cost and poor scalability of large-scale antibody generation by improving traditional hybridoma methods and developing better in vitro recombinant antibody libraries and more efficient screening technologies (10–12). For in vitro methods, continuing development of novel display technologies (13, 14) and improvements in library design and screening methods (15) has been attempted dating back 20 years. However, established synthetic antibody libraries for therapeutic antibody discovery are not yet used for large-scale reagent generation because of the concern of high cost. Despite an attempt to generate research antibodies using phage display libraries (16), it is not economical to use these resources for generating antibodies for nonclinical uses or for nonhuman proteomes.
Antibody microarray is a powerful platform for high-throughput, multiplexed protein profiling using a collection of immobilized antibodies (17, 18). By using antibody array, one can achieve low-cost and fast antibody discovery by direct array screening. In one approach, a library of ~10,000 in silico–designed and –synthesized antibody fragments was used to build an antibody array for de novo antibody discovery (19). The arrayed library was able to generate antibody leads with micromolar binding affinity for therapeutic protein targets, suggesting that a spatially addressed library comprising tens of thousands of individual antibodies should be sufficient for antibody discovery. However, this synthetic antibody library screening approach did not achieve broader impact since required additional antibody affinity maturation and engineering limited its usefulness for routine research affinity reagent development and target profiling.
Here, we present a system integrating “industrial-scale” hybridoma development, antibody microarray, and affinity proteomics to overcome previous challenges for proteome-scale antibody development and target discovery. Our technology, called the Proteome Epitope Tag Antibody Library (PETAL), takes advantage of antibody multispecificity (20, 21), an intrinsic property of antibody molecules that bind to a large number of proteins unrelated to the original antigen that the antibody was raised against with high affinity and specificity. Antibody multispecificity is exemplified by anti-p24 (HIV-1) peptide mAb (CB4-1). Its epitope sequences, consisting of key interacting residues deduced from five unrelated peptides binding to CB4-1, were identified in hundreds of heterologous proteins, and those proteins that could be obtained were shown to bind CB4-1 (22). PETAL is a mouse mAb library consisting of 62,208 mAbs made against 15,199 peptide antigens representative of 3694 proteins from 418 proteomes. PETAL has the potential to bind to a large number of proteins in nature due to antibody multispecificity. An antibody microarray was fabricated using PETAL mAbs. Using cell lysates of diverse proteomes to screen the PETAL array, we demonstrated the feasibility for proteome-scale protein targeting by mAbs. Identified antibodies are capable of broad applications, as shown for human membrane and nuclear proteins. Phenotype-specific mAb-protein pairs were identified for maize and zebrafish from respective proteome-specific mAbs. Therapeutic target candidate discovery was demonstrated by differential screening of normal versus tumor membrane proteomes. An antibody targeting CD44v9 was identified as a candidate for building an antibody-drug conjugate (ADC) for lung squamous cell carcinoma (LUSCC) in vitro and in vivo. By generating high-affinity mAbs at a small fraction of current time and cost, PETAL enables affinity reagent generation and target discovery for proteomes with available genomic sequence information.
RESULTS
The immunological basis of PETAL is antibody multispecificity (Fig. 1A). PETAL is an antipeptide antibody library of 62,208 mAbs designed to have the potential for harboring binders for a great number of proteins from diverse proteomes (Fig. 1B). When PETAL is immobilized in an array format (Fig. 1C), it enables proteome-scale antibody generation and differential target discovery (Fig. 1D).
Fig. 1. Construction and application of PETAL and its array for antibody/target discovery.
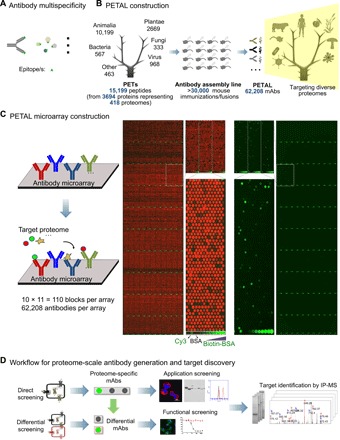
(A) Antibody multispecificity. An antibody binds to an epitope/mimotope found within a variety of proteins from different species, leading to high-affinity, specific binding of this antibody to a large number of proteins in nature. (B) PETAL construction. PETAL is a library of 62,208 mouse mAbs derived from immunization of more than 30,000 mice against 15,199 diverse peptide antigens. PETAL has the potential to recognize a great number of proteins in nature. (C) PETAL microarray construction. PETAL is printed into an antibody microarray as a high-throughput platform for antibody/target discovery (left). Right panel shows the design/layout of the array (red, visualized by a Cy5-conjugated anti-mouse antibody) and an array hybridization result using a protein sample (positive-binding mAb spots are shown as green). (D) Workflow for proteome-scale antibody generation and target discovery. Two array-screening applications are shown: direct screening to identify proteome-specific mAbs and subsequent antibody application screening and target identification or differential screening to find mAbs and their cellular targets associated with a specific phenotype.
Design and construction of PETAL
A total of 15,199 peptide antigens, called proteome epitope tags or PETs, were designed from 3694 proteins representing 418 proteomes (Fig. 1B, fig. S1A, and table S1). Within each proteome, PETs were selected from unique regions of protein sequence using the heuristic blastp algorithms optimal for short peptide sequence comparison (23, 24). Sequence analysis showed that PET sequences were random and diverse (fig. S1B).
To construct PETAL, PET antigens were synthesized and used to generate mouse mAbs by a large-scale mAb development operation modeled after an assembly line (Fig. 1B). More than 30,000 mice were immunized at an average of two mice per PET. A total of 62,208 mouse mAbs were generated (table S2). Each hybridoma cell line was used to prepare ascites containing 1 to 20 mg of mouse immunoglobulin Gs (IgGs) with varying concentrations from 0.1 to 10 mg/ml; most were in the 1- to 3-mg/ml range.
PETAL diversity
To evaluate the diversity of PETAL, particularly multiple mAbs generated using the same PET, antibody V region was sequenced for 91 randomly selected hybridomas, including 68 hybridoma clones against 24 PETs with two to five mAbs per PET and 23 hybridomas against 23 unique PETs (fig. S1, C to E). V-region sequences with ≥2 amino acid differences in the complementarity determining regions (CDR) were considered “unique,” although two antibodies with a single CDR amino acid difference were found to bind to different epitopes (25). Close to 90% of CDR sequences were unique (fig. S1D). Multiple mAbs generated against the same PET peptide antigen were mostly (80 to 100%) unique (fig. S1E), indicating that the effective library size was close the total number of PETAL mAbs since framework differences could also contribute to different binding affinity and therefore specificity.
Construction of PETAL microarray with 62,208 mAbs
To use PETAL for multiplexed screening, an antibody microarray was constructed with all 62,208 antibodies (Fig. 1C). Array printing quality was assessed using an anti-mouse secondary antibody conjugated with Cyanine 5 (Cy5) (Fig. 1C, left two panels). Antibodies of 10 to 1000 pg per spot (mostly 100 to 300 pg) were detected with a fluorescent intensity ranging from 500 to >60,000 (fig. S1F).
Typically, biotinylated antigens such as 10 to 100 μg of proteome samples (Fig. 1C, right two panels) were used for array screening. Array screening consistency was investigated using a human plasma sample in three independent replicates. An average of 20,000 antibodies showed positive binding [(signal intensity-background)/background > 3] with a coefficient of variation (CV) of 7%. More than 90% of array-positive antibodies had a fluorescent intensity of 500 to 10,000. The Pearson’s correlation coefficient (r) among triplicate experiments was 0.98 (fig. S1G). These data establish the PETAL array as a reproducible platform for screening.
To test the library/array for generating mAbs, a total of 81 recombinant proteins were used to screen the PETAL array (fig. S2 and table S3). About half (47%; 38 of 81) of the proteins produced an average of 3.7 (141/38) mAbs per antigen after enzyme-linked immunosorbent assay (ELISA) screening of array-binding mAbs using a detection limit of ≤1 μg/ml of antigen. When tested in immunoblotting assays, 31 (25 of 81) and 26% (21 of 81) of proteins were successful using recombinant proteins and endogenous samples, respectively (fig. S2 and table S3).
