

Abstract
Parkinson’s disease (PD) is a synucleinopathy that is characterized by motor dysfunction, death of midbrain dopaminergic neurons, and accumulation of alpha synuclein (α-Syn) aggregates. Evidence suggests that α-Syn aggregation can originate in peripheral tissues and progress to the brain via autonomic fibers. We tested this by inoculating the duodenal wall of mice with α-Syn preformed fibrils. Following inoculation, we observed gastrointestinal deficits and physiological changes to the enteric nervous system. We also found that α-Syn pathology is reduced by increased expression of the lysosomal enzyme glucocerebrosidase, using the AAV-PHP.S capsid to target this protein for peripheral gene transfer. Lastly, inoculation of α-Syn fibrils in aged mice, but not younger mice, resulted in progression of α-Syn histopathology to the midbrain and subsequent motor defects. Our results characterize peripheral synucleinopathy in prodromal PD and explore cellular mechanisms for the gut-to-brain progression of α-Syn pathology.
INTRODUCTION
Synucleinopathies are neurodegenerative diseases characterized by the aggregation of insoluble amyloid α-Synuclein (α-Syn) fibrils1. α-Syn accumulation in specific cell populations precipitates distinct clinical phenotypes that serve as the basis for diagnosis of synucleinopathies. For example, the appearance of α-Syn pathology in midbrain dopaminergic neurons coincides with motor dysfunction in Parkinson’s disease (PD)2. However, mounting evidence suggests that synucleinopathy diagnosis occurs late in the disease progression and that pathology may originate much earlier in the gastrointestinal (GI) tract before progressing to the brain3. Biopsies of GI tissue from both PD patients and healthy individuals have found α-Syn accumulation in the stomach, duodenum, and colon4,5. This accumulation has been observed in cells of the enteric nervous system (ENS), an organized network of interconnected ganglia comprised of neurons and enteric glial cells (EGCs) that spans the entire GI tract6. One hypothesis is that pathologic α-Syn ascends ENS-innervating vagal fibers to the nodose ganglion and brainstem nuclei7. Once in the brain, α-Syn pathology would then propagate through interconnected neurons to reach the midbrain and cause motor dysfunction. Recent studies show that α-Syn fibrils are capable of progressing through vagal fibers that innervate the gut and can cause behavioral dysfunction8–10. However, how pathologic α-Syn impacts the enteric nervous system (ENS), and the mechanisms that underlie their spread, have not been comprehensively studied.
Inflammation is linked to the peripheral etiology of PD, as pro-inflammatory cytokines are elevated in colonic biopsies and stool samples from PD patients11. In addition, in vitro work has shown that aggregated α-Syn directly activates inflammatory pathways12. Macrophages are recruited as a component of the inflammatory process and are essential for removing pathogens, including misfolded protein aggregates13. The lysosome is a major macrophagic regulator of protein homeostasis and pathogen clearance, and has been shown to interact with aggregated α-Syn14. Glucocerebrosidase (GCase) is a lysosomal enzyme and disease-associated mutations within its gene, GBA1, result in impaired lysosomal function. The connection between GCase and α-Syn was discovered in patients with Gaucher’s disease, a metabolic disorder caused by loss of GCase function, who presented with elevated levels of α-Syn in the brain15. Further studies have gone on to demonstrate that impaired GCase function also results in the abnormal accumulation of α-Syn16. Interestingly, pathologic α-Syn disrupts trafficking of GCase to the lysosome, which is hypothesized to contribute to a positive feedback loop for α-Syn aggregation17. Thus, GCase activity could be critical during prodromal PD to prevent α-Syn accumulation from reaching a pathogenic, self-propagating threshold.
In this work, we aimed to characterize the mechanisms that underlie synucleinopathy in the GI tract. We showed that inoculating duodenal intestinal lining with α-Syn preformed fibrils (PFFs) induced the formation of phosphorylated α-Syn (p-α-Syn)-containing inclusions, promoted a local inflammatory response, and disrupted ENS connectivity. We also observed progression of α-Syn histopathology from the ENS to the brainstem and reduced levels of striatal dopamine in aged, but not young mice after PFF inoculation. Finally, α-Syn pathology and ENS dysfunction is recovered by increased expression of GCase, reinforcing the role of GCase as a critical regulator of pathologic α-Syn18. Our results reveal mechanisms that contribute to peripheral, non-motor symptoms reflective of prodromal synucleinopathy and demonstrate how gut-seeded α-Syn fibrils promote the progression of α-Syn pathology to the brain in an age-dependent manner.
RESULTS
Duodenal α-Syn PFF inoculation disrupts GI function and initiates inflammatory responses
One of the most common non-motor symptoms observed in PD is GI dysfunction, which often precedes the development of motor symptoms19. However, it is not fully understood whether α-Syn pathology causes this disruption. To directly test whether pathologic α-Syn interrupts ENS control of GI function, we modeled prodromal peripheral synucleinopathy by seeding the duodenum of adult (8–10 week old), wild type (WT) mice with mouse α-Syn PFFs20 via intramuscular injection (Fig. 1a). The use of α-Syn PFFs is established as a model of idiopathic synucleinopathy in the CNS that closely represents human pathology21,22. We targeted the duodenum because of its dense innervation by vagal fibers, which is speculated to be a critical component of the gut-to-brain hypothesis of synucleinopathy etiology7. Duodenal inoculation with α-Syn PFFs resulted in time-dependent GI dysfunction through 120 days post inoculation (dpi), comparable to the phenotypes reported in transgenic models of synucleinopathy, including the ASO (Thy1-α-Syn overexpressing) line23,24 (Fig. 1b–d, Extended Data Fig. 1a–d). Interestingly, inoculation with α-Syn monomers did not produce an effect, which we observed repeatedly throughout our study.
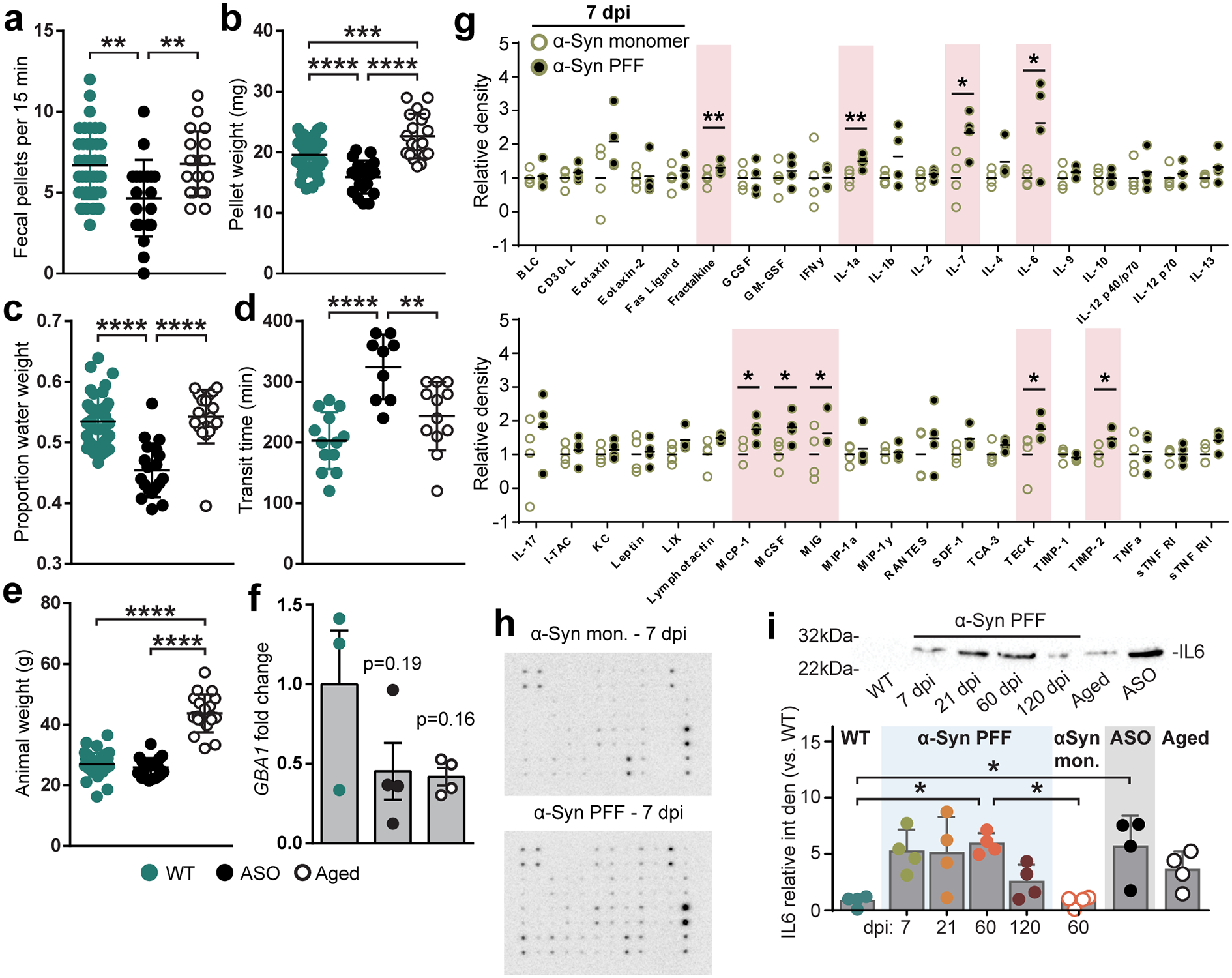
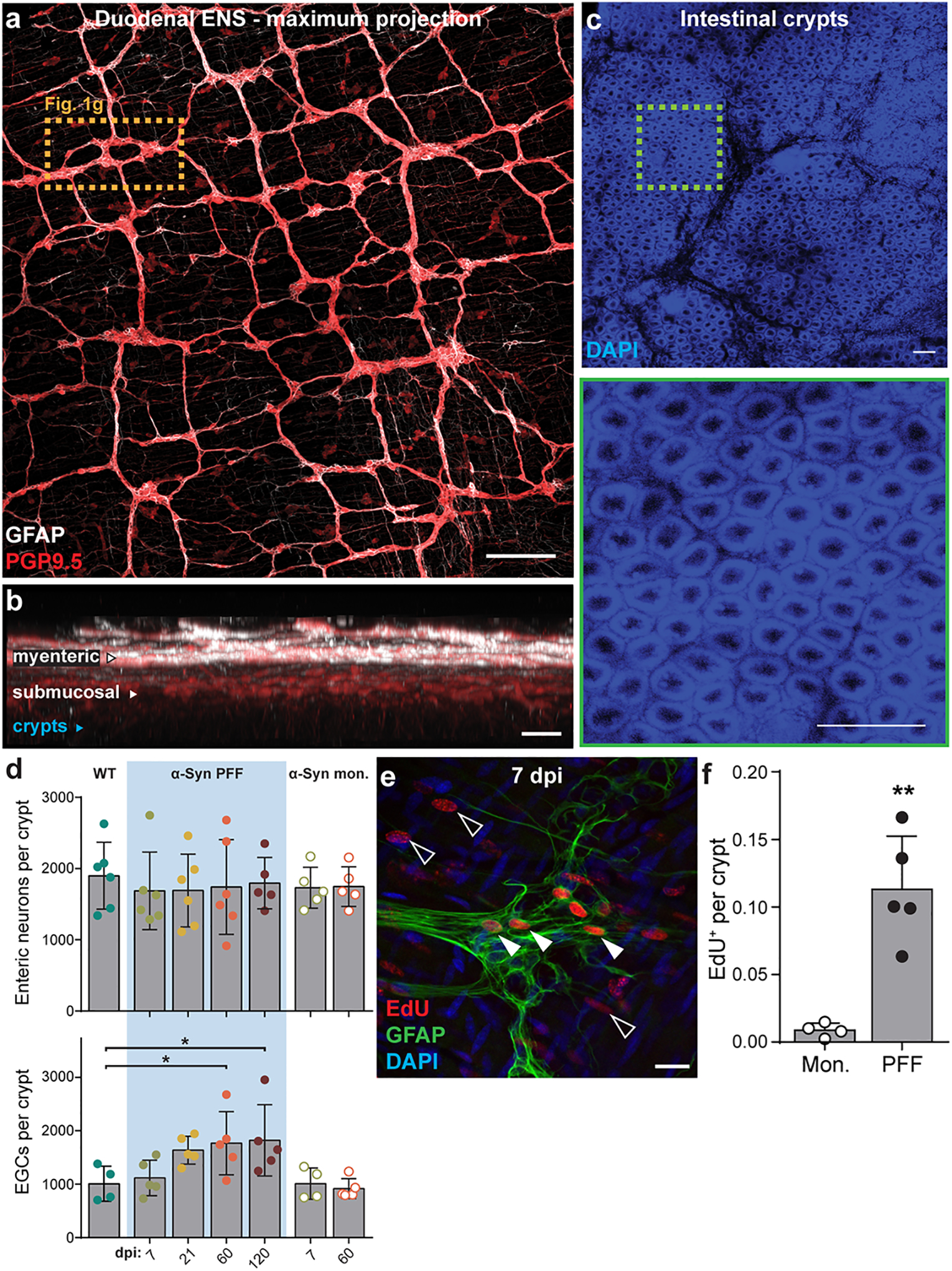
Fig. 1 |. Inoculation of duodenum with α-Syn PFF impacts the ENS and GI function.
