

Summary
Carbamylation is a post‐translational modification that can be detected on a range of proteins, including immunoglobulin (Ig)G, in several clinical conditions. Carbamylated IgG (ca‐IgG) was reported to lose its capacity to trigger complement activation, but the mechanism remains unclear. Because C1q binds with high affinity to hexameric IgG, we analyzed whether carbamylation of IgG affects binding of C1q, hexamerization and complement‐dependent cytotoxicity (CDC). Synovial tissues of rheumatoid arthritis (RA) patients were analyzed for the presence of ca‐IgG in vivo. Synovial tissues from RA patients were analyzed for the presence of ca‐IgG using mass spectrometry (MS). Monomeric or hexameric antibodies were carbamylated in vitro and quality in solution was controlled. The capacity of ca‐IgG to activate complement was analyzed in enzyme‐linked immunosorbent (ELISAs) and cellular CDC assays. Using MS, we identified ca‐IgG to be present in the joints of RA patients. Using in vitro carbamylated antibodies, we observed that ca‐IgG lost its capacity to activate complement in both solid‐phase and CDC assays. Mixing ca‐IgG with non‐modified IgG did not result in effective inhibition of complement activation by ca‐IgG. Carbamylation of both monomeric IgG and preformed hexameric IgG greatly impaired the capacity to trigger complement activation. Furthermore, upon carbamylation, the preformed hexameric IgG dissociated into monomeric IgG in solution, indicating that carbamylation influences both hexamerization and C1q binding. In conclusion, ca‐IgG can be detected in vivo and has a strongly reduced capacity to activate complement which is, in part, mediated through a reduced ability to form hexamers.
Keywords: antibodies, complement, human, rheumatoid arthritis
Carbamylation of both monomeric IgG and pre‐formed hexameric IgG greatly impaired the capacity to trigger complement activation. Upon carbamylation the pre‐formed hexameric IgG, dissociated into monomeric IgG in solution. Indicating that carbamylation does not only influence C1q binding, but also the ability to form hexamers.

Introduction
Post‐translational modifications (PTM) of proteins following biosynthesis are common in the human body, and are important in the regulation of activity, stability and folding of proteins 1, 2. Dysregulation of PTMs has been linked to inflammatory and autoimmune conditions 1. Besides dysregulation, PTMs can also cause the formation of neoepitopes on extracellular proteins during environmental exposure and ageing which subsequently give rise to autoantibodies 3, 4. The PTM carbamylation is the chemical conversion of a positively charged lysine into an uncharged homocitrulline. This conversion is mediated by cyanate, which is in equilibrium with urea, and the availability of cyanate can be increased by inflammation through the release of myeloperoxidase (MPO) from neutrophils 5. Carbamylation is therefore especially interesting in the context of inflammatory and autoimmune diseases. Carbamylated proteins are frequently targeted by autoantibodies in patients suffering from rheumatoid arthritis (RA) 6, 7. Several proteins have reported to be carbamylated in vivo, e.g. albumin 8 and alpha 1 anti‐trypsin 9, but interestingly also immunoglobulin (Ig)G 10, 11. It has been reported that carbamylation of IgG impacts upon its capacity to activate complement 10.
The complement system is a well‐described sequential autolytic cleavage system which can be activated via three different pathways: the classical pathway (CP), the lectin pathway and the alternative pathway. The CP is commonly activated upon binding of C1q to IgM or multiple copies of antigen‐bound IgG 12. One report described that, upon carbamylation of IgG, there was a dose‐dependent decrease in complement activation due to the loss of capacity to bind C1q, and hence loss of ability to initiate the CP, as measured in enzyme‐linked immunosorbent assay (ELISA)‐based and tumour opsonization assays 10. Recently, it has been demonstrated that binding of C1q requires a hexameric arrangement of monomeric IgG complexes, which assemble via non‐covalent Fc–Fc interactions 13, 14. Moreover, cryo‐electron microscopy analyses have shown that the C1 complex also binds to hexameric IgG1 complexes 15 and IgM 16. This could indicate that the reduced capacity of carbamylated IgG (ca‐IgG) to activate complement may directly or indirectly be caused by reduced hexamerization. If this is the case, then carbamylation of IgG may be utilized to dampen inflammatory responses triggered by non‐modified IgG. By the introduction of three mutations (E345R, E430G and S440Y) in the Fc domain, the ability of IgG1 to form hexamers was enhanced both in solution and on the cell surface 13, 14. This hexameric IgG is able to bind C1 in solution and activate the complement system. Here, we studied the effects of IgG carbamylation on complement binding, activation and complement‐dependent cytotoxicity (CDC) in both normal monomeric IgG1 and hexameric conformations. In addition, we investigated the possibility of using ca‐IgG to inhibit IgG‐mediated complement activation.