PETAL targets diverse proteomes for antibody and target discovery
To apply PETAL for targeting broad and diverse proteomes, 11 proteome samples of plants, animals, and bacteria were used to probe PETAL arrays (Fig. 2). The number of array-positive antibodies for each proteome was 10,000 to 20,000. A selection of ~1000 per proteome of array-positive antibodies across the fluorescent intensity scale was used to probe endogenous samples to estimate the number of antibodies suitable for immunoblotting (Fig. 2A). Typically, 20 to 30% of antibodies produced specific (single or predominant single band) immunoblotting results, giving a proteome of 2000 to 6000 potential immunoblotting mAbs. Although proteome-binding mAbs notably overlapped among proteomes (30 to 60%), the same antibodies often specifically recognized proteins of different size in different proteomes (Fig. 2B), likely to be unrelated proteins.
Fig. 2. PETAL targets diverse proteomes for antibody and target discovery.
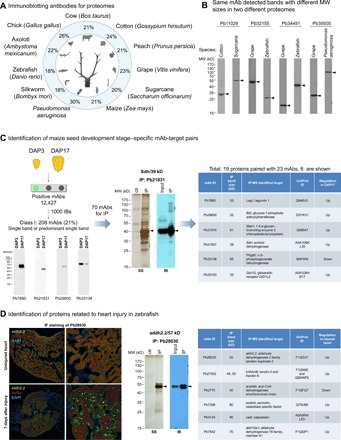
(A) Proteome-targeting PETAL mAbs for immunoblotting. Successful rates (labeled as %) of immunoblotting (producing single/predominant single bands) were shown for 11 organisms by using a panel of ~1000 proteome-binding mAbs for each organism to probe proteome samples. The total number of binding mAbs for a proteome was in the range of 10,000 to 20,000. The specific tissues for immunoblotting were cow (breast, ovary, and liver), cotton (ovule), peach (leaf or fruit), grape (nuclear fraction of seed), sugarcane (stalk), maize (seed), Pseudomonas aeruginosa (whole cell), silkworm (larva), zebrafish (embryo, heart or other tissues), axolotl (regenerating limb), and chicken (cell lines and tissues). (B) Different proteins detected by the same mAb in two different proteomes. Four examples of antibodies each recognized a specific band with different size in different proteomes upon immunoblotting. (C) Identification of maize seed development stage–specific mAb-protein pairs. PETAL array screening using a proteome sample consisting of total protein extracts from maize seeds DAP3 and DAP17 produced a total of 12,427 binding mAbs. A selection of 1000 mAbs to probe DAP3 and DAP17 samples yielded 206 class I mAbs (single or predominant single band) and additional 129 class II mAbs (multiple bands) upon immunoblotting. Seventy differentially expressed mAbs between DAP3 and DAP17 were used to IP their cellular binding proteins for MS analysis, resulting in the identification of 19 proteins paired with 23 mAbs. Six proteins are shown in the right panel. Gel pictures from left to right show class I mAb immunoblotting examples: silver staining (SS) of IP products and immunoblotting (IB) of input and IP products for Pb21831. (D) Identification of heart injury–related proteins from zebrafish. From left to right: IF staining, silver staining of IP products and immunoblotting of input and IP products for Pb28030, and summary of the identified proteins.
Proteome-specific mAbs were used to identify phenotype-specific mAbs and corresponding proteins for maize seed development and zebrafish heart regeneration. A panel of 335 immunoblotting-positive mAbs was produced from 1000 array-positive binding mAbs using a protein sample consisting of maize seeds at two developmental stages, day after pollination 3 (DAP3) and DAP17 (Fig. 2C) (26). These included 206 mAbs with single or predominant single bands (class I, 21%; 206 of 1000) and an additional 129 mAbs with more than one (typically two to three) dominant band (class II) on immunoblotting (Fig. 2C). After probing DAP3 and DAP17 protein samples separately using class I mAbs, a panel of 70 differentially expressed targets was selected for target identification by immunoprecipitation (IP) and liquid chromatography–tandem mass spectrometry (LC-MS/MS). Nineteen proteins paired with 23 mAbs were identified, and six examples are shown in Fig. 2C, including Sdh, Bt2, and Sbe1, which have previously been shown to be involved in maize seed development (26, 27).
In another example, a panel of 45 binding mAbs identified from the screening of protein lysates of zebrafish heart was further characterized by IF assays using samples from the heart before injury and 7 days after injury (Fig. 2D). Six of them showed up- or down-regulation in the injured heart. Proteins bound by these six antibodies were identified by IP and LC-MS/MS (Fig. 2D, right), including proteins with known heart injury protection function [for example, aldh2.2 in Fig. 2D (left and middle panels)] (28).
Proteome-scale antibody generation for human membrane and nuclear proteins
To apply PETAL for antibody generation for organelle proteomes, PETAL arrays were screened with total protein extracts of membrane and nucleus from human PC9, HepG2, THP-1, and Jurkat cell lines (Fig. 3A). The total number of positive mAbs for each sample was in the range of 12,000 to 18,000.
Fig. 3. Proteome-scale antibody generation for human membrane and nuclear proteomes.
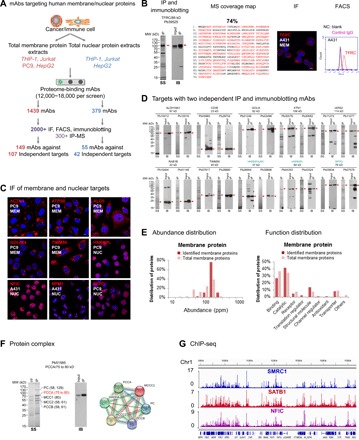
(A) Protein identification for human membrane and nuclear proteome-specific PETAL mAbs. (B) An example (TFRC/CD71) for identification of antibody binding protein. From left to right: SS for IP product, IB blot of input and after IP samples, coverage map of MS-identified peptides, and IF and FACS data. The cell line for IF was selected according to HPA data. Membrane or nuclear proteins were labeled MEM or NUC. Negative controls (NC) for FACS included staining with blank and irrelevant IgG. (C) Examples of IF data for endogenously expressed membrane and nuclear proteins. ACTN4 and ACTP5B were stained under nonpermeable conditions. Other proteins were stained under permeable staining conditions (also, see movies S1 to S6). (D) Proteins with two independent IP and immunoblotting mAbs. Panel label (SS and IB) was the same as in (B). Nuclear proteins were labeled in blue. (E) Abundance and function distribution of proteins identified from the Jurkat cell membrane proteome. (F) Protein interactome example using Pb51585 against PCCA. Protein-protein interacting map (right) analyzed by STRING with the mass-identified proteins. (G) Snapshot of the Integrative Genomics Viewer showing sequencing read density of ChIP-seq data generated with antibodies against SMRC1, SATB1, and NFIC in HepG2 cells. Chr1, chromosome 1.
To further screen for application-specific antibodies and to identify their cellular binding proteins, array-positive antibodies with high (>10,000), medium (2000 to 10,000), and low (500 to 2000) fluorescent intensity (fig. S3A) were selected. A total of 1439 positive antibodies for membrane proteomes and 379 for nuclear proteomes were subjected to immunoblotting, IF/FACS, and IP assays (Fig. 3, B and C, fig. S3B, and movies S1 to S6), and their cellular binding proteins were identified by LC-MS/MS (Fig. 3B). A total of 149 antibodies representing 107 proteins were identified from membrane proteome screening (tables S4 and S5), including known CD and RAB (small guanosine triphosphatase) molecules CD3e, CD49d, CD71 [transferrin receptor (TFRC)], CD222, CD5, CD2, CD44, RAB1B, and RAB14. For nuclear proteomes, a total of 55 antibodies representing 42 proteins were identified (tables S4 and S5), including transcriptional regulators NONO, NFIC, TRIM28, CSNK2A1, MTA2, SATB1, SFPQ, and SMARCC1. About 20% of the proteins had at least two independent antibodies that yielded similar IP and immunoblotting results, strengthening the antibody validation quality (Fig. 3D) (29, 30). The success rate of target identification was consistent over a wide fluorescent intensity range (fig. S3A), suggesting that more than 1000 proteins could be covered with 12,000 to 18,000 proteome-binding antibodies.
Only 20 to 30% of the array-positive antibodies were successful in immunoblotting assays likely due to the native protein states in the screening samples. For antibodies without successful immunoblotting data (therefore, the sizes of their target proteins were unknown), overexpression or knockdown experiments were necessary to determine their cellular binding proteins after IP and mass spectrum. For example, recognition of an antibody (Pb2795) to a multipass ion channel, PIEZO1 (31), was confirmed by the colocalization of IF staining signals with overexpressed PIEZO1–green fluorescent protein (GFP; fig. S3C).