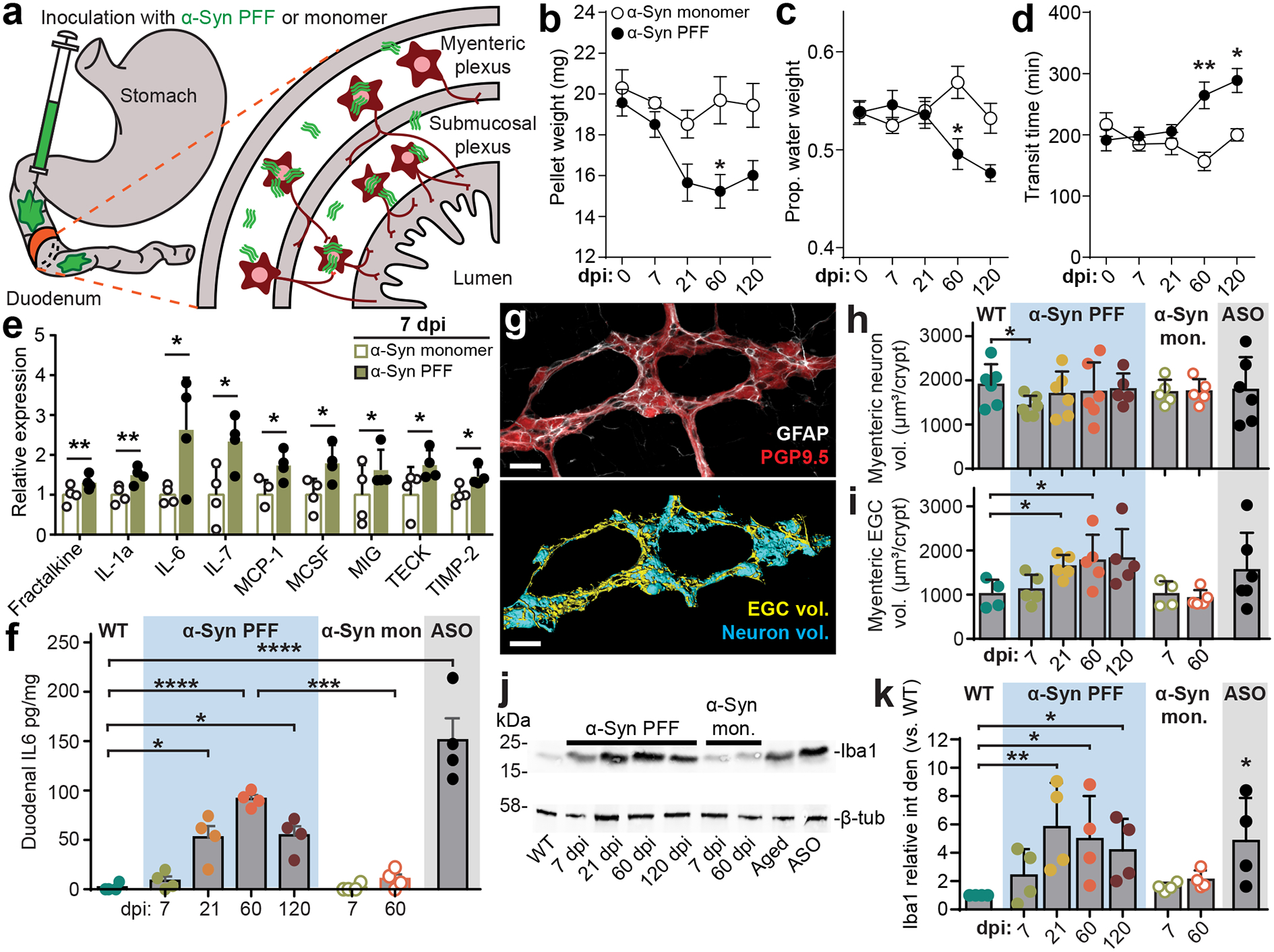
a, Schematic depicting intramuscular injection of the duodenum and interaction of injected material (α-Syn PFF, α-Syn monomer, BSA) with components of the intestinal wall, including the myenteric and submucosal plexus of the ENS, but not lumen. b-d, GI function following inoculation is characterized by determining fecal pellet weight (b; PFF vs. monomer, 60 dpi *p = 0.0187), proportion fecal water weight (c; PFF vs. monomer, 60 dpi *p = 0.0259), and whole gut fecal transit time (d; PFF vs. monomer, 60 dpi **p = 0.0012, 120 dpi *p = 0.0239). e, Densitometry analysis of cytokine expression from duodenal lysates 7 dpi with α-Syn PFF or α-Syn monomer. Values are normalized to the average of the α-Syn monomer group. (Fractalkine, **p = 0.0030; IL-1a, **p = 0.0070; IL-6, *p = 0.0415; IL-7, *p = 0.0400; MCP-1, *p = 0.0416; MCSF, *p = 0.0467; MIG, *p = 0.0412; TECK, *p = 0.0365; TIMP-2, *p = 0.0400). f, ELISA analysis of duodenal IL-6 levels (WT vs PFF: 21 dpi *p = 0.0366, 60 dpi ****p < 0.0001, 120 dpi *p = 0.0245; 60 dpi: PFF vs. monomer ***p = 0.0001; WT vs. ASO ***p < 0.0001). g, Immunohistochemical labeling of PGP9.5 (neurons) and GFAP (EGCs) visualizes myenteric ganglia (top). Three-dimensional renderings are then derived of neuronal and EGC volumes of the myenteric ganglion (bottom). The experiment was repeated in 44 independent mice, with similar results obtained. Scale bars, 30 μm. h-i, Quantification of digitally reconstructed duodenal myenteric neuronal (h; WT vs. PFF, 7 dpi *p = 0.0425) and EGC (i; WT vs. PFF, 120 dpi *p = 0.0232) volumes. j, Representative Western blots immunostained for the Iba1 microglial marker and β-tubulin control from duodenal lysates. k, Densitometry analysis of Iba1 expression from duodenal lysates, normalized to WT. ASO is statistically compared to WT (WT vs PFF, 21 dpi **p = 0.0039, 60 dpi *p = 0.0149, 120 dpi *p = 0.0467; WT vs. ASO *p = 0.0182). Data represented are mean ± s.e.m. P values were determined by two-way analysis of variance (ANOVA) (b-d), one-tailed Student’s t-test (e), or one-way ANOVA (f, h-i, k). The following n values represents number of independent animals for statistical evaluation: 1b and 1c, for 0 / 7 / 21 / 60 / 120 dpi: monomer = 9 / 9 / 9 / 8 / 8, PFF = 16 / 14 / 14 / 11 / 8; 1d, for 0 / 7 / 21 / 60 / 120 dpi: monomer = 6 / 7 / 6 / 6 / 6, PFF = 7 / 8 / 9 / 9 / 8; 1e, monomer and PFF = 4 each; 1f, all conditions = 4 each, except ASO = 5; 1h and 1i, WT = 6, ASO = 6, for 7 / 21 / 60 / 120 dpi: PFF = 6 / 6 / 5 / 5, for 7 / 60 dpi: monomer = 5 / 5; 1k, all conditions = 4 each.
To investigate the immediate physiological response to PFF seeding, we evaluated duodenal tissue lysates against a cytokine panel at 7 dpi and observed increased production of several pro-inflammatory cytokines (Fig. 1e, Extended Data Fig. 1g–h), including interleukin-6 (IL-6), which was found to be upregulated in colonic tissue from PD paients11. Additional analyses showed a time-dependent increase in duodenal IL-6 production after α-Syn PFF inoculation (Fig. 1f, Extended Data Fig. 1i). Because IL-6 has been demonstrated to promote enteric neuronal survival25, we next wanted to determine how PFF seeding impacts the ENS. Histological quantification revealed a transient decrease in neuronal volume within duodenal myenteric ganglia that recovered by 21 dpi (Fig. 1g–h, Extended Data Fig. 2a–c). To determine whether changes in volume were due to neuronal death followed by repopulation, we quantified counts of enteric neurons, however we did not observe a statistically significant decrease at 7 dpi (Extended Data Fig. 2d). Evaluation of myenteric EGCs revealed a prolonged elevation of EGC volume and an increase in EGC count over time following α-Syn PFF inoculation, indicating a reactive gliosis in response to fibril seeding (Fig. 1i, Extended Data Fig. 2d). To further understand the changes in EGC volume and cell count, we labeled with 5-ethynyl-2’-deoxyuridine (EdU), which revealed an immediate increase in cellular proliferation at 7 dpi following α-Syn PFF inoculation compared to monomer (Extended Data Fig 2e–f). While the majority of these cells were extragangliar (90.4 ± 1.9%), those that were located in myenteric ganglion were predominantly GFAP+ (91.2 ± 3.7%).
Activated EGCs, as well as enteric neurons, can mediate cytokine signaling in the gut to recruit immune cells6. Our cytokine screen also found a significant increase in fractalkine (Fig. 1e), which recruits macrophages that support enteric neurons26, and macrophage colony-stimulating factor (MCSF), which promotes macrophage differentiation and recruitment in the gut27. Subsequent analysis revealed a time-dependent increase in expression of the macrophage marker Iba1 in the duodenum following α-Syn PFF inoculation (Fig. 1j–k). Taken together, these results suggest that gut seeded α-Syn fibrils initiate an inflammatory response and activate immune cell-recruiting cytokines to maintain a healthy enteric neuronal network.
Duodenal α-Syn PFF inoculation increases α-Syn histopathology
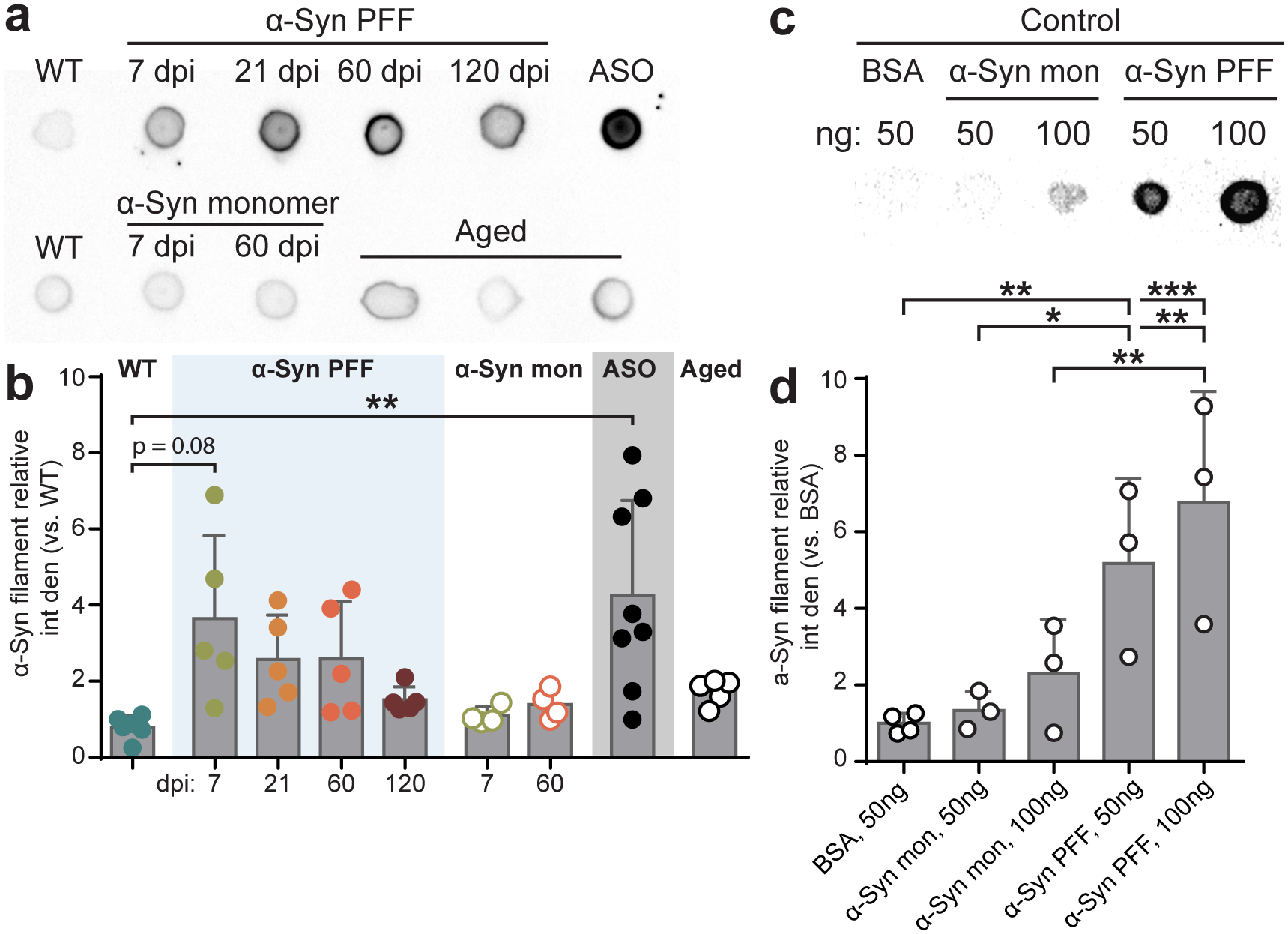
Duodenal inoculation with α-Syn PFFs induced an increase in α-Syn phosphorylation at the serine 129 (S129P) residue (Fig. 2a–d), which is a known marker of pathologic α-Syn accumulation20,28 that has neurotoxic properties whether present in soluble or insoluble aggregates29,30. We observed peak S129P signal at 60 dpi before declining at 120 dpi, suggesting that protein homeostasis mechanisms are engaged to counter the progression of α-Syn pathology. A similar trend was observed in immunoblots using a conformation-specific antibody that detects α-Syn filaments involved in fibril formation (Extended Data Fig 3). The lysosome is implicated in α-Syn homeostasis and enhanced lysosome response by increased GCase activity promotes the degradation of α-Syn aggregates18. Interestingly, pathologic α-Syn also inhibits GCase function17. Thus, we next determined the effect of PFF seeding on duodenal GCase. We observed a significant decrease in GCase production at 7 dpi, however this reversed at 21 dpi, and by 120 dpi GCase production was recovered to levels comparable to WT conditions (Fig. 2c,e).
Fig. 2 |. Pathologic α-Syn disrupts GCase and GI function and is ameliorated by GBA1 peripheral gene transfer.
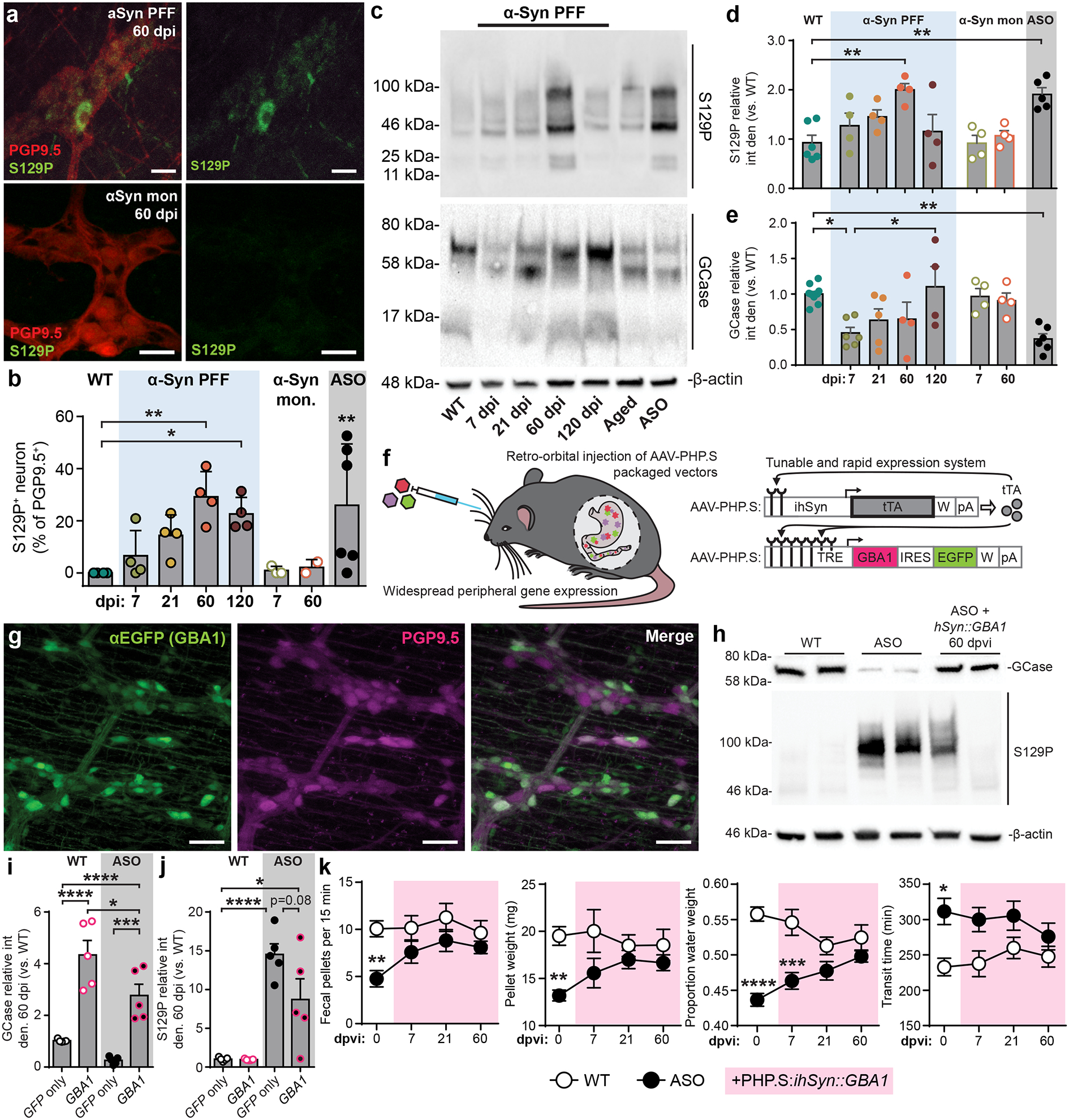
a, Immunohistochemical labeling of S129P detects p-α-Syn inclusions in PGP9.5+ myenteric neurons from α-Syn PFF (top) or α-Syn monomer (bottom) inoculated mice. Experimental images were obtained from 32 independent mice with two images taken per mouse, with similar results obtained. Scale bars, 20 μm. b, Quantification of proportion of S129P+ enteric neurons (myenteric and submucosal). ASO is statistically compared to WT (WT vs. PFF, 60 dpi ** p = 0.0026, 120 dpi *p = 0.0155; WT vs. ASO **p = 0.0032). c, Representative Western blots immunostained for S129P, GCase, and β-actin control from duodenal lysates. d-e, Densitometry analysis of GCase (d; WT vs. PFF, 60 dpi **p = 0.0042; WT vs. ASO **p = 0.0057) and S129P (e; WT vs. PFF, 7 dpi *p = 0.0202; WT vs. ASO **p = 0.0035) production from duodenal lysates. Values are normalized to WT. f, Schematic depicting two-vector scheme (ihSyn-tTA:TRE-GBA1) for rapid expression of GCase. Retro-orbital injection of AAV-PHP.S-packaged vectors that result in non-invasive, widespread delivery to peripheral organs. ihSyn – inducible human synapsin promoter, tTA – tetracycline-off transactivator, TRE – tet-responsive elements, IRES – internal ribosome entry site, W – woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), pA – human growth hormone polyadenylation (hghPA). g, Confocal image depicting efficiency of ihSyn-tTA:TRE-GBA1 vector system to transduce enteric neurons (PGP9.5). Experimental images were obtained from 6 independent mice, with similar results obtained. Scale bars, 50 μm. h, Representative Western blots immunostained for GCase, S129P, and β-actin control from duodenal lysates from virus-treated mice. i-j Densitometry analysis of GCase (i; WT/GFP vs. WT/GBA1 ****p < 0.0001; WT/GFP vs. ASO/GBA1 * p = 0.0192; ASO/GFP vs. ASO/GBA1 ***p = 0.0008; WT/GBA1 vs. ASO/GBA1 *p = 0.0410) and S129P (j; WT/GFP vs. ASO/GFP ****p < 0.0001; WT/GFP vs. ASO/GBA1 *p = 0.0139) expression from duodenal lysates 60 dpvi. Values are normalized to the WT/GFP only condition. k, GI function following peripheral GBA1 gene transfer is characterized by determining number of fecal pellets produced (0 dpi **p = 0.0078), fecal pellet weight (0 dpi **p = 0.0061), proportion fecal water weight (0 dpi ****p < 0.0001, ***p = 0.0003), and whole gut fecal transit time (0 dpi *p = 0.0123). Data depicted are mean ± s.e.m. P values were determined by one-way ANOVA (b,d-e) or two-way ANOVA (i-k). The following n values represents number of independent animals for statistical evaluation: 2b, all conditions = 4 each; 2d, all conditions = 4, except WT = 6, ASO = 5; 2e, WT = 11, ASO = 6, for 7 / 21 / 60 / 120 dpi: PFF = 6 / 5 / 4 / 4, for 7 / 60 dpi: monomer = 4 / 4; 2i, all conditions = 5 each; 2j, all conditions = 5 each; 2k for 0 / 7 / 21 / 60 dpi: WT = 17 / 12 / 11 / 8, ASO = 13 / 12 / 11 / 10.
