

Summary
Repair of DNA double-stranded breaks (DSBs) during lymphocyte development is essential for V(D)J recombination and forms the basis of immunoglobulin variable region diversity. Understanding of this process in lymphogenesis has historically been centered on the study of RAG1/2 recombinases and a set of classical non-homologous end-joining factors. Much less has been reported regarding the role of chromatin modifications on this process. Here, we show a role for the non-redundant histone H3 lysine methyltransferase, Setd2, and its modification of lysine-36 trimethylation (H3K36me3), in the processing and joining of DNA ends during V(D)J recombination. Loss leads to mis-repair of Rag-induced DNA DSBs, especially when combined with loss of Atm kinase activity. Furthermore, loss reduces immune repertoire and a severe block in lymphogenesis as well as causes post-mitotic neuronal apoptosis. Together, these studies are suggestive of an important role of Setd2/H3K36me3 in these two mammalian developmental processes that are influenced by double-stranded break repair.
Subject Areas: Biological Sciences, Molecular Biology, Immunology, Cell Biology
Graphical Abstract
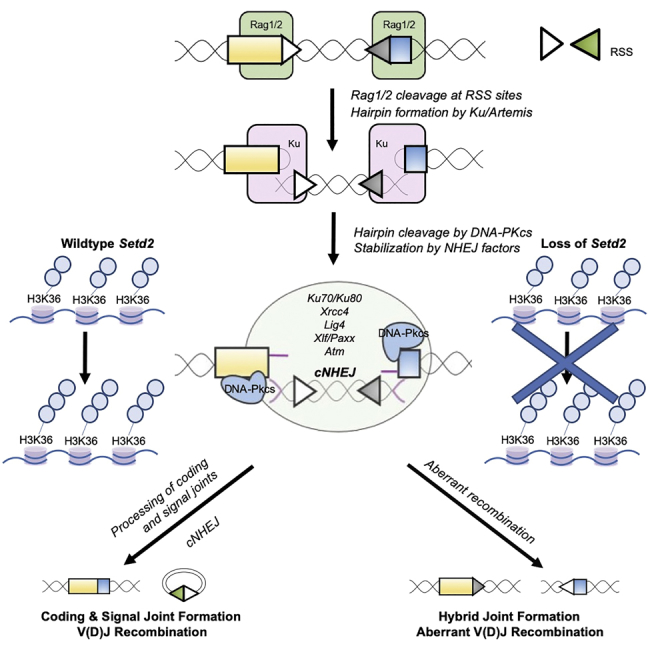
Highlights
-
•
Setd2/H3K36me3 is essential in maintaining a normal hematopoiesis
-
•
Loss of Setd2/H3K36me3 impairs lymphogenesis and V(D)J recombination
-
•
Loss of Setd2/H3K36me3 and ATM kinase activity leads to mis-repaired recombination
-
•
Setd2/H3K36me3 prevents apoptosis of post-mitotic neuronal cells
Biological Sciences; Molecular Biology; Immunology; Cell Biology
Introduction
In early normal lymphocyte development, gene segments that will eventually encode the immunoglobulin (Ig) and T cell receptor (TCR) variable regions are recombined from Variable (V), Diversity (D), and Joining (J) gene segments in a process known as V(D)J recombination (Alt et al., 2013). During the DNA recognition and cleavage stage, recombination signal sequences (RSSs) that flank the individual V, D, and J gene segments are targets of RAG1/2 endonucleases and result in the generation of hair-pinned coding ends (CEs) and blunt-ended signal ends (SEs) (Alt et al., 2013, Schatz and Swanson, 2001). In the second phase of V(D)J recombination, and-processing and end-ligation of CEs and SEs are mediated by classical non-homologous end-joining (C-NHEJ) factors and produce an imprecisely repaired coding joint (CJ) consisting of V(D)J exons and a precisely repaired but discarded circular signal joint (SJ) (Alt et al., 2013, Schatz and Swanson, 2001). A set of core C-NHEJ factors (KU70, KU80, XRCC4, and LIG4) is absolutely essential for end-joining and is evolutionarily conserved (Alt et al., 2013, Kumar et al., 2014). Loss or defects of C-NHEJ factors can impair end-processing (DNA-PKCs, ARTEMIS) or end-joining (KU proteins, XRCC4, XLF, LIG4) and results in severe immunodeficiencies in both mouse models and human disease (Alt et al., 2013; Kumar et al., 2014; Bassing et al., 2002). The DNA damage protein, ataxia telangiectasia mutated (ATM); its target, histone H2AX; and DNA damage response adaptor protein, MRI, are all also involved in the end-ligation process (Bredemeyer et al., 2006, Yin et al., 2009, Hung et al., 2018). The single loss of any of these factors, or C-NHEJ factor XLF, has only modest effects on lymphogenesis and V(D)J recombination (Bredemeyer et al., 2006, Yin et al., 2009, Hung et al., 2018, Li et al., 2008). In addition to loss of the core C-NHEJ factors, combined deficiencies of proteins non-essential for the end-joining reaction can severely impair C-NHEJ to a similar extent, as in the case of combined loss of Xlf and Atm or Xlf and Mri (Kumar et al., 2016, Hung et al., 2018, Li et al., 2008, Lescale et al., 2016, Zha et al., 2011).
Another mammalian developmental process that utilizes C-NHEJ for repair of double-strand breaks (DSBs) is embryonic neurogenesis (Frappart and McKinnon, 2008). Neural progenitors that have exited the cell cycle and are migrating out of the embryonic ventricular zones as they differentiate are thought to rely on NHEJ-mediated repair of DSBs (Frappart and McKinnon, 2008). In mice, loss of core C-NHEJ factors leads to apoptosis of post-mitotic neurons and embryonic lethality (Gao et al., 1998, Frank et al., 2000, Gu et al., 2000). Moreover, the same synthetic lethal combinations that result in severe lymphocyte developmental blocks (e.g., MRI and XLF) also display similar post-mitotic neuronal apoptosis (Hung et al., 2018, Abramowski et al., 2018), indicating the critical role of end-ligation and C-NHEJ in the repair of DSBs during neurogenesis.
Histone H3 lysine-36 tri-methylation (H3K36me3) is a histone modification that is catalyzed by the non-redundant histone methyltransferase, SETD2 (Wagner and Carpenter, 2012). H3K36me3 is associated with actively transcribed genes, and SETD2 plays important roles in the control of gene expression (Wagner and Carpenter, 2012). Loss-of-function mutations in SETD2 or dominant negative “onco-histone” mutations in the H3K36 residue itself have been described in a broad array of malignancies, particularly in hematopoietic and central nervous system (CNS) tumors (Parker et al., 2016, Zhang et al., 2012, McKinney et al., 2017, Moffitt et al., 2017, Zhu et al., 2014, Lu et al., 2016). In mammalian cells, SETD2 regulates specific steps of the DNA damage response during mismatch repair (MMR) and homologous recombination (HR) (Li et al., 2013, Pfister et al., 2014, Aymard et al., 2014). More recently, a role for Setd2 in normal thymocyte development and V(D)J recombination was described (Ji et al., 2019). Although a role for H3K36 methylation in NHEJ had been previously suggested in yeast (Fnu et al., 2011), insights into the mechanism for how this post-translation histone modification in mammalian cells may impact this mode of repair remains unknown. Thus, to determine the role, if any, of Setd2 and H3K36me3 in this mode of DNA repair in mammals, we studied its loss in two developmental pathways that utilize NHEJ. Here, we specifically show that whereas loss of Setd2/H3K36me3 does not completely abrogate repair of DSBs, loss leads to mis-repair in B-cell lymphoid development of Rag-induced DNA DSBs, especially when combined with loss of ATM kinase activity. Loss of Setd2 leads to the increased formation of aberrant hybrid joints and additionally leads to reductions in overall B cell repertoire. Finally, loss of Setd2 also leads to post-mitotic neuronal apoptosis.