To investigate whether proteins targeted by antibodies were biased toward specific types, protein abundance and function class [Gene Ontology (GO) annotations] were examined for 87 Jurkat proteins (67 membrane and 20 nuclear; Fig. 3E and fig. S3D). Abundance distribution of these proteins was similar to that of total membrane or nuclear proteins in Jurkat cells, according to the PAXdb (32). Proteins identified by antibodies were from diverse protein families and functional groups similar to total human membrane and nuclear proteins (Fig. 3E and fig. S3D) according to the HPA database, suggesting that antibodies targeted broad classes of proteins without obvious bias.
To test antibodies for additional applications, we performed protein complex enrichment and chromatin IP sequencing (ChIP-seq) with selected antibodies. Protein complex enrichment results are shown in Fig. 3F. For example, Pb51585 against Propionyl-CoA carboxylase alpha (PCCA) could pull down PCCA-interacting proteins similar to its previously mapped interactome according to Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (33) analysis.
ChIP-seq assays in HepG2 for antibodies against SMRC1, SATB1, and NFIC were carried out following previous studies (34). A total of 93.9 million sequencing reads were generated, and 53.7% were uniquely mapped to the human reference genome. These reads were further processed and yielded 46,380 peaks, representing 29,441, 3296, and 13,643 binding sites for SMRC1, SATB1, and NFIC, respectively (Fig. 3G). Further validation by comparing to commercial ChIP antibody, or with ChIP–quantitative polymerase chain reaction (PCR) assays, analyses of binding sites and enriched motifs all confirmed that antibodies against those transcription factors could be applied for ChIP-seq (fig. S3, E to H).
ADC antibody/target discovery by differential array screening
ADC selectively eliminates cancer cells by linking a toxin, for example, monomethyl auristatin E (MMAE), with an antibody targeting an internalizing tumor-associated antigen characterized by higher expression in tumors than in normal tissues (35). To identify candidate targets suitable for ADC, we screened PETAL arrays with normal and tumor cell proteomes (Fig. 4). More than 3000 antibodies of 15,000 lung membrane proteome-positive antibodies showed a fluorescent intensity fold change of >1.5 (fold change of >1.1 was considered to be significant since the CV of experiment repeats was less than 10%) between tumor and normal samples (Fig. 4A). Four antibodies were screened to show both internalization and indirect cytotoxicity from a selection of 500 antibodies with a fold change ranging from 1.5 to 5 (Fig. 4B and fig. S4A). One antibody, Pb44707, with a signal 2.4-fold higher in tumor tissue, was internalized with a half internalization time of 2.5 hours and half-maximal inhibitory concentration (IC50) of ≤100 pM for cell cytotoxicity in PC9 cells. IP and LC-MS/MS identified CD44, a putative cancer stem cell marker (36), as the most likely target protein (Fig. 4C). Target protein was further verified by peptide and small interfering RNA (siRNA) blocking experiments in which only a CD44v9-specific peptide and CD44v-targeting siRNA caused the loss of surface fluorescent signal in FACS (Fig. 4, D and E) (37). The cellular binding affinity [half-maximal effective concentration (EC50)] of Pb44707 with PC9 was 832 pM (fig. S4B) as determined by antibody titration using FACS (38).
Fig. 4. Differential array screening for ADC therapeutic target/antibody discovery.
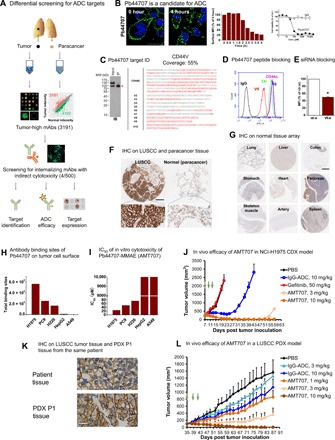
(A) Differential antibody screening for ADC targets. Inset scatter plot shows differential distribution of antibody signal intensity of NSCLC and normal lung. n = 1. More than 3000 tumor-high antibodies were identified. (B) An antibody candidate, Pb44707, for ADC. Antibody ID was labeled on the left of the IF image. IF (0 and 4 hours) image (green, antibody; blue, DAPI) time course of normalized surface fluorescence in FACS and cell cytotoxicity data are shown. Internalization half time (t1/2) and mean percent growth inhibition ± SEM (n = 3) of the antibody is labeled. IF scale bar, 50 μm. (C) Pb44707 IP and MS. MS identified multiple peptides (sequence marked in red) from CD44s and peptide sequences from variable region (shown as V number). (D) Peptide blocking of CD44v9. Graph of FACS analysis showing CD44v9 peptide but not CD44v6 peptide nor CD44s protein blocking binding of Pb44707 with PC9 cells. (E) siRNA targeting CD44v9 specifically decreases the FACS signal of Pb44707 compared to control siRNA. Normalized MFI ± SEM of PC9 cells as detected by Pb44707. (F) Representative images of the IHC staining of Pb44707 in LUSCC tumor tissue and paracancerous tissue from one patient. Scale bar, 300 μm. (G) Images of the IHC staining of Pb44707 in representative vital normal human tissues. No expression of CD44v9 is detected in these tissues. Scale bar, 400 μm. (H) Quantification of total binding sites of Pb44707 on the plasma membrane of a variety of tumor cell lines. (I) IC50 of Pb44707-ADC on tumor cell lines. n = 1 to 3 for different cell lines, respectively. (J) Growth curves of the NCI-H1975 CDX tumors of different treatment groups (n = 7 per group). Treatment with AMT707, control ADC, or gefitinib (intraperitoneal injection, dosing once a day) was initiated 7 days after tumor inoculation and administered as indicated by arrows. (K) Representative images of the IHC staining of Pb44707 in a LUSCC patient tumor tissue (top) and passage 1 (P1) PDX tumor tissue derived from the same patient. Scale bar, 50 μm. (L) Growth curves of the PDX tumors of different treatment groups (n = 6 per group). Treatment with AMT707 or control ADC was initiated 35 days after tumor inoculation and administered as indicated by arrows.
To evaluate the expression of CD44v9 in tumor and normal tissues, Pb44707 was used in immunohistochemistry (IHC) to probe tissue arrays. CD44v9 was markedly overexpressed in 60% of patients with non–small cell lung carcinoma (NSCLC) and, more notably, in close to 90% of LUSCC (Fig. 4F and fig. S4, C to E). Furthermore, mRNA of CD44v9 showed higher expression in all stages of LUSCC (fig. S4F). Most normal tissues were negatively stained with Pb44707 (Fig. 4G) except in skin, which showed both high and low expression in the population (fig. S4, E and G). To build Pb44707 into an ADC molecule, Pb44707 was conjugated to valine-citrulline (vc)-MMAE with an average drug-antibody ratio of 4.23. Pb44707-MMAE (AMT707) demonstrated in vitro cytotoxicity in lung cancer cell lines, and its potency was correlated with the CD44v9 expression level in these cells (Fig. 4, H and I). The in vivo efficacy of AMT707 was tested in both the cell line–derived xenograft (CDX) model and patient-derived xenograft (PDX) model. For the xenograft model with the gefitinib-resistant NSCLC cell line NCI-H1975 (Fig. 4J), AMT707 administered at 3 and 10 mg/kg suppressed tumor growth completely until days 47 and 57, respectively, whereas for treatment controls [i.e., phosphate-buffered saline (PBS), gefitinib, and control ADC] there was no or a much lower effect. In a CD44v9-positive LUSCC PDX model (Fig. 4, K and L), AMT707 haltered tumor growth in a dose-dependent manner, and AMT707 (10 mg/kg) suppressed tumor growth completely. Thus, by array screening, we identified multiple endocytic cell surface targets and at least one antibody and its target with a potential to build a lead ADC molecule.
DISCUSSION
To enable proteome-scale mAb development, we constructed PETAL, a hybridoma library consisting of 62,208 mAbs and its corresponding antibody array, the largest antibody microarray reported so far. Taking advantage of antibody multispecificity, PETAL may harbor binders for a large number of proteins in nature (20–22). Combining PETAL with the global screening capability of antibody microarrays, this platform enables mAb generation at a fraction of the current time and cost. High affinity and desirable specificity of selected mAbs were ensured by a fit-for-purpose workflow. We have demonstrated initial application of PETAL in large-scale antibody generation, affinity proteomics, and therapeutic target discovery.
