

Summary
Phospholipase C epsilon 1 (PLCε1) is a unique member of the phospholipase family, in that it also functions as a guanine nucleotide exchange factor (GEF) for the small GTPase Rap1. It is this function as a Rap1 GEF that gives PLCε1 an essential role in chemokine‐mediated T cell adhesion. We have utilized a syngeneic tumor model, MC38 cells in C57BL/6 mice, and observed that tumors grow larger and more quickly in the absence of PLCε1. Single‐cell analysis revealed an increased CD4+/CD8+ ratio in the spleens, lymph nodes and tumors of PLCε1 knock‐out tumor‐bearing mice. T cells isolated from PLCε1 knock‐out mice were less activated by multiple phenotypical parameters than those from wild‐type mice. We additionally noted a decrease in expression of the chemokine receptors C‐X‐C chemokine receptor type 4 (CXCR4) and C‐C motif chemokine receptor 4 (CCR4) on CD4+ T cells from the spleens, lymph nodes and tumors of PLCε1 knock‐out mice compared to wild‐type mice, and diminished migration of PLCε1‐depleted CD3+ T cells towards stromal cell‐derived factor (SDF)‐1α. Based on these results, we conclude that PLCε1 is a potential regulator of tumor‐infiltrating lymphocytes, functioning, at least in part, at the level of T cell trafficking and recruitment.
Keywords: chemokines, MC38, murine cancer model, phospholipase C epsilon 1, SDF‐1α, T cells
We discovered that tumors grow larger in the absence of the enzyme PLCε1. Importantly, and despite the fact that PLCe1 deficient T cells are intrinsically cytotoxic, they cannot migrate toward the inflammatory tumor environment, a finding that is also supported by abnormal expression of several chemokine receptors. Thus, PLCε1 is a potential regulator of tumor infiltrating lymphocytes, functioning at the level of T cell trafficking and recruitment.

Introduction
T cell trafficking is a process that is shared by fighting pathogens, autoimmune diseases and tumor rejection. The directed movement of T cells throughout the body is primarily achieved by specific expression patterns of chemokines and chemokine receptors. In the context of solid tumors, enhancing T cell infiltration is an important therapeutic strategy, given our understanding of its being a reliable predictor of patient outcome 1, 2, 3, 4. Research aimed at understanding the tumor microenvironment has gained more traction in recent years, especially as T cell checkpoint inhibitors have proved successful in extending survival of patients unresponsive to other therapies. These therapeutics, including anti‐programmed cell death 1 (PD‐1) and anti‐cytotoxic T lymphocyte antigen (CTLA)‐4 antibodies, enhance T cell activation in an effort to promote tumor cell identification and elimination 5. However, without effective T cell recruitment these therapeutics cannot work 6. The influx of immune cells into the tumor microenvironment can be exploited by the cancer cells and contribute to tumor associated inflammation, thereby promoting tumor growth, progression and metastasis 7.
As chemokine and chemokine receptor expression is regulated to maintain specificity of cellular recruitment, these signaling molecules make ideal targets for the selective intervention into T cell infiltration 6, 8, 9. Many chemokine receptor antagonists that have entered clinical trials have lacked clinical benefit 10, 11, 12, due probably to intracellular signaling redundancy among different chemokine pathways, although this redundancy is yet to be elucidated. Our previous work shows that the enzyme phospholipase C epsilon 1 (PLCε1) plays a requisite role in C‐X‐C chemokine receptor type 4 (CXCR4) signaling in T cells 13. In a model of contact sensitivity, PLCε1 knock‐out mice demonstrated major defects in T cell trafficking and migration. This protein has been implicated in the pathogenesis of numerous diseases 14, 15. The role of PLCε1 in tumor progression has primarily been explored in the context of activation state in cancer cells and the contribution to tumor progression from this perspective. The data on PLCε1 in cancer development and progression has been well reviewed 16. In this study, we aim to identify the specific contribution of PLCε1 to T cell tumor infiltration and to the anti‐tumor immune response. We report that PLCε1 plays a necessary role in T cell trafficking and activation in tumor‐bearing mice. This work contributes to our understanding of the chemokine signaling network in T cells.