Results
Loss of Setd2 Disrupts Normal Hematopoiesis, Particularly Lymphopoiesis
The complete loss of Setd2 is embryonic lethal at embryonic day 10.5 (E10.5)–E11.5 (Hu et al., 2010). Therefore, to study the role of Setd2 in normal and malignant hematopoiesis, we previously generated a conditional mouse model expressing Cre-recombinase under inducible (Mx1) or constitutive (Vav1) hematopoietic lineage-restricted promoters (Mar et al., 2017). The loss of Setd2 ablated H3K36me3 in hematopoietic tissues through excision of exon 3 of Setd2 (Figure 1A). Heterozygous mice had no overt hematopoietic phenotype (Figures S1A, S1B and S2D), whereas homozygous loss of Setd2 resulted in a significant perturbation of normal hematopoiesis, including decreased overall bone marrow cellularity (Figure 1B), significant loss of mature lymphoid cells (B220+ B cells and CD3+ T cells) in the bone marrow, and expansion of erythroid (Ter119+) cells (Figures 1C and 1D). The significant reduction in T cells in the bone marrow observed upon complete Setd2 loss was also mirrored by a severe diminution of thymic size (Figure 1E), which was concomitant with significant splenomegaly (Figure 1F). Strikingly, the splenomegaly was due to the aberrant expansion of erythroid cells and significant ablation of B-lymphoid (B220+) populations (Figure 1G). In addition, loss of Setd2 induced qualitative and quantitative defects in hematopoietic stem cells, as well as abnormal erythroid progenitor expansion in the bone marrow (Figures 1D, 1G, 1H, S1C–S1F, and S2A–S2C). These hematopoietic phenotypes are consistent with other reports on Setd2 knockout mice (Zhou et al., 2018, Zhang et al., 2018, Ji et al., 2019). Altogether, these data indicate that loss of Setd2 disrupts normal hematopoiesis and severely impacts lymphoid development.
Figure 1.

Loss of Setd2/H3K36me3 Disrupts Normal Hematopoiesis and Severely Arrests Lymphoid Development
(A) Western blot for H3K36me3, total H3, and Gapdh in bone marrow (BM), spleen (spl), and thymus (thy) of Mx1/Vav1cre Setd2Δ/Δ and Setd2f/f littermate controls.
(B) Total cell count of whole bone marrow (n = 6 for all groups).
(C) Ratio of Mx1 and Vav1 Setd2Δ/Δ to controls of total cellularity of whole bone marrow (WBM), lineage-negative bone marrow cells (LIN−), B220+ B cells in bone marrow, and thymocytes (n = 15 for all groups).
(D) Percent composition of differentiated hematopoietic cell populations in WBM, B cell (B220+), T cell (Cd3+), myeloid (Mac1+/Gr1+), and erythroid (Ter119+) (n=6 for all groups).
(E) Thymic (n = 10) and (F) spleen (n = 100) weights for Setd2Δ/Δ and Setd2f/f littermate controls.
(G) Percent composition of differentiated hematopoietic cell populations in spleen.
(H) Total cellularity of LSK (Lin−Sca1+Kit+) and SLAM (LSK Cd150+Cd48-) hematopoietic stem populations (n = 6 for all groups).
∗∗, p < 0.01 ∗∗∗, p < 0.001. See also Figures S1 and S2.
Setd2/H3K36me3 Is Important in Normal Lymphocyte Development
We and others (Zhou et al., 2018, Zhang et al., 2018, Ji et al., 2019) observed that loss of Setd2 early in hematopoiesis resulted in significant depletions of the lymphoid populations in the bone marrow, spleen, and thymus (Figures 1C, 1D, 1G and S1). To rule out that these early developmental deficiencies were not solely the result of reduced numbers of early lymphoid progenitors (Figure S1C), we crossed our knockout mice with multiple B lymphoid lineage-restricted Cre-recombinase-expressing lines and found that whereas an early and profound proB block in development was only observed upon early deletion of Setd2 (Mx1, Vav1cre) (Figure 2A), the deletion of Setd2 in later stages of B cell development (with hCD2, Mb1, and Cd19cre) resulted in abnormal lymphocytic output that was more apparent in more mature B cell populations (Figures 2B and 2C). Specifically, loss of Setd2 induced at later stages of B cell development significantly reduced detectable mature B cells (IgM+IgD+) in the bone marrow (Figures 2B and 2C) and resulted in the significant depletion of B cell lineage cells in the spleen (Figure 2C). These data suggest that Setd2/H3K36me3 is important in B lymphopoiesis at different stages, but that the severe block at the proB cell stage was only apparent with early loss in hematopoiesis.
Figure 2.

Setd2/H3K36me3 Important for B Cell Development at Different Stages
(A) Schematic of ontological expression of various B-lineage-restricted cre-recombinase mouse lines crossed to Setd2f/f mice with Igh locus rearrangement status indicated, and representative flow cytometry of B220+ early B cells progenitors (proB and preB cells) of control, Vav/Mx1, hCD2, Cd19, and Mb1cre Setd2Δ/Δ mice.
(B) (i) Representative flow cytometry of bone marrow stained for early B cell progenitors and mature and immature B cell markers (IgM and IgD). (ii) Total bone marrow cellularity. (iii) Fraction composition of B220+, proB (B220+Cd43+IgM−), preB (B220+Cd43+IgM−), immature B (B220+Cd43+IgM+), and mature B cell (B220+IgM+IgD+) populations of hCD2 (n = 7), Mb1 (n = 14), Cd19 (n = 9) Setd2Δ/Δ, and sex- and age-matched littermate controls.
(C) Spleen (i) weight (n = 14 for all groups), (ii) total cellularity (n = 3 for all groups), and (iii) percentage composition of different B cell populations in bone marrow and spleen (n = 6 for all groups).
Significance indicated as comparison with controls. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, error bars represent SD.
Setd2/H3K36me3 Is Crucial for Normal Immunoglobulin Rearrangement in Early Lymphocyte Development Recombination
The severe block in early B cell development was striking and warranted further examination. Early loss of Setd2/H3K36me3 blocked B cell development at the proB cell stage with similar total numbers of Fraction A (FrA)-defined pre-proB cells when compared with littermate controls (Figures 3A and S3A–S3E). This block at the proB cell stage was concomitant with a near-complete ablation of immature IgM+ B cells in the bone marrow and spleen (Figures 3A and S3B). Similarly, Setd2Δ/Δ mice exhibited a block in early T cell development at the double-negative stage (DN: CD4−CD8-), with an accumulation at the DN3 stage (Figure 3B). This arrest at the DN3 stage was similarly observed in Mx1-cre-driven exon 6-7 deletion Setd2 knockout mouse (Ji, et al., 2019). Thus, lymphopoiesis in Setd2Δ/Δ mice appeared to be arrested at stages wherein V(D)J recombination occurs and is reminiscent of the lymphopenia observed with deficiency of factors necessary for V(D)J recombination (Alt et al., 2013; Kumar et al., 2014; Bassing et al., 2002). In all early B/T cell progenitor populations of Setd2Δ/Δ mice, developmental blocks also coincided with increased levels of apoptosis and phospho-γH2ax (Figures 3C, S3F, and S3G), despite similar in vivo proliferation rates and cell cycle status (Figures 3D and S3H). Furthermore, Setd2Δ/Δ proB cells did not display any significant differences in the expression of factors related to V(D)J recombination at the gene or protein level (Figures S4A–S4C). The arrest in B cell, but not in T cell, development in Setd2Δ/Δ mice could be partially rescued by crossing knockout mice with a transgenic mouse expressing a fully rearranged and productive immunoglobulin heavy chain (Igh) locus (Figure 3E), indicating a role for Setd2/H3K36me3 in enforcing normal V(D)J recombination.
Figure 3.

Loss of Setd2/H3K36me3 Severely Arrests Lymphoid Development
(A) Representative flow cytometry analysis of early B cell progenitors in B220+ bone marrow and total cellularity of B cell progenitor proportions of Vav/Mx1cre Setd2Δ/Δ and controls (n = 9).
(B) Representative flow cytometric analysis of thymic cells, and total cellularity of thymic progenitor populations in Setd2Δ/Δ and Setd2f/f controls. DN: Cd4-Cd8-, DN1: DN Cd44+Cd25-; DN2: DN Cd44+Cd25+; DN3: DN Cd44-Cd25+ (n = 8).
