

Abstract
Oral tyrosine kinase inhibitors (TKIs) against epidermal growth factor receptor (EGFR) family have been introduced into the clinic to treat human malignancies for decades. Despite superior properties of EGFR-TKIs as small molecule targeted drugs, their applications are still restricted due to their low solubility, capricious oral bioavailability, large requirement of daily dose, high binding tendency to plasma albumin and initial/acquired drug resistance. Nanotechnology is a promising tool to improve efficacy of these drugs. Through non-oral routes. Various nanotechnology-based delivery approaches have been developed for providing efficient delivery of EGFR-TKIs with a better pharmacokinetic profile and tissue-targeting ability. This review aims to indicate the advantage of nanocarriers for EGFR-TKIs delivery.
Keywords: Nanotechnology, EGFR, Tyrosine kinase inhibitor, Cancer therapy
Graphical abstract
(A). Various types of EGFR-TKIs loaded nanocarriers (a) liposome, (b) polymer nanoparticle, (c) micelle, (d) nanogold particles.(B) Schematic illustration of nanocarriers are intended to exploit the enhanced permeability and retention effect to exit the blood vessels through leaky vasculature and accumulate within tumor tissues.(C) Schematic of increased targeting ability of nanocarriers via surface modification and the anti-cancer mechanism of EGFR inhibition.
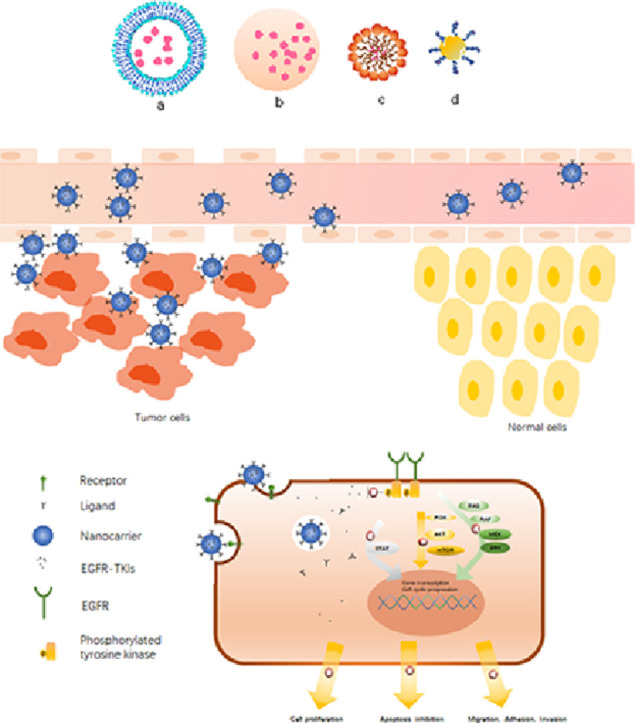
1. Introduction
The epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases (TKs) has four homologous receptors including EGFR (ErbB1/HER-1), ErbB2 (HER-2), ErbB3 (HER-3) and ERbB4 (HER-4), and these membrane receptors have intracellular domains with TK activity. The ErbB proteins function through either homo- or hetero-dimerization. Following dimerization, specific tyrosine residues can autophosphorylate, ultimately resulting in the activation of downstream signaling pathways [1], [2]. Overexpression and activating mutations of EGFR family receptors, specifically EGFR and HER2, are associated with uncontrolled cell division of many human epithelial malignancies, including non-small cell lung cancer (NSCLC), breast cancer (BC), colorectal cancer, and pancreatic cancer [3]. For instance, an activating EGFR mutation has been observed in 20%–40% of Asian patients and up to 15% of Caucasian patients with NSCLC [4].
Therefore, the inhibition of EGFR family, especially EGFR and HER2, is an important therapeutic approach in the field of cancer therapy [5]. TKIs, mostly derived from quinazoline, can block the magnesium-ATP-binding site of the intracellular tyrosine kinase domain and in vitro studies confirmed their ability to disrupt tyrosine-kinase activity followed by the reduced activation of intracellular downstream signaling.
In clinic small molecule EGFR-TKIs have shown the significant therapeutic efficacy against several cancers, which led to them as one kind of the most investigated small molecule inhibitors in cancer treatment [6], [7], [8], [9], [10].
To date, the U.S. Food and Drug Administration (FDA) has approved some TKIs targeting the EGFR family as tumor therapeutic agents and all the EGFR-TKIs available on the market are film-coated tablets for oral administration. However, the limited oral bioavailability associated with poor aqueous solubility, permeability, and extensive plasma protein binding capacity leads to restricted therapeutic efficacy [11], [12]. Since EGFR signaling pathway also plays an essential role in proliferation, differentiation, migration, and apoptosis of normal cells, such as epithelial, mesenchymal, and neuronal cells, traditional oral delivery of EGFR-TKIs is accompanied by a series of side effects including rash, erythema, diarrhea, gastrointestinal (GI) perforations, ocular lesions, and hematological disorders [13], [14]. Lipophilicity also mediates the low targeting efficiency of agents, promoting binding to unwanted targets [15]. Specially, Skin contains appreciable levels of EGFR and its inhibition may lead to skin lesions. Severe rash limits the anticancer treatment with a higher drug dosage. Additionally, for patients with GI dysfunction, like active peptic ulcer, inability to take oral medication, or prior gastrointestinal surgeries, the development and evaluation of different formulations of EGFR-TKIs are valuable.
In clinic, despite a significant initial response, the development of acquired resistance in most of the patients also limits the long-term efficacy of TKI therapy [16], [17]. With great efforts made on the development of new generations of EGFR-TKIs, multi-agent combination therapy that impacts on multiple signaling pathways is also a promising approach. It has been confirmed that by affecting different signaling pathways in cancer cells, combination therapies can provide synergistic anticancer effects, overcome mechanisms of drug resistance and minimize side effects.
To date, appropriate drug combinations have brought significant benefits to cancer patients in clinic. Due to different pharmacokinetics and biodistribution of drugs and dissimilar rates of metabolism in the body, the optimized dose ratio is hard to be maintained. One of the grand challenges of multi-agent administration is how to reach sufficient concentration with a correct ratio of drugs in the target tissues.
The nano-technology based drug delivery system (DDS) is capable of bringing hope to solve the problems mentioned above. Efficient EGFR-TKIs nanoscale delivery systems not only help to solve the fundamental shortcomings such as poor solubility and rapid degradation but also minimize the side effects via the selective accumulation at tumor tissues [18], [19], [20]. Leaky and tortuous vasculature with poor or absent lymphatic drainage is one of the characteristic features of solid tumor tissues. Nanosized carriers can pass through the leaking blood vessels and accumulation in tumor tissues with the enhanced permeability and retention (EPR) effect, so-called as “passive” targeting [21]. Meanwhile, via specific coatings, better pharmacokinetics, internalization, and targeting of nanocarriers could be achieved. Nanocarriers are also identified as excellent delivery platforms which can delivery combined agents with correct radio and well-designed release sequence to achieve the synergistic effects [22].
This review aims to discuss the nanotechnology-based DDS used for EGFR-TIKs delivery in cancer therapy. At the same time, we summarized antitumor mechanisms and clinical status of the FDA approved EGFR-TKIs for different types of cancer.
2. Clinical status of EGFR-TKIs
Since aberrant activation of EGFR signaling pathway is associated with development of variety of malignancies, numerous anti-tumor agents that primarily target EGFR family are currently at different stages of clinical development. This section provides a summary about the functional mechanisms and clinical states of the four most commonly used EGFR-TKIs approved by FDA for the therapy of solid tumors for more than 3 years. All the key properties are outlined in Table 1.
Table 1.
Properties of FDA-approved small molecule EGFR tyrosine kinase inhibitors.
| Name | Targets | The partition coefficient (log P) | Clinical dose | The FDA-approved indications (year) | Most common adverse effects |
|---|---|---|---|---|---|
| Gefitinib | EGFR | 4.50 | 250 mg once daily | Locally advanced metastatic NSCLC cancer after failure of both platinum-based and docetaxel chemotherapies (2003) | Proteinuria, diarrhea, ALT increased, decreased appetite, AST increased, and skin reactions |
| Erlotinib | EGFR | 3.14 | 150 mg once daily | NSCLC as a monotherapy after failure of at least one prior chemotherapy (2004) Advanced pancreatic cancer in combination with gemcitabine for patients who have not received previous chemotherapy (2005) |
Skin rash, diarrhea, mucositis, hyperbilirubinemia, neutropenia, and anemia |
| Lapatinib | EGFR/ErbB2 | 4.97 | 1250 mg once day | Metastatic breast cancer in combination with capecitabine whose tumors overexpress HER2 and have received prior therapy, including an anthracycline, a taxane, and trastuzumab (2006) | Diarrhea and palmar-plantar erythrodysesthesia |
| Afatinib | EGFR/ErbB2/ErbB4 | 3.97 | 40 mg once daily | First-line treatment of NSCLC with exon-19 deletions or the exon-21 L858R mutation. (2013) | Diarrhea, vomiting, dyspnea, fatigue, and hypokalemia |
2.1. Gefitinib
As orally active 4-anilino-quinazolines, gefitinib (GEF) is able to effectively and reversibly target the ATP-binding pocket of the TK domain of EGFR.
GEF originally received accelerated approval of FDA in 2003 as a monotherapy for the treatment of patients with advanced or metastatic NSCLC after a failure of both docetaxel and platinum-based chemotherapies [23]. In subsequent clinical trials, however, no evidence was observed for a correlation between the extent of expression of EGFR and patient response to GEF. FDA updated the approval and restricted the use of patients in 2005. Screening of high responders conducted to the identification of sensitivity mutations in the TK domains of the genes for EGFR: exon-19 deletions or exon-21 (L858R) substitution mutations [24], [25], [26]. Ultimately, in 2015, the FDA approved GEF for the first-line treatment of patients with metastatic NSCLC bearing such mutations
In terms of safety, the most common adverse reactions were diarrhea skin reactions, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) increase. Approximately 5% of patients treated with discontinued treatment due to adverse reactions. Bone pain, dyspnea, pulmonary toxicity and stomatitis are observed occasionally [27].
2.2. Erlotinib
Erlotinib (ETB) is another ATP-competitive quinazoline derivative that works by binding to EGFR-TK in the same manner as GEF.
