

Summary
The development of an in vitro system in which human primordial germ cell-like cells (hPGCLCs) are generated from human pluripotent stem cells (hPSCs) has been invaluable to further our understanding of human primordial germ cell (hPGC) specification. However, the means to evaluate the next fundamental steps in germ cell development have not been well established. In this study we describe a two dimensional extended culture system that promotes proliferation of specified hPGCLCs, without reversion to a pluripotent state. We demonstrate that hPGCLCs in extended culture undergo partial epigenetic reprogramming, mirroring events described in hPGCs in vivo, including a genome-wide reduction in DNA methylation and maintenance of depleted H3K9me2. This extended culture system provides a new approach for expanding the number of hPGCLCs for downstream technologies, including transplantation, molecular screening, or possibly the differentiation of hPGCLCs into gametes by in vitro gametogenesis.
Keywords: human, primordial germ cells, primordial germ cell-like cells
Graphical Abstract
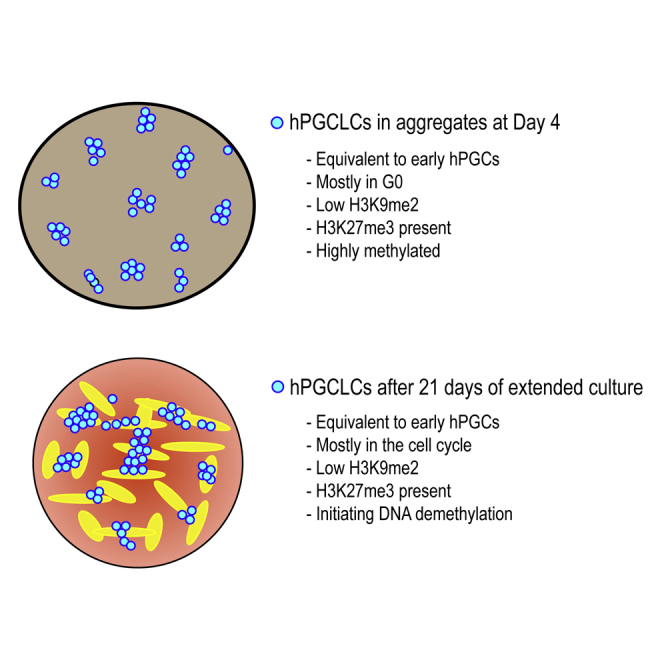
Highlights
-
•
A new technique for culturing hPGCLCs for a minimum of 21 days in extended culture
-
•
Extended culture promotes hPGCLCs to enter the cell cycle and proliferate
-
•
Extended culture promotes the initiation of DNA demethylation
In the article, Clark and colleagues describe a 2D culture system that supports the expansion of human primordial germ cell-like cells (hPGCLCs) differentiated from human pluripotent stem cells. The cultured hPGCLCs maintains germline identity, while undergoing proliferation and initiating DNA demethylation. Creating a culture system that supports hPGCLCs provides new opportunities to promote the differentiation of hPGCLCs into gametes.
Introduction
Primordial germ cells (PGCs) are the first germline embryonic progenitors in all metazoans. Once specified, PGCs are fate restricted to become mature gametes in adults. In mammals, PGC specification is followed by multiple key events as PGCs migrate from their initial site outside the embryo, into the hindgut and the genital ridges. During this time PGCs proliferate and undergo dramatic epigenetic reprogramming, including the global loss of methylated cytosines from DNA, and the dynamic loss of histone H3 lysine 9 dimethylation (H3K9me2) and gain of histone H3 lysine 27 trimethylation (H3K27me3) in PGC chromatin (Gkountela et al., 2013, Guo et al., 2015, Seisenberger et al., 2012, Seki et al., 2005, Seki et al., 2007, Tang et al., 2015). Once the PGCs have settled in the embryonic gonad, they will differentiate into male or female germ cells. Mouse models have revealed that abnormalities in PGC specification, proliferation, epigenetic reprogramming, and differentiation can lead to germ cell tumors, infertility, or the transmission of disease alleles to the next generation. Therefore, understanding PGC development is critical to understanding mechanisms responsible for mammalian fertility and to facilitate our understanding of human reproductive health.
Experimental strategies to investigate PGC proliferation, epigenetic reprogramming and differentiation in mammals has been hampered by a lack of approaches to support the long-term self-renewal of mouse (m) and human (h) PGCs ex vivo. Using the mouse, approaches for short-term culture of mPGCs or mPGC-like cells (mPGCLCs) differentiated from pluripotent stem cells have been described (Farini et al., 2005, Oliveros-Etter et al., 2015, Ohta et al., 2017). However, these approaches have limitations because removal of mPGCs from their embryonic environment results in either cell death or reversion into a pluripotent self-renewing cell type called embryonic germ cells (EGCs) (Durcova-Hills et al., 2001, Leitch et al., 2013, Matsui et al., 1992, Resnick et al., 1992). The ability of mPGCs to revert into mEGCs is age-dependent and coincident with expression of the pluripotent transcription factors Nanog, Oct4, and Sox2 during the mPGC stage beginning at embryonic day 7.5 (E7.5) (Leitch et al., 2013).
Evaluating the cell and molecular mechanisms that regulate hPGC development is challenging due to limited access to human embryonic and fetal tissue. A small number of studies have cultured hPGCs ex vivo; however, under these conditions the hPGCs either die or revert into hEGC-like cells (Hua et al., 2009, Liu et al., 2004, Shamblott et al., 1998, Turnpenny et al., 2003). Unlike the mouse, where mEGCs exhibit long-term self-renewal, the reversion of hPGCs into hEGC-like cells is extremely inefficient, and hEGC-like cells cannot be sustained in long-term self-renewing conditions (Turnpenny et al., 2006).
Given this, hPGC development is routinely modeled using the differentiation of human pluripotent stem cells (hPSCs) into hPGCLCs, with the majority of studies using three-dimensional (3D) disorganized aggregates (Irie et al., 2015, Sasaki et al., 2015, Sybirna et al., 2019). These methods create early hPGCLCs equivalent to week 3 post-fertilization of human embryo development. Recent studies using microfluidics and the generation of modeled 3D embryos from hPSCs also results in the differentiation of early hPGCLCs (Zheng et al., 2019). However, analyzing hPGCLC biology beyond specification is a challenge in this microfluidic system, as the 3D modeled embryos can only be maintained for 48–72 h before the system collapses (Zheng et al., 2019). In contrast, hPGCLCs in the 3D disorganized aggregates can be maintained for about 2 weeks in the aggregate, and during this time the hPGCLCs undergo limited epigenetic reprogramming (Irie et al., 2015, Sasaki et al., 2015, von Meyenn et al., 2016). Given the relatively undefined nature of the somatic cells in the 3D disorganized aggregates, and the relatively short time in which the modeled 3D embryo technology from stem cells can be sustained, it could be proposed that neither model has been optimized for evaluating the cell and molecular events that occur after hPGCLC specification. Given this, we tested the ability of hPGCLCs to proliferate in extended culture as a means to promote the expansion of the hPGCLC population in vitro for downstream applications.