(C) Annexin V+ of B (n = 9) and T cell (n = 5) progenitor populations.
(D) In vivo bromodeoxyuridine (BrdU) incorporation in early B and T cell progenitor compartments (n = 3). All values for BrdU were non-significant.
(E) Representative immature B cell (B220+sIgM+) flow cytometric analysis and total cellularity of immature surface IgM+ population of bone marrow cells isolated from both legs and hips of Setd2 mice crossed to mice transgenic for the Ig heavy chain complex (IgHelMD4) specific for hen egg lysozyme (HEL) (n = 4 for Setd2f/f, n = 5 for Setd2Δ/Δ MD4, and n = 3 for Setd2Δ/Δ).
∗∗∗ p<0.001, error bars represent SD. See also Figures S3 and S4.
Loss of Setd2/H3K36me3 Does Not Abrogate Chromatin Architecture or Accessibility of the Early proB Igh Locus and Causes Aberrant V(D)J Recombination
To determine the impact of loss of Setd2/H3K36me3 on the Igh locus at the proB stage, we conducted chromatin immunoprecipitation (ChIP) sequencing and found both a global loss of H3K36me3 across the genome and a focal loss on the Igh locus where a well-studied critical regulatory region near the Eμ enhancer resides (Chowdhury and Sen, 2001) (Figures 4A and S5A). As accessibility of this region is critical for B cell development (Chowdhury and Sen, 2001, Chakraborty et al., 2009), we wanted to ascertain if the loss of H3K36me3 affected local chromatin architecture or accessibility. In proB cells, ablation of H3K36me3 neither affected chromatin accessibility (Figures 4B and S5B) nor disrupted the local levels of H3K4me3 and H3K9ac (Figure 4C), two histone modifications essential for maintaining an open and actively transcribed chromatin structure at this regulatory region (Chowdhury and Sen, 2001, Chakraborty et al., 2009) and for H3K4me3, the recruitment and activation of the Rag2 protein itself (Shimazaki and Lieber, 2014, Johnson et al., 2010, Ji et al., 2010, Matheson and Corcoran, 2012, Bettridge et al., 2017). Loss of H3K36me3 did not significantly affect the methylation states of mono-, di-, or tri-methyl lysine-27 or mono- and di-methyl lysine-36 residues in this region (Figure S5C). We detected equivalent recruitment of Rag1 and Hmgb2 to this same region on the Igh locus (Shimazaki and Lieber, 2014, Johnson et al., 2010, Ji et al., 2010, Matheson and Corcoran, 2012) (Figure 4D), suggesting that the initiation phase of the V(D)J recombination reaction was intact. We were also not able to detect in sorted FrA proB cells any evidence of Rag1 recruitment or H3K36me3 at variable gene families on the Igh locus in either Setd2-deficient cells or controls (Figure S5D). In addition, the level of sterile transcription of Igh genes was only mildly reduced (Figure 4E), particularly when compared with deletion of the Eμ enhancer, which causes significant transcriptional dysregulation (Chakraborty et al., 2009). Upon closer examination of recovered V(D)J recombination products from proB cells, we observed that Setd2 deficiency resulted in aberrant recombination and, in some cases, lack of expected rearrangement products altogether (Figures 4F and S5E). Combined, these data are suggestive that the V(D)J recombination defect is not due to decreased expression or regulation of the Igh locus, but due to defects in the repair phase of the reaction.
Figure 4.

Loss of Setd2/H3K36me3 Does Not Alter Chromatin Architecture or Accessibility of the Early proB Igh Locus and Causes Abnormal V(D)J Recombination
(A) H3K36me3 ChIP sequencing of the Igh locus of Cd19−/+ proB (B220+Cd43+IgM−) cells from bone marrow of Vav Setd2Δ/Δ and controls. (n = 3 for all groups). Magnification of region of Igh with focal H3K36me3; loss of representative H3K36me3 tracks overlaid with assay for transposase-accessible chromatin (ATAC)-seq of same region from control proB cells for reference. Annotation of critical regulatory sites as indicated.
(B) Representative ATAC-seq tracks of regulatory region of the Igh locus described in (A) of sorted Cd19- proB cells from two matched control and Setd2 knockout sorted proB cells.
(C) Representative ChIP-PCR of regulatory region for histone H3 marks K36me3, K9ac, and K4me3. Data representative of n = 3 independent experiments.
(D) Representative ChIP-PCR of Rag1 and Hmgb2 of proB cells from Setd2Δ/Δ and controls at the same regulatory region with standard deviations as indicated, n = 3 independent experiments.
(E) Relative quantitation of sterile transcription of DH genes, Cμ, and enhancer RNAs of proB cells from Setd2Δ/Δ and controls. Data represented as an average of three independent ChIP-PCRs from three independently sorted proB cell populations.
(F) Schematic and representative results of PCR assay to detect V(D)J recombination products of rearrangement of the Igh locus of different VH families of sorted proB cells from Setd2Δ/Δ (KO) and controls (WT). Empty and filled triangles represent primers. No product meant germline non-rearrangement. Image representative of n = 3 independent PCR experiments.
Error bars represent SD. See also Figure S5.
Aberrant End-Joining of Rag-Induced DSB with Setd2/H3K36me3 Deficiency
To ascertain if defects in Setd2-deficient lymphogenesis could be a consequence of impaired Rag-induced DSB repair during V(D)J recombination, we generated murine Setd2-deficient (Setd2−/−), Ku80−/− (Xrcc5−/−), and Lig4 −/− late-proB v-Abelson (v-Abl)-transformed lines expressing a Bcl2 transgene by CRISPR/Cas9-mediated inactivation (Figures S6A–S6D) (Hung et al., 2018, Jacobsen et al., 2006). Loss of Setd2 in v-Abl cells neither perturbed the expression of factors involved in V(D)J recombination nor affected the cell cycle distribution of these cells (Figures S7A–S7C) in vitro. Treatment with Abl kinase inhibitor imatinib (STI) induces G1 cell-cycle arrest and Rag1/2 expression leading to κ light chain rearrangement (Hung et al., 2018, Jacobsen et al., 2006) (Figure S8A). We additionally introduced a chromosomally integrated inversion recombination substrate (pMG-INV), which can be used to assess the efficiency of V(D)J recombination by measuring GFP expression in cells and/or visualization of the repair products and intermediates by Southern blotting and PCR-based strategies (Hung et al., 2018) (Figure 5A). Inactivation of ATM kinase activity with an inhibitor (ATMi) is sufficient to induce the formation of hybrid joints (HJs, joining of CEs to SEs) (Bredemeyer et al., 2006) and could additionally be used to observe aberrant V(D)J recombination.
Figure 5.

Aberrant End-Joining of Rag-Induced DSB with Setd2/H3K36me3 Deficiency
(A) Schematic of recombination substrate pMG-INV. Unrearranged (UR) and SE and CE intermediates and resulting SJs and CJs. Long-terminal repeats (LTRs), XbaI and NheI restriction digestion sites, recombination signal sequences (RSSs), GFP, Thy1.2 cDNA, and corresponding probes shown.
(B) Representative flow cytometric analysis of GFP expression in control and Setd2−/− pMG-INV v-Abl cells treated with Abl kinase inhibitor imatinib (STI-571) and ATM kinase inhibitor (ATMi, KU55933) for 48 and 96 h.
(C) GFP expression of pMG-INV harboring v-Abl cell lines treated for 72 h with imatinib ± ATMi assessed by flow cytometry. Control, Lig4−/−, and at least four independently derived Xrcc5−/− and Setd2−/− v-Abl clones were treated in n = 4 independent induction experiments. Significance to controls to STI-571 treatment alone condition was calculated. ∗∗∗p < 0.001, error bars represent SD.
(D) Southern blot analysis of genomic DNA from induced Setd2−/− and control lines that were digested with (i and ii) XbaI and (iii) NheI hybridized with (i and iii) Thy1 or (ii) GFP probe. Hybrid Joins (HJ) indicated as well (joint of CEs and SEs).