In this study, we extend the observation that ca‐IgG is present in vivo and unable to activate complement by showing that the carbamylation impacts both on the C1q binding to the IgG Fc, but also impairs hexamerization of IgG, which is essential for effective C1q binding and classical pathway activity.
Materials and methods
Carbamylation
For carbamylation and subsequent experiments, various (therapeutic) antibodies were used: intravenous immunoglobulin (IVIg) (Nanogram; Sanquin, Amsterdam, the Netherlands), alemtuzumab (Genzyme, Cambridge, MA, USA), rituximab (MabThera) and IgG1‐dinitrophenyl (DNP) (RGY) (described below). These different antibodies were carbamylated by incubation in 0·1 M potassium cyanate (KOCN) (cat. no. 215074; Sigma‐Aldrich, St Louis, MO, USA) or incubated in phosphate‐buffered saline (PBS) as a control at 37°C for different time‐periods. After incubation, the preparations were dialysed against PBS for 48 h, or a buffer exchange was performed with PBS (pH 7.4).
Expression and purification of anti‐DNP antibodies
The IgG1‐DNP antibody consists of the variable domains of mouse monoclonal antibody (mAb) G2a2 against the hapten DNP combined with the constant domains of human IgG1 and the kappa light chain 17. A triple mutant variant of the IgG1‐DNP was produced containing three mutations (E345R, E430G and S440Y) in the Fc domain, which enhance the ability of the antibody to form hexamers both in solution and on the cell surface (designated IgG1‐DNP‐RGY) 13, 14. Gene constructs for heavy and light chains were ordered separately (Thermo Fisher Scientific GeneArt, Regensburg, Germany) and cloned into a pcDNA3.3 vector (Thermo Fisher Scientific). Antibodies were expressed by transient transfection of Expi293F™ cells with equimolar amounts of heavy and light chain plasmid, using the ExpiFectamine™ 293 transfection kit (Thermo Fisher Scientific), according to the manufacturer’s guidelines. Secreted antibodies were harvested from the supernatant 5 days post‐transfection, 0·2 µm filtered and purified on a column of protein A sepharose. Solution phase hexamerization of IgG1‐DNP‐RGY was verified using high‐pressure size exclusion chromatography (HP‐SEC) analysis, as previously described 14.
Western blot
Ten per cent Tris‐glycine gels (Biorad, Hercules, CA, USA; cat. no. 456‐1033) were loaded with equal amounts of untreated IgG and ca‐IgG under reducing conditions. Carbamylation was analyzed using anti‐carbamyl‐lysine antibody (cat. no. STA‐078; Cell Biolabs, Cambridge, UK). Next, loading was visualized by stripping Western blot and re‐probing for human IgG (Dako, Glostrup, Denmark; cat. no. P0214).
Mass spectrometry carbamylation
Mass spectrometry (MS) was carried out as described previously 9, 18, 19. Synovial fluid (SF) was centrifuged at 700 g for 5 min and the supernatant was collected and stored in aliquots at –80°C. Next, SF samples (500 μg protein) were depleted according to the instructions of the supplier for the top 12 most abundant serum proteins (Pierce/Thermo, Loughborough, UK). Subsequently, the depleted sample (50 μg) was subjected to filter‐aided sample preparation (FASP II) 20 using 13C‐urea instead of regular 12C‐urea, in order to distinguish artificial in vitro 13C carbamylation during the FASP procedure from genuine in vivo 12C carbamylation events. After FASP II procedure no in vitro carbamylation events were observed.