Results
hPGCLCs Maintain hPGC Identity when Cultured on STO Cells
In this study, hPGCLCs were differentiated in disorganized 3D aggregates from primed human embryonic stem cells (hESCs) through incipient mesoderm-like cells (iMeLCs) following the protocol first developed by Sasaki et al. (2015) with minor alterations, such as omission of stem cell factor (SCF) from the PGCLC media (Chen et al., 2017) (Figure 1A). hPGCLCs were isolated from the aggregates at day 4 (D4) of aggregate differentiation using fluorescence-activated cell sorting (FACS) for integrin alpha 6 (ITGA6) and epithelial cell adhesion molecule (EPCAM) (Chen et al., 2017, Sasaki et al., 2015). FACS-isolated hPGCLCs were maintained on a feeder layer of STO cells for up to 21 additional days (D4C21), which would correspond to a total of 26 days from the undifferentiated hESC state (Figure 1A). We first evaluated two types of culture media for maintaining hPGCLC identity on STOs. Seven-factor (7F) medium, which contains a complex recipe of cytokines and chemicals that were previously shown to be necessary for mPGC proliferation and survival (Farini et al., 2005). 7F medium has also been shown to support hPGCLC survival on polyethylene terephthalate membranes for 4 days (Gell et al., 2018). The second medium, called FR10, supports mPGCLC proliferation for 7 days on m220 feeders (Ohta et al., 2017). Both media contain the cyclic adenosine monophosphate agonist forskolin, in addition to the cytokine SCF (Table 1).
Figure 1.

hPGCLCs Cultured on STOs Maintain Germline Identity
(A) Experimental scheme for extended culture of human primordial germ cell-like cells (hPGCLCs) on STOs. Day 4 (D4) hPGCLCs are maintained in culture for additional days (X) (D4CX). hPGCLCs in this study were cultured for a maximum of 21 days (D4C21).
(B) Bright-field (left), fluorescent microscopy (middle), and merged (right) images, illustrating UCLA2-GFP D4C3 hPGCLCs in culture on STOs in FR10 media.
(C) Immunofluorescent (IF) images of UCLA2 D4C10 hPGCLCs in 7-factor (7F) (top) and FR10 (bottom) media. Germline identity was evaluated using PRDM1 (yellow), TFAP2C (magenta), and SOX17 (cyan).
(D) Quantification of IF staining in UCLA2 D4C10 hPGCLCs for triple-positive SOX17/TFAP2C/PRDM1 (S/T/P) hPGCLCs and double-positive SOX17/PRDM1 (S/P) cells, or SOX17/TFAP2C (S/T) cells.
(E) IF images of UCLA1 D4C21 hPGCLCs in FR10 media. Germline identity denoted by triple-positive PRDM1 (yellow), TFAP2C (magenta), and SOX17 (cyan) cells. Scale bars, 50 μm (B, C, and E).
Table 1.
Key Components of 7F and FR10 Media. Shown in bold are media components common to both media types.
| Media | 7F | FR10 |
|---|---|---|
| Growth factors and other additives |
|
|
hPGCLCs were isolated from four different hESC lines and sublines, UCLA1 (U1) (46, XX), UCLA2 (U2) (46, XY), a UCLA2 subline called UCLA2-GFP where GFP is driven from the ubiquitin promoter, and UCLA6 (U6) (46, XY). Using the UCLA2-GFP subline, we observed small clusters of hPGCLC by day 3 of extended culture (D4C3) in both 7F and FR10 media (Figure 1B, represents FR10). Notably, the hPGCLCs were loosely attached on top of the STOs, forming grape-like clusters rather than typical flat primed hPSC colonies.
Given this unique morphology, we next performed immunofluorescence (IF) staining of U1, U2, and U6 hPGCLCs at D4C10 in 7F and FR10 media to evaluate hPGC identity (Figures 1C, S1A, and S1B). Germline identity was evaluated by triple staining for PRDM1, TFAP2C, and SOX17, which discriminate hPGCs in vivo from somatic cells (Chen et al., 2017). Using this strategy, we discovered that the majority of hPGCLCs were triple-positive in both 7F and FR10, with a small fraction of SOX17/TFAP2C double-positive hPGCLCs, which were more apparent in 7F relative to FR10 (Figure 1D). SOX17/PRDM1 double-positive cells were never observed (Figure 1D). Given the ability to maintain a greater percent of triple-positive hPGCLCs we continued the remainder of the study with FR10 media. To determine whether extended culture could be prolonged for additional days, we cultured hPGCLCs from the U1 and U2 hESC line to 21 days (D4C21) in FR10 media (Figure 1E, represents U1) and show that SOX17/TFAP2C/PRDM1 triple-positive hPGCLCs can be sustained for at least 21 days.
Because FR10 medium supports survival of E9.5 mPGCs on m220 feeders, albeit with limited proliferation (Ohta et al., 2017), we next evaluated whether FR10 medium supports the survival of hPGCs isolated from human embryonic gonads. To achieve this, we isolated TNAP/cKIT double-positive hPGCs by FACS from male E53 embryonic testes, and cultured the resulting E53 hPGCs on STOs in FR10 medium for 10 days. In FR10 medium, the E53 hPGCs remained round and loosely attached to the STOs as individual cells. Germline identity was maintained in the E53 hPGCs at day 10 (E53D10), as evident by IF staining for VASA and TFAP2C, while being negative for SOX2 (Figure S1C). This result suggests that the culture of E53 hPGCs in FR10 on STOs does not lead to reversion into hEGC-like cells during the first 10 days of extended culture.
Extended Culture Supports a Transcriptional Identity Similar to Early hPGCLCs
Given that hPGCLCs grew in clusters in extended culture, whereas hPGCs did not, we next sought to evaluate whether the hPGCLCs were acquiring markers of hEGCs. First, we performed IF at D4C10 for SOX2 (a EGC marker) and SOX17 (an hPGC marker). Undifferentiated hESCs were used as a positive control for SOX2. These results show that hPGCLCs at D4C10 are positive for SOX17 and do not express the hEGC marker SOX2 (Figure 2A, quantified in S2A).
Figure 2.

hPGCLCs in Extended Culture Maintain a Transcriptome Similar to Specified hPGCLCs
(A) IF images of primed UCLA2 hESCs (top) and D4C10 hPGCLCs in FR10 media (bottom). Pluripotency/EGC identity was evaluated using SOX2 (magenta). SOX17 (cyan) identifies the hPGCLCs. Scale bars, 50 μm.
(B) FACS plot of UCLA2 D4C10 hPGCLCs on STOs, CD29-positive mouse cells and TRA-1-85-positive human cells form two distinct populations.
(C) Principle-component analysis (PCA) of the transcriptomes of UCLA1 (U1) naive hESCs in 5i/L/FA (N = 4); UCLA1 and UCLA2: iMeLCs (N = 2), U1 and U2 primed hESCs (N = 2), U1 and U2 D4 hPGCLCs (N = 3), U1 and U2 D4C10 hPGCLCs (N = 3); and gonadal hPGCs (N = 5). Gene expression analysis includes RNA-seq data from Pastor et al. (2016) (5i/L/FA naive hESCs) and Chen et al. (2017) (primed hESCs, iMeLCs, D4 hPGCLCs, and hPGCs). N = independent replicates.
(D) Heatmap of gene expression levels of representative genes in UCLA1 naive hESCs in 5i/L/FA; UCLA1 and UCLA2: iMeLCs, primed hESCs and D4 hPGCLCs and D4C10 hPGCLCs; gonadal PGCs. Genes evaluated are grouped as diagnostic for early hPGCs (i.e., SOX17, PRDM1, and TFAP2C) and late hPGC (i.e., DAZL, DDX4, and SYCP3). Rep, independent replicates.