GBA1 gene transfer partially rescues the pathologic α-Syn-induced GI phenotype
Our findings support the previously hypothesized positive feedback loop between pathologic α-Syn accumulation and decreased GCase function17 in the gut. Because we did not observe permanent pathologic GI effects of α-Syn PFF inoculation in adult WT mice, we evaluated GCase in the ASO mouse model due to its constitutive overproduction of α-Syn and chronically elevated p-α-Syn24. We also observed significantly decreased levels of duodenal GCase production in ASO mice (Fig. 2e), which display severe GI deficits31 (Extended Data Fig. 1a–d) and have elevated duodenal S129P (Fig. 2b,d). These findings highlight GBA1, the gene encoding GCase, as a therapeutic target for peripheral synucleinopathy. Mutations in GBA1 are among the most common risk factors for PD15 and gene transfer of GBA1 in the CNS of mice has been shown to reduce α-Syn pathology32.
To test whether GBA1 gene transfer can ameliorate peripheral synucleinopathology, we packaged a vector system that uses the tetracycline-off transactivator (tTA) to achieve rapid and robust neuronal expression of GCase (ihSyn-tTA:TRE-GBA1) in a novel viral capsid (AAV-PHP.S) that has high transduction efficiency for the peripheral nervous system and does not cross the blood brain barrier33,34 (Fig. 2f). Systemic delivery of the vector system into ASO mice transduced 49.6 ± 10.7% of enteric neurons and achieved long-term recovery of GCase production in the duodenum (Fig. 2g–h). Gene transfer of GBA1 also resulted in a reduction in duodenal p-α-Syn at 60 days post viral injection (dpvi) and partially recovered the GI phenotype observed in ASO mice (Fig. 2i–k, Extended Data Fig. 4). The incomplete functional recovery emphasizes the complexity of synucleinopathy pathology. Together, these results suggest that gut GCase helps reduce α-Syn pathology and recover gut motility.
ENS network connectivity is disrupted by α-Syn PFF inoculation and partially rescued by neuronal GBA1 gene transfer
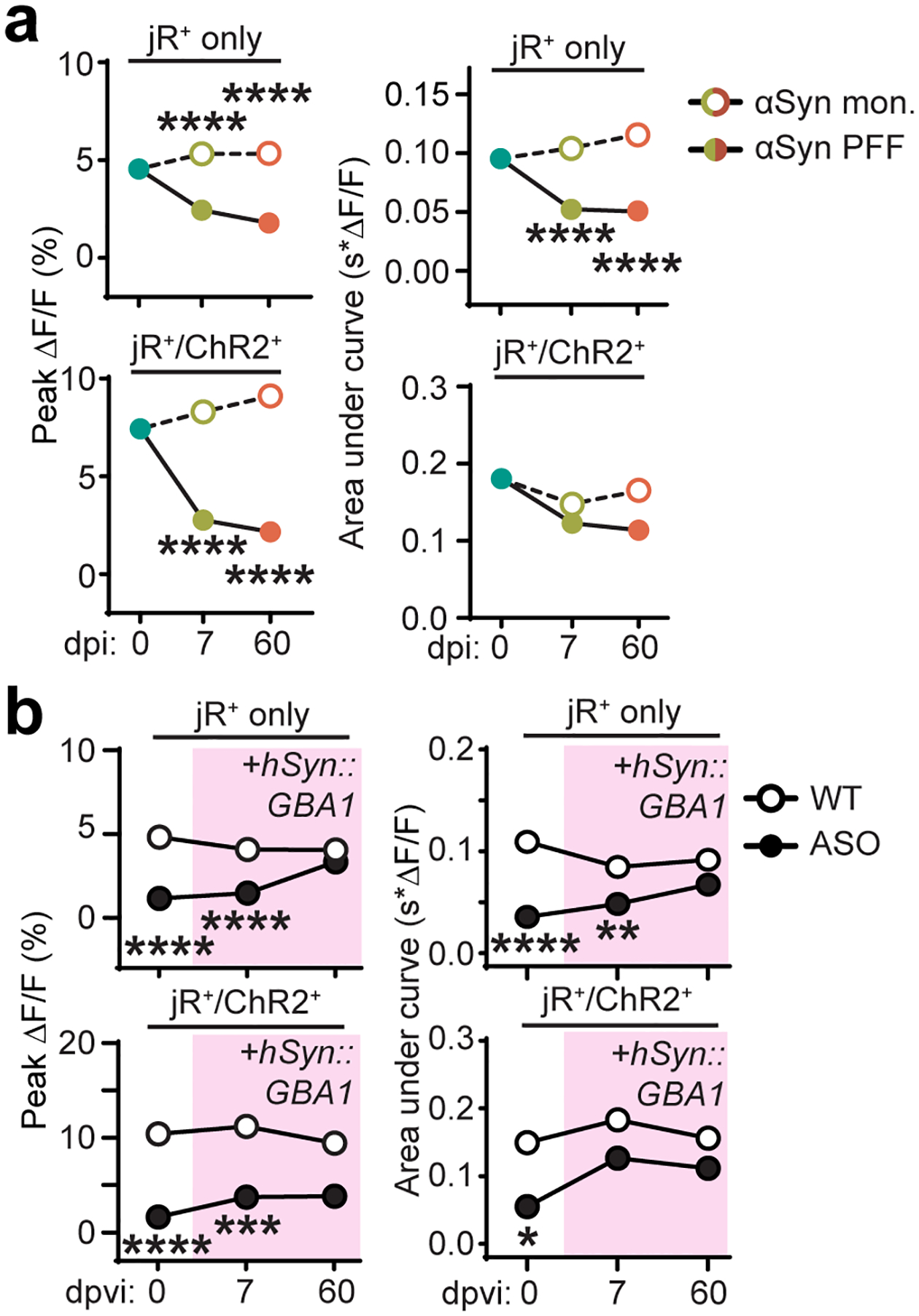
It is likely that pathologic α-Syn contributes to GI dysfunction by directly affecting ENS connectivity as α-Syn fibrils disrupt neurotransmission prior to significant accumulation35. To explore this, we used an optogenetic strategy to interrogate ENS functional connectivity. Unlike conventional electrophysiological ENS recording techniques, an optogenetic approach better maintains the integrity of the duodenal ENS in situ. We used AAV-PHP.S to deliver a tunable triple vector system where an inducer (ihSyn-tTA) drives expression of Channelrhodopsin (TRE-ChR2-EYFP) and the red-shifted calcium indicator jRGECO1a (TRE-jRGECO1a) (Fig. 3a). This strategy allowed us to activate a subpopulation of enteric neurons (i.e. ChR2-EYFP+ cells) while recording the response activity of individual cells within the ENS network (i.e. jRGECO1a+ cells). Calcium imaging has previously been used to image ENS activity36, however, controlled genetic expression of indicators and effectors in the ENS independent of transgenics has not been previously performed. We injected different viral doses of each vector to achieve abundant expression of jRGECO1a (41.6 ± 7.5% of PGP9.5+ neurons) and sparse expression of ChR2-EYFP (8.3 ± 2.9% of PGP9.5+ neurons) in the ENS for ex vivo recording (Fig. 3b–d and Supplementary Video 1). This ratio allowed us to investigate broad network activity in response to the activation of a small subset of enteric neurons as an indirect measure of ENS network health. Following α-Syn PFF seeding, the average response of jRGECO1a+-only duodenal neurons to sparse enteric network photostimulation was decreased, suggesting dysfunctional connectivity (Fig. 3e–i and Extended Data Fig. 5a). Our optogenetic strategy also allowed us to assess the firing properties of directly photoactivated enteric neurons (i.e. ChR2+/jRGECO1a+ cells), of which we observed a progressive decrease in the light-activated calcium response, indicating a reduced ability of these neurons to fire. Duodenal enteric neurons in ASO mice displayed a similar deficit in network connectivity (Fig. 3j). Following AAV-PHP.S-mediated GBA1 gene transfer in ASO mice we were able to partially recover enteric network connectivity, confirming the deleterious effect of α-Syn pathology on enteric neuronal physiology and potential for peripheral GBA1 as a therapeutic target (Fig. 3j and Extended Data Fig. 5b).
Fig. 3 |. ENS neurotransmission is disrupted by pathologic α-Syn and restored by GBA1 gene transfer.
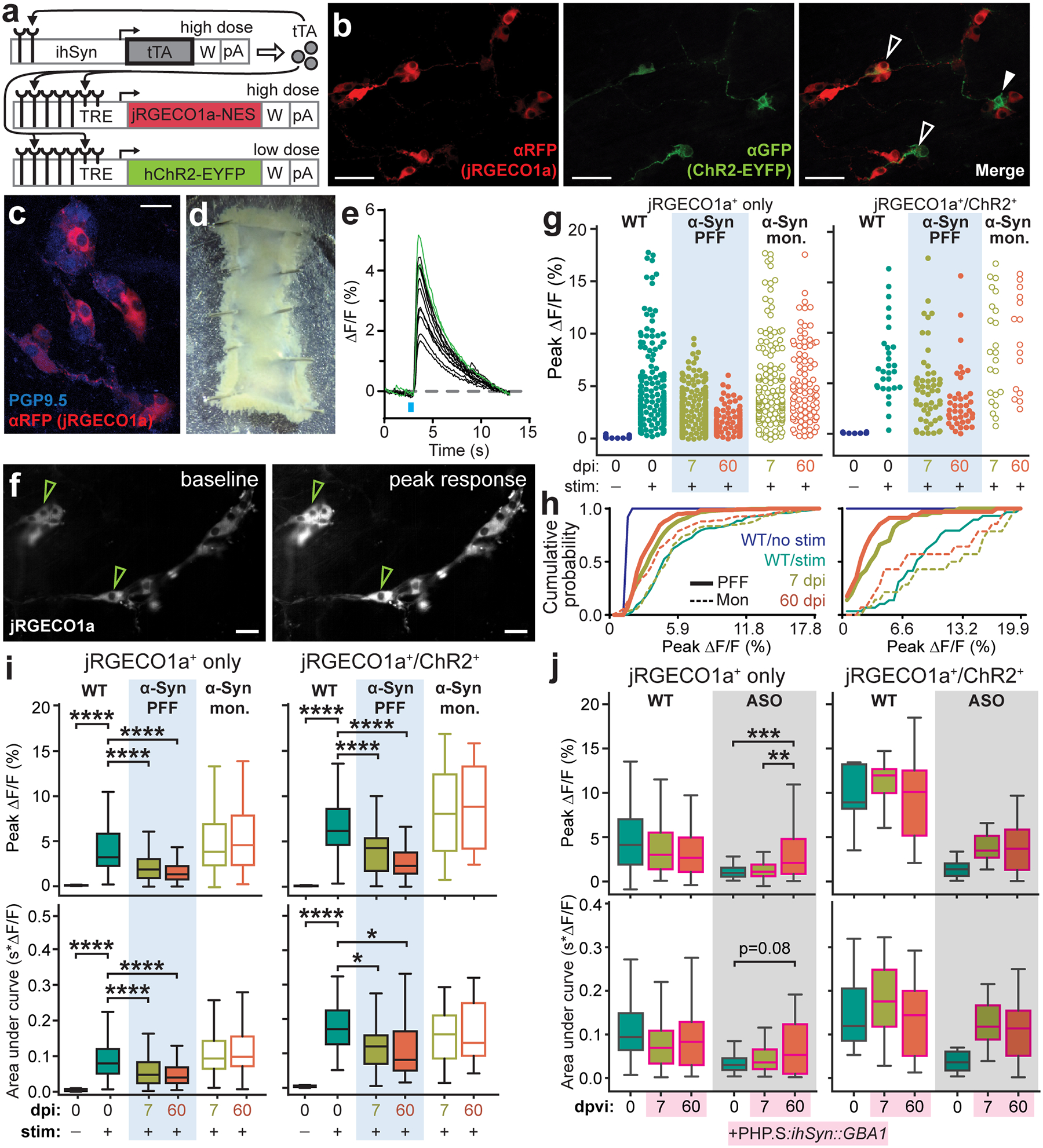
a, Triple vector scheme to express jRGECO1a calcium indicator and Channelrhodopsin (ChR2-EYFP) in peripheral neurons via packaging in AAV-PHP.S. b, Immunohistochemical labeling depicts transduction efficiency of vector system to express jRGECO1a (αRFP) and ChR2-EYFP (αGFP). Open arrows mark co-expression and closed arrow marks ChR2-EYFP expression alone. Experimental images were obtained from 6 independent mice, with similar results obtained. Scale bar, 50 μm. c, Immunohistochemical labeling depicts transduction efficiency of jRGECO1a (αRFP) in enteric neurons (PGP9.5). Experimental images were obtained from 6 independent mice, with similar results obtained. Scale bar, 25 μm. d, Representative image of an excised duodenal segment prepared for calcium imaging. e, Representative calcium trace plotted as percent change in fluorescence compared to pre-stimulation baseline fluorescence. Green traces indicate jRGECO1a+/ChR2+ neurons. f, Representative images of jRGECO1a fluorescence in duodenal enteric neurons recorded in (e) depicting baseline (left) and peak (right) calcium activity after the photostimulation pulse. Green arrows indicate jRGECO1a+/ChR2+ neurons. Experimental images were obtained from 6 independent mice, with similar results obtained. Scale bar, 50 μm. g, Peak percent change in fluorescence for individual duodenal jRGECO1a+-only or jRGECO1a+/ChR2+ neurons after photostimulation pulse. h, Cumulative probability plots of peak calcium responses shown in (g). i, Quantification of average peak percent change in fluorescence and area under the curve after photostimulation pulse for jRGECO1a+-only or jRGECO1a+/ChR2+ duodenal neurons before and after inoculation. Boxplots represent median, interquartile range, and 1.5× the interquartile range (AUC jRGECO1a+/ChR2+: WT vs. PFF, 7 dpi *p = 0.0488, 60 dpi *p = 0.0214; all ****p < 0.0001). j, Quantification of average peak percent change in fluorescence and area under the curve after photostimulation pulse for jRGECO1a+-only or jRGECO1a+/ChR2+ duodenal neurons before and after systemic delivery of AAV-PHP.S::ihSyn:GBA1. Boxplots represent median, interquartile range, and 1.5× the interquartile range (Peak ΔF/F jRGECO1a+: ASO, 0 vs. 60 dpvi ***p = 0.0007, 7 vs. 60 dpvi **p = 0.0068). P values were determined by one-way ANOVA (i) or two-way ANOVA (j). The following n values represents number of independent animals used for statistical evaluation: 3i, WT no stim = 2, WT stim = 3, PFF 7 dpi = 4, PFF 60 dpi = 3, monomer 7 dpi = 3, monomer 60 dpi = 3; 3j, all conditions = 3.