Materials and methods
Mice and tumor cell lines
Male, 6–8‐week‐old C57BL/6 (B6) wild‐type (WT) or PLCε1 knock‐out mice (KO) 13, 17 were used in all studies. Animal studies were approved by the New York University institutional animal care and use committee. The WT mice that were used for the control experiments were also used for the KO mice back‐crossing, assuring a homogeneous genetic background. Mice were housed in the same facility for 1 week before experiment to synchronize environments. The murine colon adenocarcinoma (MC38) colon carcinoma cells were a gift from Ben Neel of New York University. Prior to use, MC38 cells were authenticated by simple sequence length polymorphism (SSLP). The MC38 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with heat‐inactivated fetal bovine serum (FBS; 10%) and penicillin–streptomycin (P/S; 10 000 U/ml stock; 1%) and grown at 37°C with 5% CO2. Cells were passaged prior to storage and thawed and passaged twice prior to implantation for all described tumor experiments. All cell lines were determined to be free of mycoplasma (Lonza, Basel, Switzerland).
Tumor model
MC38 (1 × 106) cells were implanted subcutaneously in the right hind flank of mice. Tumor growth was monitored using electronic calipers and calculated according to the formula: V = length × width2 × 0·52. To analyze T cells, spleens, right inguinal lymph nodes and tumors from both groups were harvested for analysis 14 days post‐tumor implantation. For survival experiments, mice were euthanized when tumor size reached 2000 mm3 or when tumors became ulcerated. Tumor weight was measured on the day of euthanasia.
Flow cytometry
Cells freshly isolated from murine spleen, right inguinal lymph node and tumor were stained, non‐permeabilized, with fluorescently conjugated antibodies specific for CD3, CD4, CD8, PD‐1, CD44, CD62L, CD25, CXCR3, CCR4, CCR7 and CXCR4 (all from BioLegend, San Diego, CA, USA) in fluorescence activated cell sorter (FACS) buffer [phosphate‐buffered saline (PBS) without Ca+2/Mg+2 (FBS) (2%)]. Subsequently, these cells were fixed and permeabilized (True‐Nuclear Transcription Factor Buffer Set; BioLegend) to stain with a fluorescently conjugated antibody specific for forkhead box protein 3 (FoxP3) (BioLegend). Events were recorded using the LSRII (BD Biosciences, San Jose, CA, USA) with single stain compensation controls and fluorescence minus 1 controls. Compensation was performed using FACSDiva (BD Biosciences) and data were analyzed using FlowJo software (TreeStar Inc., Ashland, OR, USA). Events were first gated based on forward (FSC)‐ and side‐scatter (SSC); CD3+ events were then gated and subsequently gated into CD4+ and CD8+ populations. CD4+ and CD8+ populations were analyzed for PD‐1, CD44, CD62L, CD25, CXCR3, CCR4, CCR7 and CXCR4 expression. Jurkat T cells were stained with allophycocyanin (APC)‐conjugated mouse IgG2a κ or anti‐human CXCR4 antibody (BioLegend). Events were recorded using the LSRFortessa (BD Biosciences) and data were analyzed using FlowJo software. All flow cytometry analysis was performed in adherence with the ‘Guidelines for the use of flow cytometry and cell sorting in immunological studies’ (Cossarizza et al. 18).
In‐vitro CD8+ cytotoxicity assay
Splenocytes were isolated from 6–8‐week‐old B6 wild‐type or PLCε1 knock‐out murine spleens and red blood cells were removed by ACK lysis buffer (Lonza). Washed splenocytes were then cultured in complete RPMI‐1640 medium supplemented with β‐mercaptoethanol (55 µM), mouse interleukin (IL)‐2 (1000 IU/ml) and Staphylococcus aureus enterotoxin E (SEE; 1 µg/ml) for 72 h at a density of 2 × 107 cells/well in a 96‐well plate, 37°C with 5% CO2. Raji B cells (American Type Culture Collection, Manassas, VA, USA) were maintained in complete RPMI‐1640 media supplemented with heat‐inactivated FBS (10%) and P/S (1%) at 37°C with 5% CO2. After 72 h, CD8+ T cells were isolated from the splenocytes by negative selection (StemCell Technologies, Vancouver, Canada) and put into co‐culture with Raji cells in the presence of IL‐2 and SEE at the indicated ratios for 4 h, after which lactate dehydrogenase (LDH) activity in the supernatant was assayed (ThermoScientific, Fremont, CA, USA).
Chemotaxis assay
The chemotaxis of PLCε1‐depleted T cells towards the chemokine stromal cell‐derived factor (SDF)‐1α/CXCL12 (R&D Systems, Minneapolis, MN, USA) was assessed using 24‐well Transwell cell culture inserts with 5‐µm pores (Corning, New York, NY, USA). CD3+ T cells were isolated through negative selection enrichment kit (Miltenyi Biotech, San Diego, CA, USA).