(E) Schematic of PCR method to detect the formation of a coding joint and hybrid joint recombination product of the pMG-INV retroviral recombination substrate, and PCR result of pMG-INV coding and hybrid joints from indicated v-Abl cell clones treated for 72 h with ABLki with or without ATMki (KU55933). Il-2 gene PCR was used as a loading control. Blue arrow indicates coding joint product, and red arrow indicates hybrid joint product.
(F) PCR strategy to detect endogenous Vκ6-23 to Jκ1 coding joints (CJ) and hybrid joints (HJ) in (i) control and Setd2-deficient v-Abl lines treated for 72 h with STI-571 ± ATMi and (ii) Setd2f/f and Setd2Δ/Δ splenocytes. Il-2 gene PCR was used as a loading control and analyzed and quantified by high-sensitivity TapeStation (D1000). Blue arrow indicates CJ product, and red arrow indicates HJ product.
See also Figures S6–S9.
Comparable rearrangement efficiency of the pMG-INV substrate was observed in both wild-type (WT) and Setd2−/− v-Abl cells treated with imatinib, as indicated by GFP expression (Figure 5B). As expected, treatment of WT v-Abl cells with ATMi resulted in a modest (25%) decrement in GFP expression (Figure 5B). Strikingly, treatment of Setd2−/− cell lines with ATMi resulted in a ∼60% reduction in GFP expression (Figure 5C). This finding is reminiscent of the severe defect in GFP expression found in Xlf-deficient v-Abl cells treated with ATMi (Zha et al., 2011, Lescale et al., 2016) and suggests a functional redundancy between Setd2/H3K36me3 and Atm kinase activity in ensuring proper repair. To investigate potential effects on repair, we conducted Southern blotting, which revealed pMG-INV SJ and CJ formation without obvious accumulation of free unrepaired SEs and CEs in STI-treated WT and Setd2−/− v-Abl B cells (Figures 5D and S8B). Thus, like Xlf, Mri, and Atm, Setd2 is not essential for C-NHEJ during V(D)J recombination (Bredemeyer et al., 2006, Zha et al., 2011, Hung et al., 2018). The non-essentiality of Setd2/H3K36me3 for end-joining was further intimated by the inability of dual loss of Setd2/H3K36me3 and p53 to generate translocations leading to the development of proB-cell lymphomas (Figures S9A and S9B), unlike loss of core C-NHEJ factors in a p53-null background (Difilippantonio et al., 2000, Gao et al., 2000, Frank et al., 2000).
Despite not being required for end-joining, ATMi-treated Setd2−/− v-Abl cells exhibited significantly increased mis-repaired recombination products that corresponded to either repaired SJs but unrepaired CEs (SJ + CE) (Figures 5A, 5D(i-ii), and S8B) or the formation of hybrid joins (Figures 5A, 5D(i), and S8B), products consistent with the observed loss of GFP signal. The enhanced formation of HJs with loss of Atm kinase activity and Setd2 was further corroborated by the increased detection of aberrant HJ products of the pMG-INV substrate in ATMi-treated Setd2−/− cells (Figure 5E). Although we could detect evidence of HJ formation by PCR of the recombination substrate in Setd2−/− lines without ATMi (Figure 5E), these products were below detection by Southern blotting (Figures 5D(i) and S8B), but consistent with the modest decrease in GFP signal with STI treatment alone (Figures 5B and 5C). Furthermore, we could detect HJ formation not only from the endogenous κ light chain locus of v-Abl Setd2 knockout cells but also in Setd2Δ/Δ splenocytes (Figure 5F). Together, these data indicate a novel role for Setd2/H3K36me3, especially in combination with Atm kinase activity, in the repair phase of V(D)J recombination to ensure proper joining.
Loss of Setd2/H3K36me3 Reduces Overall B Cell Repertoire
Even without loss of Atm kinase activity, however, there was abundant evidence of aberrant and abnormal rearrangement of the endogenous Igh locus in primary Setd2Δ/Δ proB cells (Figures 4F and S5E). We sequenced a similarly sized recombination product, present in both control and Setd2Δ/Δ proB cells, for three different heavy chain Variable gene families joined to the JH4 fragment and found additional abnormalities (Figure 6A). Loss of Setd2/H3K36me3 not only reduced the overall number of unique productive Igh rearrangements (assessed by the number of unique hypervariable complementarity defining region-3 [CDR3]) but also resulted in shortening of CDR3 length and reductions in N-nucleotide additions (Figures 6A and 6B) (Bolotin et al., 2017). Similarly, when we looked at RNA-based transcripts of productive rearrangements of the Igh locus in proB cells using the hypervariable region calling algorithm TRUST (Li et al., 2017, Hu et al., 2018, Hu et al., 2019), Setd2Δ/Δ proB cells exhibited significant decreases in detectable unique CDR3 sequences and an overall decrease in overall B cell repertoire diversity (Figures 6C and 6D). Furthermore, global variable and joining gene usage was reduced (Figure 6E) in proB cells from Setd2Δ/Δ mice and coincided with evidence of increased clonality in variable gene usage, which did not appear to be dependent on the proximity to the Diversity and Joining gene segment regions on the Igh locus (Figure 6F). These observations indicate that, in addition to its role with Atm during end-joining to ensure appropriate repair, Setd2/H3K36me3 is also involved in other mechanisms that prevent mis-processing and mis-repair of broken DNA ends to ensure the fidelity of V(D)J recombination and is critical in maintaining a normal immune repertoire.
Figure 6.

Loss of Setd2/H3K36me3 Reduces CDR3 Repertoire and Variable Gene Usage in proB Cells
(A) PCR and next-generation sequencing (NGS) strategy of V(D)J recombination products of VH families to JH4; 500-bp product (blue box) was extracted and submitted for NGS amplicon sequencing. Sequencing was analyzed with MiXCR T/B cell repertoire software (Bolotin et al., 2017).
(B) (i) Number of unique CDR3 clones, (ii) fraction and total clone counts by amino acid length, (iii) number of N-nucleotide additions, (iv) total nucleotide deletions of CDR3s recovered, and (v) total sequencing reads of Setd2Δ/Δ (KO) and controls for each VH family. proB cells were sorted from n = 4 for each genotype and subjected to NGS analysis. Significance measured by Wilcoxon rank-sum test.
RNA sequencing analysis of four independently sorted control and four Setd2Δ/Δ proB cell compartments was also conducted, and de novo assembly of CDR3 sequences was conducted using the TRUST algorithm (Hu et al., 2018, Hu et al., 2019, Li et al., 2017). (C) Unique CDR3 counts for heavy and light chain Ig loci from TRUST analysis.
(D) Estimated B cell fraction of reads and B cell diversity recovered from analysis of RNA transcripts from TRUST analysis. Estimate B cell fraction was calculated by taking the fraction of number of reads mapped to BCR(IGV/IGJ/IGC) region to total read count. B cell diversity was calculated by determining the ratio of unique CDR3 count over the total number of reads mapped to BCR region.
(E) Variable (V) and Joining (J) gene usage for (i) heavy chain and (ii) kappa light chain from TRUST analysis. Boxplots with SD.
(F) Clonality of CDR3s plotted as a fraction of total reads and ordered by individual Variable gene families and distance from Diversity (D) and Joining gene loci from most proximal to most distal from right to left from TRUST analysis. Relative size of clone indicated by size of dot.
Setd2/H3K36me3 Prevents Post-mitotic Neuronal Apoptosis and Perinatal Lethality
As Setd2/H3K36me3 seemed to be important for V(D)J recombination, we next wondered if it could play a role in Rag-independent C-NHEJ repair. To first test this, we assessed the sensitivity of asynchronous WT and Setd2−/− v-Abl cells to ionizing radiation and found that Setd2−/− v-Abl lines were more sensitive to DSBs induced by ionizing radiation (Figure 7A(i)). This increased sensitivity to ionizing radiation of Setd2-deficient cells, at levels even greater than that of core NHEJ factors Xrcc5 (Ku80) and Lig4 (Figure 7A(i) and Figure S10A), could be in part due to previously described roles in HR-mediated DSB repair ascribed to Setd2/H3K36me3 (Li et al., 2013, Pfister et al., 2014, Aymard et al., 2014). To exclude the effects that may be driven by cells that may be cycling, we chose to study the sensitivity of G1-arrested v-Abl cells by inhibition with STI-571 or the Cdk4/6 inhibitor PD0332991 for 48 h before exposure to ionizing radiation. In both cell-cycle-arresting conditions, G1-arrested Setd2−/− v-Abl cells exhibited increased sensitivity cells to DSBs induced by ionizing radiation compared with WT controls (Figures 7A(ii), S10B, and S10C). Together, these data suggest that Setd2/H3K36me3 is important in the DSB repair in the G1 phase of cell cycle.