In 2004, ETB initially received FDA approval as monotherapy for the treatment of progressed NSCLC after the failure of at least one prior chemotherapy. In another Phase III trial, the combination of erlotinib (100 mg/day) with gemcitabine (1000 mg/m2, weekly) significantly prolonged survival, especially progression-free survival (PFS), in patients with advanced pancreatic cancer [8]. This pivotal clinical trial led to the FDA-approval of erlotinib combined with gemcitabine in locally advanced and metastatic pancreatic cancer in 2005. Experimental results of several preclinical and clinical studies further demonstrated the association between EGFR mutations and response to ETB [28], [29]. With this background, in 2013, ETB received approval by FDA for use as a first-line treatment of NSCLC in patients whose tumors harbor EGFR exon-19 in-frame deletions or exon-21 (L858R) mutations.
As with GEF, skin reactions and diarrhea were the main adverse events found to be associated with ETB treatment. Mucositis, anemia, neutropenia, hyperbilirubinemia, and elevated ALT have also been reported. It uncommonly produces severe skin blistering and GI perforation in which case the drug must be permanently discontinued.
2.3. Lapatinib
Approximately 15%–20% of breast cancers overexpress HER2, classified as HER2-positive subgroup. HER2- positive BC cells tend to be more aggressive due to abnormal cell growth mediated via the overexpression of the HER2 protein [30], [31]. One of critical therapeutic approaches in HER2-positive BC is inhibiting EGFR/HER2 with receptor-targeted TKIs [32].
Lapatinib (LAPA), a member of the 4-anilino-quinazoline family of TKIs, contains side chains which are different from those of GEF or ETB. It can potently and reversibly target and binds to the intracellular TK domains of EGFR and HER2 receptors. Therefore, in 2007, lapatinib was approved by the FDA as a combination with capecitabine for the treatment of advanced HER2 positive BC patients who have received prior therapy including trastuzumab and other anticancer agents. Subsequently, it was also approved by the FDA in combination with letrozole for the treatment of postmenopausal women with hormone receptor-positive advanced BC overexpressing the HER2 for whom hormonal therapy suggested [33]. A Phase III trial of 399 patients with advanced HER2 positive BC reported that compared to capecitabine alone group, combination group of lapatinib and capecitabine can prolong the median time to tumor progression (TTP) of patients [34].
Diarrhea and skin rash are the most common adverse effects attributed to the large requirement of daily oral dose
2.4. Afatinib
In clinic, most of the responders to GEF/ETB will inevitably develop acquired resistance with one or two years, and the most frequent resistance mechanism is the emergence of T790M mutation in the EGFR- tyrosine kinase domain [35], [36]. With this background, irreversibly, covalently second-generation EGFR-TKIs including afatinib (AFT) were developed.
AFT is an oral aniline-quinazoline derivative with a reactive acrylamide group. It can irreversibly inhibit EGFR-family autophosphorylation followed by downregulation of EGFR signal transduction via covalently binding to EGFR, HER2, and HER4. AFT is the first irreversible EGFR-TKIs approved by FDA for NSCLC treatment.
According to the results of two open-label, randomized, phase III trials, AFT (40 mg/d) can bring prolonged PFS to advanced NSCLC patient with EGFR mutations compared with traditional chemotherapy [37], [38]. Therefore, in 2013, oral AFT (Gilotrif™) received approval of FDA for the first-line treatment of patients with locally advanced or metastatic NSCLC who have proven EGFR mutations. The recommended dosage is 40 mg daily, and treatment should be continued until AFT is no longer tolerated or until disease progression.
The severity of common adverse events such as rash, erythema and diarrhea were found to be dose-dependent during AFT treatment. Cases of interstitial pneumonia, fulminating liver disease, severe skin blistering were also reported in a small percentage of patients, with some of these causing death.
3. Nanotechnology strategies for EGFR-TKIs delivery
3.1. Liposomes
The inherent advantages of liposomes, such as non-immunogenicity, biocompatibility, high drug loading capacity and controllable release manner, have promoted their widespread use as drug carriers in tumor therapeutics [39]. They are composed of single or multiple lipid bilayers encapsulating an aqueous space and lipid bilayers which mainly consist of cholesterol (CHOL) and phospholipids molecules [40].
The drug can be localized in lipid membrane or the internal aqueous core of liposome, depending on its lipophilicity [41]. Due to the membrane structure is similar to the cell membranes, which can facilitate cellular uptake of drugs in them, the liposomes are regarded as an ideal drug-carrier system to promising approach to improve the agents’ biodistribution.
Liposomes are divided in three subtypes, (1) conventional type: it consists of the lipid bilayer and an aqueous core (2) PEGylated type: polyethylene glycol (PEG) is attached to the surface of liposomes to extend the blood circulation time by avoiding them from recognition and uptake of the reticuloendothelial system (RES), and (3) ligand-targeted type: ligands, including antibodies and peptides, are linked to the surface of the liposomes to obtain active targeting effect to specific cells.
Numerous research groups have developed several EGFR-TKIs loaded liposomal nanocarriers.
A PEGylated ETB liposome, with an entrapment efficiency (EE) of 85.3%±1.8%, mean size of 102.4 ± 3.1 nm and zeta potential of −16 mV, was composed of PEG-1,2-distearoyl-sn‑glycero-3-phosphoethanolamine (DSPE), CHOL, Soybean phosphatidylcholine (SPC) at the ratio of 3:10:22 [42]. The cytotoxicity of blank liposomes, free-ETB and two formulations (with or without PEGylated ETB loaded liposomes) were examined in vitro studies. The results revealed that the blank liposomes had negligible cellular cytotoxicity. At the same incubation condition, liposomal ETB significantly increased the cytotoxicity against A549 NSCLC cells, compared with free ETB, which should contribute to the enhanced intracellular drug accumulation by the transportation of liposomes. In vivo, PEGylation can greatly change the pharmacokinetic behavior and improve drug bioavailability. As compared to conventional liposomes and free ETB solution, the PEGylated formulation showed longer terminal half-life, lower clearance rates and higher area under the curve 0–∞ values in pharmacokinetics analysis.
The tumor microenvironment is characterized by hypoxia, lower pH, high glutathione (GSH) levels, etc. which can be adopted as triggers to cause a release of cargo molecules from the system [43]. Through adding a kind of pH-sensitive phospholipid, cholesteryl hemisuccinate (CHEMS), Alanood et al. synthesized pH-sensitive liposomes to delivery AFT in lung cancer cells [44]. Owning to PH-responsive release and negative values of zeta potential, the anticancer efficiency of this PH-sensitive liposome was significantly higher compared with conventional and cationic liposomes containing AFT in vitro.
Encapsulating EFGR-TKIs with other anticancer agents is another formulation strategy used in cancer therapeutics. Because the synergistic effect could improve the anticancer effects and reduce the cytotoxicity toward normal cells by affecting multiple signaling pathways and releasing the drugs either simultaneously or in a predetermined sequence at target sites.
It has been proved that via prolonged EGFR inhibition, cancer cells could re-acquire a working apoptosis pathway responsive to DNA damage [45]. In detail, researchers found that EGFR inhibition dramatically sensitizes a subset of breast cancer cells to DNA damage if the drugs are given sequentially but not simultaneously. Furthermore, transcriptional, proteomic, and computational analyses of signaling networks and phenotypes in drug-treated cells revealed the enhanced treatment efficacy results via dynamic network re-wiring of an oncogenic signature maintained by active EGFR signaling to unmask an apoptotic process which involves activation of caspase-8. The increased sensitivity to chemotherapeutic agents observed required sustained inhibition of EGFR because the phenotype did not result from a rapid direct inhibition of the oncogene, but rather from modulation of an oncogene driven transcriptional network as indicated schematically. Distinct examples of multidrug delivery systems containing EGFR-TKIs for cancer therapy are depicted in Table 2.
Table 2.
Examples of nano-based multidrug delivery systems containing EGFR-TKIs for cancer therapy.
| Nanocarrier type | Drug combinations | Delivery approach | Indications | Observations | References |
|---|---|---|---|---|---|
| Liposomes | ETB and DOX | Folate to folate receptor | NSCLC | The pretreatment of ETB could enhance rewiring of apoptotic signaling networks of cancer cells, leading to improved cytotoxicity of DOX. | [46] |
| Liposomes | ETB and PFOB | Oligonucleotide Apt to EGFR | NSCLC | This approach, oxygen and ETB co-delivery, was able to ameliorate the solid tumor microenvironment, overcome Hypoxia-triggered drug resistance and thus improve anti-cancer effect in vitro and in vivo. | [48] |
| MSNs | GEF and DOX | Cetuximab to EGFR | EGFR- resistant NSCLC | The co-delivery MSNs overcame EGFR-TKI resistance NSCLC with superior targeting ability mediated by cetuximab. | [69] |
| MSNs coated by PH-responsive lipid film | ETB and DOX | Passive | NSCLC | The charge conversion at tumor pH facilitated the cellular uptake of MSNs by the cancer cells and sequentially release of drugs further enhanced antitumor activity. | [71] |
| CS-NPs | GEF and CQ | Passive | Drug resistant TNBC | CQ as an inhibitor of autophagic lysosome formation could overcome autophagy in the drug resistant BC cells and effectively reversing GEF-resistance. | [94] |
| PMs | CYP and GEF | Passive | Pancreatic cancer | GEF/CYP co-loaded PMs showed a synergistic effect against pancreatic cancer cells mainly attributed to obstruction of sonic hedgehog and EGFR signaling pathways. | [103] |
| PEG-PBC micelles | LAPA and DOX | Passive | DOX-resistant BC | The use of lapatinib not only as a dual TKI but also as an adjuvant, inhibiting efflux of P-gp, sensitized MDR cancer cells to DOX. | [109] |
| PEG–PCD micelles | LAPA and PTX | Passive | MDR prostate cancer | LAPA can minimize the efflux and enhance the intracellular accumulation of chemotherapeutic agents in MDR prostate cancer | [104] |
| PEG-PLA micelles | LAPA and PTX | Passive | HER2-positive BC | PPM-LP exhibited significantly stronger cytotoxicity (p < 0.05) to SKBr-3 cells (HER-2 positive) and showed almost no significantly different cytotoxicity (p > 0.05) to MDA-MB-231 cells (HER-2 negative) as compared with PPM PTX | [113] |
| Polymeric implants | LAPA and PTX | Passive | TNBC | This localized co-delivery system revealed steady drug accumulation in tumor sites and good synergistic effects between LAPA and PTX against TNBC | [114] |
| Nanovesicles | GEF and DOX | Passive | NSCLC | Tumor tissue or cell specific drug release mediated by reduction and pH dual-responsive nanovesicles further improved efficacy of combination therapy in lung cancer. | [118] |
| Self-assembled NPs | EBT and quercetin | Passive | NSCLC | Through combining both ETB and quercetin in one system could inhibit the phosphorylation at both upstream and downstream of EGFR signaling pathways and thereby more efficiently interrupt the EGFR signaling and eventually prevent the growth of EGFR-expressing tumors. | [121] |
Morton et al. [46] successfully designed a liposomal DDS which not only co-deliver drugs with different properties but also release them with the desired time sequence. By entrapping ETB in the hydrophobic lipid membrane and loading doxorubicin (DOX) into the hydrophilic compartment, ETB was released before DOX (Fig. 1). In brief, the liposomes containing DOX and ETB were successfully fabricated via a lipid film hydration method. Lipid vesicles were formed after hydration in an acidic citric acid buffer under sonication and high heat in the presence of the hydrophobic ETB. Subsequent pH-driven loading of DOX in the interior of the vesicles formed the final dual-drug liposomal system. Dynamic light scattering showed that the liposomes prepared in this two-step drug loading process were of uniform size on the basis of their polydispersity index (PDI), and similar results were observed via transmission electron microscope (TEM). In vitro, the sensitization effect was mediated by prolonged EGFR inhibition induced internal rewiring of apoptotic signaling networks.