With its capacity to target a large number of proteins in a proteome (Fig. 2), our technology provides a solution for proteome/subproteome-scale generation of antibodies (39). We demonstrated that antibodies targeting cell surface and nuclear proteins of cancer and immune cells were efficiently identified. Many of these antibodies were well suited for IF and IP/ChIP. From an input of 1818 antibodies, ~200 antibodies capable of immunoblotting/IF/FACS/IP targeting 149 independent membrane and nuclear proteins were identified. Given the input/protein ratio of 12:1 (1818/149), 10,000 to 20,000 array-positive antibodies would yield more than 1000 binding proteins.
Antibody-based functional proteomics has not been available for nonhuman species. Here, we demonstrated that broad proteome samples including plants, animals, and insects tested so far have all identified binding antibodies corresponding to a large number of proteins for each proteome. PETAL immediately provided immunoblotting mAbs for initial characterization of proteins in organisms with genomic sequencing information by proteome sample screening and target identification described here. Thus, identified proteome-specific mAb-protein pairs were further used to probe samples from distinctive phenotypes to find phenotype-specific mAb-protein pairs. As demonstrated here, both new and previously reported phenotype-specific proteins were found for zebrafish heart regeneration and maize seed development. For each protein found, mAbs capable of immunoblotting and IP were obtained. The overall success rate of antibody-protein deconvolution was similar to that of human membrane/nuclear screening. Thus, our technology provides a straightforward and productive path to carry out affinity proteomics for numerous proteomes currently without available affinity reagents.
When used for differential profiling of cell membrane proteomes, PETAL provides a “fit-for-purpose” approach for therapeutic target discovery. PETAL relishes the full potential of antibody-based target discovery compared to that of other functional genomics (RNA interference or CRIPSR) or MS-based approaches because it delivered the target and lead antibodies at the same time. As demonstrated in this study, differential array screening comparing tumor (NSCLC) with normal lung tissue membrane proteomes identified CD44v9-targeting antibody for building ADC molecule AMT707. The specificity of Pb44707 has been evaluated by IHC and FACS. Little nonspecific binding has been observed. Nevertheless, the specificity will be further validated using a set of comprehensive means in the future preclinical studies. On the other hand, the potential skin toxicity caused by Pb44707-based ADC may be addressed by using a more moderate payload, such as SN-38. More ADC candidate antibodies/targets are expected to be found when all 3000 “tumor-high” antibodies are subjected to the screening process.
There are some limitations of the PETAL strategy. The construction of PETAL was a substantial investment of time and effort that is difficult to repeat. Moreover, since PETAL antibodies are mouse origin, additional antibody engineering including humanization and immunogenicity evaluation is needed to move into therapeutics. However, PETAL is now a premade resource that can be readily accessed by researchers to generate mAbs for specific antigens or proteomes or to differentially screen protein samples to identify phenotype-specific protein targets (fig. S5). Although PETAL was made as an antipeptide library, it is likely that other types of antigens could produce equally useful antibody libraries (5). It is our hope that this work will not only serve as an immediate resource but also stimulate new hybridoma libraries for even more applications. For this purpose, our established industrial-scale mAb development capability can be taken advantage of to efficiently build hybridoma libraries/arrays on the scale of tens of thousands of mAbs. Current PETAL only yielded high-affinity mAbs for 20 to 30% of antigens, and this could be improved by increasing the size of the library, which in turn may also increase the success rate of application screening and target identification of PETAL mAbs.
Together, PETAL represents a notable improvement over previous antibody array and library approaches. A workflow for array screening and antibody-protein deconvolution has been established to eliminate high cost and the long development time of previous methods. We expect PETAL to accelerate functional proteomics by enabling proteome-scale antibody generation and target profiling. We will strive to make this resource accessible for broad scientific communities. We believe that PETAL will stimulate the preparation of other antibody libraries; thus, the strategy could be adopted and explored by many other researchers.
MATERIALS AND METHODS
Peptide antigen selection
A total of 15,199 peptide antigens, called PETs, were designed from 3694 proteins representing 418 proteomes. Within each proteome, PETs were selected from unique regions of protein sequence using heuristic blastp algorithms optimal for short peptide sequence comparison (23, 29). Peptide antigens representative of predicted surface epitopes from a protein sequence were selected (40, 41). Peptides were mostly 10 to 12 amino acids in length to contain two to three potential antibody epitopes (42). Predicted peptide sequences with secondary structures including alpha helix and beta sheet were omitted (43). Special sequences including transmembrane motif, signal peptide, and posttranslational modification motif were also not selected. Only disordered or surface-looped regions were selected. Hydrophobic peptides were not selected, and peptide hydrophilicity was calculated by the Hopp and Woods method (44). Last, peptides with more than one cysteine in the sequence were omitted to avoid synthesis difficulties.
All the peptide antigens were chemically synthesized by GL Biochem (Shanghai) Ltd. The purity and molecular weight of each peptide were evaluated with high-performance LC and MS.
Diversity analysis of PET library
The diversity of the PET peptide library (15,199 peptides; see table S1 for detail) is evaluated by comparing the sequence similarity of all peptides against each other. The sequence identity (%) between a peptide and its closest homologs within the library was recorded. PET sequence similarity to two random peptide libraries generated computationally was also compared. The first library was a collection of 15,199 peptide sequences randomly sampled from all species without considering amino acid preference in different species (45). The second library was constructed by randomly sampling the entire human proteome (all consensus coding sequences).
Construction of PETAL mAb library
MAbs were developed using a large-scale mAb development operation modeled after an assembly line. In the antibody assembly line, each of close to 100 highly trained technicians performs one to three discrete steps (for example, plating fusion cells onto 96-well plates or cell transfer from 96- to 384-well plates) for making hybridomas. An internally built informatics and data system (Antibody Assembler) is used for tracking materials and project status. More than 90% of all materials used are bar-coded to minimize hand labeling. Many steps have automatic data analysis and decision making (for example, clone picking). Together, antibody assembly line is scalable and cost efficient. PETAL is a premade library built by this highly efficient process. After traditional hybridoma protocol (46) and the immunization and fusion, a series of ELISA screens were performed using a peptide antigen titration from 1 × 10−7 to 1 × 10−10 M to ensure that only the hybridoma clones with the highest affinity (for example, able to detected antigen at a concentration less than 1 × 10−8 M) to peptide antigens were selected. IgG mAbs were selected using a Sigma antibody isotyping kit (no. 11493027001). Four to six IgG hybridomas per peptide antigen were selected for multiple rounds of limited dilution subcloning to ensure stability and monoclonality. Each hybridoma cell line was used to prepare milliliters of ascites containing 1 to 10 mg of mouse IgGs. Mouse strains used for immunization and ascite production were BALB/c and F1 from Shanghai Super-B&K Laboratory Animal Co. Ltd. The procedures for care and use of animals were approved by the Abmart Institutional Animal Care and Use Committee.
Hybridoma V-region sequencing
A mouse IgG primer set from Novagen (no. 69831-3) was used to amplify the IgG variable region on antibody heavy chain (VH) and variable region on antibody light chain (VL) regions from selected hybridoma clones. Briefly, 1 × 106 cells were collected for each cell line. Total RNA was extracted using TRIzol reagent (Thermo Fisher, no. 15596026). The first-strand complementary DNA was amplified using PrimeScript reverse transcription PCR kit from Takara (no. RR104A). PCR products with the expected size [an average size of about 400 base pairs (bp) for VH, and 360 bp for VL] were sequenced. The sequences of the PCR products were analyzed by IMGT/V-QUEST (www.imgt.org) (47) to define the VH or VL regions and the corresponding subelements. The uniqueness of antibody sequences was evaluated by comparing full-length V (VH and VL), frame, or CDR sequences using clustal algorithm. The homology matrixes were shown in the heat map format and that of the combined CDR sequences is shown in fig. S1C as an example.
PETAL array construction and quality evaluation
Ascites of 62,208 PETAL mAbs were prepared in 162 384-well plates and printed onto nitrocellulose-coated slides (FAST Maine Manufacturing, no. 10486111) in a high-density microarray format (named as PETAL array) using the Marathon System (Arrayjet Ltd., UK). Approximately 100 pl of ascites was printed for each antibody per spot. The array and block layout are shown in Fig. 1C. A total of 110 blocks were aligned into 10 rows and 11 columns. Each block contains a subarray of 48 × 12 = 576 individual antibody spots, except the subarrays in the last row were printed with 40 × 12 = 480 (three blocks) or 39 × 12 = 368 (eight blocks) spots. Additional control rows, including a positioning fluorescent spot (Cy3) and a biotin–bovine serum albumin (BSA) gradient (0.4 to 50 pg) of eight spots, were also printed for each block similar to previous antibody arrays (48, 49). Biotin-BSA was prepared by saturated labeling of BSA with Thermo Fisher Sulfo-NHS-LC-Biotin (no. 21336) labeling reagent. PETAL arrays were stored at −80°C.