The cartilage and synovium samples (after their extraction with hot sodium dodecyl sulphate (SDS) to remove adherent and easily soluble protein) were digested with trypsin using the following procedure: samples were incubated in 100 μl dithiothreitol (DTT) (100 mM) and NH4HCO3 (25 mM) for 20 min at 54°C. After 5 min centrifugation at maximum speed, the supernatant was saved and the pellet was incubated in 150 μl iodoacetamide (15 mM) and NH4HCO3 (25 mM) for 30 min at room temperature. After 5 min centrifugation at maximum speed, the supernatant was saved and the pellet was incubated in 200 μl 25 mM NH4HCO3 containing 10 μg trypsin for 4 h at 37°C. The supernatants from the DTT and iodoacetamide incubation were combined and concentrated on a 30‐kDa filter (Microcon; Millipore, Burlington, MA, USA), washed three times with 100 μl 25 mM NH4HCO3 and incubated with 1 μg trypsin for 4 h at 37°C. Finally, the supernatant containing digested protein from the pellet was added to the digest on the filter. The filter was washed once with 100 μl 0·5 M NaCl. Peptides were recovered from the filtrate and subjected to solid phase extraction on C18 cartridges (Oasis HLB Waters, Elstree, UK).
ELISA to detect carbamylation
Maxisorp plates (Nunc, Roskilde, Denmark) were coated with 10 µg/ml IgG in coating buffer (0·1 M Na2CO3, 0·1 M NaHCO3, pH 9.6). Plates were blocked with PBS/1% bovine serum albumin (BSA) and carbamylated signal was detected using rabbit anti‐carbamyl‐lysine antibody (cat. no. STA‐078; Cell Biolabs, Cambridge, UK) and a goat anti‐rabbit horseradish peroxidase (HRP) secondary antibody (Dako; cat. no. P0448). The substrate was added using 2,2'‐azino‐bis‐3‐ethyl benzthiazoline‐6‐suphphonic acid (ABTS) and the carbamylation signal was measured at an absorbance level of 415 nm using the Biorad iMark Microplate Absorbance Reader.
Complement activation ELISA
Maxisorp plates (Nunc) were randomly coated with 10 µg/ml (ca‐)IgG in coating buffer (0·1 M Na2CO3, 0·1 M NaHCO3, pH 9.6) In mixing experiments the final concentration was 10 µg/ml with different ratios between the ca‐IgG and non‐modified IgG, unless indicated otherwise. Plates were blocked with PBS/1% BSA and incubated with 1% normal human serum (NHS, pooled from four healthy donors) or heat‐inactivated NHS as a control (diluted in GVB++; 0·1% gelatin, 5 mM Veronal, 145 mM NaCl, 0·025% NaN3, 0·15 mM CaCl2, 0·5 mM MgCl2, pH 7.3). Complement binding or deposition was analyzed with rabbit anti‐human C1q (Dako; cat. no. A0136), goat anti‐human C4 (Quidel, San Diego, CA, USA; cat. no. A305), rabbit anti‐human C3c (Dako; cat. no. A0062) and mouse anti‐human C5b9 (Dako; cat. no. M0777) with corresponding HRP‐labelled secondary antibodies in PBS/1% BSA/0·05% Tween. Finally, the substrate was added using ABTS and absorbance measured at 415 nm using the Biorad iMark Microplate Absorbance Reader.
CDC
CDC assays with B lymphoma cell lines (Daudi and Wien‐133) were performed using 100 000 target cells opsonized with antibody concentration series in the presence of pooled NHS (20% final concentration) as a complement source (Sanquin), as previously described 21. Cells were incubated for 45 min at 37˚C and killing was calculated as the percentage of propidium idodide (PI; Sigma‐Aldrich Chemie B.V., Zwijndrecht, the Netherlands)‐positive cells determined by flow cytometry. Curves were generated using non‐linear regression (sigmoidal dose–response with variable slope) analyses within GraphPad Prism software (GraphPad Software, San Diego, CA, USA).