(E) Differentially expressed genes (DEGs) in D4C10 hPGCLCs compared to primed hESCs (left), D4 hPGCLCs (center) and gonadal hPGCs (right). Using DEGs with fold change ≥4, false discovery rate < 0.05, and average RPKM in either cell types must be >1.
(F) Dot plot depicting gene ontology (GO) terms identified for the N = 675 upregulated genes in D4C10 hPGCLCs versus D4 hPGCLCs.
Next, we developed a FACS strategy to isolate the cultured hPGCLCs from the STOs using fluorescent-labeled antibodies. This involved the use of an antibody that recognized the surface molecule, cluster of differentiation 29 (CD29) to discriminate the mouse STOs together with an antibody that recognizes TRA-1-85, which discriminates human cells. Using this approach, we identified a population of CD29-positive mouse cells and TRA-1-85-positive human cells (Figure 2B). Using FACS to isolate the TRA-1-85-positive/CD29-negative cells, we performed RNA sequencing (RNA-seq) of UCLA1 and UCLA2 D4C10 putative hPGCLCs, and compared this with RNA-seq results from previously published data, including naive hESCs (Pastor et al., 2016), primed hESCs, iMeLCs, D4 hPGCLCs (Chen et al., 2017), and hPGCs isolated at various stages of germline cell development (Chen et al., 2017). Details on the RNA-seq libraries can be found in Table S1. Principle-component analysis (PCA) revealed that D4C10 hPGCLCs clustered together with D4 hPGCLCs in both principle component 1 (PC1) and PC2 (Figure 2C), and that germline identity of the D4C10 hPGCLCs is equivalent to early hPGCs but not late PGCs that have colonized the gonad (Figure 2D). Real-time PCR was used to confirm the RNA-seq results showing that D4C10 hPGCLCs expressed equivalent levels of PRDM1, SOX17, TFAP2C, and NANOS3 to D4 hPGCLCs, and that SOX2 mRNA was not detected (Figure S2B). These results suggest that the extended culture system maintains hPGCLC identity at a stage equivalent to early hPGCs, and does not promote reversion to self-renewing pluripotent primed or naive hES-like cells.
Although the overall transcriptional identity of hPGCLCs at D4C10 was similar to D4 hPGCLCs, we identified a small number of differentially expressed genes when performing pairwise comparisons with primed hESCs, D4 hPGCLCs and hPGCs (Figure 2E). Notably, this result shows that extended culture leads to considerably more upregulated genes in the D4C10 hPGCLCs relative to downregulated genes in each pairwise comparison (Figure 2E; Chart S1). Furthermore, gene ontology (GO) (Figure 2F) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure S2C) of the 675 upregulated genes in D4C10 hPGCLCs relative to D4 hPGCLCs reveals biological terms, including extracellular matrix, endoplasmic reticulum lumen, and a variety of cell signaling pathways, indicating that the hPGCLCs in extended culture are responding to their new culture environment.
Extended Culture on STO Cells Supports hPGCLC Proliferation
Previous studies using FR10 to culture mPGCLCs resulted in 20- to 50-fold expansion in mPGCLC numbers (Ohta et al., 2017). To quantify the total number of hPGCLCs at D4C10 and D4C21 we used FACS to isolate and count the number of hPGCLCs isolated from the STOs at each time point. We compared these values with the initial number of D4 hPGCLCs plated into culture (Figure 3A). At D4C10 the UCLA1 and UCLA2 D4C10 hPGCLCs showed only modest capacity for expansion, increasing around 1.5- to 2-fold in cell number (Figure 3A). We do not think this modest increase is due to an increase in apoptosis as apoptotic genes are not differentially expressed (Figure S3A). However, by D4C21 the number of TRA-1-85 cells had increased by 25-fold (Figure 3A).
Figure 3.

hPGCLCs in Extended Culture Self-Renew and Undergo Expansion
(A) Dot plot of the fold change in FACS isolated D4C10 and D4C21 hPGCLCs, compared with the starting D4 hPGCLCs. UCLA1 D4C10 (n = 5); UCLA2 D4C10 (n = 6) and D4C21 (n = 4). N = technical replicates. UCLA1 D4C10 and UCLA2 D4C10 represent 3 independent experiments, UCLA2 D4C21 represent 2 independent experiments. Magenta dotted line represents a fold change of 1. N.S., not significant; ∗∗∗∗p ≤ 0.0001. Error bars = mean SD.
(B) IF images of Ki67 (magenta) in UCLA2 D4 aggregate hPGCLCs, marked by SOX17/TFAP2C (cyan) (top panel), D4C10 hPGCLCs, marked by SOX17 (cyan) (middle panel), and D4C21 hPGCLCs, marked by SOX17 (cyan) (bottom panel). S17/TF = SOX17/TFAP2C. Scale bars, 50 μm (top), 20 μm (middle), and 20 μm (bottom).
(C) Quantification of Ki67-positive hPGCLCs in UCLA2 D4 aggregates (D4 agg), D4C10, and D4C21 hPGCLCs in FR10 media. ∗∗∗∗p ≤ 0.0001. Error bars = mean SD for D4 agg (N = 4), D4C10 (N = 3), and D4C21 (N = 3), N, the number of independent biological replicates.
(D) IF images of 5-ethynyl-2′-deoxyuridine (EdU) (magenta) in UCLA2 D4 aggregate hPGCLCs, marked by SOX17/TFAP2C (cyan) (top panel), D4C10 hPGCLCs, marked by SOX17 (cyan) (middle panel), and D4C21 hPGCLCs, marked by SOX17 (cyan) (bottom panel). S17/TF = SOX17/TFAP2C. Scale bars, 30 μm (top), 50 μm (middle), and 50 μm (bottom).
(E) Quantification of EdU-positive hPGCLCs in UCLA2 D4 aggregates (D4 agg), D4C10, and D4C21 hPGCLCs in FR10 media. N.S., not significant. Error bars = mean SD of 9 aggregates (D4 agg), 16 colonies (D4C10), and 4 colonies (D4C21) from 3 independent biological replicates.
Using Ki67, we next quantified the percentage of hPGCLCs in cycle (Ki67+) at D4 in the aggregates and then in extended culture at D4C10 and D4C21. To identify D4 hPGCLCs in the aggregates, we quantified SOX17 and TFAP2C double positive hPGCLCs, which revealed that D4 hPGCLCs are mostly out of the cell cycle (Figure 3B, quantified in 3C). In contrast, in extended culture there was a statistically significant increase in the percentage of SOX17-positive hPGCLCs in cycle, with 60% of D4C10 and D4C21 hPGCLCs expressing Ki67 (Figure 3C). To evaluate progression through S phase, we performed a 5-ethynyl-2′-deoxyuridine staining on D4 aggregates and D4C10 and D4C21 of extended culture (Figure 3D, quantified in 3E). This result shows that, at D4, 25% of hPGCLCs are in S phase, which corresponds to the majority of cycling hPGCLCs in the aggregate. In the extended culture our results suggest that the percentage of cells in S phase remains the same at around 30%.