To evaluate PETAL array quality, the slides were blotted directly with a mixture of a Streptavidin-Cy3 (Sigma, no. S6402) and a Cy5-labeled goat anti-mouse IgG (Jackson ImmunoResearch, no. 115-175-146), both at a dilution of 1:3000 in 1× PBS. Fluorescence of Cy3 and Cy5 was recorded using 532- and 635-nm channels by the GenePix 4200A Microarray Scanner (Molecular Devices LLC). Images were analyzed using GenePix Pro 6.0 software to give fluorescent intensities of each spot and its corresponding background. Missing or distorted spots, typically controlled under 5% of the total spots, were automatically marked by the software.
Reproducibility of array experiments was evaluated by incubating a triplicate of the same sample with three PETAL arrays. The fluorescent intensity of each spot was normalized using biotin-labeled BSA signal in each block and array. The normalized fluorescent intensities were plotted between every experimental pair. Pearson product-moment correlation coefficient value (R value) was calculated for each data pair in which the r value of +1 means total positive correlation and 0 is no correlation (50).
Protein sample preparation
Cell lines [i.e., A431, A549, human embryonic kidney (HEK) 293 T, H1975, H226, Hela, HepG2, HL60, human umbilical cord endothelial cell (HUVEC), Jurkat, K562, MCF7, PC3, PC9, THP1, and U937] were purchased from the American Type Culture Collection (ATCC) or stem cell bank, Chinese Academy of Sciences (Shanghai, China). Cells were grown or maintained in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI 1640 media following the ATCC cell culture guide. When cells were grown to ~80% confluence, they were dissociated from culture plates by treatment with 1× PBS and 1 mM EDTA for 10 to 20 min. Trypsin was not used to avoid damages on cell surface proteins. Membrane or nuclear fractions of cell lysates were then prepared as described previously (51). For whole-cell lysis, 1× PBS containing 1% NP-40, 5 mM EDTA, and protease inhibitor cocktail (Calbiochem, no. 539134) was added to cells directly and incubated on ice for 30 min. An ultrasonication step was performed before collecting the supernatant. The prepared membrane fraction (MEM), nuclear fraction (NUC), and whole-cell lysate (WCL) were labeled following the cell lines, respectively. The enrichments of marker proteins in MEM, NUC, and WCL fractions were evaluated with anti-ATP5B (Abmart, no. M40013), histone 3.1 (Abmart, no. P30266), and β-tubulin (Abmart, no. M30109) mAbs, respectively.
For tissue samples from plants or animals, whole-cell lysates were prepared following a protocol described previously (52). Briefly, frozen tissues were powdered in liquid nitrogen with a pestle, suspended in 10 ml per 3 g of tissue extract protein extraction buffer [10 mM tris (pH 8.0), 100 mM EDTA, 50 mM borax, 50 mM vitamin C, 1% Triton X-100, 2% 2-mercaptoethanol, 30% sucrose] and incubated for 10 min. An equal volume of tris-HCl (pH 7.5)–saturated phenol was then added and vortex-mixed for 10 min at room temperature. The phenolic phase separated by centrifugation was recovered and reextracted twice with 10 ml of extraction buffer. Proteins in the final phenolic phase were precipitated overnight at −20°C with 5× volumes of saturated ammonium acetate in methanol. Protein pellets collected by centrifugation were washed twice with ice-cold methanol and once with ice-cold acetone. Pellets were then dried and dissolved with 500 mM triethylammonium bicarbonate containing 0.5% SDS (pH 8.5). Bacteria lysates were prepared using an ultrasonic apparatus.
Patient samples
All tissue microarray chips were purchased from Shanghai Outdo Biotech Co. Ltd. Tumor and paracancerous tissues (normal) were freshly excised from a patient with NSCLC undergoing surgery. Tumor tissue and matched paracancerous tissue were homogenized (53). Briefly, the specimens were cut into 0.5-mm sections before digestion with 0.1% collagenase IV (Gibco, no. 17104019) for 1 hour at 37°C. The cells were then passed through a 70-μm cell strainer (BD, no. 352350) and collected by centrifugation for 15 min at 400g. Plasma membrane proteome extracts were prepared from single-cell suspensions of tissues.
Screening PETAL array with recombinant protein antigens
Recombinant protein antigens were first labeled with biotin using EZ-Link NHS-LC-Biotin reagent (Thermo Fisher, no. 21366) and then hybridized with the PETAL array. Array-bound proteins were incubated with Streptavidin-Cy3. The fluorescent intensity of mAb spots was then recorded by the GenePix 4200A Microarray Scanner. Array-positive spots were defined as (signal-background)/background > 3. Protein-binding PETAL mAbs selected from array experiments were further screened through protein-mAb ELISA using a detection limit of 1 μg/ml of protein antigen. The ELISA-positive mAbs were then validated on immunoblotting assays with recombinant proteins or endogenous samples.
Screening PETAL array with proteomic antigens
Proteomic antigens including membrane, nuclear, or whole-cell lysates were labeled with biotin using EZ-Link NHS-LC-Biotin reagent and incubated with the PETAL arrays. Following a similar procedure as described above, antibody spots positive in three independent experiments were then ranked by the averaged fluorescence intensities. A limited number of array-positive antibodies (1000 to 2000 according to the expected output of each screening) with high (>10,000), medium (2000 to 10,000), and low (500 to 2000) fluorescent intensity were selected as candidate antibodies for further validation assays.
Immunoblotting assays of PETAL mAbs
For recombinant protein immunoblotting, selected mAbs were used to probe 50, 10, 2, and 0.4 ng of recombinant protein antigens. For immunoblotting of endogenous human protein samples by PETAL mAbs, cell lines were selected according to the protein expression profile from HPA and UniProt databases. For membrane or nuclear proteins, corresponding cellular fractions were prepared for immunoblotting. Typically, 20 μg of protein was loaded onto each lane. Support-positive immunoblotting results were evaluated following the criteria described by Antibodypedia (http://antibodypedia.com/text/validation_criteria#western_blot) and HPA (29). Basically, an antibody was qualified as immunoblotting positive when the size of a single or predominant single band on immunoblotting matched or was within 10% of the predicted antigen molecular weight. In some cases, an immunoblotting-positive conclusion was enhanced when the same predicted protein band was detected in two or more different cell lysates. Some antibodies detected multiple bands with different sizes, but the predicted size protein band was also detected.
IF and FACS validation
A cell line with target protein expression (HPA data) was selected for IF and FACS assays. Known/predicted subcellular localization of the target protein was also obtained from HPA or UniProt (table S4). For cell surface proteins, IF and FACS assays were performed under nonpermeable conditions without detergent in the buffers. For intracellular proteins, the permeable condition with 0.1% Triton added to the buffers was used throughout. Antibody binding signal was detected using Alexa Fluor 488 and 594 goat anti-mouse IgG secondary antibodies (Jackson ImmunoResearch, no. 115-545-003 and no. 115-585-003). Briefly, cells attached on coverslips (IF assays) or suspended in 1× PBS (FACS assays) were first fixed in 4% paraformaldehyde (PFA) for 10 min. PFA was then removed, and cells were rinsed three times with 1× PBS. Cells were blocked overnight at 4°C in blocking buffer (1× PBS containing 10% normal goat serum, 0.1% Triton was added for intracellular proteins). After removing the blocking buffer, cells were incubated with primary antibody (dilution in the blocking buffer at 1:100 to 1000) for 3 hours at room temperature. Cells were rinsed six times in 1× PBS before being incubated with fluorescence-labeled secondary antibody (diluted in blocking buffer at 1:500 dilution ratio with 1:10,000 Hoechst 33258; Sigma, no. 94403) for 1 hour. Last, cells were rinsed three times with 1× PBS. IF images were recorded with a Nikon confocal system A1Si. The three-dimensional reconstruction of the IF results was performed in ImageJ [National Center for Biotechnology Information, NIH (NCBI) free software]. IF staining patterns were compared with HPA data to confirm the subcellular localization of the target proteins. The FACS data were collected using a BD Accuri C6 Plus system. A control sample without primary antibody and another sample with isotype control antibody were used.