PLCε1 depletion was achieved by introducing inhibitory RNA (Dharmacon, Lafayette, CO, USA), as previously described 8. Control cells were transfected with a scrambled, non‐targeting siRNA (Dharmacon). At the start of the assay, T cells were stained with 1 µM carboxyfluorescein succinimidyl ester (CFSE; BioLegend) and added to the apical compartment of the Transwell insert (100 µl RPMI‐1640 media containing 10% FBS, 1·5 × 105 cells/insert). The basolateral compartment was filled with 600 µl of RPMI‐1640 containing 10% FBS and 75 ng/ml SDF‐1α/CXCL12. Chemotaxis was allowed for 3 h at 37°C with 5% CO2. The media and cells from the basolateral compartment were then collected, cells were pelleted and resuspended in 130 µl of FACS buffer to be transferred to a 96‐well plate. Cells that moved into the basolateral compartment were quantified by flow cytometry using uniform volume acquisition on the LSRFortessa (BD Biosciences) and analysis was performed using FlowJo software. Cells were gated based on FSC versus SSC morphology of primary T cells then on carboxyfluorescein succinimidyl ester (CFSE) positivity. The number of CFSE+ T cells was used to determine the percentage chemotaxis by dividing the number of CFSE+ siScramble or siPLCε1 CD3+ T cells by the number of CFSE+ siScramble cells, therefore normalizing siScramble chemotaxis to 100%.
Statistics
Values are reported as means ± standard error of the mean (s.e.m.). Statistical analyses were performed using Student’s t‐test in GraphPad Prism (version 7.0c).
Results
PLCε1 limits MC38 tumor growth and contributes to T cell subset distribution
Our previous work shows that the enzyme PLCε1 plays a requisite role in CXCR4 signaling in T cells 13. Within the colorectal cancer samples included in The Cancer Genome Atlas (TCGA) (http://www.oncolnc.org/kaplan/?lower%A0=%A033%26upper%A0=%A033%26cancer%A0=%A0READ%26gene_id%A0=%A051196%26raw%A0=%A0plce1%26species%A0=%A0mRNA), high PLCε1 expression correlates with greater 5‐year survival (Supporting information, Fig. S1a). We implanted MC38 cells into C57BL/6 wild‐type (WT) or PLCε1 knock‐out (KO) mice and observed that tumors grew larger in the KO mice (Fig. 1a and Supporting information, Fig. S1b). We next isolated cells from the tumors, right inguinal lymph nodes (LN) and spleens of mice 14 days after MC38 cell implantation or spleens of non‐tumor‐bearing mice and analyzed by flow cytometry. In the spleens of non‐tumor‐bearing mice, CD4+/CD8+ ratios were equivalent between WT and KO mice (Fig. 1b). Following 14 days of tumor growth, the CD4+/CD8+ ratio in the spleens decreased in WT mice, although not in KO mice (Fig. 1c). Similarly, the CD4+/CD8+ ratio was higher in the LN and tumors of KO mice compared to the same sites in WT mice (Fig. 1d,e). At all sites, the increase in CD4+ T cells could not be accounted for by regulatory T cells, as there was no difference observed in this subset (Supporting information, Fig. S2).
Figure 1.