Fig. 1.

Characterization of the combination therapeutic–loaded liposomal system. (A) Cryogenic transmission electron micrograph of dual drug–loaded liposomes. Scale bar, 100 nm. (B) Schematic of dual loading of a small-molecule inhibitor (ETB, blue) into the hydrophobic, vesicular wall compartment and of a cytotoxic agent (DOX, green) into the aqueous, hydrophilic interior.
In order to further improve the therapeutic effect of EGFR-TKIs, great efforts have been made to develop innovative liposomes with targeting ability. Li et al. [47]. prepared liposomes anchored with anti-EGFR aptamer (Apt)-conjugated chitosan (Cs) and subsequently loaded ETB into these liposomal complexes (Apt-CL-E). After adjusting the ratio of lecithin, CHOL and CS, the drug loading (DL) of ETB has been optimized to 8% with nearly 100% EE and relatively stable mean particle size of 185 nm. in vitro, Apt-CL-E had specific cell recognition, increased cellular uptake and better antiproliferative effect in H1975 (EGFR-TKI-resistant) NSCLC cell lines, as compared with regular ETB-loaded liposomes without anti-EGFR aptamer conjugated. The results in vitro demonstrated that aptamer played an important role in increasing ETB intracellular accumulation and inducing more cell cycle arrest and apoptosis to overcome acquired EGFR-TKI resistance in NSCLC.
A similar attempt has been made for targeting delivery oxygen and ETB by Apt-modified liposomal complexes to reverse hypoxia-induced drug resistance [48] (Fig. 2). Novel multifunctional liposomes anchored with anti-EGFR Apt-conjugated CS (ACLEP) were formulated to co-delivery ETB and an artificial blood substitute, perfluorooctyl bromide (PFOB), for reversing hypoxia-induced drug resistance. Using thin film hydration method, ACLEP exhibited suitable particle sizes (184.8 ± 5.87 nm), narrow PDI (0.200 ± 0.012), applicable zeta potentials (33.55 ± 0.64 mV), superior oxygen content (0.406 ± 0.14 mg/ml) and acceptable biostability.
Fig. 2.

Schematic illustration of the mechanisms of action of ACLEP to overcome hypoxia-triggered erlotinib resistance. ACLEP could achieve oxygen and drug co-delivery and regulate the expression of HIF-1a in hypoxic microenvironment in both EGFR-wild and EGFR-mutated NSCLC.
Hypoxia-inducible factor-1α (HIF-1α), a subunit of HIF-1, could be responsively up-regulated when tumor cells are exposed to a hypoxic environment, promoting angiogenesis and tumor growth [49], [50]. Previous investigations have reported that hypoxia significantly increased the population of tumor cells resistant to EGFR-TKIs in NSCLC with up-regulated HIF-1α and EGFR expression [51], [52]. The 50% inhibitory concentration (IC50) values of ETB against A549, PC-9 and H1975 NSCLC cell lines were 5.65-,6.12- and 3.28- fold increase in hypoxic condition. in vitro studies, compared to ordinary liposomal ETB this novel formulation, ACLEP, significantly inhibited cell proliferation, induced apoptosis, and down-regulated the expression of the protein EGFR, p-EGFR andHIF-1α in these NSCLC cells. Authors suggested that the enhanced anti-tumor efficacy was mainly associated with specific binding to overexpressing EGFR receptor of NSCLC cells guided by the anti-EGFR Apt and thereby efficiently delivery ETB and oxygen into hypoxic cells.
Manifested anti-tumor efficacy of ACLEP was evidenced by reduced tumor growth in A549-bearing mice. The in vitro and in vivo results consistently evidenced that the drug and oxygen co-delivery approach could be an effective method to modulate the solid tumor microenvironment and improve the efficacy of chemotherapy.
3.2. Protein-based nanocarriers
Protein-based DDS have been exploited and utilized for decades due to their biocompatible, non-immunogenic and biodegradable properties coupled with low toxicity [53], [54].
In biological systems ferritin (FT) is used to store iron, and apoferritin (apo-FT) is formed when the iron atoms are removed from FT. FT/apo-FT protein cage is composed of 24 subunits which assemble into a 12 nm diameter cage with an internal 8 nm hollow span. This protein cage also has 14 channels to allow the exchange of contents between the interior and exterior environments [55], [56]. In many malignant tumor cells, such as brain and breast cancer cells, transferrin receptors (TfR) highly express, which can mediate membrane-specific endocytosis. Also, FT and apo-FT can disassemble into subunits at low pH (< 2) allowing the release of cargo, and reassemble at higher pH (> 8.5) [57]. Due to its particular structure, reversible assembly/disassembly behavior and targeting ability, FT/ apo-FT has been studied as an interesting nanocarrier.
In a study by Anchalam et al. [58], ten molecules of GEF were passively loaded in each apo-FT molecule with a small particle size of 12 nm. This nanocarrier was utilized to target TfR positive SKBR3 cell lines. Using the fluorescent property of GEF, they observed facilitated intracellular drug accumulation in the apo-FT-encapsulated-GEF group from confocal microscopy and flow cytometry. in vitro, the cytotoxicity of apo-FT-encapsulated-GEF was much higher than that of GEF alone, which resulted from increased endocytosis and PH-responsive controlled drug release.
In recent years, albumin also attracts substantial interest as an attractive carrier due to its properties including high stability, nontoxicity, biocompatibility and biodegradability. It can also be preferentially taken up by tumors cells as a nutrient. In addition, the existence of charged functional groups, like amino and carboxylic groups, offers albumin with chances to interact with a wide variety of therapeutic agents [59], [60], [61].
Wan et al. [62] prepared LAPA-loaded human serum albumin (HSA) nanoparticles (LHNP) with a narrow size distribution of 140 nm and good EE of nearly 85% for intravenous administration by employing nanoparticle albumin-bound technology (Nab™). Nab™ developed by American Bioscience, Inc. is an innovative technique for preparing hydrophobic drugs loaded albumin nanoparticles (NPs) by passing through the aqueous mixture of therapeutic drug molecules and albumin under high pressure through a jet [63]. The LHNPs displayed enhanced stimulation of apoptosis in 4T1 monolayer cells and facilitated penetration and inhibitory efficacy in tumor spheroids relative to LAPA solution. Intravenous injection of LHNPs in mice led to 16-fold increased tumor accumulation compared to Tykerb™, the commercial tablets of LAPA for oral administration, and prevented lung metastasis at one-tenth dose of Tykerb™.
Noorani and co-workers developed ETB-loaded albumin nanoparticles (E-ANPs), by desolvation method with a mixed solvent followed by thermal cross-linking for stabilization, which is one of the best approached to achieve stable biodegradable structure [64], [65]. The in vitro cytotoxicity of E-ANPs against ASPC-1 and PANC-1 pancreatic cancer was higher than ETB solution at 2.9 and 3.0-fold, respectively. Authors suggested that albumin encapsulation boosted the specific endocytic uptake of ETB, which thereby promoted antiproliferative effects.
E-ANPs seem to be a promising therapeutic agent for pancreatic cancer, and in vivo tests for further evaluation are needed.
3.3. Inorganic nanocarriers
In past decades, inorganic nanocarriers such as silica, gold, graphene and carbon nanotubes have been exploited as drug delivery vehicles, on account of their versatile physicochemical properties including biocompatibility, low cytotoxicity, readily availability, easily functionalization and ability to accumulate in cancer cells without P-Glycoprotein (P-gp) recognition, causing increased intracellular concentration of drugs [66], [67]. Though their delivery efficiency appears to be lower than that of cationic carriers and viral at present, inorganic nanocarrier with modified surface employing bio-functional molecules like antibodies, aptamers and proteins can effectively reduce the gap [43].
Silica exists in many forms such as silica particle, nanotube, hollow silica particle, mesoporous silica nanoparticle (MSN), and hollow mesoporous silica nanoparticle (HMSN) of which the latter two are more promising in the field of nano-DDS. Large surface areas, high pore volume, high chemical stability, high drug-loading capability and easily functionalized surface are the main advantages of HMSN/MSN in the field of nanocarriers [68].
Wang et al. [69] selected cetuximab, the anti-EGFR monoclonal antibody, as targeting ligand to maximum specific uptake of drug loaded MSN by EGFR overexpressing tumor cells. Subsequently, they developed cetuximab-modified MSNs (CET-capped MSNs) through the cross-linking of disulfide bond. This unique MSNs were designed as drug delivery platforms to simultaneously, specifically release DOX and GEF to targeting tumor cells with high EGFR expression. The researchers reported that in PC9 cells, a GEF resistant NSCLC cell line, CET-capped MSNs facilitated the endocytosis and reached high intracellular accumulation in a time-dependent manner, which was confirmed through flow cytometry analyses. in vitro and in vivo, CET-capped GEF/DOX MSN was showed a superior inhibitory effect for PC9 NSCLC cells relative to ordinary co-loaded MSNs. All the findings corroborate that this unique DDS might serve as a great strategy to overcome EGFR-TKI resistance and be a potential drug candidate for the management of EGFR-mutant lung cancer. After being endocytosed, CET-capped MSN could release loaded drugs via the cleavage of disulfide bond through the interaction with the high level of GSH in cytoplasm of cancer cells.
As compared with the approach of active targeting ligand modification, the nano-systems using pH value of tumor microenvironment for improved cellular internalization is easy to prepare and can be applied for the treatment of all kinds of tumor [70].
With this background, He et al. [71] successfully developed pH-responsive charge-reversal MSNs to delivery synergistic ETB and DOX combination with sequential release manner for the treatment of NSCLC (Fig. 3). Besides, the study by Lee et al. emphasized the importance of a fixed period of time between the administration of each drug for maximizing the synergistic effect of combination chemotherapy [45].