Duodenal α-Syn PFF inoculation in aged mice promotes progression of α-Syn histopathology to the brain
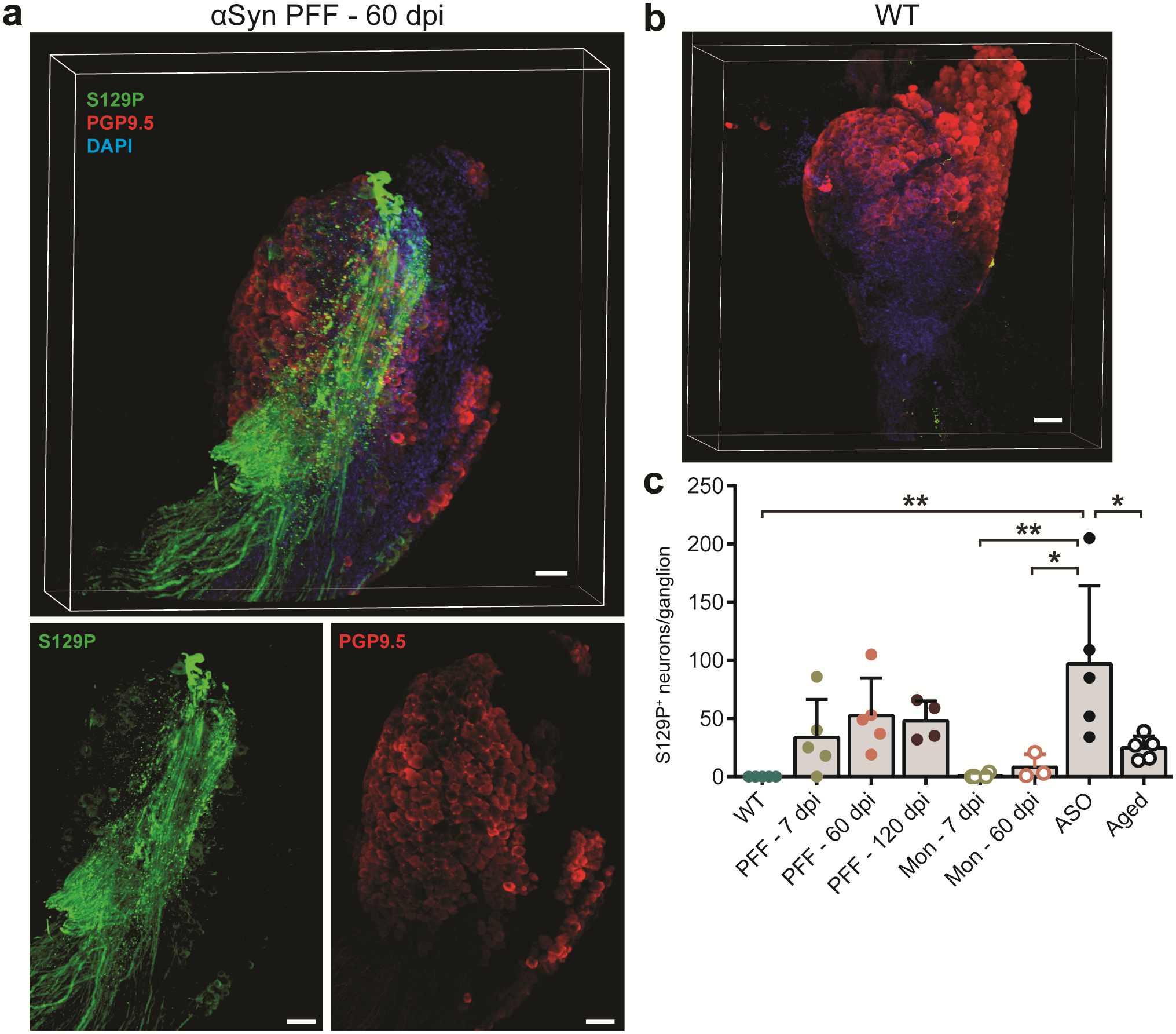
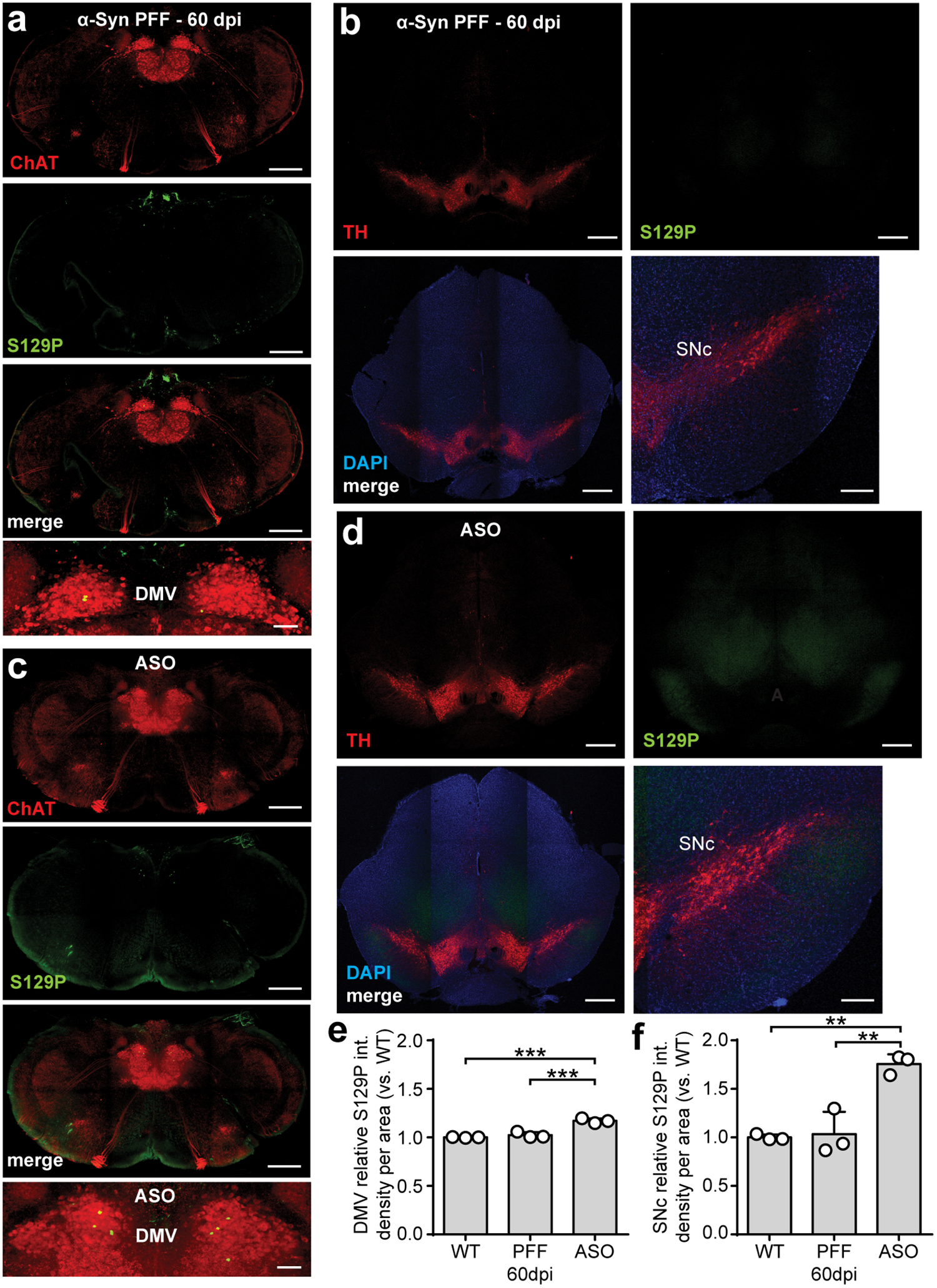
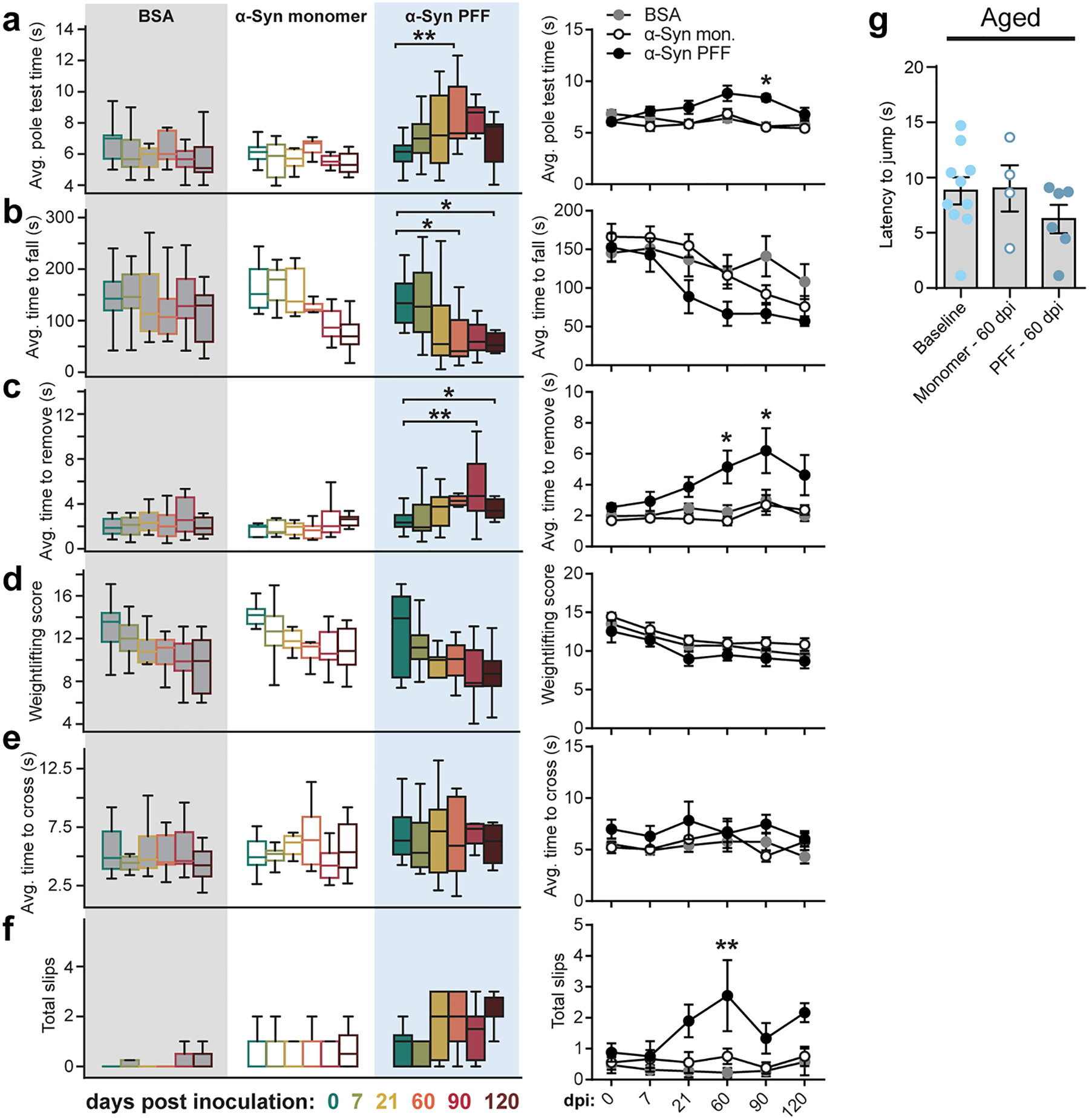
Finally, we determined if PFF-induced seeding of pathologic α-Syn in the duodenum resulted in progression of α-Syn pathology to the CNS. We first evaluated p-α-Syn in PACT cleared37 nodose ganglia, and though the number of S129P+ neurons had increased, this was not statistically significant compared to pre-inoculation (Extended Data Fig. 6). Likewise, we did not observe marked increases in p-α-Syn in the brainstem or SNc following duodenal α-Syn PFF inoculation (Extended Data Fig. 7a,c,e–f). This was in contrast to tissues from ASO mice, where p-α-Syn was more pronounced in each region (Extended Data Fig. 7b,d,e–f). We also characterized sensorimotor behaviors after α-Syn PFF inoculation, and did observe behavioral impairments in the adhesive removal, pole descent, and beam traversal paradigms at 60 dpi and 90 dpi in the PFF-inoculated cohort, however these effects reverted by 120 dpi (Extended Data Fig. 8a–f).
Given that α-Syn histopathology is not observed in the brain after duodenal PFF inoculation and that pathological progression peaked at 60 dpi, protein homeostasis mechanisms may be preventing gut-to-brain progression of α-Syn pathology. Aging is considered the principal risk factor for neurodegenerative disease as cellular mechanisms are unable to efficiently mitigate protein pathology38. In the duodenum of aged mice (16-month old) we observed decreased GCase production compared to younger adult WT mice (8–10 weeks old), and a statistically not significant decrease in levels of GBA1 transcript (Fig. 2c, 4a, and Extended Data Fig. 1f). These findings implicate a reduced capacity of GCase to degrade pathologic α-Syn in the duodenum of aged mice. Indeed, a greater proportion of the aged cohort presented with p-α-Syn+ duodenal enteric neurons compared to the younger cohort, though the quantified levels detected were intermediate to those observed in younger adults and ASO mice (Fig. 2c, 4b–c). The lack of significant difference in GI function suggests that a diminished capacity for α-Syn homeostasis alone does not result in GI pathology (Extended Data Fig. 1a–d).