Antibody binding
Antibody binding assays with B lymphoma cell lines (Daudi and Wien‐133) were performed using 100 000 target cells opsonized with antibody concentration series and incubated for 30 min at 4˚C. Next, cells were washed and incubated for 30 min at 4°C with R‐phycoerythrin (PE)‐conjugated goat‐anti‐human IgG F(ab)2 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA; cat. no. 109‐116‐098). Cells were washed and analyzed by determining the mean fluorescent intensity (MFI) using flow cytometry. Binding curves were generated using non‐linear regression (sigmoidal dose–response with variable slope) analyses within GraphPad Prism software (GraphPad Software).
Complement‐mediated liposomal lysis assay
Liposomes were prepared using dimyristoylphosphatidylcholine (DMPC), dimyristoylphosphatidylglycerol (DMPG), cholesterol and DNP‐cap‐PE, purchased from Avanti Polar Lipids (Alabaster, AL, USA). Lipid films were composed of DMPC–DMPG–cholesterol–DNP‐cap‐PE (45 : 5 : 49 : 1 mol%). Components were dissolved in chloroform–methanol (9 : 1 v/v) before drying under nitrogen gas and desiccation overnight. Films were rehydrated at 37°C for 30 min with a self‐quenching concentration of sulforhodamine B (20 mM; S1402 from Sigma Aldrich, St Louis, MO, USA) in PBS to a final lipid concentration of 0·8 mg/ml. The sulforhododamine B‐liposome mixture was sonicated for 5 min at 37°C in a water bath. Purification of liposomes was performed through size‐exclusion chromatography using a prepacked NAP‐25 column (17‐0852‐01; GE Healthcare, Little Chalfont, UK).
To analyze complement activity via membrane attack pore membrane attack complex (MAC)‐mediated dye leakage, purified liposomes were diluted ×10 in PBS and mixed with NHS (10% v/v final concentration) from Complement Technologies (Tyler, TX, USA). Sulforhodamine B fluorescence was measured with an excitation wavelength of 565 nm and emission wavelength of 585 nm using a CLARIOstar microplate reader (BMG Labtech, Offenburg, Germany). Fluorescence was measured at 21°C for 100 s before different antibodies (IgG1‐DNP and IgG1‐DNP‐RGY, both non‐modified and carbamylated) were added to final concentrations of 4·35 μg/ml/ml before assaying for a further 10 min. Total lysis was performed by adding 70% ethanol after assay. Experiments were performed in triplicate.
Statistics
Statistical analysis was performed using GraphPad Prism version 7.02. Statistical differences were determined using t‐tests; a P‐value of < 0·05 was considered statistically significant. Data are either representative for multiple experiments or the mean with standard deviation (s.d.) is shown.
Results
Identification of in vivo‐occurring ca‐Ig(G) using mass spectrometry
Previously, ca‐IgG was detected in synovial fluid from two RA patients 10. Therefore, we investigated various tissue samples from RA patients. In both synovium and synovial fluid from either the knee or hip, carbamylated immunoglobulin peptides were found using MS (Table 1). Interestingly, the same carbamylated immunoglobulin peptides were found in independent donors; for example, VGVETTkpSkQSNNkYAASSYLSLTPEQWK (k = carbamylation on lysine, p = oxidation on proline) found in two of 14 donors (14%). Analysis of the relative abundance of the carbamylated immunoglobulin peptides revealed that, at a reasonable coverage of the immunoglobulin proteins (on average 63%, ranging from 25 to 100%), we detected the carbamylated peptides at a roughly 1000‐fold lower abundance compared to the abundance of the protein from which the peptide is derived. Moreover, in various osteoarthritis tissue samples (×2 synovial fluid, ×2 synovium) no carbamylated Ig peptides were found. These data indicate that carbamylation of immunoglobulins, including IgG, is occurring in vivo, as exemplified using samples from RA patients.
Table 1.