Extended Culture Maintains Partial Histone Reprogramming
Given that hPGCLCs are proliferating in extended culture, we next sought to evaluate epigenetic reprogramming. In the mouse embryo, H3K9me2 is one of the earliest histone modifications to be depleted from chromatin soon after mPGC specification (Kurimoto et al., 2015, Prokopuk et al., 2017, Seki et al., 2005). To evaluate H3K9me2, we compared H3K9me2 IF intensity in D4 aggregate hPGCLCs and at D4C10 and D4C21 of extended culture. Fluorescence intensity was normalized relative to the D4 aggregate somatic cells (Figure 4A, quantified in 4B). Consistent with previous reports (Sasaki et al., 2015), H3K9me2 was reduced in hPGCLCs in the aggregate compared with the somatic cells, and these reduced levels were maintained in extended culture. Because hPGCs are enriched in H3K27me3 (Gkountela et al., 2013, Guo et al., 2015, Tang et al., 2015), we analyzed H3K27me3 in hPGCLCs at D4, D4C10, and D4C21 relative to D4 somatic cells (Figure 4C, quantified in 4D). These results revealed an enrichment in H3K27me3 in D4 hPGCLCs relative to somatic cells. However, with extended culture to day D4C21, H3K27me3 levels were reduced to similar levels found in somatic cells of the aggregate. We also evaluated H3K9me2 and H3K27me3 in D4C10 hPGCLCs from the UCLA1 and UCLA6 hESC lines, which gave similar results (Figures S4A and S4B).
Figure 4.

Partial Chromatin Remodeling Is Maintained in Extended Culture hPGCLCs
(A) IF images of H3K9me2 in aggregates containing UCLA2 D4 hPGCLCs, marked by SOX17/TFAP2C (cyan) (top panel), D4C10 hPGCLCs, marked by SOX17 (cyan) (middle panel), and D4C21 hPGCLCs, marked by SOX17 (cyan) (bottom panel). S17/TF = SOX17/TFAP2C. Scale bars, 40 μm (top), 30 μm (middle), and 50 μm (bottom).
(B) Quantification of H3K9me2 in UCLA2 aggregates containing D4 hPGCLCs (D4 agg), D4C10 hPGCLCs, and D4C21 hPGCLCs in FR10 medium, as compared with H3K9me2 in D4 agg somatic cells, relative to DAPI fluorescence intensity. N.S., not significant; ∗∗∗∗p < 0.0001. Error bars = mean SD of three independent biological replicates. Numbers in parentheses are equal to the total number of cells analyzed.
(C) IF images of H3K27me3 in UCLA2 aggregates containing D4 hPGCLCs, marked by SOX17/TFAP2C (cyan) (top panel), D4C10 hPGCLCs, marked by SOX17 (cyan) (middle panel), and D4C21 hPGCLCs, marked by SOX17 (cyan) (bottom panel). S17/TF = SOX17/TFAP2C. Scale bars, 50 μm (top), 20 μm (middle), and 50 μm (bottom).
(D) Quantification of H3K27me3 levels in D4 hPGCLCs in the aggregate (D4 agg), U2 D4C10 hPGCLCs, and U2 D4C21 hPGCLCs in FR10 medium, as compared with H3K27me3 levels of D4 agg somatic cells, relative to DAPI fluorescence intensity. ∗∗∗∗p < 0.0001. Error bars = mean SD of three independent biological replicates. Numbers in parentheses are equal to the total number of cells analyzed.
Extended Culture Supports Heterogeneous DNA Demethylation of hPGCLCs
Given that reduced H3K9me2 levels were maintained in hPGCLCs during extended culture, we next evaluated DNA methylation. In the mouse, loss of DNA methylation in mPGCs is hypothesized to be due to repression of UHRF1 protein and loss of replication-coupled DNA methylation maintenance (Kagiwada et al., 2013). Furthermore, UHRF1 protein is also repressed in hPGCs (Gkountela et al., 2015, Tang et al., 2015). Using IF, we found that UHRF1 protein is not detectable in hPGCLCs at D4 as reported previously (Sasaki et al., 2015) and remains repressed during extended culture (Figure 5A).
Figure 5.

hPGCLCs in Extended Culture Undergo Partial Genome-Wide Demethylation
(A) IF images of UHRF1 expression in UCLA2 (U2) D4 aggregate hPGCLCs, marked by SOX17/TFAP2C (cyan) (top left panel), U2 D4C10 hPGCLCs, marked by SOX17 (cyan) (bottom left panel), and U2 D4C21 hPGCLCs, marked by SOX17 (cyan) (top right panel). Scale bars, 50 μm.
(B) Bar graph of average percent CG methylation in UCLA2: hESCs (gray), D4 hPGCLCs (blue), and D4C10 hPGCLCs (yellow).
(C) Boxplot of CG methylation percentages in U2 hESCs, D4 hPGCLCs, and D4C10 hPGCLCs over primary imprints (N = 31). U2 hESCs (gray), U2 D4 hPGCLCs (blue), and U2 D4C10 hPGCLCs (yellow).
(D) Boxplot of CG methylation percentages over long terminal repeats (LTRs). U2 hESCs (gray), U2 D4 hPGCLCs (blue), and U2 D4C10 hPGCLCs (yellow). ∗p < 2.2e16.
(E) Metaplot of percent CG methylation over protein coding genes and flanking 2-kb regions in U2 hESCs (gray), U2 D4 hPGCLCs (blue), and U2 D4C10 hPGCLCs (yellow). TSS, transcription start site; TTS, transcription termination site.
Replicates for (B–E) are independent replicates. R1, replicate 1; R2, replicate 2.
(F) t-SNE plot based of the strand bias of single-cell U2 D4 aggregate hPGCLCs (dots) and U2 D4C10 hPGCLCs cultured in FR10 (triangles). Cluster 1, low variance; cluster 2, high variance. Black circle indicate cells with the highest strand bias variance.
(G) Dot plot of each individual U2 D4 hPGCLC (blue) and U2 D4C10 hPGCLC (red), ordered by variance in strand bias.
Given the repression of UHRF1 protein in hPGCLCs, we next evaluated expression of the DNA methyltransferases (DNMT) (Figure S5A) and ten-eleven translocation 1-3 (TET1-3) genes (Figure S5B). We discovered that DNMT1 and DNMT3A mRNA are expressed by hPGCs and D4C10 hPGCLCs, whereas DNMT3B and DNMT3L levels are reduced. This result suggests a reduction in de novo DNA methylation activity in the hPGCLCs during extended culture. In addition, our results show that the TET genes are expressed at similar levels in D4C10 hPGCLCs relative to the levels in hPGCs. These results suggest the potential for some loss of DNA methylation in the hPGCLCs during extended culture.
To quantify DNA methylation, we performed whole-genome bisulfite sequencing (WGBS) of UCLA2 hESCs, hPGCLCs at D4 and hPGCLCs cultured to D4C10. Averaging all CG methylation in each biological replicate revealed that undifferentiated hESCs had on average 80% CG DNA methylation, with comparable levels in D4 hPGCLCs (Figure 5B). In contrast the average CG DNA methylation at D4C10 was reduced to around 60% (Figure 5B). Consistent with the RNA-seq data showing repression of DNMT3L and DNMT3B in hPGCLCs with extended culture, we also found that non-CG methylation was reduced in hPGCLCs relative to undifferentiated hESCs (Figures S5C–S5E).