Fig. 4 |. Inoculating the duodenum of aged mice with α-Syn PFF promotes progression of p-α-Syn to the brain.
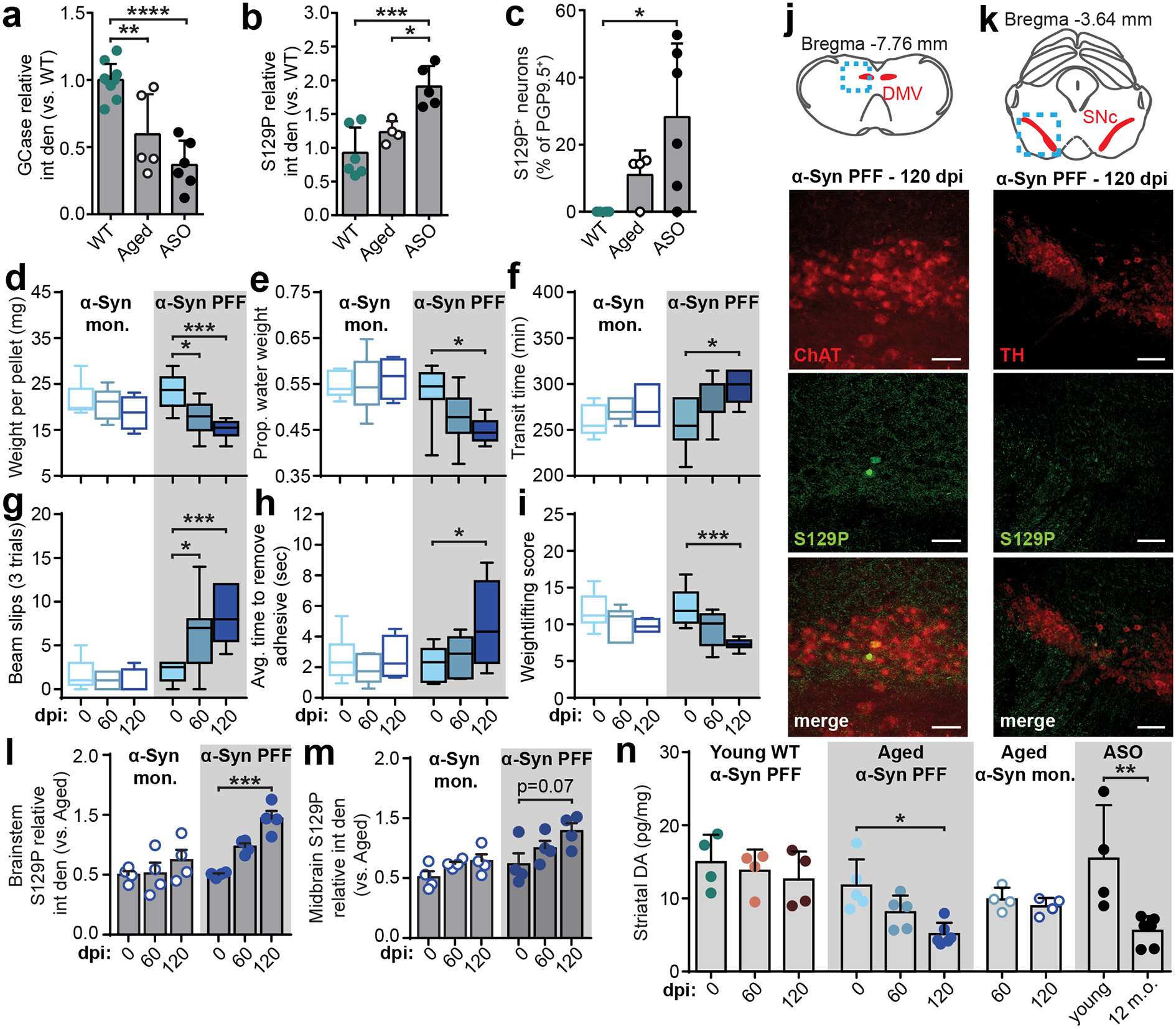
a, Duodenal GCase expression determined by densitometry analysis of Western blots from adult WT, aged, and adult ASO mice (WT vs. Aged **p = 0.0022, WT vs. ASO ****p < 0.0001). b-c, Duodenal α-Syn pathology determined by densitometry analysis of Western blots (b; WT vs. ASO ***p = 0.0006, Aged vs. ASO *p = 0.0202) and histological quantification of proportion of S129P+ neurons (c; WT vs. ASO *p = 0.0468). d-f, GI function in aged mice is characterized by fecal pellet weight (d; PFF 0 vs. 60 dpi *p = 0.0300, 0 vs. 120 dpi ***p = 0.0007), proportion fecal water weight (e; PFF 0 vs. 120 dpi *p = 0.0209), and whole gut fecal transit time (f; PFF 0 vs. 120 dpi *p = 0.0343). Boxplots represent median, interquartile range, and 1.5× the interquartile range. g-i, Sensorimotor performance in aged mice is characterized by weight-independent tasks: total slips during beam cross (g; PFF 0 vs. 60 dpi *p = 0.0110, 0 vs. 120 dpi ***p = 0.0002), average time to remove adhesive from nasal bridge (h; PFF 0 vs. 120 dpi *p = 0.0442), and weightlifting task (I; PFF 0 vs. 120 dpi ***p = 0.0006). Boxplots represent median, interquartile range, and 1.5× the interquartile range. j-k, Immunohistochemical labeling in aged mice of S129P in brainstem cholinergic (ChAT – choline acetyltransferase) neurons of the dorsal motor nucleus of vagus (DMV) (j) or midbrain dopaminergic (TH – tyrosine hydroxylase) neurons of the SNc (k) after α-Syn PFF inoculation 120 dpi. Experimental images were obtained from 8 independent mice, with similar results obtained. Scale bars, 50 μm. l-m, Densitometry analysis of S129P in the brainstem (l; PFF 0 vs. 120 dpi ***p = 0.0005) or midbrain (m). n, ELISA analysis of striatal dopamine levels (Aged PFF 0 vs. 120 dpi *p = 0.0487; ASO young vs. 12 m.o. **p = 0.0017). Data depicted are mean ± s.e.m. P values were determined by one-way ANOVA (a-c,n) or two-way ANOVA (d-i,l-m). The following n values represents number of independent animals used for statistical evaluation: 4d-i, for 0 / 60 / 120 dpi: monomer = 9 / 6 / 4, PFF = 10 / 7 / 6; 4l-m, all conditions = 4 each; 4n, ASO young = 4, ASO 12 m.o. = 6, for 0 / 60 / 120 dpi: WT = 4 / 4 / 4, Aged PFF = 5 / 5 / 6, Aged monomer = 0 / 4 / 4.
Based on the decrease in GCase production, we hypothesized that seeding α-Syn PFFs in the gut of aged mice would exacerbate α-Syn-induced pathology compared to younger mice. Duodenal inoculation with α-Syn PFFs in aged mice resulted in a decline in GI function (Fig. 4d–f) as well as sensorimotor deficits up to 120 dpi (Fig. 4g–i). We employed animal weight-independent paradigms, as aged mice are significantly heavier than adults (Extended Data Fig. 1e). Additional behavioral testing determined these deficits were not caused by a heightened pain response (Extended Data Fig. 8g). Next, we explored the gut-to-brain progression of α-Syn pathology and observed a significant increase in brainstem p-α-Syn at 120 dpi in α-Syn PFF-inoculated aged mice compared to pre-inoculation (Fig. 4j,l). However, the increase in the midbrain was not statistically significant and the number of tyrosine hydroxylase-positive cells was unchanged (Fig. 4k,m, Extended Data Fig. 9). Because neurotransmission deficits precede accumulation of pathologic α-Syn35,39, we evaluated total striatal dopamine levels to determine SNc activity40. We found a significant decrease in striatal dopamine in aged mice inoculated with α-Syn PFFs, but not in α-Syn PFF-inoculated younger WT mice nor in monomer-inoculated aged mice (Fig. 4n). At 120 dpi, striatal dopamine levels in α-Syn PFF-seeded aged mice were comparable to that observed in 12-month-old ASO mice, but not younger ASO mice, which further implicates age-related factors in the progression of α-Syn pathology.
DISCUSSION
Approximately 90% of PD diagnoses are idiopathic41, yet the factors that underlie pathogenesis remain unclear. Interactions between genetic and environmental factors, such as toxins, likely trigger pathogenesis by initiating α-Syn oligomerization, aggregation, and propagation42. This is supported by observations of α-Syn-containing inclusions in peripheral tissue from PD-patients and otherwise healthy individuals4,5. Understanding the impact of pathologic α-Syn on the peripheral nervous system is critical for developing novel diagnostic and therapeutic tools that target early synucleinopathy. Such tools could allow the progression of pathology to be halted early in disorders such as PD before the onset of motor symptoms. However, the progression and mechanisms of α-Syn pathology in the periphery has not been thoroughly characterized. In this work, we determined that α-Syn fibrils inoculated in the duodenum activate an inflammatory response and disrupt ENS physiology and GCase function. We also demonstrated age-dependent progression of α-Syn histopathology from the gut to the brain. Finally, increasing levels of GCase reduced α-Syn-induced pathology and functional deficits, suggesting viral-based gene transfer as a potential therapeutic strategy for idiopathic PD. Our results propose mechanisms that may underlie the etiology of sporadic PD and highlight GBA1 as a therapeutic target for prodromal, peripheral synucleinopathy.
Our work reinforces the role of the lysosomal enzyme GCase as a critical regulator of α-Syn pathology in the periphery. It is known that oligomeric α-Syn disrupts lysosomal trafficking17, however the comorbidity of Gaucher’s disease and PD is only 10%43, suggesting that GCase deficiency alone may not be sufficient to initiate α-Syn aggregation. Here, we show that GCase production is significantly reduced in aged mice, which correlates with an elevation in p-α-Syn (Fig. 4a–c). Interestingly, the average age of PD symptom onset in Gaucher’s patients is up to 6 years earlier than in idiopathic PD patients alone44. Thus, a proposed mechanism for PD pathogenesis is a feedforward loop between GCase and α-Syn that results in a gradual accumulation of amyloid α-Syn and diminution of GCase until a pathological threshold is crossed17. The lower levels of duodenal GCase we observed in the aged mice cohort may implicate a susceptibility to the progression of α-Syn pathology following gut α-Syn fibril seeding. Whether this interaction occurs in the GI tract has not been determined. Colonic dysmotility has been observed in a double mutant mouse line (hSNCAA53T/GBAL444P/+), however protein interactions within the tissue were not described16. Here, we reported that duodenal α-Syn PFF inoculation decreases GCase production (Fig. 2e), which may be a contributing factor in the peripheral progression of synucleinopathy. We did observe recovery of GCase production at later time points, suggesting that otherwise healthy adult mice possess some resilience to pathological insults. It may be likely that the α-Syn PFF dose with which we chose to inoculate allowed this effect to be observed, as other studies that utilized much higher amounts for inoculation reported more prominent pathology9,10. That we see diminished baseline GCase production in the duodenum of aged mice suggests that an aged population may be more readily susceptible to α-Syn pathology in the GI tract.
In the duodenum of ASO mice, we observed similar pathology to that of α-Syn PFF-inoculated mice, demonstrating the utility of the ASO mouse line in studying peripheral synucleinopathy. Additionally, because the effects of PFF inoculation reverted after 60 dpi, we believed the chronicity of α-Syn-induced pathology in ASO mice represented a better opportunity to study peripheral GBA1 gene transfer as a therapeutic intervention. Indeed, we were able to use systemic delivery of AAVs packaged with the AAV-PHP.S capsid33 to produce broad and uniform non-invasive peripheral gene transfer in ASO mice that recovered pathologic α-Syn-induced GI dysfunction (Fig. 2f–k). This is in line with previous studies that found that GBA1 gene transfer reduced levels of pathologic α-Syn in the brains of A53T transgenic mice32,45. We were unable to completely restore GI function, indicating that α-Syn pathology targets multiple mechanisms or that in the ASO mouse line, genetic intervention must occur before α-Syn accumulates beyond an irreversible pathologic threshold. An effective therapy may require combinatorial treatments, such as the inclusion of GCase chaperone proteins, and precise timing with respect to progression of pathology. Additionally, our strategy increased GCase production solely in peripheral neurons; it may be necessary to target additional cell types such as EGCs and macrophages for a greater effect. It is also possible that GCase function in the CNS plays a role in gut homeostasis.
After duodenal α-Syn PFF inoculation, we observed a disruption in ENS connectivity and activation of an inflammatory response, both of which may serve as indicators of prodromal PD. The role of inflammation in synucleinopathies is complex, though evidence suggests it is an initiating or propagating factor rather than protective46. In the ENS, cytokines can be released by EGCs47, and a positive correlation between pro-inflammatory cytokines and glial markers was observed in colonic biopsies from PD patients11. Our findings are in line with these studies and support α-Syn fibril-mediated increases in EGC tone and inflammation (Fig. 1e–k). Regarding synaptic connectivity, studies have shown that dopaminergic signaling from the SNc is diminished in early synucleinopathy, prior to the formation of Lewy aggregates35,39. We corroborated these findings as we observed decreased striatal dopamine in aged mice without significantly increased p-α-Syn in the SNc following α-Syn PFF inoculation in the duodenum (Fig. 4m–n). The effect of pathologic α-Syn on ENS connectivity is not as well understood. Here, we show that α-Syn PFF inoculation decreased ENS network signaling (Fig. 3g–i), which likely played a role in the accompanying dysfunctional GI phenotype (Fig. 1b–d). Because α-Syn pathology is capable of progressing transsynaptically, it may also be likely that propagation to innervating autonomic fibers and the distal GI tract is responsible for altered nutrient and water homeostasis, leading to the observed change in fecal composition. The presence of α-Syn aggregates may also cause dysfunction in enteroendocrine cells as these cell types also produce α-Syn and modulate gut motility and absorption48. These peripheral measurements may be valuable diagnostic markers the for early detection of synucleinopathies.
In this work, we validated previous studies by showing that duodenal α-Syn PFF inoculation in adult mice advanced pathology towards the CNS. However, in younger mice, we observed termination of this progression in the brainstem (Extended Data Fig. 7). Findings from Uemura et al., mirror our own as they observed an initial increase in brainstem pathology at 45 dpi followed by a steady decline at 4, 8, and 12 months post-injection9. More recent work by Kim et al., published in June 2019, found that intramuscular injection of α-Syn PFF in the stomach and duodenum resulted in significant midbrain pathology and sensorimotor deficits at 7 months post-injection10. Major differentiating factors between these studies and ours are different injection locations (duodenum alone in our work; stomach alone in Uemura et al.; upper duodenum and pyloric stomach in Kim et al.), the time points observed (4 months post-inoculation in our work; 12 months post-inoculation in Uemura et al.; 10 months post-inoculation in Kim et al.), and, importantly, the amount of α-Syn PFF injected into the GI wall. While Kim et al. introduced 25 μg of material, we selected a modest 6 μg injection quantity to ask how a small “seeding” dose would specifically affect enteric physiology, and whether it was sufficient to promote gut-to-brain progression of α-Syn pathology. Comparing our results from younger adult and aged mice suggests that as the protein homeostatic function in the gut declines during aging49, so does the ability to eliminate α-Syn aggregates, promoting CNS pathology and sensorimotor deficits (Fig. 4). Interestingly, Uemura et al., introduced 48 μg of α-Syn PFF that did result in brainstem α-Syn pathology, but not a further caudo-rostral progression. A possible factor underlying this discrepancy is their selection of the stomach for inoculation, which has a much larger surface area versus the duodenum and differential innervation by autonomic fibers both compared to the duodenum and between different gastric regions50. The parameters we chose for this study allowed us to better understand the dynamic relationship between α-Syn and GCase in the peripheral nervous system.