Figure 7.

Post-mitotic Neuronal Apoptosis and Perinatal Lethality with Loss of Setd2/H3K36me3
(A) (i) Asynchronous and (ii) STI-571 G1-arrested WT, Xrcc5, and two different Setd2−/− v-Abl cells subjected to different doses of ionizing radiation and serially diluted five times in triplicate to assess survival. Values plotted as a mean of each dose as a percent of non-irradiated controls; bars represent standard deviation. Data are representative for three different independent treatment experiments. Viability was assessed at 72 h. No viable cells were detected beyond the 0.5 Gy dose for G1-arrested Xrcc5 v-Abl clones.
(B) Expected and observed genotype distributions of matings between Nestin-cre Setd2Δ/+ mice to Setd2f/+ mice (n = 15 litters) and Nestin-cre Setd2Δ/+ mice to Setd2f/f mice (n = 16 litters) 21 days post-partum (dpp).
(C) Embryo genotypes at different embryonic stages (n = 5 litters for each stage).
(D) Litter sizes of E14.5, E16.5, E18.5, and 0.25 dpp from Nestin-cre Setd2Δ/+ mice to Setd2f/f breedings. Significance to 0.25 dpp was measured (n = 5 litters for each stage except 0.25 dpp, n = 11).
(E) Cleaved caspase 3 immunohistochemistry of E18.5 embryos. Sagittal sections show staining of regions of the diencephalic, telencephalic, and mesencephalic regions with magnifications indicated by corresponding colored boxes.
(F) TUNEL assay of 2-h post-partum control and Nestin-cre Setd2Δ/Δ pups. Coronal sections of the lateral ventricle with magnifications indicated by corresponding red and green boxes.
∗∗p < 0.01 ∗∗∗p < 0.001, error bars represent SD. See also Figures S10 and S11.
In addition to lymphogenesis, core C-NHEJ factors are critical for enforcing normal neurogenesis, where differentiating post-mitotic neurons migrating out of the ventricular zone do not have HR available for repair of DNA DSBs and instead rely on C-NHEJ (Frappart and McKinnon, 2008). Given our findings for a role for Setd2/H3K36me3 in the repair phase of V(D)J recombination, we tested its potential functions in neurogenesis. Consistent with the defective CNS development and embryonic lethality observed in knockout mouse models of core C-NHEJ factors (Gao et al., 1998, Frank et al., 2000, Gu et al., 2000), we observed post-mitotic neuronal apoptosis and perinatal lethality in mice with homozygous deletion of Setd2 in neural progenitor-restricted Nestin-cre-expressing mice, where deletion has been demonstrated to occur before post-mitotic neurogenesis (Liang et al., 2012). The severity of this phenotype is demonstrated by the complete absence of Nestin-cre Setd2Δ/Δ pups at 21 days post-partum (dpp) (Figures 7B and 7C) with perinatal lethality by 0.25 dpp (Figure 7D). Histopathological analysis of embryos revealed evidence of apoptosis of post-mitotic neurons as early as E14.5 (Figures S11A and S11B), as well as more widespread apoptosis in the developing brain in E18.5 embryos and in 2-h post-partum pups (Figures 7E, 7F, S11C, and S11D) suggesting a role for Setd2/H3K36me3 in embryonic neurogenesis that is reminiscent of deficiency for C-NHEJ factors.
Discussion
The preservation of a genome is predicated on proper repair of DNA DSB and a balance between HR, which is a highly accurate and also slower form of repair that is restricted to the availability of a sister chromatid strand, and NHEJ, which is highly efficient but intrinsically error prone (Takata et al., 1998). Although there have been some studies highlighting the importance of chromatin accessibility, nucleosome positioning, DNA looping, and recognition of histone modifications (e.g., H3K4me3 by RAG2) (Shimazaki and Lieber, 2014, Johnson et al., 2010, Bettridge et al., 2017, Matthews et al., 2007, Matheson and Corcoran, 2012) in the regulation of the V(D)J recombination process and phosphorylation of γH2AX in repair (Yin et al., 2009, Celeste et al., 2003), little has been described in terms of the impact of other chromatin modifications, especially in the end-joining phase of the V(D)J recombination reaction.
Here, we discovered a strong dependency of normal hematopoiesis and, in particular, lymphogenesis on Setd2/H3K36me3 phenotypes consistent with previously published reports of three distinctly engineered knockout mouse models (Zhou et al., 2018, Zhang et al., 2018, Ji et al., 2019). Early loss of Setd2/H3K36me3 leads to severely impaired B and T cell development that could be partially rescued, in the case of B lymphogenesis, with the expression of a fully rearranged Igh locus, pointing to a role for Setd2/H3K36me3 in V(D)J recombination. This role of Setd2 in V(D)J recombination is further corroborated by the partial rescue of T lymphopoiesis with the over-expression of a rearranged TCR in Lck-cre Setd2Δ/Δ mice (Ji, et al., 2019). Ji et al. also report decreased Rag1 occupancy across the Igh locus in B220+Cd19+ cells from Cd19cre Setd2Δ/Δ mice (Ji et al., 2019), including at variable region family genes. In sorted proB cells (B220+Cd43+IgM−) where we induced loss of Setd2/H3K36me3 early in hematopoiesis, however, we could not detect a significant effect on the recruitment of Rag1 to the critical regulatory region near where the Eμ enhancer resides and where Rag1 has previously been shown to bind in vivo at the proB cell stage of development (Ji et al., 2010). The observed Rag1 occupancy differences observed by Ji et al., (2019) in populations of lymphoid cells are likely impacted by their inclusion of more mature B cell populations (B220+Cd19+ includes Cd43+ late proB and more mature Cd43-preB cells) that are depleted in Setd2Δ/Δ mice, as well as by incomplete and leaky deletion of Setd2 via the use cre-expressing mouse lines (Cd19cre) that delete after the proB cell developmental stage of interest (Rickert et al., 1997, Hobeika et al., 2006, Kraus et al., 2004, Siegemund et al., 2015).
We wanted to understand the nature of the defect in repair with early loss of Setd2 in proB cells, especially with respect to end-ligation, as we could not detect significant differences in the initiation phase of V(D)J recombination and as we could detect recombination of the Igh locus, albeit abnormal recombination. We therefore turned to an inducible system to measure the repair defect and uncovered new roles for Setd2/H3K36me3 in the fidelity of the V(D)J recombination reaction, especially in combination with ATM kinase activity. We determined that Setd2/H3K36me3 is not absolutely required for end-ligation by the lack of detectable unrepaired CEs and SEs of an ectopically integrated recombination substrate, pMG-INV. This non-essentiality for repair is consistent with our ability to detect recombination products in proB cells of Setd2Δ/Δ mice, albeit abnormal ones, and the lack of proB lymphomas as a consequence of translocation events in dual Setd2Δ/Δ p53Δ/Δ mice. It is noteworthy that unlike dual loss of ATM and XLF, loss of Setd2/H3K36me3 and Atm kinase activity did not result in complete abolishment of proper end-joining, but did lead to increased abnormal joining (e.g., SJ + CE joins and HJs), suggesting that Setd2/H3K36me3's role in end-ligation is functionally distinct from C-NHEJ XRCC4 paralogs XLF and PAXX (Lescale et al., 2016, Kumar et al., 2016). Although we could detect robust and efficient conversion of fully and properly recombined SJ + CJ products of the pMG-INV recombination substrate in induced Setd2-deficient v-Abl cells, we could also detect, with combined loss of Atm kinase activity, an aberrant SJ + CE recombination product. This raises the possibility of a CE-specific hairpin opening/repair defect, which would certainly require further study as CEs are thought be efficiently processed and repaired at a much higher rate than SEs and CE-only defects have not previously been reported (Ramsden and Gellert, 1995, Schlissel et al., 1993, Roth et al., 1992, Canela et al., 2016, Meek et al., 2016).