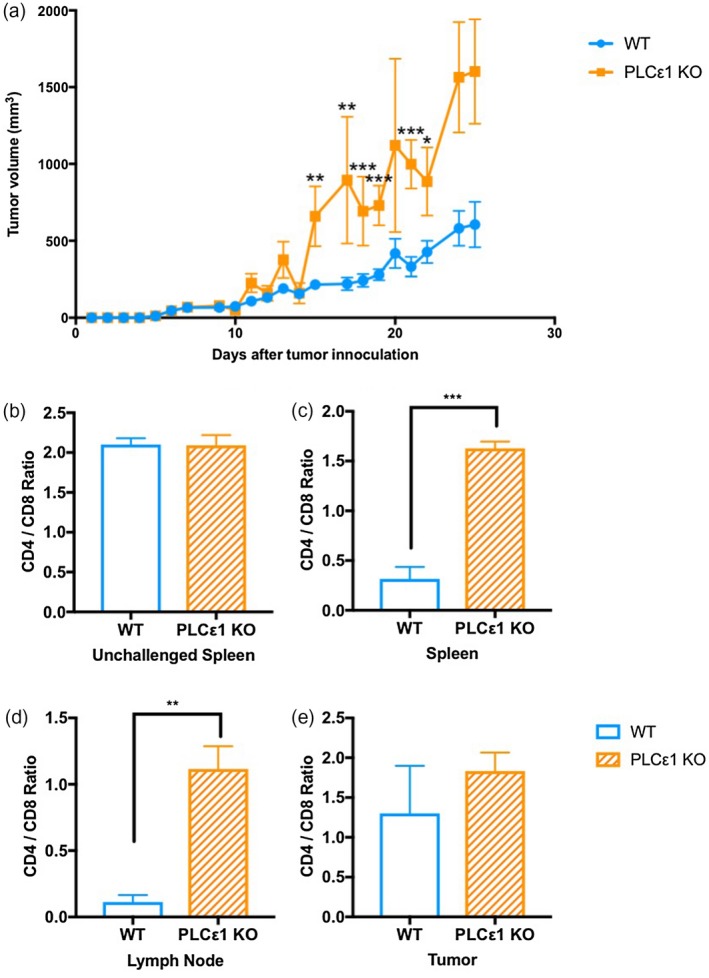
Phospholipase C epsilon 1 (PLCε1) limits murine colon adenocarcinoma (MC38) tumor growth and contributes to T cell subset distribution. (a) MC38 tumor volume calculated daily; n = 14 PLCε1 knock‐out (KO) mice from three independent experiments with n ≥ 3 mice per experiment, n = 22 wild‐type (WT) mice from four independent experiments with n ≥ 5 mice per experiment; points represent mean ± SEM. (b–e) Cells from spleens of normal (b) and tumor‐bearing (c) mice along with the right inguinal lymph nodes (d) and tumors (e) from tumor‐bearing mice were isolated and stained for CD3, CD4 and CD8 then analyzed by flow cytometry. Events were gated for live CD3+ events then CD4+ or CD8+; populations shown are total CD3+, CD3+ CD4+CD8−, CD3+CD4−CD8+; n = 4 WT and PLCε1 KO mice from two independent experiments with two mice per experiment per group; bars represent mean ± standard error of the mean (s.e.m.). *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001, ****P ≤ 0·0001, P > 0·05 not indicated.
Activation of tumor‐infiltrating T cells requires PLCε1
To assess the activation state of the CD4+ and the CD8+ cells, we determined surface expression of CD44, CD62L and PD‐1. Overall, significantly fewer KO CD4+ T cells expressed CD44 and PD‐1 in the spleens, LN and tumors (Fig. 2a and Supporting information, Fig. S3). Additionally, there was an increased presence of naive CD4+ T cells (naive; CD62L+CD44−) at all three sites (Fig. 2b). Fewer CD44+ CD8+ T cells were observed isolated from the spleens, LN and tumors of KO mice (Fig. 2c). The majority of CD8+ T cells isolated from all sites in KO mice were naive, with a near absence of central memory (Tcm; CD62L+CD44+) and effector memory (Tem; CD62L−CD44+) populations (Fig. 2d), demonstrating overall lower activation in the absence of PLCε1. Unexpectedly, and unlike the CD4+‐infiltrating T cells, many of the CD8+‐infiltrating T cells were negative for PD‐1 (Fig. 2d).
Figure 2.
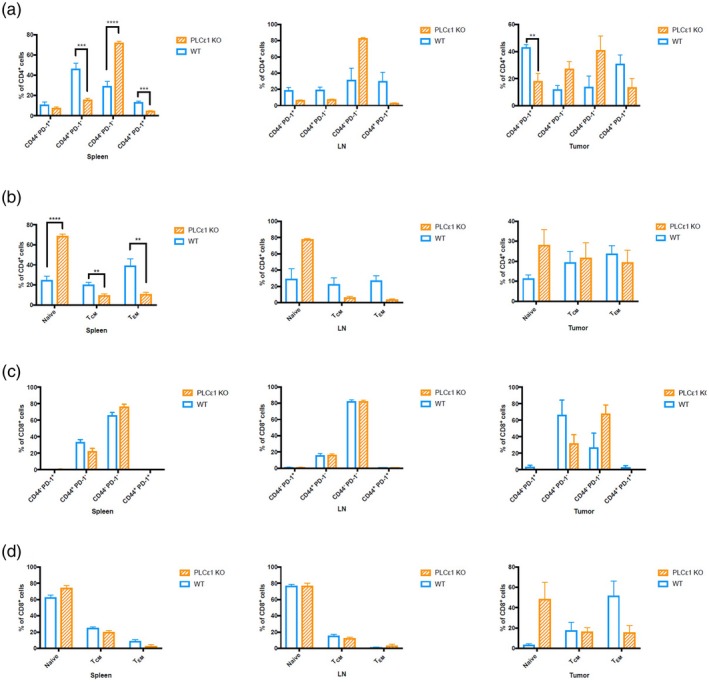
Activation of tumor infiltrating T cells requires phospholipase C epsilon 1 (PLCε1). Cells from spleens, lymph nodes and tumors from tumor‐bearing mice were isolated and stained for CD3, CD4, CD8, CD44, PD‐1 and CD62L then analyzed by flow cytometry. Events were gated for live CD3+ events then CD4+CD8− (a,b) or CD4−CD8+ (c, d); n = 4 of wild‐type (WT) and PLCε1 knock‐out (KO) mice from two independent experiments with two mice per experiment per group. LN = lymph nodes. Bars represent mean ± standard error of the mean (s.e.m.). *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001, ****P ≤ 0·0001, P > 0·05 not indicated.