Fig. 3.

Schematic illustration of preparation of ETB/DOX combination co-delivery nanocarriers and synergistic therapy of ETB and DOX. After administration, MSNs were positively charged at extracellular environment leading to an easy internalization by tumor cells. As ETB was loaded in the exterior lipid bilayer and the controlled releaseability of MSN, after entering into tumor cells ETB released faster than DOX.
This pH-sensitive charge-reversal lipid film consisted of SPC/ 1, 5-Dioctadecyl-l-glutamyl 2-histidyl-hexahydrobenzoic acid (HHG2C18) /Chol at a weight ratio of 3.75:1.25:1, which were able to reverse surface zeta potential from negative to positive at tumor acidic extracellular microenvironment, then facilitate the internalization for the targeted cancer cells. In this platform, ETB was released faster than DOX intracellularly, because the lipid membrane containing ETB was coated on the surface of DOX-loaded MSN. In vivo data showed that this ETB/DOX co-loaded pH-responsive charge-reversal MSNs inhibited tumor growth effectively with negligible systemic toxicity.
Gold nanoparticles (AuNPs), a kind of metallic nanoparticles, in sizes of 1–100 nm are extensively used for drug and gene delivery on account of its inertness, excellent biocompatibility and lower toxicity than other metal material. Besides, bioavailability, targeting ability and endocytosis of AuNP can be greatly improved with a functionalized surface [72], [73], [74]. In this context, one of the most commonly used compounds for modification of AuNPs is PEG which can be covalently bound to the surface atoms of AuNPs.
In a recent study, a DDS based on PEGylated AuNPs conjugated with AFT (PEG-AuNPs-A) was designed with a mean size of 41 nm, low polydispersity index PDI, zata potential of −35 mV and high store stability [75]. Coelho et al. tested the drug activity against pancreatic cancer cells (S2-013) and NSCLC (A549) by the evaluation of in vitro cytotoxicity and further identify the mechanisms via confocal imaging and flow cytometry. The results showed that 5 and 20 times AFT alone are required than PEG-AuNPs- A to induce 50% cell survival in S2-013 and A549 cells with a higher cellular uptake of PEG-AuNPs-A observed by cancer cells. All their findings reinforced that uptake and cytotoxicity of AFT in pancreatic and lung cancer cells can be highly enhanced by conjugated with PEG-AuNPs.
Smart stimuli-responsive DDS has provided a profound impact on preclinical and clinical applications. Of these systems, thermo-responsive DDS can achieve controllable drug release by applying heat whenever necessary. Poly(N-isopropylacrylamide) (PNIPAAm) regarding as thermo-sensitive polymers is generally used in the development of nanocarriers [76], [77], [78].
ETB encapsulated CS copolymer-gold hybrid (CGH) NPs as smart thermo-responsive nanocarriers was synthesized via autoreduction of auric cations [79]. In brief, oleic acid (OA) and (N-isopropylacrylamid) (NIPAAm) were copolymerized on the modified CS to obtain desired thermo-responsive copolymer and then CGH—NPs were formed through the reduction ability of amino functional groups of CS copolymer. ETB can be facilely loaded in CGH—NPs with the EE of 30% since PNIPAAm chains are hydrophobic. The ETB was released from the CGH—NPs in a thermo-responsive manner. Subsequently, flow cytometry analysis corroborated high cellular uptake (85.81%) of CGH—NPs by A549 lung cancer cells and the cytotoxicity evaluations proved (the) high anti-tumor activity of the ETB loaded CGH NPs with an excellent cytocompatibility.
3.4. Polymeric nanoparticles
Polymeric nanoparticles (NPs) can effectively provide more accumulation of drugs within the solid tumor tissues because of nanoscale induced EPR and greater adsorption to cells. They are also able to improve the pharmacodynamic and pharmacokinetic properties of various bioactive molecules to overcome part of the limitations in traditional formulations [80].
Poly (D,L-lactic-co-glycolic acid) (PLGA) is a copolymer approved by the FDA for medical applications because it can undergo hydrolysis in the body and produce the biodegradable metabolite monomers, glycolic acid and lactic acid [81]. PLGA-NP is one of the excellent approaches for improving bioavailability and enhancing the treatment efficacy of EGFR-TKIs. A study in rat showed that ETB loaded PLGA-NPs enhanced the oral bioavailability and reduced subacute toxicity of ETB, and this can be attributed to the improved pharmacokinetics, sustained release and a longer residence time [82].
Recently, an original synthetic method to obtain ETB-loaded PLGA-NPs was developed by Vaidya et al. [83]. The authors suggested that by preparing PLGA-NPs with multiple emulsion solvent evaporation methods with water-soluble cyclodextrin complex, the entrapment of ETB in the NPs increased almost 3-fold than reported single emulsion method. The obtained PLGA-encapsulated ETB NPs (PLGA-ETB) showed a mean size of 210 ± 8 nm, DL of 5% and sustained release profile. Developed NPs also demonstrated improved efficacy against NSCLC cells in terms of increased apoptosis, low IC50 values,and autophagy inhibition.
Polymeric NPs made from poly(ε-caprolactone)-poly(ethyleneglycol)-poly(ε-caprolactone) (PCEC) has also been utilized as a biocompatible nanocarrier to effectively delivery hydrophobic EGFR-TKIs. In the research by Xiao et al. [84], GEF-loaded PCEC (PCEC-GEF) NPs were developed via a solid dispersion method, with an average size of 24 nm, zeta potential of −18 mV, DL of 9% and EE of 92%. This PCEC-GEF NPs could slowly release GEF and within 5 days 80% cargo molecules were released in controlled and sustained behavior. This might be the reason why PCEC-GEF has higher IC50 against lung cancer cells (A549) in vitro after 24 and 48 incubation than GEF solution. In vivo, the experimental results showed that the intravenous injection of PCEC-GEF NPs led to a significantly longer delay on tumor growth, lower GEF-related side effects and an increased median survival time of approximately 23 d compared with GEF treated group. Subsequent flow cytometry and evaluation of the expression of Ki-67 and CD31 was conducted in order to further investigated the mechanism of anticancer action of PCEC-GEF NPs in vivo. The results suggested this GEF-NPs could effectively increase cellular apoptosis, reduce proliferation and inhibit angiogenesis of solid tumor in a xenograft mouse model induced by lung cancer cells.
Natural polymers such as chitosan, a hydrophilic polysaccharide, is a natural polymer [85]. Similar to other biopolymers, chitosan has validated biodegradability, biocompatibility, non-immunogenicity, high mucoadhesion, fungistatic and antimicrobial activity [86], [87].
Autophagy, a basic catabolic mechanism, involves cell degradation of dysfunctional or unnecessary cellular components through the actions of lysosomes. During the autophagic process, the autophagosome and lysosome were fused to develop autophagic lysosome, degrading deformed molecules in cytoplasm and organelles, and then recycling the degraded product to provide nutritive material for the survival of cells [88], [89], [90]. According to recent investigations, autophagy may take dominated part in promoting acquired drug resistance of cancer cells. Autophagy facilitates cancer cells to survive in the adverse environment by accelerating the growth of cancer cells and preventing antitumor agents from killing cells [91], [92], [93].
CS-NPs entrapping both GEF and chloroquine (CQ), a known inhibitor of autophagic lysosome formation, were fabricated by Zhao et al. and their ability to enhance the delivery of anticancer agents against acquired MDR tumor cells was validated [94]. Western blot analysis also confirmed that the ratio of LC3I to LC3II as an autophagosome marker was decreased and expression of caspase-3 protein as the main apoptosis relevant protein was elevated.
3.5. Polymeric micelles
Polymeric micelles (PMs), a fast-growing area in the field of drug delivery, are composed of hydrophilic and hydrophobic functional groups which can self-assemble in aqueous media when the concentration of amphiphilic copolymers exceeds critical micelle concentration (CMC) [95], [96]. Also, micelles are the core-shell structure where the core is formed by the hydrophobic part and the shell is formed by the hydrophilic part. This structure is more stable than liposomes with a smaller size range and provides the possibility to simultaneously encapsulate multiple therapeutic agents with different physicochemical properties. Concomitantly, the PEGylated shell prevents the uptake of PMs by (the) reticuloendothelial system (RES). Drugs may passively be loaded or covalently attached to the PMs depending on the preparation. These intrinsic and modifiable properties of PMs make them primely suited for drug delivery applications [97].
An injectable formulation, AFT-loaded PM, consisted of AFT/ MPEG-PCL / Mal-PEG-PCL at a ratio of 1:9:1 was successfully fabricated by film hydration method [98]. In vitro, the experimental findings indicated that AFT-encapsulated PMs showed more cytotoxicity than free AFT solution did. The improved pharmacokinetics, tissue-distribution as well as the enhanced anticancer activity of AFT-encapsulated PMs were confirmed with HER2-overexpressing HCT-15 cell line induced colon tumor model.
Fathi et al. [99] synthesized CS based micelles grafted with PNIPAm as a temperature-sensitive monomer and oleic acid as hydrophobic segments. The PMs were modified with folic acid and loaded with ETB for cancer-specific targeting drug delivery. Folate-(PNIPAAm-co-OA)-g-CS micelles were obtained through a self-assembly process resulting in stable core-shell nanostructure with a mean diameter of 100 nm and zeta potential of −10 mV. The ETB loaded PMs showed good EE of nearly 40%, excellent biocompatibility,and a temperature-responsive release profile. In cellular uptake and cytotoxicity assays in vitro, a specific targeting behavior and enhanced cytotoxic efficacy of ETB-loaded folate-(PNIPAAm-co-OA)-g-CS micelles were observed in the folate-positive OVCAR-3 ovarian cells, which indicated the potential application of this micellar system as an injectable formulation in targeting therapy of folate positive cancer cells.
Several evidences have indicated that aberrant activation and cross-talk in sonic Hedgehog and EGFR signaling pathways play major roles in pancreatic carcinogenesis, disease progression, metastasis, and chemoresistance [100], [101], [102]. However, the low aqueous solubility of GEF and cyclopamine (CYP), a hedgehog pathway inhibitor, greatly limited their effective clinical translations as a promising approach. To resolve this issue, Chitkara et al. co-loaded GEF and CYP into PEG-b-poly(carbonate-co-lactic acid) (PEG-b-p(CB-co-LA)) micelle efficiently using the nanoprecipitation method [103]. The micelles displayed uniformed spheres with an average diameter of 54 nm and low PDI. In vitro, compared with free GEF and CYP, GEF/CYP co-loaded PMs showed a synergistic effect against pancreatic cancer cells and specifically increased Caspase 3/7 activity, indicating apoptotic cell death, compared with free GEF and CYP. Also, this co-delivery micellar system decreased tumor growth rate in the xenograft mouse model induced by pancreatic cancer cells.