IP and mass spectrum assays to identify antibody binding protein
IP assays were performed using cyanogen bromide (CNBr)–activated Sepharose 4B (GE, no. 17-0430-02) by following the user’s manual. Briefly, 200 μg of purified PETAL mAbs was cross-linked to 20 μl of hydrolyzed CNBr beads and used to pull-down target protein from 1 mg of cell membrane or nuclear protein samples. Typically, an excessive amount of antibodies was used. A similar procedure was developed following the instructions described previously (54) to identify the binding proteins of the tested antibodies.
Essentially, target identification for immunoblotting-successful (yielding single or predominant single band) mAbs used for IP was done by comparing the silver staining result of the IP product with immunoblotting data on samples before and after IP. Expected size band (matched on silver staining and immunoblotting) was selected for MS analysis. For some mAbs, more than one band on SDS–polyacrylamide gel electrophoresis (PAGE) was selected for MS identification; several identified proteins could be the binding targets of an antibody. For antibodies that failed in the immunoblotting assay, their IP products separated on silver-stained SDS-PAGE were compared to IP products from other mAbs. One predominant specific band or several stoichiometric-specific bands were selected for MS analysis.
Once one or more bands were selected for MS, 20 μl of IP product was separated in an SDS-PAGE gel and stained with Coomassie blue R-250. The selected bands were excised and sent to MS facilities (Instrumental Analysis Center of Shanghai JiaoTong University or Biological Mass Spectrometry Facility at Robert Wood Johnson Medical School and Rutgers, The State University of New Jersey) for target identification using LC-MS/MS on Thermo Q Exactive HF or Thermo Orbitrap-Velos Pro.
Mascot distiller (version 2.6, Matrix Science) or Protein Discovery software (version 2.2) was used to convert raw to mgf or mzML format for downstream analysis. The LC-MS/MS data were searched against UniProt human (557,992 proteins) for the human cell culture sample, UniProt zebrafish (61,675) for zebrafish tissue sample, or UniProt maize (137,106) for corn tissue sample. Enzyme specificity was set as C terminal to Arg and Lys and allowed for two missed cleavages. Furthermore, ±10 ppm and 0.02 Da (Thermo Q Exactive HF) or 1 Da (Thermo Orbitrap-Velos Pro) were used as tolerance for precursor (MS) and product ions (MS/MS), respectively. Carbamidomethylated cysteine was set as complete modifications. N-terminal protein acetylation and oxidation of methionine were set as potential modifications. Deamidation at asparagine and glutamine and oxidation at methionine and tryptophan were specified as variable modifications.
To ensure MS data quality, we used a threshold of 20 total identified peptide number or five nonredundant peptide number to achieve high confidence of the identified protein. In analyzing the MS result for the antibody, the identified protein list was first prioritized using the total identified peptide number. Proteins that were identified in multiple different antibodies were excluded. For most antibodies in this study, a unique protein with the highest total identified peptide number and matched protein size detected on the silver staining and immunoblotting was selected as the target protein.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRoteomics IDEntifications (PRIDE) (55) partner repository with the dataset identifier PXD011629 (reviewer account details: username, reviewer41517@ebi.ac.uk; password, 7ZqfVOM8).
Abundance distribution and molecular function analysis
The identified membrane, nuclear, and other proteins were from the reference database (Nucleoplasm protein database and Nuclear membrane plus Plasma membrane protein database from HPA). Expression abundance information of human proteins was obtained from the PAXdb. Function distributions were clustered using the PANTHER classification system (56) depending on the molecular function.
ChIP-seq assay
The ChIP and input DNA libraries were prepared as previously described (34, 57). Briefly, 10 million HepG2 cells were cross-linked with 1% formaldehyde for 10 min at room temperature and then quenched with 125 mM glycine. The chromatin was fragmented and then immunoprecipitated with Protein A + G magnetic beads coupled with antibodies against SMRC1, SATB1, and NFIC. After reverse cross-linking, ChIP and input DNA fragments were used for library construction with NEBNext Ultra Ligation Module (NEB, no. E7445). The DNA libraries were amplified and subjected to deep sequencing with an Illumina sequencer. The ChIP-seq data processing was performed as we reported recently (57). Cis-regulatory sequence elements that mediate the binding of SMRC1, SATB1, or NFIC were predicted with MEME-ChIP (58).
Internalization assay
For the IF assay, live PC9 cells were cultured on coverslips and incubated with mAbs (10 μg/ml) for 1 hour on ice before being washed three times with PBS. Cells were then cultured at 37°C for 0, 2, or 4 hours before fixation with 4% PFA. Antibodies were then labeled with Alexa Fluor 488–conjugated anti-mouse antibody. Images were acquired by Nikon confocal system A1Si.
For the FACS assay, live PC9 cells were incubated with mAbs (10 μg/ml) for 0.5 hour on ice before being washed three times with PBS. Cells were then cultured at 37°C for up to 4 hours before fixation with 4% PFA. Cells were then stained with Alexa Fluor 488–conjugated anti-mouse antibody and analyzed with FACS. Surface mean fluorescence intensity (MFI) was calculated. Surface mean fluorescent intensity (MFI), which represented surface localization of mAbs, was measured by FACS.
Indirect cytotoxicity assay
PC9 cells were cultured in 96-well plates at 2000 per well confluence overnight. Cells were treated with serial dilution of mAbs together with MMAE-conjugated secondary goat anti-mouse IgG antibody (2 μg/ml) for 72 hours. Cell number was then calculated by Cell Counting Kit-8 (CCK8; Dojindo, no. CK04-20). Antibody-drug conjugation services were provided by Levena Biopharma, Nanjing.
In vivo tumor models
For the CDX model, 5 × 106 NCI-H1975 cells were suspended in Matrigel (BD Biosciences, no. 354234) and injected subcutaneously to the right flank of female BALB/c nude mice (jsj-lab). For studies with the PDX model, the tumor fragments from patients with LUSCC were passaged twice in nonobese diabetic–severe combined immunodeficient mice (Beijing Vital River Laboratory Animal Technology Co. Ltd.). Tumor fragments obtained from in vivo passage were then implanted subcutaneously in the right flank of female BALB/c nude mice (jsj-lab). Body weight and tumor volume (0.5 × length × width2) were measured every 3 days. Mice were randomized into control and treatment groups on the basis of the primary tumor sizes (median tumor volume of approximately 100 mm3). Pb44707-ADCs and control ADCs were administered intravenously every third day and repeated for a total of three times (Q3Dx3). Gefitinib (Selleck, ZD1839) was administered intraperitoneally every day.
siRNA knockdown and overexpression
PC9 was transfected with siRNA targeting human CD44V9 (sense, 5′-CUACUUUACUGGAAGGUUAtt-3′; antisense, 5′-UAACCUUCCAGUAAAGUAGtt-3′), which has been reported previously (37) or control siRNA (sense, 5′-UUCUCCGAACGUGUCACGUtt-3′; antisense, 5′-ACGUGACACGUUCGGAGAAtt-3′) by Lipofectamine 2000 (Thermo Fisher, no. 11668019) 48 hours before performing experiment.
PIEZO1-GFP plasmid used for overexpression validation was a gift from D. Beech, which was described previously (59). PIEZO1-GFP was transfected into HUVEC cells by Lipofectamine 2000. Cells were fixed and stained with anti-PIEZO1 antibody following IF procedure described above.
Peptide-blocking assay
Pb44707 (1 mg/ml) was preincubated with CD44 recombinant protein (1 mg/ml; Abcam, no. ab173996) or CD44V peptide in 1:1 ratio at 4°C overnight before used in FACS analysis.
Antibody cellular binding site quantification
The antibody binding sites on cell lines were determined with the QIFIKIT (Dako, no. K0078) according to the manufacturer’s instructions.