Infiltrating T cells express different chemokine receptors in the absence of PLCε1
This observation of lower T cell activation could be due to a role for PLCε1 in T cell receptor (TCR) signaling or to improper localization with other cells in the microenvironment. Accordingly, we isolated CD8+ T cells and assessed non‐specific cytotoxic activity. Nevertheless, PLCε1 KO T cells could induce Raji cell cytotoxicity to the same extent as WT CD8+ T cells in the presence of superantigen SEE (Fig. 3a). This suggests that PLCε1 does not contribute to T cell effector function, and leaves open the possibility that cellular mislocalization to or within the tumor is responsible for the observed lack of activation.
Figure 3.
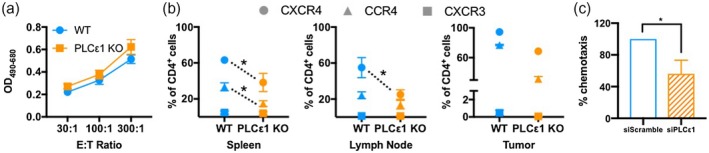
Infiltrating CD4+ T cells express different chemokine receptors in the absence of phospholipase C epsilon 1 (PLCε1). (a) Splenocytes from wild‐type (WT) and PLCε1 knock‐out (KO) mice were stimulated with Staphylococcus aureus enterotoxin E (SEE) for 72 h. Then CD8+ T cells were isolated and co‐incubated with Raji cells and SEE for 4 h, after which lactate dehydrogenase (LDH) activity was quantified. Biological replicates were from n = 3 WT mice, n = 5 PLCε1 KO mice stimulated independently; points represent mean ± standard error of the mean (s.e.m.). (b) Cells from spleens, right inguinal lymph nodes and tumors were isolated and stained for CD3, CD4, CD8, CXCR4, CCR4 and CXCR3 then analyzed by flow cytometry. Events were gated for live CD3+ events then CD4+CD8−; n = 3–4 WT and PLCε1 KO mice from two independent experiments with two mice per experiment per group; points represent mean ± s.e.m; *P ≤ 0·05 based on comparison between WT and KO for individual chemokine receptors (indicated by dotted line), P > 0·05 not indicated. (c) Primary murine CD3+ T cells transfected with siRNA non‐targeting (siScramble) or PLCε1 (siPLCε1) were added to the apical chamber of Transwell inserts with stromal cell‐derived factor (SDF)‐1α containing media in the basolateral chamber. After 3 h cells in the basolateral chamber were quantified; n = 3 independent experiments, each performed in duplicate. Bars indicate mean ± s.e.m. *P ≤ 0·05.
When assessing the expression levels of various chemokine receptors on the surface of T cells isolated from the spleens, LN and tumors, we found that fewer KO CD4+ T cells isolated from the spleens and tumors expressed CCR4 (Fig. 3b). Within the LN, fewer CD4+ T cells expressed CCR4 in KO mice although differences were less pronounced due to overall lower CCR4 positivity in both genotypes. Additionally, fewer CD4+ T cells expressed CXCR4 in the spleens, LN and tumors of KO mice (Fig. 3b), suggesting that CCR4 and CXCR4 are critically contributing to T cell recruitment in this model. No consistent difference in the low level of CXCR3 expression on CD4+ T cells was observed. Analysis of CXCR4, CCR4 and CXCR3 expression on CD8+ T cells did not uncover any differences (Supporting information, Fig. S4). This finding is consistent with the increased CD4+ T cell presence observed in KO mice (Fig. 1). No differences were observed in the expression of CCR7. Mechanistically, we also identified a defect in the PLCε1‐deficient CD4+ T cells migrating towards the CXCR4 ligand SDF‐1α (Fig. 3c). Importantly, no change in CXCR4 surface expression was detected when PLCε1 was knocked down (Supporting information, Fig. S5), suggesting that the defect in migration is due to signaling differences downstream of CXCR4. These data, combined with the observation of fewer CCR4+ and CXCR4+ T cells in the study sites of PLCε1 KO mice, might suggest that these cells are unable to utilize these pathways to traffic to these sites. Overall, our data indicate a potential role for CCR4 and CXCR4 pathways in T cell localization within the tumor, and a role for PLCε1 in these signaling cascades.