In summary, our findings suggest that age-related declines in protein homeostasis, including diminished GCase function, may promote susceptibility to α-Syn pathology in the ENS and support the gut-to-brain hypothesis of synucleinopathy etiology. Understanding vulnerabilities in peripheral systems to pathologic α-Syn will advance the development of early detection techniques and therapeutics. Towards this, we demonstrated that α-Syn fibrils directly disrupt ENS connectivity, which may signify a physiological signature of prodromal synucleinopathy. We also highlighted the potential of using AAV-mediated peripheral gene transfer for intervention by restoring GCase expression in enteric neurons. Taken together, our work shifts the focus of neurodegenerative disease etiology to the peripheral nervous system and expands our understanding of the role the ENS plays in prodromal synucleinopathy.
METHODS
Animals.
Wild type (WT) C57BL/6N mice were obtained from Charles River (Hollister, CA) and bred in-house. Adult mice were 8–10 weeks old. Aged mice were WT mice allowed to age to 16 months before experimental manipulation or C57BL/6N mice obtained through the NIH/NIA Aged Rodent Colony. To generate Thy1-α-synuclein overexpressing (ASO) mice, female BDF1/Thy1-ASO animals heterozygous for the Thy1-α-Syn transgene on the X chromosome were crossed with WT male BDF1 mice24,51. Male BDF1 were bred by crossing C57BL/6N females with DBA/2 males (Charles River). Unless otherwise stated, ASO mice used in experiments were 6 months old. Male mice were used in all experiments because of insertion of the Thy1-α-Syn transgene into the X chromosome, which results in significantly masked phenotype and pathology24. Animals were housed on a reverse light-dark cycle. Care and experimental manipulation of animals were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Caltech Institutional Animal Care and Use Committee. For each time course experiment, 1–3 cohorts of animals were used, with littermates randomized to the appropriate groups prior to surgical manipulation. Mice from each condition were chosen at random and sacrificed at the indicated time points to collect tissue for analysis and comparison. Data collection and analysis were not performed blind to the conditions of the experiments.
Tests of gastrointestinal and sensorimotor function.
All assessment was performed between hours 7 and 9 of the dark cycle in laminar-flow biosafety cabinets within the animal facility. Tests were performed over 2 days. On day one, the fecal output, beam traversal, pole descent, and wire hang tests were performed. On day two, the weightlifting, adhesive removal, hind limb tests, and whole gut transit assay were performed. Animals were sacrificed immediately after the last test for tissue collection.
Fecal output was performed by placing mice in covered 12 cm diameter × 25 cm height translucent cylinders for 15 min. Following conclusion of the test, fecal pellets were blotted on paper towels to absorb urine that may have been expelled during the task. Pellets were weighed, desiccated at room temperature over 3 days, then reweighed to determine total weight and water weight per pellet.
Whole gut transit assay was performed as previously described52.
Beam traversal was performed as previously described53 with minor modifications. Animals were trained over 2 days and 8 trials. Mice in the first 2 training trials were guided along the beam by placing the home cage close to the position of the mouse and encouraged to traverse by experimenter intervention. By the end of training, animals were adept beam traversal and did not require additional training during time course experiments.
Pole descent was performed by placing a 0.5 m long, 1 cm diameter ring stand vertically in the animal’s home cage. The pole was covered in rubber shelf liner to facilitate grip. Mice were trained over 6 trials. For the first 3 training trials, animals were placed on the pole, head towards the cage at 10 cm, 20 cm, and 30 cm heights above the cage floor and allowed to navigate back to the home cage independently. For the remaining 3 training trials, mice were placed on the rod with heads directed toward the ceiling and allowed to turn and descend. Mice were considered trained when, upon being placed on the pole, they immediately turned and descended. If this criterion was not met, additional training trials were performed with assistance from the experimenter to encourage descent. The next day, animals performed 3 trials in a row with 1 min between trials.
Inverted wire hang was performed as previously described54 with minor modifications. Animals were placed on a steel mesh screen and timed until they released their grip or remained on the screen for 5 min. Mice performed 3 trials, allowing at least 15 min between each trial.
Weightlifting paradigm was performed as previously described54.
Adhesive removal was performed as previously described53. Mice performed 2 trials with 5 min between each trial.
Hot plate test.
Evaluation of nociception was adapted from previous work55,56. Mice were habituated to the unheated hot plate apparatus for 5 min. Hot plate was then heated to 55°C and verified by probe thermometer. Mice were placed one at a time into the chamber, timed, and recorded for playback. Each trial initiated when all paws touched the surface. Trial was terminated and time recorded upon first occurrence of the following endpoint behaviors: hindpaw licking, vocalizing, or jumping. A cutoff time of 30 sec was established as the endpoint in the event the mouse did not exhibit these behaviors.
α-Syn PFF and monomer preparation.
Purification of recombinant, untagged mouse α-Syn and PFF generation was performed as previously described20. α-Syn monomers were subjected to a Pierce Limulus Amebocyte Lysate (LAL) high capacity endotoxin removal resin. Levels of endotoxin were measured to be 0.017 Units/μg of protein as determined using a Pierce LAL endotoxin quantitation kit.
Duodenal intramuscular injections.
Mice were anesthetized with 1–4% isoflurane and placed in a supine position on a self-regulating heating pad. The hair over the abdomen was removed and a 2 cm incision was made along the midline. The duodenal intestinal lining was directly injected at 2 sites, 1 cm apart, with a 10 μl Hamilton syringe equipped with a 36 GA beveled needle (World Precision Instruments, Sarasota, FL). Each site was injected with 3 μl of saline containing 1 μg/μl of protein (α-Syn PFF, α-Syn monomer, or BSA). Injection into the gastric lining was confirmed with 0.2% Fast Green FCF (Sigma-Aldrich, F7252). Once injections were made, the duodenum was carefully replaced and the skin was sutured. Mice were sacrificed at 7, 21, 60, and 120 days post inoculation.
Duodenal histology.
Prior to sacrifice, mice were injected intraperitoneally with 200 units of heparin in saline. After 15 min, mice were anesthetized with pentobarbital and transcardially perfused with cold phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. The gastrointestinal tract was removed and post-fixed for 12–16 h in 4% PFA at 4°C, then washed and stored in PBS with 0.05% sodium azide. A 2 cm segment of the duodenum was excised just after the pylorus and cleared of connective tissue. Antigen retrieval was performed by boiling the tissue in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0) at 95–100°C for 20 min followed by a 10 min wash under running tap water at room temperature. Two lengthwise cuts were made on each duodenum, one along the mesentery and the other along the opposite side, and the two pieces were blocked using the Mouse on Mouse kit (Vector Labs, Burlingame, CA) diluted in PBS with 0.1% Triton X-100 (TX100). Whole duodenal sections were incubated in primary antibodies (Supplementary Table 1) diluted in PBS with 3% normal donkey serum (NDS) and 0.1% TX100 at room temperature overnight on an orbital shaker. Individual duodenal pieces were washed in 4 × 10 ml PBS buffer exchanges across 6 h before incubation in secondary antibodies diluted in PBS with 3% NDS at room temperature overnight on an orbital shaker. Duodenal pieces were again washed in 4 × 10 ml PBS buffer exchanges for 6 h and then incubated in a refractive index matching solution (RIMS) overnight at room temperature on an orbital shaker. Tissue was mounted serosa facing up on a slide affixed with a 0.5 mm spacer (SunJin Labs, Hsinchu City, Taiwan) and chamber filled with additional RIMS, and coverslipped for confocal imaging (LSM 880, Zeiss, Oberkochen, Germany).
EdU staining.
Adult mice received a single intraperitoneal injection of 5-Ethynyl-2′-deoxyuridine (EdU, Sigma Aldrich, 900584) at a concentration of 100 mg/kg, 24 hours after inoculation with α-Syn PFF or monomer. At 7 dpi, mice were sacrificed and tissue was fixed as described above. Incorporation of EdU was visualized using Click-iT EdU Cell Proliferation Kit for Imaging (ThermoFisher, C10337).
Nodose ganglia tissue clearing and histology.
Nodose ganglia were removed from transcardially-perfused mice and podst-fixed for 12–16 h in 4% PFA at 4°C. The passive CLARITY technique (PACT) was performed on nodose ganglia as previously described37. Overnight incubations in 3% NDS and 0.1% TX100 in PBS at room temperature were performed for blocking and primary antibody (Supplementary Table 1) steps. Ganglia were washed in 3 × 10 mL PBS over 2 h before incubation in secondary in PBS with 3% NDS. Ganglia were again washed in 3 × 10 ml PBS over 2 h before incubated in RIMS overnight at room temperature on an orbital shaker. Ganglia were mounted on a slide affixed with a 0.5 mm spacer and chamber filled with additional RIMS, and coverslipped for confocal imaging.
Brain section histology.
Brains were extracted from transcardially perfused mice and post-fixed in 4% PFA in PBS for 16–20 h at 4°C. Serial sections 70–80 μm thick were cut in sets of 4 on a vibratome (Leica Biosystems, VT1200, Wetzlar, Germany) in PBS. For each of the brainstem or substantia nigra, three sections were mounted on Superfrost slides and allowed to dry for antibody labeling. Staining was performed directly on the slides in a slide moisture chamber. Slides were blocked with 3% NDS and 0.1% TX100 in PBS for 2 h at room temperature and incubated in primary antibody (Supplementary Table 1) in the same buffer overnight at 4°C. Slides were washed 3× with PBS and then incubated with secondary antibodies diluted in PBS with 3% NDS. Slides were again washed 3× in PBS, mounted with Vectashield (Vector Labs), and coverslipped for confocal imaging.
Stereological quantification.
The number of TH-positive cells in midbrain sections was determined manually from maximum projection confocal images. Boundaries of the SNc were determined by TH signal threshold detection using ImageJ and manually excluding non-SNc regions within the section. Density was determined by quantifying number of cells per SNc volume (area multiplied by the thickness of the slice).
Tissue lysates and protein quantification.
Tissues were placed in RIPA buffer (Millipore) diluted in PBS with a protease inhibitor cocktail (Thermo Fisher, A32963) and homogenized in 2 ml Lysing Matrix D tubes (MP Biomedicals, Santa Ana, CA) using a Mini-Beadbeater (Biospec, Bartlesville, OH). Homogenates were centrifuged and the protein concentration of the supernatant was determined with a BCA assay (Thermo Fisher). The cyotkine array (Abcam, ab133999) was performed according to manufacturer’s instructions using 250μg of total protein from fresh lysate. ELISA assays for interleukin-6 (IL-6) (Thermo Fisher) and dopamine (Eagle Biosciences, Nashua, NH) were performed according to manufacturers’ instructions from fresh lysate. For Western blots, 35 μg of protein from fresh or previously frozen lysate was separated by 4–20% SDS-PAGE (Bio-Rad, Hercules, CA) and transferred to PVDF membranes (Bio-Rad). For dot blot quantification of α-Syn filament, 1 μg of tissue homogenate was spotted in 1 μL volume aliquots onto 0.45 μm nitrocellulose membrane. Tissue from 1–2 WT mice was always included for intra-membrane comparisons. On orbital shakers, membranes were blocked with 5% blocking solution (Bio-Rad) in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 2 h at room temperature then incubated with primary antibodies (Supplementary Table 1) in TBS-T at 4°C overnight. Membranes were washed 3× in TBS-T and incubated with fluorescent or horseradish peroxidase (HRP)-conjugated secondaries in TBS-T for 2 h at room temperature on an orbital shaker. Membranes were again rinsed 3× in TBS-T. A Bio-Rad ChemiDoc MP was used to detect fluorescence or HRP-conjugated (via Clarity Max chemiluminescent substrate [Bio-Rad]) secondaries. Densitometry analysis was performed in ImageJ or Image-Lab (Bio-Rad), as described below.
RNA quantification.
Duodenal samples in RNAlater were homogenized in TRI Reagent (Zymo Research) and a Tissue LyserII (Retsch) and RNA was isolated with DirectZol RNA extraction kit (Zymo Research) via manufacturer’s instructions. cDNA was synthesized using a RevertAid first strand cDNA synthesis kit (Thermo Fischer Scientific) according to manufacturer’s instructions.
Real-time quantitative PCR was performed using SybrGreen qPCR master mix (Applied Biosystems) on an ABI 7900 Prism qPCR instrument. cDNA was probed with the following primers from Harvard PrimerBank: GAPDH – Forward 5′ TGG CCT TCC GTG TTC CTA C 3′ Reverse 5′ GAG TTG CTG TTG AAG TCG CA 3′; GBA1 – Forward 5′ GCC AGG CTC ATC GGA TTC TTC 3′ Reverse 5′ CAC GGG GTC AAG AGA GTC AC 3′. Target genes were normalized to GAPDH and fold change calculated by ΔΔCT relative to adult control samples. Data is graphed as the average of technical triplicates.
Viral constructs.
The pAAV-ihSyn-tTA construct was developed in the Gradinaru lab33 and is available on Addgene (99120, Cambridge, MA). The pAAV-TRE-GBA1-IRES-EGFP, pAAV-TRE-jRGECO1a-NES, and pAAV-TRE-hChR2-EYFP plasmids were cloned as described below. The pAAV-TRE-EGFP was obtained from Addgene (89875). All viruses were packaged with the AAV-PHP.S capsid (pUCmini-iCAP-PHP.S, Addgene, 103006) in-house, as previously described34.
pAAV-TRE-GBA1-IRES-EGFP was created by isolating the GBA1 gene transcript variant 1 (NM_008094) from a plasmid obtained from Origene (MC200608, Rockville, MD) via restriction digest with AsiSI and BsiWI. The IRES-EGFP site was PCR cloned to add 5’ BsiWI and 3’ EcoRI sites from a plasmid obtained from Addgene (20672). The TRE promoter was PCR cloned from an Addgene plasmid (99113) to add a 5’ XbaI and 3’ AsiSi restriction sites. The pAAV plasmid backbone from pAAV-ihSyn-tTA (Addgene, 99120) was restriction digested with XbaI and EcoRI. The pAAV backbone, TRE promoter, GBA1 gene, and IRES-EGFP insert were ligated using T4 DNA Ligase (M0202L, New England BioLabs [NEB], Ipswich, MA) and transformed into NEB Stable competent cells (C3040H, NEB). We also created pAAV-TRE-GBA1 for use in calcium imaging experiments by PCR cloning a WPRE site to add a 5’ BsiWI site and retaining the 3’ AfeI site. We digested pAAV-TRE-GBA1-IRES-EGFP with BsiWI and AfeI and inserted the cloned WPRE fragment to remove the IRES-EGFP sequence. The plasmid products were restriction digested and sequence verified before use in viral preparations. The following primers were used: TRE: forward 5’-ATCTAGATTACTTGTACAGCTCGTCCATGC-3’ and reverse 5’-TCAGTCGCGATCGCAGGCTGGATCGGTCC-3’; IRES-EGFP forward 5’-ACGTACGGGATCAATTCCGCC-3’ and reverse 5’-ACTGGAATTCTTACTTGTACAGCTCGTCCATGC-3’; WPRE forward 5’-ACGTACGGCTTATCGATAATCAACCTCTGG-3’ and reverse 5’-ACTGACAGCGCTCGGTATCGAT-3’.
pAAV-TRE-jRGECO1a and pAAV-TRE-ChR2-EYFP were constructed by digesting pAAV-TRE-mTurquoise2 (Addgene, 99113) with KpnI and EcoRI and the pAAV-TRE backbone was isolated. jRGECO1a-NES was PCR cloned out from an Addgene plasmid (61563) to add a 5’ KpnI site and a 3’ EcoRI site. A plasmid containing ChR2-EYFP was obtained from Addgene (26973) and digested with KpnI and EcoRI to isolate the ChR2-EYFP fragment. Plasmids were constructed by ligation of the inserts into the pAAV-TRE backbone using T4 DNA Ligase (NEB) then transformed into NEB Stable competent cells (NEB). The plasmid products were restriction digested and sequence verified before use in viral preparations. The following primers were used to amplify jRGECO1a-NES: forward 5’-CAGGTACCATGCTGCAGAACGAGCTT-3’ and reverse 5’-ACTGGAATTCCTACTTCGCTGTCATCATTTGT-3’.