Identification of in vivo‐occurring ca‐IgG using mass spectrometry
| Diagnosis | Sex | Age | Location | Tissue | Protein group accessions | Sequence | Protein descriptions |
|---|---|---|---|---|---|---|---|
| RA | Female | 76 | Knee | Synovium | A0A087X130; A0A075B6H6 | HkVYAcEVTHQGLSSPVTK | Ig kappa chain C region |
| P01857; A0A087WYE1; A0A087WYC5 | KVEPkScDkTHTcppcpAPELLGGPSVFLFPPKPK | Ig gamma‐1 chain C region | |||||
| RA | Male | 56 | Hip | Synovium | A0A087X130; A0A087WZW8; A0A075B6H6 | HkVYAcEVTHQGLSSPVTK | Ig kappa chain C region |
| P01876 | TFTcTAAYPESkTPLTATLSK | Ig alpha‐1 chain C region | |||||
| A0A087X130; A0A087WZW8; A0A075B6H6 | VYAcEVTHQGLSSPVTkSFNRGEc | Ig kappa chain C region | |||||
| A0A087X130; A0A087WZW8; A0A075B6H6 | ADYEkHKVYAcEVTHQGLSSPVTK | Ig kappa chain C region | |||||
| A0A087WYE1; A0A087WYC5 | kVEPkScDKTHTcPpcpApELLGGPSVFLFPPKPK | Ig gamma‐1 chain C region | |||||
| P01861 | VDkRVESkYGppcpScpApEFLGGPSVFLFPPKPK | Ig gamma‐4 chain C region | |||||
| RA | Male | 49 | Knee | Synovial fluid | A0M8Q6 | VGVETTkpSkQSNNkYAASSYLSLTPEQWK | Ig lambda‐7 chain C region |
| RA | Male | 67 | Knee | Synovial fluid | A0M8Q6 | VGVETTkpSkQSNNkYAASSYLSLTPEQWK | Ig lambda‐7 chain C region |
Carbamylated immunoglobulins (ca‐IgG) detected in vivo in samples from rheumatoid arthritis (RA) patients, as analyzed by mass spectrometry, the carbamylated lysines are annotated (k); k = carbamyl, c = carbamidomethyl, p = oxidation.
Successful carbamylation of IgG preparations
To study the biology of carbamylation of antibodies, different IgG antibodies (alemtuzumab, rituximab, IgG1‐DNP, IgG1‐DNP‐RGY) and IVIg were carbamylated by 0·1 M KOCN (cyanate) at 37°C for different time‐points (1, 3, 6 and 24 h). Carbamylation of all antibody preparations were successful as detected by Western blot and an ELISA‐based assay. Both the Western blot and the ELISA show a time‐dependent increase in carbamylation for all antibodies, as exemplified by rituximab (Fig. 1a,b). Moreover, the 24‐h carbamylated rituximab sample was analyzed with MS to identify the carbamylated lysines (Fig. 1c). MS analysis showed extensive presence of carbamylation; several of the carbamylated lysines were identical to the carbamylated lysines found in the in vivo samples.
Figure 1.
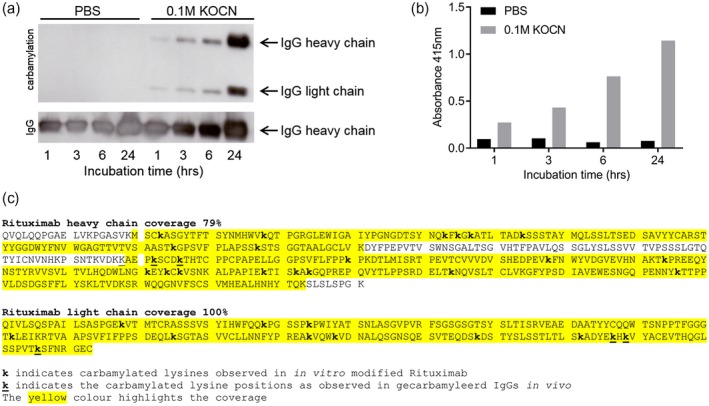
Successful carbamylation (ca) of rituximab. The results from rituximab are shown and are representative for all carbamylated antibodies [intravenous immunoglobulin [(IVIg), alemtuzumab, dinitrophenyl (DNP) variants]. Antibodies were either incubated in phosphate‐buffered saline (PBS) as a control (buffer only) or in 0·1 M potassium cyanate (KOCN). (a) Western blot analysis of 2 µg (ca)‐rituximab, carbamylation was visualized using a carbamyl–lysine‐specific antibody. Equal loading of the preparations was shown by detection of human IgG. Incubation time is indicated in hours. (b) Carbamylation of rituximab as analyzed by enzyme‐linked immunosorbent assay (ELISA), where an equal amount of antibody (10 µg/ml) was used to coat the well. Detection was performed using a carbamyl–lysine‐specific antibody. (c) Mass spectrometry analysis of rituximab that was carbamylated for 24 h. The carbamylated lysines are annotated (k), coverage is indicated with yellow highlight.