Paradoxically, whereas the end-ligation defect of Setd2/H3K36me3 loss, as determined by the assessment of a recombination substrate in transformed v-Abl cells, may appear subtle, in mice, its loss severely arrests normal B/T lymphocyte development, similar to what is observed in mice with loss of C-NHEJ factors (Alt et al., 2013; Kumar et al., 2014) and in contrast to loss of factors involved in end-ligation, such as Atm, Xlf, Paxx, or Mri individually (Bredemeyer et al., 2006, Kumar et al., 2016, Hung et al., 2018, Li et al., 2008, Lescale et al., 2016, Zha et al., 2011). This difference suggests that there are other determinants that contribute to the severe block in B (and T) lymphoid development in Setd2 knockout mice. For example, certain factors present at the Igh locus in proB cells may not be fully recapitulated in the assessment of rearrangement of an ectopic recombination substrate in transformed v-Abl cells, potentially affecting both the efficiency and accuracy of repair.
Likewise, it is also possible that there are other roles of ATM, or ATM targets, and SETD2 in end-joining that are not directly related and are mediated by distinct processes that, when combined, exacerbate mis-repair overall. Specifically, the loss of Setd2/H3K36me3 could influence the recruitment of factors associated with NHEJ in DNA DSB that recognize H3K36me3 (e.g., PHRF1 and PHF1) that could impact end-joining efficiency (Chang et al., 2015, Hong et al., 2008, Musselman et al., 2012), rather than playing a direct role in the stabilization of broken chromosomal ends, a function that has been ascribed to ATM (Bredemeyer et al., 2006). Additionally, we did not find any overt gene expression differences in alternative-NHEJ (A-NHEJ) factors and were not able to detect higher rates of deletions or translocations thought to accompany some forms of this A-NHEJ (Corneo et al., 2007) in either our CDR3 repertoire analysis, products of recombination of the recombination substrate in v-Abl cells, or in mice with dual loss of Setd2 and p53. These correlative lines of evidence however, do not fully preclude the possibility of rare forms of A-NHEJ contributing to the observed phenotype, warranting further investigation.
When we examined the abnormal recombination in proB cells of Setd2Δ/Δ mice more closely, we additionally noted significant loss of B cell repertoire and reduction of variable gene usage, characterized by less diversity in re-arranged, productive CDR3 sequences, as well as other abnormalities. Shortening of CDR3 length, without concomitant reduction of immune repertoire, has been previously connected to end-ligation factor, XLF (IJspeert et al., 2016). In addition, shortening of CDR3 sequences of Ig heavy chain rearrangements has previously been observed in Pol X family knockouts (pol λ, μ), which with terminal deoxynucleotidyl transferase participates in nucleotide end-processing of heavy chain junctions during V(D)J recombination (Bertocci et al., 2006). Thus, it is possible that Setd2/H3K36me3 may play additional roles in the end-processing of junctions, in addition to preventing the mis-repair in end-ligation we observed.
We also found that loss of Setd2/H3K36me3 increases sensitivity of cells to DSBs induced by ionizing radiation in both asynchronous and G1-arrested v-Abl cells, suggesting that Setd2/H3K36me3 may play a role in DSB repair activities in cells where HR would not be available, due to the lack of a sister chromatid. This result seemed to be supported by our discovery that neural-specific deletion of Setd2/H3K36me3 resulted in post-mitotic neuronal apoptosis. Although it is possible that other factors may be contributing to the neurogenesis defect, the temporal and spatial localization of apoptotic neuronal cells are highly suggestive of a role for Setd2/H3K36me3 in post-mitotic neurogenesis. The perinatal lethality we observed in Nestin-cre Setd2Δ/Δ mice is in stark contrast to Nestin-cre-driven conditional loss of core C-NHEJ factors, Xrcc4 or Lig4, where mice survive to adulthood (Frappart et al., 2009, Yan et al., 2006). Instead, it parallels the severity of the developmental arrest we observed in lymphogenesis, despite Setd2/H3K36me3 not being essential for C-NHEJ repair.
Previous studies have indicated a role for H3K36me3 in not only the recruitment of components on DNA damage repair in both MMR (MSH2/6) (Li et al., 2013) and HR (CtIP) (Pfister et al., 2014, Aymard et al., 2014) but also a role in a myriad of other cellular processes (Wagner and Carpenter, 2012). Although we have not exhaustively ruled out every potential role of Setd2/H3K36me3 in DSB repair, transcription, and splicing, we have attempted to study the role of Setd2/H3K36me3 in two systems that utilize NHEJ-mediated DSB repair by taking advantage of the non-redundancy of Setd2 in two independent mammalian developmental processes. Given the high frequency of SETD2 mutations in B and T cell lymphomas (Parker et al., 2016; Zhang et al., 2012, McKinney et al., 2017, Moffitt et al., 2017), primary human immunodeficiency (Ji et al., 2019), and neurological/developmental disorders (e.g., autism, intellectual disability, high-grade pediatric glioma) (Fontebasso et al., 2013, D'Gama et al., 2015, Tlemsani et al., 2016, Lelieveld et al., 2016), our findings demonstrating a role of Setd2/H3K36me3 in normal lymphogenesis and neurogenesis are especially noteworthy.
Limitations of Studies
Limitations of our studies include our inability to assess localization of critical NHEJ factors (e.g., Rag2, Ku70, Ku80, Xrcc4, Lig4, and others) due to the lack of readily available antibodies to perform ChIP of these proteins. In addition, although the use of v-Abl-transformed late-proB cell lines with an ectopically expressed recombination substrate can determine the essentiality of factors for end-joining in NHEJ, the use of such a system fails to recapitulate the factors or genomic structure present at the endogenous immunoglobulin loci, which may also influence the V(D)J recombination process during early lymphocyte development. Thus, it is possible that the severity of the block in normal lymphocyte development observed in vivo could be influenced by factors and mechanisms beyond the role of Setd2/H3K36me3 in end-joining during the repair phase of V(D)J recombination. Also, we were unable to assess if there was a non-enzymatic role for the Setd2 protein itself in any of the NHEJ processes in lymphocyte development, as expression of a full-length Setd2 (∼2,500 amino acids, ∼8-kb nucleotide coding sequence) in primary murine cells was unable to be achieved (by us and also not reported by other groups with genetically engineered Setd2 knockout mouse models, including but limited to Zhou et al., 2018, Zhang et al., 2018, Ji et al., 2019). We also did not fully and exhaustively investigate the role of A-NHEJ factors or the potential contribution of other roles that Setd2 and H3K36me3 play in normal cellular processes in both V(D)J recombination or post-mitotic neurogenesis settings.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank the Armstrong Laboratory for helpful feedback and fruitful scientific suggestions. We thank Dr. Fred W. Alt for experimental discussions, feedback, and critical review. We would also like to thank Charlie Hatton for help with GEO submissions. We thank the flow cytometry cores at Memorial Sloan-Kettering Cancer Center and Dana Farber Cancer Institute (Hematologic Neoplasia Core) for assistance in cell sorting, MGH Center for Computation and Integrative Biology DNA core for complete amplicon NGS sequencing, and the Dana-Farber Harvard Medical School rodent pathology core and Servicebio Inc. for support in histological analyses for these studies. This work was supported by National Institutes of Health, National Cancer Institute grants R01 CA176745 and 5P01 CA066996(SAA). S.H.C. is a Damon Runyon-Sohn Pediatric Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-5-13). V.K. is supported by an NCI 5F30CA189740-05. J.Z. was funded by the Chinese Scholarship Council. Funding. The Nussenzweig Laboratory is supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, Center for Cancer Research, and a Alex’s Lemonade Stand Foundation Award. The Sleckman laboratory is supported by NIH grants R01s AI047829 and AI074953.