In clinic, the prolonged use of chemotherapeutic agents inevitably induces multiple-drug resistance (MDR) and eventually lead to therapy failures [104]. This is mainly because of the overexpression of MDR transporters, the ATP-binding cassette (ABC) superfamily, which can increase drug efflux and decrease drug accumulation in tumor cells [105], [106]. To date, three subtypes of MDR efflux pumps in cancer have been characterized: P-glycoprotein (P-gp; MDR1), breast cancer resistance protein (BCRP) and multidrug resistance-associated protein 1 (MRP1). Researches have indicated that LAPA could inhibit the function of ABC transporters and thus sensitize MDR tumor cells to chemotherapeutic drugs [107], [108].
With this background, Wang et al. [109] synthesized Poly(ethylene-glycol)-blockpoly(2-methyl-2-benzoxycarbony-lpropylene carbonate) (PEG-PBC) polymers for fabricating LAPA and DOX co-loaded PMs by film dispersion method. The low critical micelle concentration (CMC) of PEG-PBC (1.5 mg/L) with an average diameter of 100 nm implied its high dynamic stability. MCF-7/ADR cells is a drug resistant BC cell line with overexpressed P-gp. In vitro, researchers observed almost no intracellular accumulation of DOX after 4 h incubation with free Dox solution in MCF-7/ADR cells. Cytotoxicity tests also demonstrated that this co-delivery system notably increased anticancer activity of DOX in drug resistant BC cell lines. Moreover, the DOX-LAPA-PMs showed a significant decrease in tumor growth compared to DOX mono-therapy in the xenograft mouse model.
Similarly, Li et al. [110] synthesized the di-block polymer, poly(ethylene glycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate-graft-dodecanol) (PEG–PCD) to develop LAPA and PTX co-encapsulated PMs for the treatment of MDR prostate cancer. The obtained PMs showed a mean size of 60 nm with nearly 100% EE of both drugs. Results from experiments confirmed that polymer PEG-PCD efficiently delivered a combination of LAPA and PTX to prostate cancer cells in vitro and in vivo with negligible cellular and systemic toxicity. In addition, with the inhibitory effect of LAPA on P-gp function, the toxicity of PTX dramatically enhanced in P-gp overexpressing prostate cancer cells.
The overexpressed HER2 in BC is associated with poor prognosis and chemotherapy resistance, thus targeted therapy aiming at HER2 is regarded as an important strategy for BC treatment. Combining LAPA with other traditional chemotherapies such as PTX has revealed a higher antitumor efficacy in recent years, for example, two phase III studies were conducted to investigate the combination effect of LAPA plus PTX against HER2-positive metastatic BC, and ultimately the combination therapy offered a significant survival advantage over PTX mono-therapy [111], [112].
Wei et al. designed a PMs to better simulate clinical administration, long-term release of LAPA and short-term release of PTX, against HER2-positive BC [113]. In order to enhance the incorporation of LAPA in micelles, researchers firstly synthesized LAPA-conjugated poly (ethylene glycol) (PEG) and poly (lactic acid) (PLA) (L-PEG-PLA). Subsequently, micelles were prepared by thin film hydration method, entrapping PTX during the process, with a high drug loading capacity. Findings of cytotoxicity studies, employing HER2-positive BC cell line (SKBr-3), exhibited the enhancement of anticancer efficacy of PTX with sustaining action of LAPA, as evidenced by the reduction in IC50 value of co-delivery of PMs. Also, the results of apoptosis assay were consistent with findings of in vitro cytotoxicity, as increased apoptosis index was observed in cells in combined PMs
3.6. Polymeric implants
Polymeric implants, capable of encapsulating small molecule substances in a polymeric matrix, have exhibited notable potential for delivery of various bioactive molecules including therapeutic drugs, proteins, and gene with a controlled rate. Polymeric implants can protect encapsulated drugs from degradation and provide in situ treatment to specific anatomic sites with a continuous sustained release of drugs and minimized systemic exposure [114], [115]. LAPA is characterized by poor oral bioavailability and high binding tendency to plasma albumin resulting in a meager potion of oral dose to reach the desired site. For avoiding the risk and cost from the high oral dose, localized delivery of LAPA is expected to obtain the same anticancer efficacy with much less dose of LAPA.
Hu et al. [116] designed another DDS to imitate the clinical administration of oral LAPA and injectable PTX. PTX-NPs and microparticles of LAPA (LAPA-MPs) were encapsulated in a thermosensitive injectable hydrogel made from Pluronic F127. The results of cytotoxicity assays revealed the most synergistic effect between LAPA and PTX against the HER2 and P-gp overexpressing BC cell lines. Furthermore, localized sustained drug release and promising combinational effects with very low dose of LAPA brought impressing anti-tumor effect in vivo. In addition, gel group showed less toxicity, which caused less body weight loss and less tissue injury. Daily oral administration of high dose of LAPA also resulted in more drug accumulation in some key tissues like liver, heart and lung. The drug concentration in tumors tissues was detected between gel and oral groups at different time point. PTX in both groups had a faster release than LAPA at first, and the high concentration maintained less than one week. However, the changing pattern of LAPA residue in tumor site was quite different between these two combinational groups. Although the dose of oral LAPA was very high, the amount of LAPA in oral group was very low at beginning, and increased slowly with time. While in gel group with very low dose of LAPA, the primary amount of LAPA was relatively high and kept at a steady level during the detection time.
3.7. Other nano-formulations
Agrawal et al. [117] designed HA-coated lapatinib nanocrystals via high-pressure homogenization technique. Hyaluronic acid (HA), a natural hydrophilic polysaccharide, not only extends the circulation time by evading RES-mediated clearance but also participates in tumor targeting. Cluster of differentiation 44 (CD44), a marker of a wide range of malignant cells. is a critical cell surface receptor for HA. [118], [119] In vivo trials, these HA-coated nanocrystals showed enhanced endocytosis. It also reported that the higher uptake of LAPA encapsulated HA coated nanocrystals (LAPA-HA-NCs) resulted in higher apoptosis and disruption of mitochondrial membrane potential in MDA-MB-231 cells in concentration and time dependent manner. One of the reasons for apoptosis induction is the dysfunction of mitochondria which results due to the opening of mitochondrial permeability pore and releases apoptogenic factors causing loss of oxidative phosphorylation which result in cell death (Fig. 4). In this study, sufficient evidences were provided, which supported and demonstrated the magnificent tumor targeting capability of LAPA-HA-NCs against Triple-Negative Breast Cancer (TNBC). Overall, the administration of LAPA-HA-NCs can be an effective approach in the treatment of TNBC and its metastasis.
Fig. 4.

Enhanced delivery of LAPA in the form of HA coated nanocrystals formulation against triple negative breast cancer. External coating of HA actively targeted extracellular CD44 receptor thereby causing preferential accumulation of LAPA around the tumor cells. Intracellularly, LAPA led to programmed cell death, apoptosis, through the series of activation of apoptosis markers.
In a similar manner to liposomes, polymeric nanovesicles also can physically encapsulate hydrophilic agents in the aqueous cores and hydrophobic agents in the laminar membranes. As shown in Fig. 5, Chen et al. [120] designed a smart pH- and redox-responsive polymeric nanovesicle based on a copolymer of PEG and a polypeptide derivative, P(Asp(DBA-coMEA)-b-Phe. Lipophilia GEF and hydrophilic DOX can be simultaneously encapsulated in the shell and core of these nanovesicles via double-emulsion solvent evaporation method and the vesicles incorporating dual drugs displayed prominent pH/redox sensitivities to trigger the release of GEF and DOX inside cancer cells. This approach manifested a synergistic anticancer effect both in vitro and in vivo. Particularly, a remarkable therapeutic effect was observed in vivo in N2a cell line induced neuroblastoma model.
Fig. 5.

Illustrative preparation of nanovesicle as well as the dual sensitive release of DOX and GEF inside tumor cell.
To overcome the limitations of both polymeric and lipid nanocarriers, Mandal et al. designed core-shell type lipid-polymer hybrid nanoparticles (LPNs) for the delivery of ETB [121]. In this innovative DDS, lipid membranes can improve permeation and cellular internalization, and the polymeric cores are able to control the drug release behavior. ETB-encapsulated LPNs (E-LPNs) were prepared by single-step sonication method as the polymeric core and phospholipid-shell structure consisting of 1,2-distearoyl-sn‑glycero-3-phosphoethanolamine-N-[methoxy polyethyleneglycol)−2000] (DSPE-PEG2000) and hydrogenated soy phosphatidylcholine (HSPC). The optimized E-LPNs were obtained with a mean size of about 170 nm, narrow PDI below 0.2, drug EE of about 66% and excellent storage stability. Cellular uptake and colony formation assays were conducted to evaluate the efficacy of E-LPNs in NSCLC cells. As compared with ETB solution, the results revealed enhanced cellular internalization and anticancer effects of E-LNPs. Therefore, LNPs might be a potential tool for ETB delivery in cancer treatment.
Li and co-workers reported an innovative dual drug delivery platform based on disulfide-bridged quercetins (QSSQ) which was synthesized by modification of multiple phenolic hydroxyl groups on quercetin [122]. Some research has manifested that linking two hydrophobic monomers with disulfide bridge can facilitate the self-assembly of molecules into NPs. In this study, QSSQ successfully self-assembled into NPs and meanwhile encapsulated ETB during the process to form ETB-encapsulated NPs (E-QSSQ). As a natural product of flavonoids with diverse biological activities, quercetin exhibits antitumor effects through modulating several key elements located downstream of EGFR signaling pathways [123], [124], [125]. Additionally, the overexpressed glutathione (GSH) in tumor tissue can trigger the release of erlotinib and quercetin via cleaving the disulfide bond in QSSQ. In vitro, a lower IC50 value (4.1 × 10−6 M) and a higher percentage of apoptosis (28.5%–75.6%) against A549 cells for E-QSSQ than free ETB (8.7 × 10−6 M, 13.1%–58.0%), respectively. E-QSSQ, meanwhile, showed a high antitumor activity in a xenograft model of NSCLC in vivo. These results consistently corroborated that E-QSSQ was able to block the autophosphorylation at both upstream and downstream of EGFR signaling pathways, and therefore interrupt the EGFR signaling more potently, and eventually prevent the growth of EGFR-overexpressing tumor cells.
4. Conclusion and outlook
Over the past 20 years, great efforts have been made to design various inhibitors targeting EGFR family in cancer treatment. These unique small molecular inhibitors for EGFR have brought clinical benefits in various types of human malignancies.