In previous studies analyzing imprint demethylation in hPGCLCs differentiated from naive hESCs could not be performed because naive hESCs have eroded imprints (Pastor et al., 2016, von Meyenn et al., 2016). In the primed state, we identified 31 germline imprinted regions with an average CG DNA methylation of ∼50% (Figure 5C). In D4 and D4C10 hPGCLCs, we discovered that the average CG methylation over these primary imprinting control regions was equivalent (Figure 5C). Therefore, although in bulk WGBS we can detect a modest reduction in global DNA demethylation, imprinting control regions remain methylated.
Given that almost 50% of the genome is composed of transposons we next evaluated average DNA methylation at long interspersed nuclear elements (LINEs), short interspersed nuclear elements, and long terminal repeats (Figures 5D and S5F–S5I). The DNA methylation levels of major transposon classes were equivalent to the genome average for all samples. An exception to this is the “escaper” LINE1 human-specific (L1HS) retrotransposon family, which is more resistant to DNA demethylation in hPGCs (Gkountela et al., 2015). Our results show that in D4C10, L1HS retrotransposons have similar DNA methylation levels to D4 hPGCLCs, whereas an evolutionary older descendant, L1PA8, has significantly reduced DNA methylation relative to D4 hPGCLCs (Figures S5H and S5I). Protein coding genes also exhibited DNA demethylation in D4C10 hPGCLCs at the promoter and along the gene body compared with D4 hPGCLCs (Figure 5E). However, the transcription start site (TSS) was similarly demethylated in all samples. Together, these data show that, in the extended culture, hPGCLCs undergo modest genome-wide DNA demethylation. However, imprinted genes and L1HS remain methylated.
Because UHRF1 protein is not detectable in hPGCLCs and around 30% of hPGCLCs in extended culture are in S phase, we evaluated DNA methylation in single cells, with the hypothesis that hPGCLCs at D4C10 are heterogeneously demethylating, meaning that some cells are initiating DNA demethylation while other cells are not. Utilizing a strand-specific, enzymatic-based method of sequencing, we compared the 5mC content of the plus strand relative to the whole chromosome in n = 84 single hPGCLCs at D4 and n = 68 single hPGCLCs at D4C10. This calculation is denoted as strand bias (f) (f = 5 mC on + strand/total 5 mC on chromosome). Calculation of strand bias in individual cells allowed for evaluation of replication-coupled DNA (demethylation with 5mC maintenance represented by a strand bias value of 0.5. A failure to maintain 5 mC during replication would cause an increase in strand bias variance at individual chromosomes. This experiment revealed that a small number of single cells at both D4 and D4C10 exhibit strand bias variance deviating from 0.5, with a trend for more D4C10 hPGCLCs exhibiting strand bias variance, and therefore a failure to maintain DNA methylation during DNA replication (Figures 5F and 5G). Using a tSNE plot, the single-cell data are displayed as D4 hPGCLCs (dots) and D4C10 hPGCLCs (triangles) (Figure 5F). Cluster 1 represents cells with low strand bias variance with 5mC maintained on both strands (Figures 5F and S5J). Cluster 2 represents cells with higher strand bias variance, such that the cells with the highest strand bias variance represent those cells that have lost 5mC on one strand while retaining 5 mC on the other (black circle) (Figures 5F and S5K). A scatterplot illustrating each individual D4 (blue) and D4C10 (red) hPGCLC further illustrates that increased variance in strand bias occurs in the D4C10 hPGCLCs (Figure 5G). Taken together, culturing D4 hPGCLCs in extended culture for 10 days leads to heterogeneous replication-coupled DNA demethylation.
Discussion
In this paper we sought to develop an in vitro culture system with the capability to maintain hPGCLC identity while promoting proliferation. Previous attempts to culture hPGCs from fetal gonads have failed to establish cell lines that maintain germline identity, and instead resulted in the formation of hEGC-like cells that cannot be maintained in culture (He et al., 2007, Hua et al., 2009, Liu et al., 2004, Shamblott et al., 1998, Turnpenny et al., 2003). Promoting hPGCLCs to proliferate while maintaining germline identity provides the opportunity for future applications, such as molecular screening, transplantation, or molecular analyses, where larger numbers of cells are required. Previous reports have suggested that D8 hPGCLCs are capable of some epigenetic reprogramming (Sasaki et al., 2015). Indeed, our results confirm that the loss of H3K9me2 is initiated by D4 while the hPGCLCs are still in the aggregate. However, a distinct advantage of using extended culture is the maintenance of hPGCLCs for at least 3 weeks. In particular, extended culture could be used downstream of 3D modeled embryos from stem cells cultured microfluidic devices which only last 48–72 h (Zheng et al., 2019).
RNA-seq of D4C10 hPGCLCs revealed a transcriptome similar to D4 hPGCLCs, verifying the maintenance of germline identity rather than reversion to pluripotency as expected for EGCs. Furthermore, lack of SOX2 protein in the cultured hPGCLCs serves as an alternate approach for showing that extended culture does not promote the generation of hEG-like cells. One of the clear conclusions from the RNA-seq analysis is that the D4C10 hPGCLCs are not progressing into late stage hPGCs equivalent to hPGCs isolated from the fetal gonad. In vivo, the progression from early to late hPGCs in primates occurs around week 4–5 of embryo development, and is preceded by genome-wide DNA demethylation (Sasaki et al., 2016). It is anticipated that until genome-wide DNA demethylation is achieved, the early to late hPGCLC transition will not occur. Thus, an experimental approach for promoting additional DNA methylation reprogramming is required for germ cell development to progress in an orderly manner.
In this study, the hPGCLCs in extended culture had reduced H3K9me2, even after 21 days, combined with a partial loss of DNA methylation. The bulk levels of DNA methylation in our study were consistent with the work of von Meyenn et al. (2016) in which differentiation of hPGCLCs from naive hESCs through a re-primed intermediate showed a modest loss of DNA methylation by around 20%. In our study, we were able to extend this work by evaluating DNA methylation at germline imprints, which we found were protected from genome-wide DNA demethylation at D4C10. Consistent with the mouse data, this result suggests that the removal of DNA methylation from imprinted genes may be different from the mechanism that drives the initial stages of DNA demethylation in hPGCs (Hackett et al., 2013, Hargan-Calvopina et al., 2016, Hill et al., 2018, Vincent et al., 2013). Regarding the partial genome-wide loss of DNA methylation, our data suggest that this is due to heterogeneous replication-coupled DNA demethylation in some cells but not others. Future studies are necessary to capture the full DNA demethylation expected of hPGCs in vivo.
In summary, we report an extended culture system for hPGCLCs that retains germline identity while also recapitulating the earliest stages of epigenetic reprogramming. This 2D culture platform is an important new tool as it allows for new innovations in understanding imprint erasure in the human germline and efficient establishment of the late-stage gonocyte program, which has so far eluded stem cell biologists interested in germline cell differentiation. Achieving these two events will be critical before initiating sex-specific differentiation toward eggs and sperm.