Discussion
Overall, this study is descriptive and preliminary. While it does not provide definitive proof of mechanism, it should be regarded as hypothesis‐generating. We hypothesize that further development of this work will demonstrate a role for the enzyme PLCε1 in T cell distribution throughout multiple anatomical sites in tumor‐bearing mice. Our finding that CD4+ T cells are more abundant, less activated and express lower levels of CXCR4 and CCR4 in the spleens, LN and tumors of PLCε1 KO mice suggests to us that the overall trafficking of these cells relies upon PLCε1 function. While both CD4+ and CD8+ T cell frequencies are altered in the absence of PLCε1, the observed chemokine receptor differences were preferentially within the CD4+ subset. Two possible explanations for this distinction are that either the disruption of CD4+ T cell migration and activation precedes and precludes CD8+ T cell activation and proliferation or that other PLCε1‐dependent chemokine receptors not quantified in this study may be preferentially expressed on CD8+ T cells. To address these hypotheses, more extensive and conclusive analysis of these pathways must be conducted.
We propose that PLCε1 is active downstream of CCR4, as we have shown previously for CXCR4 13, and that these pathways may play critical roles in the proper localization of CD4+ T cells within the tumor microenvironment. We have shown previously that the impact of PLCε1 loss on inflammation was T cell intrinsic 13, although the indirect role for PLCε1 may be more widespread. For example, one ligand for CCR4 is CCL2, which also binds the receptor CCR2, a chemokine receptor that is expressed highly on M2‐like macrophages. In fact, CCL2–CCR2 engagement on macrophages has been shown to contribute to anti‐inflammatory polarization of macrophages with higher IL‐10 production 19, poor antigen presentation and promotion of new blood vessel growth 20. Each of these outcomes could lead to enhanced tumor growth. Lower levels of CCR4 on T cells within the same tumor microenvironment could lead to higher local levels of CCL2 available to interact with CCR2 on macrophages. Accordingly, we cannot rule out that PLCε1 also plays a role in other types of immune cells.
This work is not free of limitations. Flow cytometry provides phenotype data and does not prove mechanism; this leaves open multiple mechanistic possibilities, as discussed and explored through in‐vitro studies. While the mechanistic studies provided in this manuscript are not exhaustive, the cytotoxicity assay demonstrates that PLCε1 expression in T cells does not contribute to cytotoxic function and the chemotaxis assay demonstrates that, in the absence of PLCε1, chemotaxis is significantly decreased in both human and murine T cells. Overall, the data included in this manuscript shows that MC38 tumors are larger in mice that do not express PLCε1, the T cell ratio is altered in multiple anatomical compartments in tumor‐bearing PLCε1 knock‐out mice, T cell activation is decreased in multiple anatomical compartments in tumor‐bearing PLCε1 knock‐out mice, PLCε1 knock‐out T cells maintain cytotoxic function and PLCε1‐depleted T cells do not respond to chemotactic stimuli to the same extent as wild‐type T cells. From these data collectively, as well as the known literature on PLCε1, we conclude that PLCε1 expression in T cells may contribute to an anti‐tumor immune response.
It would be of interest to assess the combined impact of PLCε1 activation with checkpoint inhibition within a tumor model, given that enhanced tumor infiltration is critical to T cell‐mediated tumor clearance. A recent study published in Immunity 21 took the first steps in linking the chemokine network with anti‐PD‐1 treatment. Their work concludes that CXCR3 expression on CD8+ T cells is critical for proper localization within the tumor and anti‐PD‐1 efficacy. With further work to uncover the mechanism underlying the role for PLCε1 in these chemokine pathways, we suggest that local activation of PLCε1 within T cells may be a viable therapeutic strategy in the treatment of solid tumors.
Disclosures
None.