Supplementary Material
Acknowledgments
The authors wish to thank all Abmart employees for building the industrialized mAb assembly line that generated PETAL. Funding: This project was supported in part by a special Endowed Professorship grant from Northwest University (Xi’an, China) to X.M., the National Key Research and Development Project of China Grant (2016YFA0500600), the National Natural Science Foundation of China Grants 31670831 and 36370813 to S.-c.T., the Life Science Research Foundation (LSRF; Astellas Pharma) to M.I.P., the National Natural Science Foundation of China Grant 31770310 to K.W., the National Natural Science Foundation of China Grant 31560415 to D.H., and the National Natural Science Foundation of China Grant 31570126 to L.Z.M. Author contributions: X.M. and S.-C.T. developed the conceptual ideas and designed the study. Zhaohui Wang, Yang Li, B.H., M.I.P., M.W., G.C., W.W., Yuemeng Wang, Yiqiang Wang, Y.T., X.X., R.P., F.L., N.W., Z.C., S.W., L.Z.M., X.D., Yangrui Li, D.H., J.L., Zhiqiang Wang, W.Z., Ying Zhang, Ying Lin, Z.L., Q.X., J.G., H.D., Y.Y., Z.Y., Q.N., X.Z., K.W., F.C., Q.Z., Yuxian Zhu, S.Z., and K.D.P. performed the experiments, X.-d.Z. and J.Y. analyzed the ChIP-seq data, X.M., S.-C.T., Zhaohui Wang, Yang Li, and B.H. wrote the manuscript with suggestions from the other authors. Competing Interests: X.M. is cofounder and shareholder of Abmart Inc. Zhaohui Wang, B.H., Yuemeng Wang, M.W., G.C., W.W., Yiqiang Wang, Y.T., X.X., R.P., and F.L. are employees of Abmart Inc. is the owner and responsible for the maintenance of PETAL library. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The mass spectrometry data are deposited at the PRIDE database under the accession number PXD011629. The ChIP-seq data in this study are deposited at the Gene Expression Omnibus under the accession number GSE108514. Additional data related to this paper may be requested from authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaax2271/DC1
Fig. S1. PETAL antigens, antibody diversity, and array performance.
Fig. S2. PETAL screen by protein antigens.
Fig. S3. Proteome-scale antibody generation for human membrane and nuclear proteins.
Fig. S4. Differential array screening for ADC therapeutic target/antibody.
Fig. S5. Two approaches to access PETAL library/array.
Table S1. Peptide antigens used for PETAL library construction.
Table S2. The list of the 62,208 antibodies and their locations on the microarray.
Table S3. Summary for PETAL screening on recombinant protein targets.
Table S4. PETAL-screened protein targets from human cell lines.
Table S5. Summary table for mAbs screened from human cell membrane and nuclear extractions.
Movie S1. Movie showing cellular localization of TFRC in A431.
Movie S2. Movie showing cellular localization of ACTN4 in PC9.
Movie S3. Movie showing cellular localization of TIMM50 in PC9.
Movie S4. Movie showing cellular localization of GOLIM4 in PC9.
Movie S5. Movie showing cellular localization of NPM1 in A431.
Movie S6. Movie showing cellular localization of SFPQ in PC9.
Reference (60)
REFERENCES AND NOTES
- 1.Koepfli K.-P., Paten B.; Genome 10K Community of Scientists, O'Brien S. J., The genome 10K project: A way forward. Annu. Rev. Anim. Biosci. 3, 57–111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter P., Smith L., Ryan M., Identification and validation of cell surface antigens for antibody targeting in oncology. Endocr. Relat. Cancer 11, 659–687 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Hellstrom I., Garrigues H. J., Garrigues U., Hellstrom K. E., Highly tumor-reactive, internalizing, mouse monoclonal antibodies to Le(y)-related cell surface antigens. Cancer Res. 50, 2183–2190 (1990). [PubMed] [Google Scholar]
- 4.Israeli R. S., Powell C. T., Fair W. R., Heston W. D., Molecular cloning of a complementary DNA encoding a prostate-specific membrane antigen. Cancer Res. 53, 227–230 (1993). [PubMed] [Google Scholar]
- 5.Hosen N., Matsunaga Y., Hasegawa K., Matsuno H., Nakamura Y., Makita M., Watanabe K., Yoshida M., Satoh K., Morimoto S., Fujiki F., Nakajima H., Nakata J., Nishida S., Tsuboi A., Oka Y., Manabe M., Ichihara H., Aoyama Y., Mugitani A., Nakao T., Hino M., Uchibori R., Ozawa K., Baba Y., Terakura S., Wada N., Morii E., Nishimura J., Takeda K., Oji Y., Sugiyama H., Takagi J., Kumanogoh A., The activated conformation of integrin β7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 23, 1436–1443 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Benicky J., Hafko R., Sanchez-Lemus E., Aguilera G., Saavedra J., Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell. Mol. Neurobiol. 32, 1353–1365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michel M. C., Wieland T., Tsujimoto G., How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch. Pharmacol. 379, 385–388 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Szigyarto C. A.-K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Thul P. J., Åkesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Björk L., Breckels L. M., Bäckström A., Danielsson F., Fagerberg L., Fall J., Gatto L., Gnann C., Hober S., Hjelmare M., Johansson F., Lee S., Lindskog C., Mulder J., Mulvey C. M., Nilsson P., Oksvold P., Rockberg J., Schutten R., Schwenk J. M., Sivertsson Å., Sjöstedt E., Skogs M., Stadler C., Sullivan D. P., Tegel H., Winsnes C., Zhang C., Zwahlen M., Mardinoglu A., Pontén F., von Feilitzen K., Lilley K. S., Uhlén M., Lundberg E., A subcellular map of the human proteome. Science 356, eaa13321 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Jin A., Ozawa T., Tajiri K., Obata T., Kondo S., Kinoshita K., Kadowaki S., Takahashi K., Sugiyama T., Kishi H., Muraguchi A., A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat. Med. 15, 1088–1092 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Di Cristina M., Nunziangeli L., Giubilei M. A., Capuccini B., d’Episcopo L., Mazzoleni G., Baldracchini F., Spaccapelo R., Crisanti A., An antigen microarray immunoassay for multiplex screening of mouse monoclonal antibodies. Nat. Protoc. 5, 1932–1944 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Reddy S. T., Ge X., Miklos A. E., Hughes R. A., Kang S. H., Hoi K. H., Chrysostomou C., Hunicke-Smith S. P., Iverson B. L., Tucker P. W., Ellington A. D., Georgiou G., Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat. Biotechnol. 28, 965–969 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Feldhaus M. J., Siegel R. W., Opresko L. K., Coleman J. R., Feldhaus J. M. W., Yeung Y. A., Cochran J. R., Heinzelman P., Colby D., Swers J., Graff C., Wiley H. S., Wittrup K. D., Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 21, 163–170 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Harvey B. R., Georgiou G., Hayhurst A., Jeong K. J., Iverson B. L., Rogers G. K., Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc. Natl. Acad. Sci. U.S.A. 101, 9193–9198 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaffitzel C., Hanes J., Jermutus L., Plückthun A., Ribosome display: An in vitro method for selection and evolution of antibodies from libraries. J. Immunol. Methods 231, 119–135 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Mersmann M., Meier D., Mersmann J., Helmsing S., Nilsson P., Gräslund S.; Structural Genomics Consortium, Colwill K., Hust M., Dübel S., Towards proteome scale antibody selections using phage display. N. Biotechnol. 27, 118–128 (2010). [DOI] [PubMed] [Google Scholar]
- 17.de Wildt R. M., Mundy C. R., Gorick B. D., Tomlinson I. M., Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 18, 989–994 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Wingren C., Antibody-based proteomics. Adv. Exp. Med. Biol. 926, 163–179 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Mao H., Graziano J. J., Chase T. M. A., Bentley C. A., Bazirgan O. A., Reddy N. P., Song B. D., Smider V. V., Spatially addressed combinatorial protein libraries for recombinant antibody discovery and optimization. Nat. Biotechnol. 28, 1195–1202 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Mariuzza R. A., Multiple paths to multispecificity. Immunity 24, 359–361 (2006). [DOI] [PubMed] [Google Scholar]
- 21.James L. C., Roversi P., Tawfik D. S., Antibody multispecificity mediated by conformational diversity. Science 299, 1362–1367 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Kramer A., Keitel T., Winkler K., Stöcklein W., Höhne W., Schneider-Mergener J., Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell 91, 799–809 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berglund L., Andrade J., Odeberg J., Uhlén M., The epitope space of the human proteome. Protein Sci. 17, 606–613 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabat E. A., Wu T. T., Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J. Immunol. 147, 1709–1719 (1991). [PubMed] [Google Scholar]