Experimental Procedures
hPGCLCs or hPGCs Cultured on STOs
Human ESCs were cultured as previously described (Gell et al., 2018). Human fetal tissue was acquired and staged as previously described (Gkountela et al., 2013, Gkountela et al., 2015). Sorted hPGCLCs or hPGCs were cultured in either 7F or FR10 medium as indicated. 7F media contents were prepared as described previously, with key components given in Table 1 (Gell et al., 2018, Oliveros-Etter et al., 2015). FR10 medium (Ohta et al., 2017) contains 10% KSR, 2.5% FBS (Thermo Fisher Scientific, SH3007003), 1× NEAA (Gibco, 11140-050), 1 mM sodium pyruvate (Gibco, 11360070), 2 mM L-glutamine (Gibco, 25030081), 0.1 mM 2-mercaptoethanol (Gibco, 21985-023), 1× penicillin streptomycin (Gibco, 15140-122), 100 ng/mL SCF (PeproTech, 250-03), 10 μM forskolin (Sigma, F6886), 10 μM rolipram (Sigma, R6520), and 50 ng/mL primocin in Glasgow's MEM (Gibco, 11710-035). hPGCLCs were plated onto 200,000 cells/cm2 of STOs. hPGCLCs were cultured on either 4-well glass chamber slides (Falcon, 08-774-209), 12- or 24-well plates. hPGCLCs were plated at a density of 1,000–3,000 cells per well for chamber slides and 24-well plates, and 10,000 cells per well for 12-well plates. Media were changed daily. For D4C21 hPGCLCs in chamber slides, STOs were repleted at a density of 100,000 cells/cm2 on day 10 of culture. Chamber slides were used for IF. Twelve- and 24-well plates were used for downstream experiments. All hESC experiments were reviewed and approved by the UCLA Embryonic Stem Cell Research Oversight Committee. All human fetal tissue research was reviewed and approved by the University of Washington IRB, no identifiers or codes accompanied the fetal tissue to UCLA. Therefore, the UCLA IRB determined that the fetal tissue research experiments at UCLA were not subject to additional human subjects review.
Quantification of hPGCLC Expansion in Extended Culture
D4 hPGCLCs were plated at a density of 1,000 cells in a 24-well plate containing STOs, this is denoted as culture day 0 (D4C0). For D4C21 cell counts, hPGCLCs were split on D4C10, using 0.05% trypsin for 5 min in 37°C. Cells were collected and centrifugation at 1.5k rpm for 5 min. Cells were resuspended in FR10 medium and passed through a 100-μM strainer. Cells were split 1 well to 2 wells. The total number of human cells in culture at D4C10 and D4C21 was quantified by collecting all TRA-1-85-positive cells by FACS (see further details in Supplemental Experimental Procedures). Fold change was calculated. Prism GraphPad was used to generate graphs and statistical analysis, with an unpaired t test.
Immunofluorescence Quantification
Image analyses and quantifications were performed with the image analysis software Imaris 9.3.1 (Bitplane). Fluorescence intensity values for epigenetic markers (H3K9me2 and H3K27me3) were extracted using the surface-rendering feature to build surfaces for hPGCLCs and somatic cells. D4 hPGCLC were quantified by building surfaces for all TFAP2C/SOX17 double-positive nuclei, implementing the co-localization feature to make an hPGCLC co-localization channel, which excludes all single positive or negative for SOX17 and TFAP2C. For the somatic cell population, surfaces were built for all DAPI-positive nuclei that were not included in the hPGCLC group. To quantify hPGCLCs in extended culture, surfaces were built for all SOX17 cells that overlapped with DAPI signals. Mean fluorescence intensity (MFI) values were collected using the mask channel option on H3K9me2, H3K27me3, and DAPI channels. Relative intensity values for individual nuclei were calculated by dividing the MFI for the respective epigenetic mark by the MFI for DAPI. To quantify the proportion of Edu-positive and Ki67-positive cells, a similar procedure building surfaces was used to quantify hPGCLCs and somatic cells. The proportion of Edu-positive or Ki-67-positive hPGCLCs was obtained by dividing the number of hPGCLCs positive for either mark by the total number of DAPI-positive nuclei. Quantification of each group was based on confocal images of at least three individual aggregates or clusters of hPGCLCs for each time point. All IF experiments were performed as three independent replicates. Prism GraphPad was used to generate graphs and statistical analysis. Ordinary one-way ANOVA was used to compare the means between groups for statistical significance. To determine which groups differed from each other a Tukey's multiple comparison post hoc test was run.
Library Preparation and Sequencing
RNA was extracted using the RNeasy Micro Kit (QIAGEN) and quantified using a NanoDrop ND-1000 (NanoDrop). RNA sequencing libraries were prepared using the Nugen RNA-seq System V2 with 5–100 ng starting material. DNA was extracted using the Quick gDNA MiniPrep Kit (Zymo) and quantified using the Qubit dsDNA High-Sensitivity Kit (Life Technologies). Bisulfite-sequencing libraries were prepared using the Ovation Ultralow Methyl-Seq Library System (NuGEN). Unmethylated Lambda phage DNA (NEB) was spiked in at 0.25% input DNA quantity to determine conversion efficiency, which was 99.3%–99.5% for all libraries. Libraries were sequenced on Illumina HiSeq instruments. Details on the RNA-seq and WGBS libraries can be found in Tables S1 and S2.
RNA-Seq Analysis
Analysis of Individual Gene Expression
RNA-seq data were analyzed as described previously (Chen et al., 2017). In general, raw paired end sequencing reads were first mapped to hg19 genome with TopHat (Trapnell et al., 2009) allowing up to two mismatches and one maximal multi-hit (-g 1). Read counts for each individual gene were calculated by HTseq (Anders et al., 2015) with default parameters. Expression levels of individual genes were calculated as RPKM (reads per kilobase of exons per million aligned reads) in R. For published datasets (GSE76970 and GSE93126) (Chen et al., 2017, Pastor et al., 2016), processed data of the raw read counts for each gene were utilized, while the downstream analysis was processed in the same way as RNA-seq data generated from this study.
Heatmap
For plotting the heatmap of RNA-seq data in this study log2 (RPKM+1) values for selected genes were used and plotted in R using the ComplexHeatmap package (Gu et al., 2016).
PCA
RPKM values for each gene across different samples were used as input to calculate the principal component. PCA analysis (prcomp function in R) was performed on all genes across different samples. PCA plots were then plotted with ggplot2 package in R (http://ggplot2.org).
Differential Expressed Genes
To define differential expressed genes, the DESeq package was utilized in R (Anders and Huber, 2010). Genes with average fold change > 4, adjusted p value < 0.05, and mean RPKM > 1 across replicates in either cell types for the comparison, were defined as differential expressed genes.
GO Term and KEGG Pathway Enrichment Analysis
To identify enriched GO term and KEGG pathway, analyses were performed using R package clusterProfiler with differential expressed genes called before (Yu et al., 2012).
WGBS Analysis
Raw reads of WGBS data were first aligned to hg19 genome using BSMAP (Xi and Li, 2009) by allowing up to two mismatches (-v2), maximal one equal best hit to count (-w 1) and mapping to both strands (-n 1). When calculating methylation level over each cytosine, only reads uniquely mapped were kept and PCR duplicates were removed. Methylation levels at CG sites were then calculated by #C/(#C+#T).
Metaplot of WGBS Data
For the metaplot of CG methylation over protein coding genes, all gene coordinates were utilized with 2 kb upstream TSS and 2 kb downstream of TSS. Then the 2-kb flanked genic regions were divided into 100-bp bins, and methylation level were calculated within each bin. Metaplots were then plotted in R with customized scripts.
Analysis on Transposons
To calculate CG methylation over transposons, transposons type and coordinates were downloaded from RepeatMasker for hg19 (http://repeatmasker.org/). The Wilcoxon rank-sum test was used for statistical analysis.
Analysis on Imprints
Coordinates for stable primary imprints and transient imprints were obtained from previously published data (Okae et al., 2014, Pastor et al., 2016, Smith et al., 2014).