Author Contributions
S.H.C. and S.A.A. designed experiments and wrote the paper. S.H.C., J.R.C., C.N.M., J.C.M., Y.X., and B.-R.C. performed experiments. J.Z. performed MiXCR and TRUST analyses, and R.P.K. performed bioinformatics analyses. V.K., E.C., P.J.H., Z.F., X.S.L., J.C., A.N., and B.P.S. provided experimental support and reagents for these studies as well as critical review of this paper.
Declaration of Interests
S.A.A. has been a consultant and/or shareholder for Epizyme Inc, Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, Syros Pharmaceuticals, OxStem Oncology, Accent Therapeutics, and Mana Therapeutics. S.A.A. has received research support from Janssen, Novartis, and AstraZeneca. S.H.C. is currently an employee at Beam Therapeutics. The authors have no additional financial interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100941.
Data and Code Availability
RNA-seq, ATAC-seq, ChIP-seq, TRUST, and MIXCR data generated and analyzed during the current study have been deposited into the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). The accession numbers for the RNA-seq, ATAC-seq, ChIP-seq, TRUST, and MIXCR data reported in this paper are GSE130904, GSE131588, GSE131608. These are unified under SuperSeries GSE131690.
Supplemental Information
References
- Abramowski V., Etienne O., Elsaid R., Yang J., Berland A., Kermasson L., Roch B., Musilli S., Moussu J.P., Lipson-Ruffert K. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2018;25:444–452. doi: 10.1038/cdd.2017.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt F.W., Zhang Y., Meng F.L., Guo C., Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;52:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F., Bulger B., Schmid C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing C.H., Swat W W., Alt F.W. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109:S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- Bertocci B., De Smet A., Weill J.C., Reynaud C.A. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006;25:31–41. doi: 10.1016/j.immuni.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Bettridge J., Na C.H., Pandey A., Desiderio S. H3K4me3 induces allosteric conformational changes in the DNA-binding and catalytic regions of the V(D)J recombinase. Proc. Natl. Acad. Sci. U S A. 2017;114:1904–1909. doi: 10.1073/pnas.1615727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin D.A., Poslavsky S., Davydov A.N., Frenkel F.E., Fanchi L., Zolotareva O.I., Hemmers S., Putintseva E.V., Obraztsova A.S., Shugay M. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 2017;35:908–911. doi: 10.1038/nbt.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer A.L., Sharma G.G., Huang C.Y., Helmink B.A., Walker L.M., Khor K.C., Nuskey B., Sullivan K.E., Pandita T.K., Bassing C.H., Sleckman B.P. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Canela A., Sridharan S., Sciascia N., Tubbs A., Meltzer P., Sleckman B.P., Nussenzweig A. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell. 2016;63:1–14. doi: 10.1016/j.molcel.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A., Difilippantonio S., Difilippantonio M.J., Fernandez-Capetillo O., Pilch D.R., Sedelnikova O.A., Eckhaus M., Ried T., Bonner W.M., Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T., Perlot T., Subrahmanyam R., Jani A., Goff P.H., Zhang Y., Ivanova I., Alt F.W., Sen R. A 220-nucleotide deletion of the intronic enhancer reveals an epigenetic hierarchy in immunoglobulin heavy chain locus activation. J. Exp. Med. 2009;206:1019–1027. doi: 10.1084/jem.20081621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.F., Chu P.C., Wu P.Y., Yu M.Y., Lee J.Y., Tsai M.D., Chang M.S. PHRF1 promotes genome integrity by modulating non-homologous end-joining. Cell Death Dis. 2015;6:e1716. doi: 10.1038/cddis.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D., Sen R. Stepwise activation of the immunoglobulin mu heavy chain gene locus. EMBO J. 2001;20:6394–6403. doi: 10.1093/emboj/20.22.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneo B., Wendland R.L., Deriano L., Cui X., Klein I.A., Wong S.Y., Arnal S., Holub A.J., Weller G.R., Pancake B.A. Rag mutations reveal robust alternative end joining. Nature. 2007;449:483–486. doi: 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- D'Gama A.M., Pochareddy S., Li M., Jamuar S.S., Reiff R.E., Lam A.N., Sestan N., Walsh C.A. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 2015;88:910–917. doi: 10.1016/j.neuron.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio M.J., Zhu J., Chen H.T., Meffre E., Nussenzweig M.C., Max E.E., Ried T., Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fnu S., Williamson E.A., De Haro L.P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S.H., Nickoloff J.A., Hromas R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. U S A. 2011;108:540–545. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso A.M., Schwartzentruber J., Khuong-Quang D.A., Liu X.Y., Sturm D., Korshunov A., Jones D.T., Witt H., Kool M., Albrecht S. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125:659–669. doi: 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank K.M., Sharpless N.E., Gao Y., Sekiguchi J.M., Ferguson D.O., Zhu C., Manis J.P., Horner J., DePinho R.A., Alt F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Frappart P.O., McKinnon P.J. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair (Amst) 2008;7:1051–1060. doi: 10.1016/j.dnarep.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart P.O., Lee Y., Russell H.R., Chalhoub N., Wang Y.D., Orii K.E., Zhao J., Kondo N., Baker S.J., McKinnon P.J. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc. Natl. Acad. Sci. U S A. 2009;106:1880–1885. doi: 10.1073/pnas.0806882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Sun Y., Frank K.M., Dikkes P., Fujiwara Y., Seidl K.J., Sekiguchi J.M., Rathbun G.A., Swat W., Wang J. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Gao Y., Ferguson D.O., Xie W., Manis J.P., Sekiguchi J., Frank K.M., Chaudhuri J., Horner J., DePinho R.A., Alt F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- Gu Y., Sekiguchi J., Gao Y., Dikkes P., Frank K., Ferguson D., Hasty P., Chun J., Alt FW F.W. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc. Natl. Acad. Sci. U S A. 2000;97:2668–2673. doi: 10.1073/pnas.97.6.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E., Thiemann S., Storch B., Jumaa H., Nielsen P.J., Pelanda R., Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. U S A. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z., Jiang J., Lan L., Nakajima S., Kanno S., Koseki H., Yasui A. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 2008;36:2939–2947. doi: 10.1093/nar/gkn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Sun X.J., Zhang Y.L., Kuang Y., Hu C.Q., Wu W.L., Shen S.H., Du T.T., Li H., He F. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc. Natl. Acad. Sci. U S A. 2010;107:2956–2961. doi: 10.1073/pnas.0915033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Zhang J., Liu J.S., Li B., Liu X.S. Evaluation of immune repertoire inference methods from RNA-seq data. Nat. Biotechnol. 2018;36:1034. doi: 10.1038/nbt.4294. [DOI] [PubMed] [Google Scholar]
- Hu X., Zhang J., Wang J., Fu J., Li T., Zheng X., Wang B., Gu S., Jiang P., Fan J. Landscape of B cell immunity and related immune evasion in human cancers. Nat. Genet. 2019;51:560–567. doi: 10.1038/s41588-018-0339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung P.J., Johnson B., Chen B.R., Byrum A.K., Bredemeyer A.L., Yewdell W.T., Johnson T.E., Lee B.J., Deivasigamani S., Hindi I. MRI is a DNA damage response adaptor during classical non-homologous end joining. Mol. Cell. 2018;71:332–342. doi: 10.1016/j.molcel.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJspeert H., Rozmus J., Schwarz K., Warren R.L., van Zessen D., Holt R.A., Pico-Knijnenburg I., Simons E., Jerchel I., Wawer A. XLF deficiency results in reduced N-nucleotide addition during V(D)J recombination. Blood. 2016;128:650–659. doi: 10.1182/blood-2016-02-701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E.A., Ananieva O., Brown M.L., Chang Y. Growth, differentiation, and malignant transformation of pre-B cells mediated by inducible activation of v-Abl oncogene. J. Immunol. 2006;176:6831–6838. doi: 10.4049/jimmunol.176.11.6831. [DOI] [PubMed] [Google Scholar]
- Johnson K., Chaumeil J., Skok J.A. Epigenetic regulation of V(D)J recombination. Essays Biochem. 2010;48:221–243. doi: 10.1042/bse0480221. [DOI] [PubMed] [Google Scholar]