AAV-PHP.S production and intravenous administration.
AAV packaging, purification, and delivery were performed as previously described34. Intravenous administration of AAVs was performed by injection into the retro-orbital sinus of mice. For in vivo GCase rescue experiments, 3.5 × 1011 GC (genome copies) of both AAV-PHP.S:ihSyn-tTA and AAV-PHP.S:TRE-GBA1-IRES-EGFP were delivered. For in vitro GCase rescue experiments (in HEK293T), 1 × 109 GC of virus was applied to each 6 cm dish. For calcium-imaging experiments 3.5 × 1011 GC of both AAV-PHP.S:ihSyn-tTA and AAV-PHP.S:TRE-jRGECO1a and 1 × 1011 GC of AAV-PHP.S:ChR2-EYFP were delivered. For calcium-imaging experiments following GCase rescue, 3.5 × 1011 GC of each of AAV-PHP.S:ihSyn-tTA, AAV-PHP.S:TRE-jRGECO1a, and AAV-PHP.S:TRE-GBA1, and 1 × 1011 GC of AAV-PHP.S:ChR2-EYFP were delivered. For calcium-imaging experiments, injection of α-Syn occurred 7 days after viral injection.
Calcium imaging of enteric neurons.
Mice were intracardially perfused with ice cold Kreb’s solution (composed of, in mM: NaCl 118, K Cl 4.8, NaH2PO4 1, NaHCO3 25, MgCl2 1.2, CaCl2 2.5, glucose 11) adjusted to pH 7.4 with NaOH and 310 mOsm with NaCl. The duodenum was quickly excised and placed in oxygenated (95% O2, 5% CO2), ice cold Kreb’s solution for 1 h followed by 15 min at room temperature. After incubation, the duodenum was placed into Kreb’s solution in a Sylgard-coated dish and cut in half. One segment was returned to the oxygenated Kreb’s solution at room temperature for later recording while the other was unraveled via a cut along the mesentery line. To prevent movement artifacts, the duodenal segment was pinned taut and flat, mucosa-side down, on a custom Sylgard-lined recording chamber (PH1, Warner. Segments were perfused with oxygenated room temperature Kreb’s solution during recording. Fluorescent calcium signals were recorded with an Andor Neo sCMOS camera (Oxford Instruments, Abingdon, England) attached to an upright Zeiss microscope (Examiner D1) using illumination from a Spectra X light engine (Lumencor, Beaverton, OR) filtered through a custom cube (part nos. FF01–464/547–25, FF01–607/36–25, DI03-R561-T1–25X36; AVR Optics, Rochester, NY). Excitation protocols were executed with a Digidata 1440A and pCLAMP software (Molecular Devices, San Jose, CA), and the fluorescent signal was captured in Andor Solis software. ImageJ was used to analyze the calcium signals, as described below. For the duration of the protocol, the field of view was illuminated with 561 nm light and photostimulation was performed with 15 ms pulses of 473 nm light. Light power was measured to be 0.1 mW mm−2 for 473 nm and 561 nm at the focal plane.
Quantification and statistical analysis.
Power analyses were conducted using G*Power (University of Dusseldorf) to estimate the number of animals needed to attain >90% power for each experiment. This was based on expected effect sizes of 0.7 calculated from our preliminary studies. Power analyses were performed using a SAS based model for multifactorial ANOVA designs. Statistical analysis of datasets was performed in Python 3 (pandas) and GraphPad Prism 6 software and is summarized in Supplementary Table 2. Data distribution was assumed to be normal but this was not formally tested. Pairwise comparisons were made using Student’s t-test. Group comparisons were made using one- or two-way ANOVA with Bonferroni post-hoc analysis. Measurements were made from distinct samples and statistics assume Gaussian distribution. The annotation for significance values is: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data were visualized in GraphPad Prism 6 or Python 3 (seaborn). Bar graph and column data are depicted as the mean and S.E.M. Boxplot data are depicted as the median, interquartile range (25th/75th percentiles), and 1.5× the interquartile range, which effectively covers 99% of the data points. Outliers are not included in boxplots, but are quantified.
Analysis of Western blots was performed in ImageJ or Image Lab (Bio-Rad). Regions of interest (ROI) were drawn around bands of interest or the length of the lane (for S129P) and the integrated density was measured. Relative integrated densities were calculated against the average of the WT samples on each membrane and normalized to housekeeping genes (β-actin, β-tubulin) or total protein (Stain Free Gels, Bio-Rad).
Analysis of immunohistochemical signals was performed in ImageJ. ROIs were drawn as delineated by presence of neurotransmitter signal specific to the region (e.g. TH for SNc, ChAT for DMV) and the integrated density was measured. Integrated densities were calculated per unit area and normalized to the average from WT samples.
Analysis of calcium signals was performed in ImageJ. Acquired video signals were loaded as image stacks and ROIs were drawn around each cell exhibiting jRGECO1a signal. An integrated density measurement was taken of the entire image stack for each cell. Percent change in fluorescence after photostimulation pulse was calculated by dividing the integrated density signal by the average of the 20 frames (2 s) that preceded the blue light pulse. Data were analyzed in GraphPad Prism to determine the area under the curve.
Data availability.
No datasets were generated or analyzed during the current study. The data that support the findings of this study are available from the corresponding author upon request.
Extended Data
Extended Data Fig. 1. Effect of αSyn pathology on GI health and cytokine expression.
a-d, GI function in WT, ASO, and aged mice is characterized by fecal pellet production (a; WT vs. ASO **p = 0.022, ASO vs. Aged **p = 0.0075), fecal pellet weight (b; WT vs. ASO ****p < 0.0001, WT vs. Aged ***p = 0.0009, ASO vs. Aged ****p < 0.0001), proportion fecal water weight (c; WT vs. ASO ****p < 0.0001, ASO vs. Aged ****p < 0.0001), and whole gut fecal transit time (d; WT vs. ASO ****p < 0.0001, ASO vs. Aged **p = 0.0038). e, Animal weight (WT vs. Aged ****p < 0.0001, ASO vs. Aged ****p < 0.0001). f, qPCR analysis of GBA1 expression in duodenum. Dots represent average of technical triplicates from a single animal. g, Densitometry analysis of cytokine panel from duodenal lysates after inoculation with α-Syn PFF or α-Syn monomer 7 dpi (Fractalkine **p = 0.0030, IL-1a **p = 0.0070, IL-6 *p = 0.0415, IL-7 * p = 0.0400, MCP-1 *p = 0.0416, MCSF *p = 0.0467, MIG *p = 0.0412, TECK *p = 0.0365, TIMP-2 *p = 0.0400). h, Representative cytokine panels detected by chemiluminescence. i, Representative Western blot immunostained for IL-6 and densitometry analysis normalized to WT (WT vs. PFF 60 dpi *p = 0.0327, 60 dpi PFF vs. monomer *p = 0.0480, WT vs. ASO *p = 0.0304). Data depicted are mean ± s.e.m. P values were determined by one-way ANOVA (a-f,i). The following n values represents number of independent animals used for statistical evaluation: e1a-c, WT = 42, ASO = 20, Aged = 19; e1d, WT = 13, ASO = 9, Aged = 12; e1e, WT = 42, ASO = 20, Aged = 19; e1f, WT = 3, ASO = 4, Aged = 4; e1g, monomer and PFF = 4 each; e1i, all conditions = 4 each.
Extended Data Fig. 2. Visualization of the duodenal ENS and cell proliferation.
a, Maximum intensity projection confocal image of duodenum whole-mount stained for PGP9.5 (neurons) and GFAP (EGCs). Higher magnification of dashed box shown in Fig. 1g. Scale bar, 200 μm. b, Virtual cross-section of the duodenal wall after optical clearing distinguishes myenteric and submucosal plexuses. Intestinal crypt layer is marked in blue. Scale bar, 100 μm. c, DAPI nuclear labeling visualizes the intestinal crypt layer used for histological analysis of the ENS (top). High-magnification of dashed box shows individual crypts (bottom). Scale bars, 100 μm. Experimental images for (a-c) were obtained from 8 independent mice, with similar results obtained. d, Quantification of number of myenteric neurons per crypt (top) or myenteric EGCs per crypt (bottom; WT vs. PFF 60 dpi *p = 0.0329, WT vs. PFF 120 dpi *p = 0.0232) e, Immunohistochemical labeling of EdU detects recently proliferated extraganglionic (open arrows) or myenteric ganglion cells (closed arrows). EGCs are co-labeled with GFAP. Scale bar, 20 μm. f, Quantification of total EdU+ cells per crypt (Monomer vs. PFF *p = 0.0071). Data depicted is mean ± s.e.m. P values were determined by One-Way ANOVA (d) or one-tailed Student’s t-test (f). The following n values represents number of independent animals used for statistical evaluation: e2d enteric neurons per crypt, WT = 6, PFF 0 dpi = 6, PFF 7 dpi = 6, PFF 21 dpi = 6, PFF 60 dpi = 6, PFF 120 dpi = 5, monomer conditions = 5 each; e2d EGCs per crypt, WT = 4, all PFF = 5 each, monomer 7 dpi = 4, monomer 60 dpi = 5; e2f, monomer = 4, PFF = 5.
Extended Data Fig. 3. Inoculation of duodenum with αSyn PFF promotes formation of αSyn filaments.
a, Representative dot blot images of duodenal homogenates immunostained with an aggregation-specific α-Syn antibody (α-Syn filament) show an increase after α-Syn PFF inoculation and in ASO mice. b, Densitometry analysis of α-Syn filament dot blots of duodenal homogenates normalized to pre-injection WT mice (WT vs. ASO **p = 0.0092). c, Representative dot blot image of controls immunostained with α-Syn filament antibody: 50 ng BSA, 50 ng and 100 ng α-Syn monomer, 50 ng and 100 ng α-Syn PFF. d, Densitometry analysis of control blots normalized to 50 ng BSA (BSA vs. PFF 50ng **p = 0.0080, BSA vs. PFF 100ng ***p = 0.0009, monomer 50ng vs. PFF 50ng *p = 0.0178, monomer 100ng vs. PFF 100ng **p = 0.0078). Data depicted is mean ± s.e.m. P values were determined by one-way ANOVA (a, d). The following n values represents number of independent animals used for statistical evaluation: e3b, WT = 8, all PFF = 5 each, all monomer = 4 each, ASO = 8, Aged = 5; e3d, BSA = 4, all other groups = 3 each.
Extended Data Fig. 4. GBA1 gene transfer increases GCase function and improves GI health.
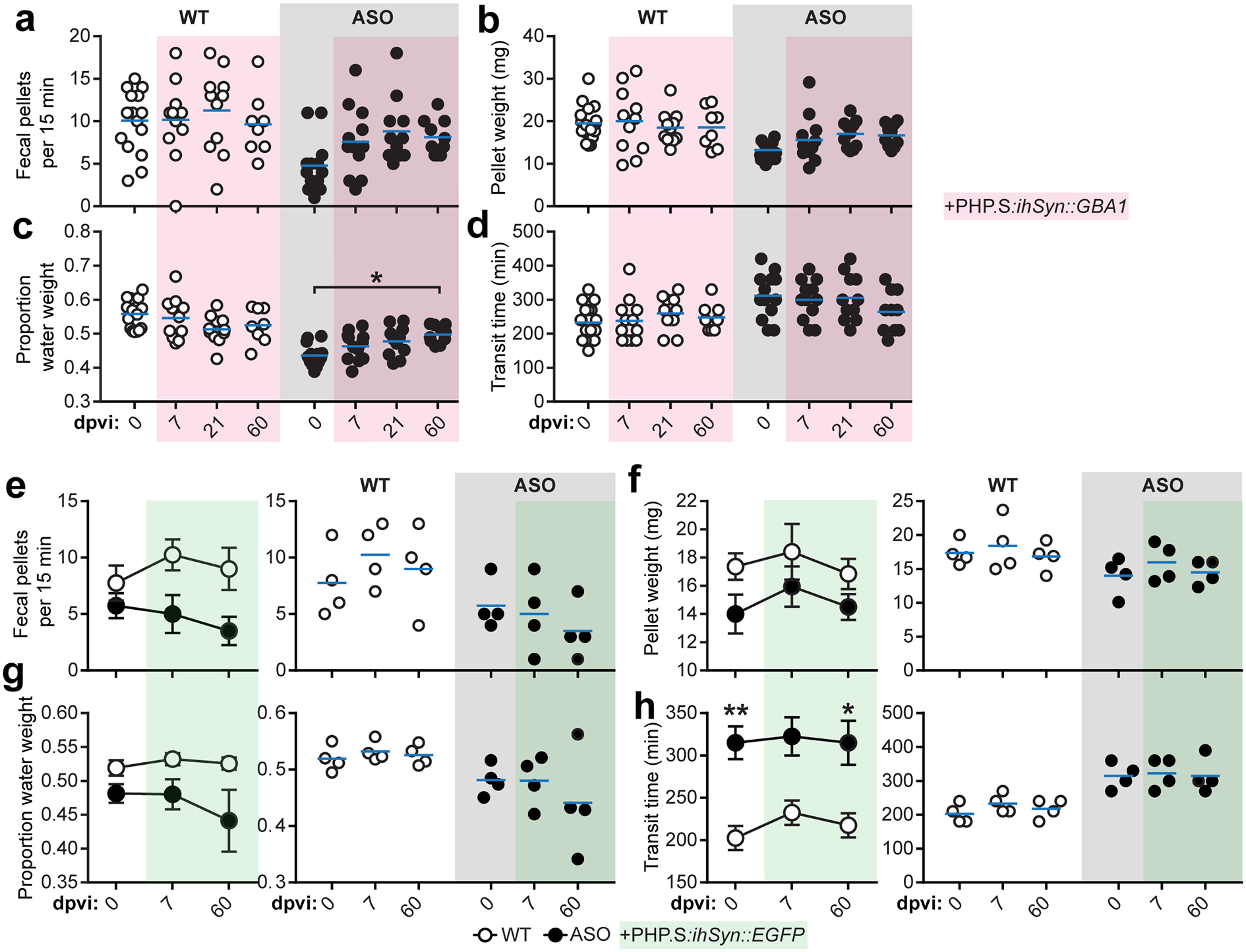
a-d, Characterization of GI function before and after peripheral GBA1 gene transfer by quantifying fecal pellet production (b), fecal pellet weight (c; ASO 0 vs. 60 dpvi *p = 0.0265), proportion fecal water weight (d), and whole gut fecal transit time (e). e-h, Characterization of GI function before and after peripheral EGFP gene transfer by quantifying fecal pellet production (e), fecal pellet weight (f), proportion fecal water weight (g), and whole gut fecal transit time (h; 0 dpi WT vs. ASO **p = 0.0085, 60 dpi WT vs. ASO *p = 0.0293). Data depicted are mean ± s.e.m. P values were determined by two-way ANOVA (a-h). The following n values represents number of independent animals used for statistical evaluation: e4a-d, for 0 / 7 / 21 / 60 dpi: WT = 17 / 12 / 11 / 8, ASO = 13 / 12 / 11 / 10; e4e-h, all conditions = 4 each.