Whole-Genome Single-Cell 5mC Sequencing (scMspJI-Seq)
Samples were prepared as described in scMspJI-seq with minor modifications (Sen et al., 2019). TRA-1-85-positive single hPGCLCs we sorted into each reaction well of a 384-well plate containing 4 μL of Vapor-Lock (QIAGEN) and 200 nL of lysis buffer (0.0875% IGEPAL CA-630) and were stored at −80°C until use. Cells were prepared as described by Sen et al. (2019) with some minor adjustment in reagent volumes. The ds-adaptor sequences used are described in scMspJI-seq (Sen et al., 2019). Excluding the Vapor-Lock, all reagent dispenses were performed using the Nanodrop II liquid handling robot (BioNex Solutions). Each plate was pooled into 4 libraries containing 96 uniquely barcoded cells. For each library, DNA clean-up was then performed with 1× Agencourt Ampure XP beads and eluted in 30 μL of nuclease-free water. The samples were vacuum centrifuged to a volume of 6.4 μL, and 9.6 μL of in vitro transcription mix was added (1.6 μL of each ribonucleotide, 1.6 μL of T7 buffer, 1.6 μL T7 enzyme mix [MEGAscript T7 Transcription Kit, Ambion]) and incubated at 37°C for 13 h. Library preparation steps were performed as described previously (Mooijman et al., 2016, Rooijers et al., 2019). The identification of 5 mC sites in the human genome from the sequencing data is described in scMspJI-seq (Sen et al., 2019).
Analysis on scMspJI-Seq
MspJI recognizes 5 mC sites and cuts gDNA 16 bp downstream of the site leaving a 4-bp 5′ overhang, allowing both position-specific and strand-specific detection of 5 mC. The strand-specific distribution of 5 mC on a chromosome is quantitatively described by a metric called strand bias, as described previously (Sen et al., 2019, Mooijman et al., 2016). Ninety-six U2 D4 hPGCLCs and 94 U2 D4C10 hPGCLCs cells were sequenced, along with 2 negative control empty reaction wells. Poorly sequenced cells containing less than 10,000 unique 5 mCG sites were removed (median detection 60,597 unique 5 mCG sites per cell), leaving 84 U2 D4 hPGCLCs and 68 U2 D4C10 hPGCLCs passing quality control. A 5 mCG strand bias was calculated for each chromosome in all cells. The variance in the strand bias of chromosomes within each cell was used as a measure of DNA methylation maintenance. Mean silhouette scores were used to identify two populations of cells from k-means clustering on the 5 mCG strand bias, a population with low variance in the strand bias distribution, and another population with high variance in the strand bias distribution (Sen et al., 2019).
Author Contributions
J.J.G. conceived and performed the experiments and wrote the manuscript. W.L. analyzed the sequencing data. Y.T. preformed preparation of the WGBS libraries. E.S., G.H., and G.B. performed the IF experiments. E.S. quantified the IF experiments. S.E.W. quantified the hPGCLCs in culture. A.C. and S.S.D. performed and analyzed the single-cell 5 mC sequencing. A.T.C. conceived the experiments, wrote the manuscript, and maintained the Embryonic Stem Cell Research Oversight for hESCs.
Acknowledgments
This project was funded with supported from the Pablove Foundation 20183715 (to J.J.G.), R01HD079546 (to A.T.C.), and supplement to R01 R01HD058047 (to A.T.C.), and UC Cancer Research Coordinating Committee (CTN-19-585462) grant to S.S.D. G.H. acknowledges the support of the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Reserach at UCLA Training Program. The authors thank the following UCLA Broad Stem Cell core facilities: The Imaging core, Flow cytometry core, Genomics core, and the Stem Cell Banking core. The authors acknowledge the use of the Biological Nanostructures Laboratory within the California NanoSystems Institute for Illumina sequencing, supported by the University of California, Santa Barbara and the University of California, Office of the President. Human fetal tissue procurement in this research was supported by 5R24HD000836. Requests for fetal tissues can be made to Ian Glass at the University of Washington Birth Defects Laboratory.
Published: February 13, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.01.009.
Accession Numbers
All data mentioned in the paper are deposited in the NCBI GEO. RNA-seq and WGBS under accession number GSE139115, scMspJI-seq under accession number GSE138982.
Supplemental Information
References
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq––a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Liu W., Lukianchikov A., Hancock G.V., Zimmerman J., Lowe M.G., Kim R., Galic Z., Irie N., Surani M.A. Germline competency of human embryonic stem cells depends on eomesodermin. Biol. Reprod. 2017;97:850–861. doi: 10.1093/biolre/iox138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcova-Hills G., Ainscough J., McLaren A. Pluripotential stem cells derived from migrating primordial germ cells. Differentiation. 2001;68:220–226. doi: 10.1046/j.1432-0436.2001.680409.x. [DOI] [PubMed] [Google Scholar]
- Farini D., Scaldaferri M.L., Iona S., La Sala G., De Felici M. Growth factors sustain primordial germ cell survival, proliferation and entering into meiosis in the absence of somatic cells. Dev. Biol. 2005;285:49–56. doi: 10.1016/j.ydbio.2005.06.036. [DOI] [PubMed] [Google Scholar]
- Gell J.J., Zhao J., Chen D., Hunt T.J., Clark A.T. PRDM14 is expressed in germ cell tumors with constitutive overexpression altering human germline differentiation and proliferation. Stem Cell Res. 2018;27:46–56. doi: 10.1016/j.scr.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkountela S., Li Z., Vincent J.J., Zhang K.X., Chen A., Pellegrini M., Clark A.T. The ontogeny of cKIT+ human primordial germ cells proves to be a resource for human germ line reprogramming, imprint erasure and in vitro differentiation. Nat. Cell. Biol. 2013;15:113–122. doi: 10.1038/ncb2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkountela S., Zhang K.X., Shafiq T.A., Liao W.W., Hargan-Calvopina J., Chen P.Y., Clark A.T. DNA demethylation dynamics in the human prenatal germline. Cell. 2015;161:1425–1436. doi: 10.1016/j.cell.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Guo F., Yan L., Guo H., Li L., Hu B., Zhao Y., Yong J., Hu Y., Wang X., Wei Y. The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell. 2015;161:1437–1452. doi: 10.1016/j.cell.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Hackett J.A., Dietmann S., Murakami K., Down T.A., Leitch H.G., Surani M.A. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports. 2013;1:518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargan-Calvopina J., Taylor S., Cook H., Hu Z., Lee S.A., Yen M.R., Chiang Y.S., Chen P.Y., Clark A.T. Stage-specific demethylation in primordial germ cells safeguards against precocious differentiation. Dev. Cell. 2016;39:75–86. doi: 10.1016/j.devcel.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Wang Y., Li Y.L. Fibroblast-like cells derived from the gonadal ridges and dorsal mesenteries of human embryos as feeder cells for the culture of human embryonic germ cells. J. Biomed. Sci. 2007;14:617–628. doi: 10.1007/s11373-007-9185-z. [DOI] [PubMed] [Google Scholar]