- Ji Y., Resch W., Corbett E., Yamane A., Casellas R., Schatz D.G. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z., Sheng Y., Miao J., Li X., Zhao H., Wang J., Cheng C., Wang X., Liu K., Zhang K. The histone methyltransferase Setd2 is indispensable for V(D)J recombination. Nat. Commun. 2019;10:3353. doi: 10.1038/s41467-019-11282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus M., Alimzhanov M.B., Rajewsky N., Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117:787–800. doi: 10.1016/j.cell.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Kumar V., Alt F.W., Frock R.L. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc. Natl. Acad. Sci. U S A. 2016;113:10619–10624. doi: 10.1073/pnas.1611882113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Alt F.W., Oksenych V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst). 2014;16:11–22. doi: 10.1016/j.dnarep.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld S.H., Reijnders M.R., Pfundt R., Yntema H.G., Kamsteeg E.J., de Vries P., de Vries B.B., Willemsen M.H., Kleefstra T., Löhner K. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016;19:1194–1196. doi: 10.1038/nn.4352. [DOI] [PubMed] [Google Scholar]
- Lescale C., Abramowski V., Bedora-Faure M., Murigneux V., Vera G., Roth DB D.B., Revy P., de Villartay J.P., Deriano L. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat. Commun. 2016;7:10529. doi: 10.1038/ncomms10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Li T., Wang B., Dou R., Zhang J., Liu J.S., Liu X.S. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA–seq data. Nat. Genet. 2017;49:482–483. doi: 10.1038/ng.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Mao G., Tong D., Huang J., Gu L., Yang W., Li G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Alt FW F.W., Cheng H.L., Brush J.W., Goff P.H., Murphy M.M., Franco S., Zhang Y., Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol. Cell. 2008;31:631–640. doi: 10.1016/j.molcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H., Hippenmeyer S., Ghashghaei H.T. A Nestin-cre transgenic mouse is insufficient for recombination in early embryonic neural progenitors. Biol. Open. 2012;1:1200–1203. doi: 10.1242/bio.20122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Jain S.U., Hoelper D., Bechet D., Molden R.C., Ran L., Murphy D., Venneti S., Hameed M., Pawel B.R. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–849. doi: 10.1126/science.aac7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar B.G., Chu S.H., Kahn J.D., Krivtsov A.V., Koche R., Castellano C.A., Kotlier J.L., Zon R.L., McConkey M.E., Chabon J. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood. 2017;130:2631–2641. doi: 10.1182/blood-2017-03-775569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson L.S., Corcoran A.E. Local and global epigenetic regulation of V(D)J recombination. Curr. Top. Microbiol. Immunol. 2012;356:65–89. doi: 10.1007/82_2011_137. [DOI] [PubMed] [Google Scholar]
- Matthews A.G., Kuo A.J., Ramón-Maiques S., Han S., Champagne K.S., Ivanov D., Gallardo M., Carney D., Cheung P., Ciccone D.N. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney M., Moffitt A.B., Gaulard P., Travert M., De Leval L., Nicolae A., Raffeld M., Jaffe E.S., Pittaluga S., Xi L. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7:369–379. doi: 10.1158/2159-8290.CD-16-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek K., Xu Y., Bailie C., Kefei Y., Neal J.A. The ATM kinase restrains joining of both VDJ signal and coding ends. J. Immunol. 2016;197:3165–3174. doi: 10.4049/jimmunol.1600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt A.B., Ondrejka S.L., McKinney M., Rempel R.E., Goodlad J.R., Teh C.H., Leppa S., Mannisto S., Kovanen P.E., Tse E. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J. Exp. Med. 2017;214:1371–1386. doi: 10.1084/jem.20160894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C.A., Avvakumov N., Watanabe R., Abraham C.G., Lalonde M.E., Hong Z., Allen C., Roy S., Nuñez J.K., Nickoloff J. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012;19:1266–1272. doi: 10.1038/nsmb.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker H., Rose-Zerilli M.J., Larrayoz M., Clifford R., Edelmann J., Blakemore S., Gibson J., Wang J., Ljungström V., Wojdacz T.K. Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia. 2016;30:2179–2186. doi: 10.1038/leu.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S.X., Ahrabi S., Zalmas L.P., Sarkar S., Aymard F., Bachrati C.Z., Helleday T., Legube G., La Thangue N.B., Porter A.C., Humphrey T.C. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006–2018. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden D.A., Gellert M. Formation and resolution of double-strand break intermediates in V(D)J rearrangement. Genes Dev. 1995;9:2409–2420. doi: 10.1101/gad.9.19.2409. [DOI] [PubMed] [Google Scholar]
- Rickert R.C., Roes J., Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nuclei Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth D.B., Menetski J.P., Nakajima P.B., Bosma M.J., Gellert M. V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:983–991. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- Schatz F.G., Swanson P.G. V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 2001;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Schlissel M., Constantinescu A., Morrow T., Baxter M., Peng A. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 1993;7:2520–2532. doi: 10.1101/gad.7.12b.2520. [DOI] [PubMed] [Google Scholar]
- Siegemund S., Shepherd J., Xiao C., Sauer K. hCD2-iCre and Vav-iCre mediated gene recombination patterns in murine hematopoietic cells. PLoS One. 2015;10:e0124661. doi: 10.1371/journal.pone.0124661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki N., Lieber M.R. Histone methylation and V(D)J recombination. Int. J. Hematol. 2014;100:230–237. doi: 10.1007/s12185-014-1637-4. [DOI] [PubMed] [Google Scholar]
- Takata M., Sasaki M.S., Sonoda E., Morrison C., Hashimoto M., Utsumi H., Yamaguchi-Iwai Y., Shinohara A., Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlemsani C., Luscan A., Leulliot N., Bieth E., Afenjar A., Baujat G., Doco-Fenzy M., Goldenberg A., Lacombe D., Lambert L. SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. J. Med. Genet. 2016;53:743–751. doi: 10.1136/jmedgenet-2015-103638. [DOI] [PubMed] [Google Scholar]
- Wagner E.J., Carpenter P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012;23:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.T., Kaushal D., Murphy M., Zhang Y., Datta A., Chen C., Monroe B., Mostoslavsky G., Coakley K., Gao Y. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc. Natl. Acad. Sci. U S A. 2006;103:7378–7383. doi: 10.1073/pnas.0601938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin B., Savic V., Juntilla M.M., Bredemeyer A.L., Yang-Iott K.S., Helmink B.A., Koretzky G.A., Sleckman B.P., Bassing C.H. Histone H2AX stabilizes broken DNA strands to suppress chromosome breaks and translocations during V(D)J recombination. J. Exp. Med. 2009;206:2625–2639. doi: 10.1084/jem.20091320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S., Guo C., Boboila C., Oksenych V., Cheng H.L., Zhang Y., Wesemann D.R., Yuen G., Patel H., Goff P.H. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469:250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Ding L., Holmfeldt L., Wu G., Heatley S.L., Payne-Turner D., Easton J., Chen X., Wang J., Rusch M. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.L., Sun J.W., Xie Y.Y., Zhou Y., Liu P., Song J.C., Xu C.H., Wang L., Liu D., Xu A.N. Setd2 deficiency impairs hematopoietic stem cell self-renewal and causes malignant transformation. Cell Res. 2018;28:476–790. doi: 10.1038/s41422-018-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Yan X., Feng X., Bu J., Dong Y., Lin P., Hayashi Y., Huang R., Olsson A., Andreassen P.R. Setd2 regulates quiescence and differentiation of adult hematopoietic stem cells by restricting RNA polymerase II elongation. Hematologica. 2018;103:110–1123. doi: 10.3324/haematol.2018.187708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., He F., Zeng H., Ling S., Chen A., Wang Y., Yan X., Wei W., Pang Y., Cheng H. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat. Genet. 2014;46:287–293. doi: 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ATAC-seq, ChIP-seq, TRUST, and MIXCR data generated and analyzed during the current study have been deposited into the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). The accession numbers for the RNA-seq, ATAC-seq, ChIP-seq, TRUST, and MIXCR data reported in this paper are GSE130904, GSE131588, GSE131608. These are unified under SuperSeries GSE131690.