- 26.Jiang L., Yu X., Qi X., Yu Q., Deng S., Bai B., Li N., Zhang A., Zhu C., Liu B., Pang J., Multigene engineering of starch biosynthesis in maize endosperm increases the total starch content and the proportion of amylose. Transgenic Res. 22, 1133–1142 (2013). [DOI] [PubMed] [Google Scholar]
- 27.de Sousa S. M., del Giúdice Paniago M., Arruda P., Yunes J. A., Sugar levels modulate sorbitol dehydrogenase expression in maize. Plant Mol. Biol. 68, 203–213 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Budas G. R., Disatnik M.-H., Mochly-Rosen D., Aldehyde dehydrogenase 2 in cardiac protection: A new therapeutic target? Trends Cardiovasc. Med. 19, 158–164 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berglund L., Björling E., Oksvold P., Fagerberg L., Asplund A., Al-Khalili Szigyarto C., Persson A., Ottosson J., Wernérus H., Nilsson P., Lundberg E., Sivertsson Å., Navani S., Wester K., Kampf C., Hober S., Pontén F., Uhlén M., A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell Proteomics 7, 2019–2027 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., Rimm D. L., Rodriguez H., Hiltke T., Snyder M., Yamamoto T., A proposal for validation of antibodies. Nat. Methods 13, 823–827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romac J. M.-J., Shahid R. A., Swain S. M., Vigna S. R., Liddle R. A., Piezo1 is a mechanically activated ion channel and mediates pressure induced pancreatitis. Nat. Commun. 9, 1715 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geiger T., Wehner A., Schaab C., Cox J., Mann M., Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell Proteomics 11, M111.014050 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., von Mering C., The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–d368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X. D., Han X., Chew J. L., Liu J., Chiu K. P., Choo A., Orlov Y. L., Sung W.-K., Shahab A., Kuznetsov V. A., Bourque G., Oh S., Ruan Y., Ng H.-H., Wei C.-L., Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1, 286–298 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Beck A., Goetsch L., Dumontet C., Corvaia N., Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 16, 315–337 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Orian-Rousseau V., CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 46, 1271–1277 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi K., Matsumoto H., Matsuyama H., Fujii N., Inoue R., Yamamoto Y., Nagao K., Clinical significance of CD44 variant 9 expression as a prognostic indicator in bladder cancer. Oncol. Rep. 36, 2852–2860 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Willuda J., Linden L., Lerchen H. G., Kopitz C., Stelte-Ludwig B., Pena C., Lange C., Golfier S., Kneip C., Carrigan P. E., Mclean K., Schuhmacher J., von Ahsen O., Müller J., Dittmer F., Beier R., el Sheikh S., Tebbe J., Leder G., Apeler H., Jautelat R., Ziegelbauer K., Kreft B., Preclinical antitumor efficacy of BAY 1129980-a novel auristatin-based anti-C4.4A (LYPD3) antibody-drug conjugate for the treatment of non-small cell lung cancer. Mol. Cancer Ther. 16, 893–904 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Stoevesandt O., Taussig M. J., Affinity proteomics: The role of specific binding reagents in human proteome analysis. Expert Rev. Proteomics 9, 401–414 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Hopp T. P., Woods K. R., Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. U.S.A. 78, 3824–3828 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welling G. W., Weijer W. J., van der Zee R., Welling-Wester S., Prediction of sequential antigenic regions in proteins. FEBS Lett. 188, 215–218 (1985). [DOI] [PubMed] [Google Scholar]
- 42.Getzoff E. D., Tainer J. A., Lerner R. A., Geysen H. M., The chemistry and mechanism of antibody binding to protein antigens. Adv. Immunol. 43, 1–98 (1988). [DOI] [PubMed] [Google Scholar]
- 43.Chou P. Y., Fasman G. D., Prediction of protein conformation. Biochemistry 13, 222–245 (1974). [DOI] [PubMed] [Google Scholar]
- 44.Hopp T. P., Use of hydrophilicity plotting procedures to identify protein antigenic segments and other interaction sites. Methods Enzymol. 178, 571–585 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Moura A., Savageau M. A., Alves R., Relative amino acid composition signatures of organisms and environments. PlOS One 8, e77319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de StGroth S. F., Scheidegger D., Production of monoclonal antibodies: Strategy and tactics. J. Immunol. Methods 35, 1–21 (1980). [DOI] [PubMed] [Google Scholar]
- 47.Giudicelli V., Brochet X., Lefranc M.-P., IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011, 695–715 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Delfani P., Dexlin Mellby L., Nordström M., Holmér A., Ohlsson M., Borrebaeck C. A. K., Wingren C., Technical advances of the recombinant antibody microarray technology platform for clinical immunoproteomics. PlOS One 11, e0159138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borrebaeck C. A. K., Wingren C., Design of high-density antibody microarrays for disease proteomics: key technological issues. J. Proteomics 72, 928–935 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Cohen J., Statistical Power Analysis. Curr. Dir. Psychol. Sci. 1, 98–101 (2016). [Google Scholar]
- 51.J. V. Hagen, U. Michelsen, in Methods in enzymology, L. Jon, Ed. (Academic Press, 2013), vol. 533, pp. 25–30. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Wang W., Cai J., Zhang Y., Qin G., Tian S., Tomato nuclear proteome reveals the involvement of specific E2 ubiquitin-conjugating enzymes in fruit ripening. Genome Biol. 15, 548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang D.-G., Jiang A.-G., Lu H.-Y., Zhang L.-X., Gao X.-Y., Isolation, cultivation and identification of human lung adenocarcinoma stem cells. Oncol. Lett. 9, 47–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcon E., Jain H., Bhattacharya A., Guo H., Phanse S., Pu S., Byram G., Collins B. C., Dowdell E., Fenner M., Guo X., Hutchinson A., Kennedy J. J., Krastins B., Larsen B., Lin Z. Y., Lopez M. F., Loppnau P., Miersch S., Nguyen T., Olsen J. B., Paduch M., Ravichandran M., Seitova A., Vadali G., Vogelsang M. S., Whiteaker J. R., Zhong G., Zhong N., Zhao L., Aebersold R., Arrowsmith C. H., Emili A., Frappier L., Gingras A.-C., Gstaiger M., Paulovich A. G., Koide S., Kossiakoff A. A., Sidhu S. S., Wodak S. J., Gräslund S., Greenblatt J. F., Edwards A. M., Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. Methods 12, 725–731 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., Hermjakob H., 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mi H., Muruganujan A., Casagrande J. T., Thomas P. D., Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X. L., Wu J., Wang J., Shen T., Li H., Lu J., Gu Y., Kang Y., Wong C.-H., Ngan C. Y., Shao Z., Wu J., Zhao X., Integrative epigenomic analysis reveals unique epigenetic signatures involved in unipotency of mouse female germline stem cells. Genome Biol. 17, 162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Machanick P., Bailey T. L., MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J., Hou B., Tumova S., Muraki K., Bruns A., Ludlow M. J., Sedo A., Hyman A. J., McKeown L., Young R. S., Yuldasheva N. Y., Majeed Y., Wilson L. A., Rode B., Bailey M. A., Kim H. R., Fu Z., Carter D. A., Bilton J., Imrie H., Ajuh P., Dear T. N., Cubbon R. M., Kearney M. T., Prasad R. K., Evans P. C., Ainscough J. F., Beech D. J., Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gertz J., Savic D., Varley K. E., Partridge E. C., Safi A., Jain P., Cooper G. M., Reddy T. E., Crawford G. E., Myers R. M., Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell 52, 25–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/11/eaax2271/DC1
Fig. S1. PETAL antigens, antibody diversity, and array performance.
Fig. S2. PETAL screen by protein antigens.
Fig. S3. Proteome-scale antibody generation for human membrane and nuclear proteins.
Fig. S4. Differential array screening for ADC therapeutic target/antibody.
Fig. S5. Two approaches to access PETAL library/array.
Table S1. Peptide antigens used for PETAL library construction.
Table S2. The list of the 62,208 antibodies and their locations on the microarray.
Table S3. Summary for PETAL screening on recombinant protein targets.
Table S4. PETAL-screened protein targets from human cell lines.
Table S5. Summary table for mAbs screened from human cell membrane and nuclear extractions.
Movie S1. Movie showing cellular localization of TFRC in A431.
Movie S2. Movie showing cellular localization of ACTN4 in PC9.
Movie S3. Movie showing cellular localization of TIMM50 in PC9.
Movie S4. Movie showing cellular localization of GOLIM4 in PC9.
Movie S5. Movie showing cellular localization of NPM1 in A431.
Movie S6. Movie showing cellular localization of SFPQ in PC9.
Reference (60)