Extended Data Fig. 5. ENS neurotransmission is disrupted by αSyn pathology and restored by GBA1 gene transfer.
a, Quantification of average peak percent change in fluorescence and area under the curve after photostimulation pulse for jRGECO1a+-only or jRGECO1a+/ChR2+ duodenal neurons before and after inoculation (all ****p < 0.0001). b, Quantification of average peak percent change in fluorescence and area under the curve after photostimulation pulse for jRGECO1a+-only or jRGECO1a+/ChR2+ duodenal neurons before and after systemic delivery of AAV-PHP.S::ihSyn:GBA1 (Peak ΔF/F jRGECO1a+ 0 dpvi WT vs. ASO ****p < 0.0001, 7 dpvi WT vs. ASO ****p < 0.0001; Peak ΔF/F jRGECO1a+/ChR2+ 0 dpvi WT vs. ASO ****p < 0.0001, 7 dpvi WT vs. ASO ***p = 0.0006; AUC jRGECO1a+ 0 dpvi WT vs. ASO ****p < 0.0001, 7 dpvi WT vs. ASO **p = 0.0031; AUC jRGECO1a+/ChR2+ 0 dpi WT vs. ASO *p = 0.0398). Data depicted are mean ± s.e.m. P values were determined by two-way ANOVA. The following n values represents number of independent animals used for statistical evaluation: e5a, 0 dpi = 3, PFF 7 dpi = 4, PFF 60 dpi = 3, monomer 7 dpi = 3, monomer 60 dpi = 3; e5b, all conditions = 3.
Extended Data Fig. 6. Inoculation of duodenum with αSyn PFF in adult mice does not progress αSyn pathology to the nodose ganglion.
a, Maximum intensity 3D projection confocal image of a whole nodose ganglion from an α-Syn PFF inoculated WT mouse 60 dpi. Ganglion was PACT cleared and immunolabeled for PGP9.5 (neurons), S129P, and DAPI (nuclei). Experimental images were obtained from 14 independent mice, with similar results obtained. Scale bars, 100 μm. Z-stack depth, 500 μm. b, Maximum intensity 3D projection confocal image of a nodose ganglion from a WT mouse. Experimental images were obtained from 5 independent mice, with similar results obtained Scale bar, 100 μm. Z-stack depth, 450 μm. c, Quantification of S129P+ nodose ganglion neurons (WT vs. ASO **p = 0.0011, monomer 7 dpi vs. ASO **p = 0.0027, monomer 60 dpi vs. ASO *p = 0.0176, ASO vs. Aged *p = 0.0323). Data depicted is mean ± s.e.m. P values were determined by one-way ANOVA. The following n values represents number of independent animals used for statistical evaluation: e6c, WT = 5, ASO = 5, Aged = 5, for 7 / 60 / 120 dpi, PFF = 5 / 5 / 4, monomer = 4 / 3 / 0.
Extended Data Fig. 7. Inoculation of duodenum with αSyn PFF in adult mice does not progress αSyn pathology to the brain.
a-b, Maximum intensity projection confocal images of the brainstem (approximately −7.76 mm from Bregma) from an α-Syn PFF-inoculated 8 week old WT mouse 60 dpi (a) and an ASO mouse (b), immunolabeled for cholinergic neurons (ChAT) and S129P. Scale bars, 500 μm. Experimental images for (a-b) were obtained from 3 independent mice, with similar results obtained. c-d, Maximum intensity projection confocal images of the midbrain (approximately −3.64 mm from Bregma) from an α-Syn PFF-inoculated WT mouse 60 dpi (c) and an ASO mouse (d), immunolabeled for dopaminergic neurons (TH), S129P, and nuclei (DAPI). Scale bars: 500 μm. Experimental images for (c-d) were obtained from 3 independent mice, with similar results obtained. e-f, Densitometry analysis of S129P signal in the DMV (e; WT vs. ASO ***p = 0.0003, PFF 60 dpi vs. ASO ***p = 0.0007) and SNc (f; WT vs. ASO **p = 0.0011, PFF 60 dpi vs. ASO **p = 0.0014) per unit area, normalized to WT. Data depicted is mean ± s.e.m. P values were determined by one-way ANOVA (a, d). The following n values represents number of independent animals used for statistical evaluation: e7e-f, all conditions = 3 each.
Extended Data Fig. 8. Duodenal inoculation with αSyn PFF in adult mice does not produce sensorimotor deficits.
a-f, Sensorimotor behaviors after inoculation were quantified using average time to turn and descend a pole (a; PFF 0 vs. 60 dpi **p = 0.0012, 90 dpi PFF vs. monomer *p = 0.0265), average time before falling during the inverted wire-hang paradigm (b; PFF 0 vs. 60 dpi *p = 0.0306, PFF 0 vs. 120 dpi *p = 0.0285), average time to remove adhesive from nasal bridge (c; PFF 0 vs. 90 dpi **p = 0.0014, PFF 0 vs. 120 dpi *p = 0.0285, 60 dpi PFF vs. monomer *p = 0.0193, 90 dpi PFF vs. monomer *p = 0.0342), average weightlifting paradigm score (d), average time to cross a narrowing beam (e), and total number of slips incurred during 3 trials of the narrowing beam paradigm (f; 60 dpi PFF vs. BSA **p = 0.0023). Boxplots represent median, interquartile range, and 1.5× the interquartile range. g, Hot plate test did not reveal any changes in nociception at 60 dpi. Data depicted is mean ± s.e.m. P values were determined by two-way ANOVA (a-f) or one-way ANOVA (g). The following n values represents number of independent animals used for statistical evaluation: e8a-c, for 0 / 7 / 21 / 60 / 90 / 120 dpi, PFF = 16 / 14 / 14 / 11 / 9 / 8, monomer = 9 / 9 / 9 / 8 / 8 / 8, BSA = 17 / 16 / 11 / 9 / 7 / 7; e8d, for 0 / 7 / 21 / 60 / 90 / 120 dpi, PFF = 8 / 8 / 12 / 8 / 8 / 8, monomer = 9 / 9 / 9 / 8 / 8 / 8, BSA = 17 / 16 / 11 / 9 / 7 / 7; e8e-f, for 0 / 7 / 21 / 60 / 90 / 120 dpi, PFF = 8 / 8 / 10 / 7 / 6 / 6, monomer = 9 / 9 / 9 / 8 / 8 / 8, BSA = 17 / 16 / 11 / 9 / 7 / 7; e8g, baseline = 10, monomer = 4, PFF = 6.
Extended Data Fig. 9. Duodenal αSyn PFF inoculation in aged mice does not result in SNc dopaminergic cell loss.
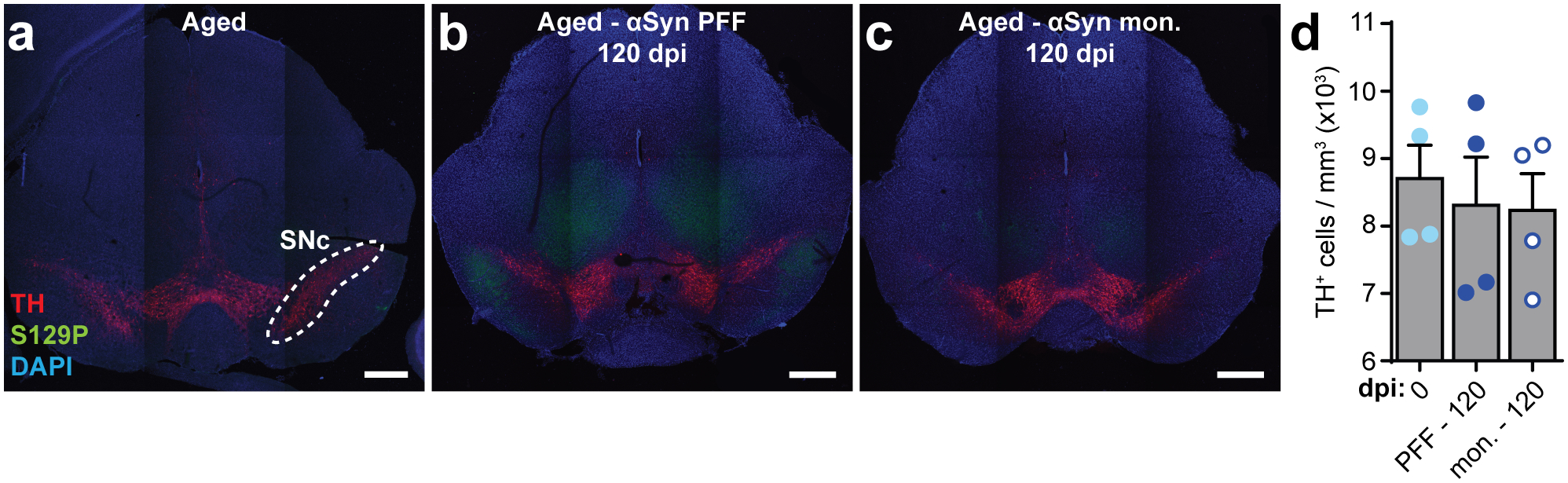
a-c, Representative images of the midbrain immunostained for dopaminergic neurons (TH), S129P, and nuclei (DAPI) from aged mice pre-inoculation (a), α-Syn PFF-inoculated aged mice 120 dpi, and α-Syn-monomer inoculated aged mice 120 dpi. Scale bars, 500 μm. Experimental images for (a-c) were obtained from 12 independent mice, with similar results obtained. d, Quantification of dopaminergic cell density within the boundary of the SNc as determined by TH+ signal (example shown in [a]). Data depicted is mean ± s.e.m. P values were determined by one-way ANOVA. The following n values represents number of independent animals used for statistical evaluation: e9d, all conditions = 4.
Supplementary Material
Acknowledgments.
We thank Claire Bedbrook for help with calcium imaging, and Keith Beadle and Elisha Mackey for additional technical support. This work was supported by grants to V.G.: NIH Director’s New Innovator IDP20D017782-01 and PECASE; NIH/NIA R01AG047664-01; NIH BRAIN 1U01NS090577; Heritage Medical Research Institute; Pew Charitable Trust, and CZI Neurodegeneration Challenge Network. C.C. was supported by NIH/NIA F32AG054101. S.K.M. was supported by NIH/NINDS R01NS085910. L.A.V. was supported by NINDS R01NS102257 and Morris K. Udall Centers of Excellence for Parkinson’s Disease Research P50NS108675. T.R.S was supported by the Larry L. Hillblom Foundation. B.B.Y. was supported by NIH/NIGMS 5T32GM007616 and the Caltech Center for Environmental Microbial Interactions (CEMI).
Footnotes
Declaration of Interests. All authors declare no competing interests.
References
- 1.McCann H, Stevens CH, Cartwright H & Halliday GM α-Synucleinopathy phenotypes. Parkinsonism Relat. Disord 20, S62–S67 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Lees AJ, Hardy J & Revesz T Parkinson’s disease. Lancet 373, 2055–66 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Hawkes CH, Del Tredici K & Braak H A timeline for Parkinson’s disease. Parkinsonism Relat. Disord 16, 79–84 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Sánchez-Ferro Á et al. In vivo gastric detection of α-synuclein inclusions in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc 30, 517–524 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Shannon KM et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc 27, 709–715 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Yoo BB & Mazmanian SK The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 46, 910–926 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, Rüb U, Gai WP & Del Tredici K Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm 110, 517–36 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Holmqvist S et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. (Berl.) 128, 805–20 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Uemura N et al. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener 13, 21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim S et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron (2019) doi: 10.1016/j.neuron.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devos D et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis 50, 42–48 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Codolo G et al. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLOS ONE 8, e55375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amor S, Puentes F, Baker D & Valk PVD Inflammation in neurodegenerative diseases. Immunology 129, 154–169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu Y, Dodiya H, Aebischer P, Olanow CW & Kordower JH Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis 35, 385–98 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Neumann J et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fishbein I, Kuo Y-M, Giasson BI & Nussbaum RL Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 137, 3235–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazzulli JR et al. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 146, 37–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sardi SP et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci 108, 12101–12106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Sullivan SS et al. Nonmotor symptoms as presenting complaints in Parkinson’s disease: A clinicopathological study. Mov. Disord 23, 101–106 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Volpicelli-Daley LA, Luk KC & Lee VM-Y Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc 9, 2135–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luk KC et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volpicelli-Daley LA et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hallett PJ, McLean JR, Kartunen A, Langston JW & Isacson O Alpha-synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol. Dis 47, 258–267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chesselet M-F et al. A Progressive Mouse Model of Parkinson’s Disease: The Thy1-aSyn (“Line 61”) Mice. Neurotherapeutics 9, 297–314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schafer K-H, Mestres P, Marz P & Rose-John S The IL-6/sIL-6R Fusion Protein Hyper-IL-6 Promotes Neurite Outgrowth and Neuron Survival in Cultured Enteric Neurons. J. Interferon Cytokine Res. 19, 527–532 (1999). [DOI] [PubMed] [Google Scholar]
- 26.De Schepper S et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 175, 400–415.e13 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Sehgal A et al. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun 9, 1272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrenschee M et al. Distinct pattern of enteric phospho-alpha-synuclein aggregates and gene expression profiles in patients with Parkinson’s disease. Acta Neuropathol. Commun 5, 1–1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujiwara H et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol 4, 160–164 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Grassi D et al. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci 115, E2634–E2643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampson TR et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morabito G et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther (2017) doi: 10.1016/j.ymthe.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan KY et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci 20, 1172–1179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Challis RC et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc 1 (2019) doi: 10.1038/s41596-018-0097-3. [DOI] [PubMed] [Google Scholar]
- 35.Froula JM et al. α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol. Commun 6, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boesmans W, Hao MM & Berghe PV Optical Tools to Investigate Cellular Activity in the Intestinal Wall. J. Neurogastroenterol. Motil 21, 337–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treweek JB et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc 10, 1860–1896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reeve A, Simcox E & Turnbull D Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev 14, 19–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kordower JH et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain J. Neurol 136, 2419–2431 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salvatore MF, Pruett BS, Dempsey C & Fields V Comprehensive Profiling of Dopamine Regulation in Substantia Nigra and Ventral Tegmental Area. JoVE J. Vis. Exp e4171–e4171 (2012) doi: 10.3791/4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nalls MA et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet 46, 989–993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanner CM et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect 119, 866–872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenbloom B et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells. Mol. Dis 46, 95–102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sidransky E & Lopez G The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rocha EM et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis 82, 495–503 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Whitton PS Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol 150, 963–976 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neunlist M et al. Enteric glial cells: recent developments and future directions. Gastroenterology 147, 1230–7 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Chandra R, Hiniker A, Kuo Y-M, Nussbaum RL & Liddle RA α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douglas PM & Dillin A Protein homeostasis and aging in neurodegeneration. J. Cell Biol 190, 719–729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fox EA, Phillips RJ, Martinson FA, Baronowsky EA & Powley TL Vagal afferent innervation of smooth muscle in the stomach and duodenum of the mouse: morphology and topography. J. Comp. Neurol 428, 558–576 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Rockenstein E et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res 68, 568–578 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Li Z et al. Essential Roles of Enteric Neuronal Serotonin in Gastrointestinal Motility and the Development/Survival of Enteric Dopaminergic Neurons. J. Neurosci 31, 8998–9009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fleming SM, Ekhator OR & Ghisays V Assessment of sensorimotor function in mouse models of Parkinson’s disease. J. Vis. Exp e50303–e50303 (2013) doi: 10.3791/50303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deacon RMJ Measuring the strength of mice. J. Vis. Exp e2610–e2610 (2013) doi: 10.3791/2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bannon AW & Malmberg AB Models of Nociception: Hot-Plate, Tail-Flick, and Formalin Tests in Rodents. Curr. Protoc. Neurosci 41, 8.9.1–8.9.16 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Park J et al. Pain perception in acute model mice of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Mol. Pain 11, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analyzed during the current study. The data that support the findings of this study are available from the corresponding author upon request.