However, the drawbacks related to the inherent property of these small molecule agents couldn't be ignored, including poor specificity, unexpected pharmacokinetics and biodistribution, which generally exert more serious damage to normal tissues, causing increased toxicity and side effects.
Supported by the rapid development of nanotechnology, EGFR-TKIs loaded nanocarriers show substantial effects on increasing solubility, prolonging systemic circulation, elevating accumulation in tumor site and reducing distribution in the normal tissues, which lead to significant enhancement on therapeutic efficacy. And more co-delivery systems were designed for simultaneously encapsulating multiple anticancer agents and controlling drug release in a predictable manner to maximize the synergistic effects. Though current results on EGFR-TKIs nanosized formulation to date seems hopeful in preclinical researches, there are still many challenges that are required to be addressed in order to attain clinical potential.
First of all, the ability of nanocarriers to hold the cargoes and maintain the dosage ratio from the administration site to the final targets is also challenging. The difference in the encapsulation capacities of the nanocarriers results in inconsistent tendencies in premature leakage during the delivery process. Accordingly, for the future development of EGFR-TKIs DDS detecting the final drug concentration at the target site is a prerequisite. Secondly, the multifunctionality of nanocarriers ensure more efficient of targeting delivery, while it brings about complexity of material synthesis and difficulty of biodegradation. This suggests that the future material design should focus on multifunctionality, optimized synthesis, biodegradation and safety of primary/secondary metabolites etc. Thirdly, challenge for nanocarriers is targeting delivery EGFR-TKIs to a specific cell. The difficulty lies in the efficient recognition of a targeted cell among thousands of untargeted cells. To date, most of research efforts at present mainly focused on the delivery efficiency in one or two cell lines. However, the cellular delivery targeting single cell line in a mixture of different cell types is desired and should be pursued. Lastly, multifarious tumor cell-associated endogenous stimuli have already been applied for controlled drug release in a programmed manner. However, these stimuli, such as redox potential or acidity change due to individual differences. The most efficacious stimuli should be taken into account for clinical practice.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Acknowledgments
This work was financially supported by the National Natural Science Foundation (31525009 and 31771096), The National Key Research and Development Program of China (2017YFC1103502), Sichuan Innovative Research Team Program for Young Scientists (2016TD0004), Distinguished Young Scholars of Sichuan University (2011SCU04B18), and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajps.2019.06.001.
Appendix. Supplementary materials
References
- 1.Roskoski R., Jr The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Wang X., Batty K.M., Crowe P.J., Goldstein D., Yang J.L. The potential of panHER inhibition in cancer. Front Oncol. 2015;5:2. doi: 10.3389/fonc.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yewale C., Baradia D., Vhora I., Patil S., Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- 4.Carpenter R.L., Lo H.W. Dacomitinib, an emerging HER-targeted therapy for non-small cell lung cancer. J Thorac Dis. 2012;4(6):639–642. doi: 10.3978/j.issn.2072-1439.2012.10.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patra C.R., Cao S., Safgren S., Hattacharya R., Ames M.M., Shah V. Intracellular fate of a targeted delivery system. J Biomed Nanotechnol. 2008;4(4):508–514. [Google Scholar]
- 6.Herbst R.S., Giaccone G., Schiller J.H., Natale R.B., Miller V., Manegold C. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 2. J Clin Oncol. 2004;22(5):785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 7.Thatcher N., Chang A., Parikh P., Rodrigues Pereira J., Ciuleanu T., von Pawel J. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa survival evaluation in lung cancer) Lancet. 2005;366(9496):1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 8.Moore M.J., Goldstein D., Hamm J., Figer A., Hecht J.R., Gallinger S. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of canada clinical trials group. J Clin Oncol. 2007;25(15):1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 9.Senderowicz A.M., Johnson J.R., Sridhara R., Zimmerman P., Justice R., Pazdur R. Erlotinib/gemcitabine for first-line treatment of locally advanced or metastatic adenocarcinoma of the pancreas. Oncology (Williston Park) 2007;21(14):1696–1706. discussion 1706-1699, 1712, 1715. [PubMed] [Google Scholar]
- 10.Di Leo A., Gomez H.L., Aziz Z., Zvirbule Z., Bines J., Arbushites M.C. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol. 2008;26(34):5544–5552. doi: 10.1200/JCO.2008.16.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budha N.R., Frymoyer A., Smelick G.S., Jin J.Y., Yago M.R., Dresser M.J. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–213. doi: 10.1038/clpt.2012.73. [DOI] [PubMed] [Google Scholar]
- 12.Burris H.A., 3rd, Taylor C.W., Jones S.F., Koch K.M., Versola M.J., Arya N. A phase I and pharmacokinetic study of oral lapatinib administered once or twice daily in patients with solid malignancies. Clin Cancer Res. 2009;15(21):6702–6708. doi: 10.1158/1078-0432.CCR-09-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roskoski R., Jr Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395–411. doi: 10.1016/j.phrs.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Aggarwal S., Yadav S., Gupta S. EGFR targeted PLGA nanoparticles using gemcitabine for treatment of pancreatic cancer. J Biomed Nanotechnol. 2011;7(1):137–138. doi: 10.1166/jbn.2011.1238. [DOI] [PubMed] [Google Scholar]
- 15.Meanwell N.A. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24(9):1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- 16.Tartarone A., Lerose R. Clinical approaches to treat patients with non-small cell lung cancer and epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance. Ther Adv Respir Dis. 2015;9(5):242–250. doi: 10.1177/1753465815587820. [DOI] [PubMed] [Google Scholar]
- 17.Gazdar A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28 Suppl 1:S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naseri N., Valizadeh H., Zakeri-Milani P. Solid lipid nanoparticles and nanostructured lipid carriers: structure, preparation and application. Adv Pharm Bull. 2015;5(3):305–313. doi: 10.15171/apb.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marslin G., Sheeba C.J., Kalaichelvan V.K., Manavalan R., Reddy P.N., Franklin G. Poly(D,L-lactic-co-glycolic acid) nanoencapsulation reduces erlotinib-induced subacute toxicity in rat. J Biomed Nanotechnol. 2009;5(5):464–471. doi: 10.1166/jbn.2009.1075. [DOI] [PubMed] [Google Scholar]
- 20.Hatziagapiou K., Bethanis K., Lambrou G.I., Yannakopoulou K., Karpusas M., Braoudaki M. Enhanced gefitinib cytotoxicity in the presence of Cyclodextrins: in vitro and biophysical studies towards potential therapeutic interventions for cancer. J Biomed Nanotechnol. 2017;13(5):522–533. [Google Scholar]
- 21.Matsumura Y., Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 22.Da Silva C.G., Peters G.J., Ossendorp F., Cruz L.J. The potential of multi-compound nanoparticles to bypass drug resistance in cancer. Cancer Chemother Pharmacol. 2017;80(5):881–894. doi: 10.1007/s00280-017-3427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen M.H., Williams G.A., Rajeshwari S., Gang C., W David M., David M. United States food and drug administration drug approval summary: gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10(4):1212–1218. doi: 10.1158/1078-0432.ccr-03-0564. [DOI] [PubMed] [Google Scholar]
- 24.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 25.Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paez J.G., Janne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 27.Thongprasert S., Duffield E., Saijo N., Wu Y.L., Yang J.C., Chu D.T. Health-related quality-of-life in a randomized phase III first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients from asia with advanced NSCLC (IPASS) J Thorac Oncol. 2011;6(11):1872–1880. doi: 10.1097/JTO.0b013e31822adaf7. [DOI] [PubMed] [Google Scholar]
- 28.Zhou C., Wu Y.L., Chen G., Feng J., Liu X.Q., Wang C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):710–711. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 29.Rosell R., Carcereny E., Gervais R., Vergnenegre A., Massuti B., Felip E. Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 30.Gullick W.J., Srinivasan R. The type 1 growth factor receptor family: new ligands and receptors and their role in breast cancer. Breast Cancer Res Treat. 1998;52(1–3):43–53. doi: 10.1023/a:1006107016969. [DOI] [PubMed] [Google Scholar]
- 31.Waks A.G., Winer E.P. Breast cancer Treatment: a review. JAMA. 2019;321(3):288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 32.Kumler I., Tuxen M.K., Nielsen D.L. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev. 2014;40(2):259–270. doi: 10.1016/j.ctrv.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Qin R., Ibrahim A., Cohen M.H., Johnson J., Ko C., Sridhara R. FDA drug approval Summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. 2008;13(10):1114–1119. doi: 10.1634/theoncologist.2008-0816. [DOI] [PubMed] [Google Scholar]
- 34.Cameron D., Casey M., Oliva C., Newstat B., Imwalle B., Geyer C.E. Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial. Oncologist. 2010;15(9):924–934. doi: 10.1634/theoncologist.2009-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan C.S., Cho B.C., Soo R.A. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. doi: 10.1016/j.lungcan.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Lee J.K., Shin J.Y., Kim S., Lee S., Park C., Kim J.Y. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013;24(8):2080–2087. doi: 10.1093/annonc/mdt127. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y.L., Zhou C., Hu C.P., Feng J., Lu S., Huang Y. Afatinib versus cisplatin plus gemcitabine for first-line treatment of asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 38.Sequist L.V., Yang J.C., Yamamoto N., O'Byrne K., Hirsh V., Mok T. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 39.Bozzuto G., Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975–999. doi: 10.2147/IJN.S68861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroeter A., Engelbrecht T., Neubert R.H.H., Goebel A.S.B. New nanosized technologies for dermal and transdermal drug Delivery. A review. J Biomed Nanotechnol. 2010;6(5):511–528. doi: 10.1166/jbn.2010.1149. [DOI] [PubMed] [Google Scholar]