Author contributions
Conception and design: M. S. and A. M. Development of methodology: M. S., K. A., A. S., S. L., and A. M. Acquisition of data: M. S., A. S. and A.M. Analysis and interpretation of data: M. S. and A. M. Writing, review and/or revision of the manuscript: M. S. and A. M. Administrative, technical or material support: M. S. and K. A. Study supervision: M. S. and A. M.
Supporting information
Fig. S1. The top 33% of PLCε1 expression (high) in rectum adenocarcinoma samples from the TCGA correlated with increased survival compared to the lowest 33% of PLCε1 expression (low). Log rank P‐value = 0.0772 (http://www.oncolnc.org/kaplan/?lower=33&upper=33&cancer=READ&gene\_xml:id=51196&raw=plce1&species=mRNA) (a) Tumors were excised and weighed from mice that survived to day 25 post tumor inoculation (b) n = 4 PLCε1 KO mice, n = 12 WT mice from 3 independent experiments; bars represent min ‐ max. ****P ≤ 0.0001.
Fig. S2. CD3+ CD4+ CD8‐ cells were analyzed for expression of CD25 and FoxP3, with double positive (CD25+ FoxP3+) cells being regarded as regulatory T cells (Treg). Bars represent the mean % of CD4+ cells that were Treg ± SEM.
Fig. S3. Cells from spleens, lymph nodes, and tumors from tumor‐bearing mice were isolated and stained for CD3, CD4, CD8, CD44, PD‐1, and CD62L then analyzed by flow cytometry. Events were gated for live CD3+ events then CD4+ CD8‐ (a,b) or CD4‐ CD8+ (c,d). Plots representative of n = 4 WT and PLCε1 KO mice from 2 independent experiments with 2 mice per experiment per group.
Fig. S4. Cells from spleens, right inguinal lymph nodes, and tumors from tumor‐bearing mice were isolated and stained for CD3, CD4, CD8, CXCR4, CCR4, and CXCR3 then analyzed by flow cytometry. CD3+ CD4‐ CD8+ cells are included in this analysis. n = 4 WT and PLCε1 KO mice from 2 independent experiments with 2 mice per experiment per group; points represent mean ± SEM.
Fig. S5. (a) Jurkat T cells expressing shRNA non‐targeting (shScramble) or PLCε1 (shPLCε1) were added to the apical chamber of transwell inserts with SDF‐1α containing media in the basolateral chamber. After 3 hours cells in the basolateral chamber were quantified. n = 3 independent experiments, each performed in duplicate. Bars indicate mean ± SEM; *P ≤ 0.05. (b) Jurkat T cells expressing shScramble or shPLCε1 were stained with APC‐conjugated isotype or CXCR4 antibody and analyzed by flow cytometry to assess CXCR4 expression.
Acknowledgements
This research was supported by grants to A. M. (NIH AI125640, NIH CA231277 and the Cancer Research Institute) and to A. V. S. (NIH R35GM127303). Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056.
References
- 1. Khan H, Pillarisetty VG, Katz SC. The prognostic value of liver tumor T cell infiltrates. J Surg Res 2014; 191:189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peske JD, Woods AB, Engelhard VH. Control of CD8 T‐cell infiltration into tumors by vasculature and microenvironment. Adv Cancer Res 2015; 128:263–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stanton SE, Disis ML. Clinical significance of tumor‐infiltrating lymphocytes in breast cancer. J Immunother Cancer 2016; 4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Ruiter EJ, Ooft ML, Devriese LA, Willems SM. The prognostic role of tumor infiltrating T‐lymphocytes in squamous cell carcinoma of the head and neck: A systematic review and meta‐analysis. Oncoimmunology 2017; 6:e1356148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348:56–61. [DOI] [PubMed] [Google Scholar]
- 6. Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol 2014; 178:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atretkhany KN, Drutskaya MS, Nedospasov SA, Grivennikov SI, Kuprash DV. Chemokines, cytokines and exosomes help tumors to shape inflammatory microenvironment. Pharmacol Ther 2016; 168:98–112. [DOI] [PubMed] [Google Scholar]
- 8. Strazza M, Mor A. Consider the chemokines: a review of the interplay between chemokines and T cell subset function. Discov Med 2017; 24:31–9. [PMC free article] [PubMed] [Google Scholar]
- 9. Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol 2011; 11:355–63. [DOI] [PubMed] [Google Scholar]