- Hill P.W.S., Leitch H.G., Requena C.E., Sun Z., Amouroux R., Roman-Trufero M., Borkowska M., Terragni J., Vaisvila R., Linnett S. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature. 2018;555:392–396. doi: 10.1038/nature25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J., Yu H., Liu S., Dou Z., Sun Y., Jing X., Yang C., Lei A., Wang H., Gao Z. Derivation and characterization of human embryonic germ cells: serum-free culture and differentiation potential. Reprod. Biomed. Online. 2009;19:238–249. doi: 10.1016/s1472-6483(10)60079-x. [DOI] [PubMed] [Google Scholar]
- Irie N., Weinberger L., Tang W.W., Kobayashi T., Viukov S., Manor Y.S., Dietmann S., Hanna J.H., Surani M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell. 2015;160:253–268. doi: 10.1016/j.cell.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S., Kurimoto K., Hirota T., Yamaji M., Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–353. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K., Yabuta Y., Hayashi K., Ohta H., Kiyonari H., Mitani T., Moritoki Y., Kohri K., Kimura H., Yamamoto T. Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell. 2015;16:517–532. doi: 10.1016/j.stem.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Leitch H.G., Nichols J., Humphreys P., Mulas C., Martello G., Lee C., Jones K., Surani M.A., Smith A. Rebuilding pluripotency from primordial germ cells. Stem Cell Reports. 2013;1:66–78. doi: 10.1016/j.stemcr.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Liu H., Pan Y., Tang S., Xiong J., Hui N., Wang S., Qi Z., Li L. Human embryonic germ cells isolation from early stages of post-implantation embryos. Cell Tissue Res. 2004;318:525–531. doi: 10.1007/s00441-004-0990-7. [DOI] [PubMed] [Google Scholar]
- Matsui Y., Zsebo K., Hogan B.L. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- Mooijman D., Dey S.S., Boisset J.C., Crosetto N., van Oudenaarden A. Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat. Biotechnol. 2016;34:852–856. doi: 10.1038/nbt.3598. [DOI] [PubMed] [Google Scholar]
- Ohta H., Kurimoto K., Okamoto I., Nakamura T., Yabuta Y., Miyauchi H., Yamamoto T., Okuno Y., Hagiwara M., Shirane K. In vitro expansion of mouse primordial germ cell-like cells recapitulates an epigenetic blank slate. EMBO J. 2017;36:1888–1907. doi: 10.15252/embj.201695862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H., Chiba H., Hiura H., Hamada H., Sato A., Utsunomiya T., Kikuchi H., Yoshida H., Tanaka A., Suyama M. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 2014;10:e1004868. doi: 10.1371/journal.pgen.1004868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros-Etter M., Li Z., Nee K., Hosohama L., Hargan-Calvopina J., Lee S.A., Joti P., Yu J., Clark A.T. PGC reversion to pluripotency involves erasure of DNA methylation from imprinting control centers followed by locus-specific re-methylation. Stem Cell Reports. 2015;5:337–349. doi: 10.1016/j.stemcr.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor W.A., Chen D., Liu W., Kim R., Sahakyan A., Lukianchikov A., Plath K., Jacobsen S.E., Clark A.T. Naive human pluripotent cells feature a methylation landscape devoid of blastocyst or germline memory. Cell Stem Cell. 2016;18:323–329. doi: 10.1016/j.stem.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopuk L., Stringer J.M., Hogg K., Elgass K.D., Western P.S. PRC2 is required for extensive reorganization of H3K27me3 during epigenetic reprogramming in mouse fetal germ cells. Epigenetics Chromatin. 2017;10:7. doi: 10.1186/s13072-017-0113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick J.L., Bixler L.S., Cheng L., Donovan P.J. Long-term proliferation of mouse primordial germ cells in culture. Nature. 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- Rooijers K., Markodimitraki C.M., Rang F.J., de Vries S.S., Chialastri A., de Luca K.L., Mooijman D., Dey S.S., Kind J. Simultaneous quantification of protein-DNA contacts and transcriptomes in single cells. Nat. Biotechnol. 2019;37:766–772. doi: 10.1038/s41587-019-0150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K., Yokobayashi S., Nakamura T., Okamoto I., Yabuta Y., Kurimoto K., Ohta H., Moritoki Y., Iwatani C., Tsuchiya H. Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell. 2015;17:178–194. doi: 10.1016/j.stem.2015.06.014. [DOI] [PubMed] [Google Scholar]
- Sasaki K., Nakamura T., Okamoto I., Yabuta Y., Iwatani C., Tsuchiya H., Seita Y., Nakamura S., Shiraki N., Takakuwa T. The germ cell fate of cynomolgus monkeys is specified in the nascent amnion. Dev. Cell. 2016;39:169–185. doi: 10.1016/j.devcel.2016.09.007. [DOI] [PubMed] [Google Scholar]
- Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y., Hayashi K., Itoh K., Mizugaki M., Saitou M., Matsui Y. Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Dev. Biol. 2005;278:440–458. doi: 10.1016/j.ydbio.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Seki Y., Yamaji M., Yabuta Y., Sano M., Shigeta M., Matsui Y., Saga Y., Tachibana M., Shinkai Y., Saitou M. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134:2627–2638. doi: 10.1242/dev.005611. [DOI] [PubMed] [Google Scholar]
- Sen M., Mooijman D., Boisset J.-C., Chialastri A., Popovic M., Heindryckx B., de Sousa Lopes S.M.C., Dey S.S., van Oudenaarden A. Strand-specific single-cell methylomics reveals distinct modes of DNA demethylation dynamics during early mammalian development. bioRxiv. 2019 doi: 10.1038/s41467-021-21532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblott M.J., Axelman J., Wang S., Bugg E.M., Littlefield J.W., Donovan P.J., Blumenthal P.D., Huggins G.R., Gearhart J.D. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc. Natl. Acad. Sci. U S A. 1998;95:13726–13731. doi: 10.1073/pnas.95.23.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Chan M.M., Humm K.C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sybirna A., Tang W.W.C., Dietmann S., Gruhn W.H., Surani A.M. A critical but divergent role of PRDM14 in human primordial germ cell fate revealed by inducible degrons. bioRxiv. 2019 doi: 10.1038/s41467-020-15042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R., Hackett J.A., Chinnery P.F., Surani M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnpenny L., Brickwood S., Spalluto C.M., Piper K., Cameron I.T., Wilson D.I., Hanley N.A. Derivation of human embryonic germ cells: an alternative source of pluripotent stem cells. Stem Cells. 2003;21:598–609. doi: 10.1634/stemcells.21-5-598. [DOI] [PubMed] [Google Scholar]
- Turnpenny L., Spalluto C.M., Perrett R.M., O'Shea M., Hanley K.P., Cameron I.T., Wilson D.I., Hanley N.A. Evaluating human embryonic germ cells: concord and conflict as pluripotent stem cells. Stem Cells. 2006;24:212–220. doi: 10.1634/stemcells.2005-0255. [DOI] [PubMed] [Google Scholar]
- Vincent J.J., Huang Y., Chen P.Y., Feng S., Calvopina J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F., Berrens R.V., Andrews S., Santos F., Collier A.J., Krueger F., Osorno R., Dean W., Rugg-Gunn P.J., Reik W. Comparative principles of DNA methylation reprogramming during human and mouse in vitro primordial germ cell specification. Dev. Cell. 2016;39:104–115. doi: 10.1016/j.devcel.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y., Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Xue X., Shao Y., Wang S., Esfahani S.N., Li Z., Muncie J.M., Lakins J.N., Weaver V.M., Gumucio D.L. Controlled modelling of human epiblast and amnion development using stem cells. Nature. 2019;573:421–425. doi: 10.1038/s41586-019-1535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.