- 41.Doktorova M., Heberle F.A., Eicher B., Standaert R.F., Katsaras J., London E. Preparation of asymmetric phospholipid vesicles for use as cell membrane models. Nat Protoc. 2018;13(9):2086–2101. doi: 10.1038/s41596-018-0033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou X., Tao H., Shi K.H. Development of a nanoliposomal formulation of erlotinib for lung cancer and in vitro/in vivo antitumoral evaluation. Drug Des Devel Ther. 2018;12:1–8. doi: 10.2147/DDDT.S146925. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Siafaka P.I., Ustundag Okur N., Karavas E., Bikiaris D.N. Surface modified multifunctional and stimuli responsive nanoparticles for drug Targeting: current status and uses. Int J Mol Sci. 2016;17(9):1440. doi: 10.3390/ijms17091440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Almurshedi A.S., Radwan M., Omar S., Alaiya A.A., Badran M.M., Elsaghire H. A novel pH-sensitive liposome to trigger delivery of afatinib to cancer cells: impact on lung cancer therapy. J Mol Liq. 2018;259:154–166. [Google Scholar]
- 45.Lee M.J., Ye A.S., Gardino A.K., Heijink A.M., Sorger P.K., MacBeath G. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149(4):780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morton S.W., Lee M.J., Deng Z.J., Dreaden E.C., Siouve E., Shopsowitz K.E. A nanoparticle-based combination chemotherapy delivery system for enhanced tumor killing by dynamic rewiring of signaling pathways. Sci Signal. 2014;7(325):ra44. doi: 10.1126/scisignal.2005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F., Mei H., Xie X., Zhang H., Liu J., Lv T. Aptamer-Conjugated chitosan-anchored liposomal complexes for targeted delivery of erlotinib to EGFR-Mutated lung cancer cells. AAPS J. 2017;19(3):814–826. doi: 10.1208/s12248-017-0057-9. [DOI] [PubMed] [Google Scholar]
- 48.Li F., Mei H., Gao Y., Xie X., Nie H., Li T. Co-delivery of oxygen and erlotinib by aptamer-modified liposomal complexes to reverse hypoxia-induced drug resistance in lung cancer. Biomaterials. 2017;145:56–71. doi: 10.1016/j.biomaterials.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 49.Preethy P., Gordijo C.R., Abbasi A.Z., Azusa M., Angela I., Andrew Michael R. Correction to multifunctional Albumin-MnO2nanoparticles modulate solid tumor microenvironment by attenuating Hypoxia, Acidosis, vascular endothelial growth factor and enhance radiation response. ACS Nano. 2014;8(6):3202–3212. doi: 10.1021/nn405773r. [DOI] [PubMed] [Google Scholar]
- 50.Song X., Feng L., Liang C., Yang K., Liu Z. Ultrasound triggered tumor oxygenation with oxygen-shuttle nanoperfluorocarbon to overcome hypoxia-associated resistance in cancer therapies. Nano Lett. 2016;16(10):6145–6153. doi: 10.1021/acs.nanolett.6b02365. [DOI] [PubMed] [Google Scholar]
- 51.Minakata K., Takahashi F., Nara T., Hashimoto M., Tajima K., Murakami A. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012;103(11):1946–1954. doi: 10.1111/j.1349-7006.2012.02408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franovic A., Gunaratnam L., Smith K., Robert I., Patten D., Lee S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci USA. 2007;104(32):13092–13097. doi: 10.1073/pnas.0702387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawkins M.J., Soon-Shiong P., Desai N. Protein nanoparticles as drug carriers in clinical medicine. Adv Drug Deliv Rev. 2008;60(8):876–885. doi: 10.1016/j.addr.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y., Wang J., Wang J., Wang L., Tan X., Tu K. Aptamer functionalized cisplatin-albumin nanoparticles for targeted delivery to epidermal growth factor receptor positive cervical cancer. J Biomed Nanotechnol. 2016;12(4):656–666. doi: 10.1166/jbn.2016.2203. [DOI] [PubMed] [Google Scholar]
- 55.Maham A., Tang Z., Wu H., Wang J., Lin Y. Protein-based nanomedicine platforms for drug delivery. Small. 2009;5(15):1706–1721. doi: 10.1002/smll.200801602. [DOI] [PubMed] [Google Scholar]
- 56.Bhushan B., Kumar S.U., Matai I., Sachdev A., Dubey P., Gopinath P. Ferritin nanocages: a novel platform for biomedical applications. J Biomed Nanotechnol. 2014;10(10):2950–2976. doi: 10.1166/jbn.2014.1980. [DOI] [PubMed] [Google Scholar]
- 57.Truffi M., Fiandra L., Sorrentino L., Monieri M., Corsi F., Mazzucchelli S. Ferritin nanocages: a biological platform for drug delivery, imaging and theranostics in cancer. Pharmacol Res. 2016;107:57–65. doi: 10.1016/j.phrs.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Kuruppu A.I., Zhang L., Collins H., Turyanska L., Thomas N.R., Bradshaw T.D. An Apoferritin-based drug delivery system for the tyrosine kinase inhibitor gefitinib. Adv Healthc Mater. 2015;4(18):2816–2821. doi: 10.1002/adhm.201500389. [DOI] [PubMed] [Google Scholar]
- 59.Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132(3):171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Elzoghby A.O., Samy W.M., Elgindy N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. doi: 10.1016/j.jconrel.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 61.Neumann E., Frei E., Funk D., Becker M.D., Schrenk H.H., Muller-Ladner U. Native albumin for targeted drug delivery. Expert Opin Drug Deliv. 2010;7(8):915–925. doi: 10.1517/17425247.2010.498474. [DOI] [PubMed] [Google Scholar]
- 62.Wan X., Zheng X., Pang X., Zhang Z., Zhang Q. Incorporation of lapatinib into human serum albumin nanoparticles with enhanced anti-tumor effects in HER2-positive breast cancer. Colloids Surf B Biointerfaces. 2015;136:817–827. doi: 10.1016/j.colsurfb.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 63.Green M.R., Manikhas G.M., Orlov S., Afanasyev B., Makhson A.M., Bhar P. Abraxane, a novel Cremophor-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann Oncol. 2006;17(8):1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- 64.Noorani M., Azarpira N., Karimian K., Heli H. Erlotinib-loaded albumin nanoparticles: a novel injectable form of erlotinib and its in vivo efficacy against pancreatic adenocarcinoma ASPC-1 and PANC-1 cell lines. Int J Pharm. 2017;531(1):299–305. doi: 10.1016/j.ijpharm.2017.08.102. [DOI] [PubMed] [Google Scholar]
- 65.Storp B., Engel A., Boeker A., Ploeger M., Langer K. Albumin nanoparticles with predictable size by desolvation procedure. J Microencapsul. 2012;29(2):138–146. doi: 10.3109/02652048.2011.635218. [DOI] [PubMed] [Google Scholar]
- 66.Casals E., Puntes V.F. Inorganic nanoparticle biomolecular corona: formation, evolution and biological impact. Nanomedicine (Lond) 2012;7(12):1917–1930. doi: 10.2217/nnm.12.169. [DOI] [PubMed] [Google Scholar]
- 67.Xu Z.P., Zeng Q.H., Lu G.Q., Yu A.B. Inorganic nanoparticles as carriers for efficient cellular delivery. Chem Eng Sci. 2006;61(3):1027–1040. [Google Scholar]
- 68.Slowing I.I., Vivero-Escoto J.L., Wu C.W., Lin V.S. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Deliv Rev. 2008;60(11):1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y., Huang H.Y., Yang L., Zhang Z., Ji H. Cetuximab-modified mesoporous silica nano-medicine specifically targets EGFR-mutant lung cancer and overcomes drug resistance. Sci Rep. 2016;6:25468. doi: 10.1038/srep25468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee E.S., Gao Z., Bae Y.H. Recent progress in tumor pH targeting nanotechnology. J Control Release. 2008;132(3):164–170. doi: 10.1016/j.jconrel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He Y., Su Z., Xue L., Xu H., Zhang C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release. 2016;229:80–92. doi: 10.1016/j.jconrel.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 72.Nicol J.R., Dixon D., Coulter J.A. Gold nanoparticle surface functionalization: a necessary requirement in the development of novel nanotherapeutics. Nanomedicine (Lond) 2015;10(8):1315–1326. doi: 10.2217/nnm.14.219. [DOI] [PubMed] [Google Scholar]
- 73.Coelho S.C., Rocha S., Juzenas P., Sampaio P., Almeida G.M., Silva F.S. Gold nanoparticle delivery-enhanced proteasome inhibitor effect in adenocarcinoma cells. Expert Opin Drug Deliv. 2013;10(10):1345–1352. doi: 10.1517/17425247.2013.827659. [DOI] [PubMed] [Google Scholar]
- 74.Rosi N.L., Giljohann D.A., Thaxton C.S., Lytton-Jean A.K., Han M.S., Mirkin C.A. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312(5776):1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 75.Coelho S.C., Almeida G.M., Pereira M.C., Santos-Silva F., Coelho M.A. Functionalized gold nanoparticles improve afatinib delivery into cancer cells. Expert Opin Drug Deliv. 2016;13(1):133–141. doi: 10.1517/17425247.2015.1083973. [DOI] [PubMed] [Google Scholar]
- 76.Karimi M., Eslami M., Sahandi-Zangabad P., Mirab F., Farajisafiloo N., Shafaei Z. pH-Sensitive stimulus-responsive nanocarriers for targeted delivery of therapeutic agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8(5):696–716. doi: 10.1002/wnan.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karimi M., Sahandi Zangabad P., Ghasemi A., Amiri M., Bahrami M., Malekzad H. Temperature-Responsive smart nanocarriers for delivery of therapeutic Agents: applications and recent advances. ACS Appl Mater Interfaces. 2016;8(33):21107–21133. doi: 10.1021/acsami.6b00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L., Wang L., Guo B., Ma P.X. Cytocompatible injectable carboxymethyl chitosan/N-isopropylacrylamide hydrogels for localized drug delivery. Carbohydr Polym. 2014;103:110–118. doi: 10.1016/j.carbpol.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 79.Fathi M., Sahandi Zangabad P., Barar J., Aghanejad A., Erfan-Niya H., Omidi Y. Thermo-sensitive chitosan copolymer-gold hybrid nanoparticles as a nanocarrier for delivery of erlotinib. Int J Biol Macromol. 2018;106:266–276. doi: 10.1016/j.ijbiomac.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 80.Sahoo S.K., Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discov Today. 2003;8(24):1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- 81.Mir M., Ahmed N., Rehman A.U. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf B Biointerfaces. 2017;159:217–231. doi: 10.1016/j.colsurfb.2017.07.038. [DOI] [PubMed] [Google Scholar]
- 82.Marslin G., Sheeba C.J., Kalaichelvan V.K., Manavalan R., Reddy P.N., Franklin G. Poly(D,L-lactic-co-glycolic acid) nanoencapsulation reduces Erlotinib-induced subacute toxicity in rat. J Biomed Nanotechnol. 2009;5(5):464–471. doi: 10.1166/jbn.2009.1075. [DOI] [PubMed] [Google Scholar]