- 10. Gladue RP, Brown MF, Zwillich SH. CCR1 antagonists: what have we learned from clinical trials. Curr Top Med Chem 2010; 10:1268–77. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Romero J, Chan A et al Biarylsulfonamide CCR9 inhibitors for inflammatory bowel disease. Bioorg Med Chem Lett 2015; 25:3661–4. [DOI] [PubMed] [Google Scholar]
- 12. Feagan BG, Sandborn WJ, D’Haens G et al Randomised clinical trial: vercirnon, an oral CCR9 antagonist, vs. placebo as induction therapy in active Crohn’s disease. Aliment Pharmacol Ther 2015; 42:1170–81. [DOI] [PubMed] [Google Scholar]
- 13. Strazza M, Azoulay‐Alfaguter I, Peled M et al PLCepsilon1 regulates SDF‐1alpha‐induced lymphocyte adhesion and migration to sites of inflammation. Proc Natl Acad Sci USA 2017; 114:2693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalwa H, Storch U, Demleitner J et al Phospholipase C epsilon (PLCepsilon) induced TRPC6 activation: a common but redundant mechanism in primary podocytes. J Cell Physiol 2015; 230:1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sadowski CE, Lovric S, Ashraf S et al A single‐gene cause in 29.5% of cases of steroid‐resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26:1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tyutyunnykova A, Telegeev G, Dubrovska A. The controversial role of phospholipase C epsilon (PLCepsilon) in cancer development and progression. J Cancer 2017; 8:716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H, Oestreich EA, Maekawa N et al Phospholipase C epsilon modulates beta‐adrenergic receptor‐dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 2005; 97:1305–13. [DOI] [PubMed] [Google Scholar]
- 18. Cossarizza A, Chang HD, Radbruch A et al Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol 2017; 47:1584–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sierra‐Filardi E, Nieto C, Dominguez‐Soto A et al CCL2 shapes macrophage polarization by GM‐CSF and M‐CSF: identification of CCL2/CCR2‐dependent gene expression profile. J Immunol 2014; 192:3858–67. [DOI] [PubMed] [Google Scholar]
- 20. Weagel ESC, Liu PG, Robison R, O’Neill K. Macrophage polarization and its role in cancer. J Clin Cell Immunol 2015; 6:338. [Google Scholar]
- 21. Chow MT, Ozga AJ, Servis RL et al Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity 2019; 50:1498–512.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The top 33% of PLCε1 expression (high) in rectum adenocarcinoma samples from the TCGA correlated with increased survival compared to the lowest 33% of PLCε1 expression (low). Log rank P‐value = 0.0772 (http://www.oncolnc.org/kaplan/?lower=33&upper=33&cancer=READ&gene\_xml:id=51196&raw=plce1&species=mRNA) (a) Tumors were excised and weighed from mice that survived to day 25 post tumor inoculation (b) n = 4 PLCε1 KO mice, n = 12 WT mice from 3 independent experiments; bars represent min ‐ max. ****P ≤ 0.0001.
Fig. S2. CD3+ CD4+ CD8‐ cells were analyzed for expression of CD25 and FoxP3, with double positive (CD25+ FoxP3+) cells being regarded as regulatory T cells (Treg). Bars represent the mean % of CD4+ cells that were Treg ± SEM.
Fig. S3. Cells from spleens, lymph nodes, and tumors from tumor‐bearing mice were isolated and stained for CD3, CD4, CD8, CD44, PD‐1, and CD62L then analyzed by flow cytometry. Events were gated for live CD3+ events then CD4+ CD8‐ (a,b) or CD4‐ CD8+ (c,d). Plots representative of n = 4 WT and PLCε1 KO mice from 2 independent experiments with 2 mice per experiment per group.
Fig. S4. Cells from spleens, right inguinal lymph nodes, and tumors from tumor‐bearing mice were isolated and stained for CD3, CD4, CD8, CXCR4, CCR4, and CXCR3 then analyzed by flow cytometry. CD3+ CD4‐ CD8+ cells are included in this analysis. n = 4 WT and PLCε1 KO mice from 2 independent experiments with 2 mice per experiment per group; points represent mean ± SEM.
Fig. S5. (a) Jurkat T cells expressing shRNA non‐targeting (shScramble) or PLCε1 (shPLCε1) were added to the apical chamber of transwell inserts with SDF‐1α containing media in the basolateral chamber. After 3 hours cells in the basolateral chamber were quantified. n = 3 independent experiments, each performed in duplicate. Bars indicate mean ± SEM; *P ≤ 0.05. (b) Jurkat T cells expressing shScramble or shPLCε1 were stained with APC‐conjugated isotype or CXCR4 antibody and analyzed by flow cytometry to assess CXCR4 expression.