- 83.Vaidya B., Parvathaneni V., Kulkarni N.S., Shukla S.K., Damon J.K., Sarode A. Cyclodextrin modified erlotinib loaded PLGA nanoparticles for improved therapeutic efficacy against non-small cell lung cancer. Int J Biol Macromol. 2019;122:338–347. doi: 10.1016/j.ijbiomac.2018.10.181. [DOI] [PubMed] [Google Scholar]
- 84.Ni X.L., Chen L.X., Zhang H., Yang B., Xu S., Wu M. In vitro and in vivo antitumor effect of gefitinib nanoparticles on human lung cancer. Drug Deliv. 2017;24(1):1501–1512. doi: 10.1080/10717544.2017.1384862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nagpal K., Singh S.K., Mishra D.N. Chitosan nanoparticles: a promising system in novel drug delivery. Chem Pharm Bull. 2010;58(11):1423–1430. doi: 10.1248/cpb.58.1423. [DOI] [PubMed] [Google Scholar]
- 86.Younes I., Rinaudo M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar Drugs. 2015;13(3):1133–1174. doi: 10.3390/md13031133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J.J., Zeng Z.W., Xiao R.Z., Xie T., Zhou G.L., Zhan X.R. Recent advances of chitosan nanoparticles as drug carriers. Int J Nanomed. 2011;6:765–774. doi: 10.2147/IJN.S17296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Levine B., Packer M., Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lieberman A.P., Puertollano R., Raben N., Slaugenhaupt S., Walkley S.U., Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8(5):719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klionsky D.J., Baehrecke E.H., Brumell J.H., Chu C.T., Codogno P., Cuervo A.M. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7(11):1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dupere-Richer D., Kinal M., Menasche V., Nielsen T.H., Del Rincon S., Pettersson F. Vorinostat-induced autophagy switches from a death-promoting to a cytoprotective signal to drive acquired resistance. Cell Death Dis. 2013;4:e486. doi: 10.1038/cddis.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhai B., Hu F., Jiang X., Xu J., Zhao D., Liu B. Inhibition of akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13(6):1589–1598. doi: 10.1158/1535-7163.MCT-13-1043. [DOI] [PubMed] [Google Scholar]
- 93.Hassan B., Akcakanat A., Sangai T., Evans K.W., Adkins F., Eterovic A.K. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget. 2014;5(18):8544–8557. doi: 10.18632/oncotarget.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao L., Yang G., Shi Y., Su C., Chang J. Co-delivery of gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J Nanobiotechnology. 2015;13:57. doi: 10.1186/s12951-015-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Croy S.R., Kwon G.S. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12(36):4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 96.Wang Z., Wen J., Liu H., Zheng L., Luo H., Liu Z. Biodegradable interlayer-crosslinked polymer micelles with reduction sensitivity for non-small cell lung cancer therapy. J Biomed Nanotechnol. 2018;14(7):1225–1238. doi: 10.1166/jbn.2018.2538. [DOI] [PubMed] [Google Scholar]
- 97.Howard M.D., Jay M., Dziublal T.D., Lu X. PEGylation of nanocarrier drug delivery systems: state of the art. J Biomed Nanotechnol. 2008;4(2):133–148. [Google Scholar]
- 98.Guan S.S., Chang J., Cheng C.C., Luo T.Y., Ho A.S., Wang C.C. Afatinib and its encapsulated polymeric micelles inhibits HER2-overexpressed colorectal tumor cell growth in vitro and in vivo. Oncotarget. 2014;5(13):4868–4880. doi: 10.18632/oncotarget.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fathi M., Zangabad P.S., Aghanejad A., Barar J., Erfan-Niya H., Omidi Y. Folate-conjugated thermosensitive O-maleoyl modified chitosan micellar nanoparticles for targeted delivery of erlotinib. Carbohydr Polym. 2017;172:130–141. doi: 10.1016/j.carbpol.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 100.Berman D.M., Karhadkar S.S., Maitra A., Montes De Oca R., Gerstenblith M.R., Briggs K. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 101.Thayer S.P., di Magliano M.P., Heiser P.W., Nielsen C.M., Roberts D.J., Lauwers G.Y. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang Z., Sengupta R., Banerjee S., Li Y., Zhang Y., Rahman K.M. Epidermal growth factor receptor-related protein inhibits cell growth and invasion in pancreatic cancer. Cancer Res. 2006;66(15):7653–7660. doi: 10.1158/0008-5472.CAN-06-1019. [DOI] [PubMed] [Google Scholar]
- 103.Chitkara D., Singh S., Kumar V., Danquah M., Behrman S.W., Kumar N. Micellar delivery of cyclopamine and gefitinib for treating pancreatic cancer. Mol Pharm. 2012;9(8):2350–2357. doi: 10.1021/mp3002792. [DOI] [PubMed] [Google Scholar]
- 104.Luqmani Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2014;14(Suppl.1):35–48. doi: 10.1159/000086183. [DOI] [PubMed] [Google Scholar]
- 105.Chen Z., Shi T., Zhang L., Zhu P., Deng M., Huang C. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 2016;370(1):153–164. doi: 10.1016/j.canlet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 106.Fletcher J.I., Williams R.T., Henderson M.J., Norris M.D., Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Update. 2016;26:1–9. doi: 10.1016/j.drup.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 107.Dai C.L., Tiwari A.K., Wu C.P., Xiao-Dong S., Si-Rong W., Dong-Geng L. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily b member 1 and g member 2. Cancer Res. 2008;68(19):7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Collins D.M., Crown J., O'Donovan N., Devery A., O'Sullivan F., O'Driscoll L. Tyrosine kinase inhibitors potentiate the cytotoxicity of MDR-substrate anticancer agents independent of growth factor receptor status in lung cancer cell lines. Invest New Drugs. 2010;28(4):433–444. doi: 10.1007/s10637-009-9266-0. [DOI] [PubMed] [Google Scholar]
- 109.Wang H., Li F., Du C., Wang H., Mahato R.I., Huang Y. Doxorubicin and lapatinib combination nanomedicine for treating resistant breast cancer. Mol Pharm. 2014;11(8):2600–2611. doi: 10.1021/mp400687w. [DOI] [PubMed] [Google Scholar]
- 110.Li F., Danquah M., Singh S., Wu H., Mahato R.I. Paclitaxel- and lapatinib-loaded lipopolymer micelles overcome multidrug resistance in prostate cancer. Drug Deliv Transl Res. 2011;1(6):420–428. doi: 10.1007/s13346-011-0042-2. [DOI] [PubMed] [Google Scholar]
- 111.Angelo D.L., Gomez H.L., Zeba A., Zanete Z., Jose B., Arbushites M.C. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol. 2008;26(34):5544–5552. doi: 10.1200/JCO.2008.16.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhongzhen G., Binghe X., Desilvio M.L., Zhenzhou S., Wichit A., Zhongsheng T. Randomized trial of lapatinib versus placebo added to paclitaxel in the treatment of human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. J Clin Oncol. 2013;31(16):1947. doi: 10.1200/JCO.2011.40.5241. [DOI] [PubMed] [Google Scholar]
- 113.Wei Y., Xu S., Wang F., Zou A., Zhang S., Xiong Y. A novel combined micellar system of lapatinib and paclitaxel with enhanced antineoplastic effect against human epidermal growth factor Receptor-2 positive breast tumor in vitro. J Pharm Sci. 2015;104(1):165–177. doi: 10.1002/jps.24234. [DOI] [PubMed] [Google Scholar]
- 114.Wadee A., Pillay V., Choonara Y.E., du Toit L.C., Penny C., Ndesendo V.M. Recent advances in the design of drug-loaded polymeric implants for the treatment of solid tumors. Expert Opin Drug Deliv. 2011;8(10):1323–1340. doi: 10.1517/17425247.2011.602671. [DOI] [PubMed] [Google Scholar]
- 115.Iqbal S., Rashid M.H., Arbab A.S., Khan M. Encapsulation of anticancer drugs (5-Fluorouracil and Paclitaxel) into polycaprolactone (PCL) nanofibers and in vitro testing for sustained and targeted therapy. J Biomed Nanotechnol. 2017;13(4):355–366. doi: 10.1166/jbn.2017.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hu H., Lin Z., He B., Dai W., Wang X., Wang J. A novel localized co-delivery system with lapatinib microparticles and paclitaxel nanoparticles in a peritumorally injectable in situ hydrogel. J Control Release. 2015;220(Pt A):189–200. doi: 10.1016/j.jconrel.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 117.Agrawal S., Dwivedi M., Ahmad H., Chadchan S.B., Arya A., Sikandar R. CD44 targeting hyaluronic acid coated lapatinib nanocrystals foster the efficacy against triple-negative breast cancer. Nanomedicine. 2018;14(2):327–337. doi: 10.1016/j.nano.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 118.Ye T.T., Zhang W.J., Sun M.S., Rui Y., Shuangshuang S., Yuling M. Study on intralymphatic-targeted hyaluronic acid-modified nanoliposome: influence of formulation factors on the lymphatic targeting. Int J Pharm. 2014;471(1–2):245–257. doi: 10.1016/j.ijpharm.2014.05.027. [DOI] [PubMed] [Google Scholar]
- 119.Shen H., Shi S., Zhang Z., Gong T., Sun X. Coating solid lipid nanoparticles with hyaluronic acid enhances antitumor activity against melanoma Stem-like cells. Theranostics. 2015;5(7):755–771. doi: 10.7150/thno.10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen Y., Li X., Hong X., Xiao J., Bo L., Chen X. Reduction and pH dual-sensitive nanovesicles co-delivering doxorubicin and gefitinib for effective tumor therapy. RSC Adv. 2018;8(4):2082–2091. doi: 10.1039/c7ra12620d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mandal B., Mittal N.K., Balabathula P., Thoma L.A., Wood G.C. Development and in vitro evaluation of core-shell type lipid-polymer hybrid nanoparticles for the delivery of erlotinib in non-small cell lung cancer. Eur J Pharm Sci. 2016;81:162–171. doi: 10.1016/j.ejps.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 122.Li B.W., Xin X., Chen Z.L., Zhan C.Y., Fang Z., Wu S.Z. Tumor inhibition achieved by targeting and regulating multiple key elements in EGFR signaling pathway using a self assembled nanoprodrug. Adv Funct Mater. 2018;28(22):1800692. [Google Scholar]
- 123.Du L., Hao M., Li C., Wu W., Wang W., Ma Z. Quercetin inhibited epithelial mesenchymal transition in diabetic rats, high-glucose-cultured lens, and SRA01/04 cells through transforming growth factor-beta2/phosphoinositide 3-kinase/Akt pathway. Mol Cell Endocrinol. 2017;452:44–56. doi: 10.1016/j.mce.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 124.Granato M., Rizzello C., Gilardini Montani M.S., Cuomo L., Vitillo M., Santarelli R. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J Nutr Biochem. 2017;41:124–136. doi: 10.1016/j.jnutbio.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 125.Rodriguez-Mateos A., Vauzour D., Krueger C.G., Shanmuganayagam D., Reed J., Calani L. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: an update. Arch Toxikol. 2014;88(10):1803–1853. doi: 10.1007/s00204-014-1330-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.