Carbamylation of IgG completely blocks the complement activating potential of IgG
Complement activation assays were performed using 10 µg/ml (ca‐)IgG‐coated ELISA plates. Binding of C1q and deposition of C4, C3c and C5b9 was significantly decreased upon carbamylation of IVIg, rituximab and alemtuzumab when compared to the non‐modified counterpart (Fig. 2). We analyzed whether carbamylation affected the antibody’s immobilization in the ELISA plate and, although we observed some differences, these were too small to explain the observed biological effects (Supporting information, Fig. S1). Previously, it has been demonstrated that optimal complement activation via IgG is achieved when the IgGs are in a hexameric arrangement. Therefore, we wondered whether adding ca‐IgG to IgG would inhibit this process by limiting the possibilities to form productive hexamers. Mixing experiments were performed to analyze whether the presence of ca‐IgG indeed affects the ability of non‐modified IgG to activate the complement system. Mixing ca‐IgG antibodies with non‐modified antibodies, while maintaining a similar end concentration, resulted in a decrease in complement activation (at the levels of C1q, C4, C3c and C5b9) (Fig. 3), which was proportional to the decreasing concentration of coated non‐modified IgG. Although ca‐IgG was unable to activate complement by itself, we did not observe an inhibitory effect of ca‐IgG on the complement activation induced by non‐modified IgG other than the simple dose–response effect of diluting the non‐modified IgG. As the coated antibodies in ELISA plates are known to form a high density surface of IgGs that bind C1q without a need to form hexamers 14, the data indicate that the carbamylation on lysine residues in the Fc impact directly on the binding of C1q.
Figure 2.
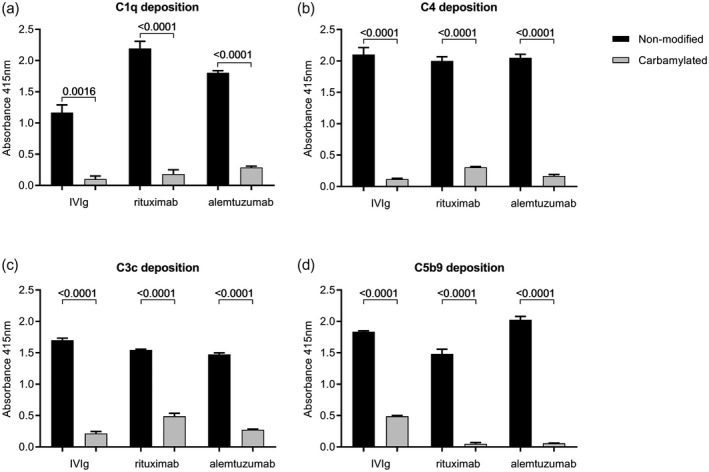
Complement deposition of C1q, C4, C3c and C5b9 on non‐modified and carbamylated immunoglobulin (Ig)G. Complement deposition enzyme‐linked immunosorbent assays (ELISAs) were performed using 10 μg/ml plate‐bound non‐modified or carbamylated (ca) intravenous immunoglobulin [(IVIg), rituximab and alemtuzumab. C1q (a), C4 (b), C3c (c) and C5b9 (d) deposition after incubation for 1 h with 1% normal human serum (NHS) was measured at an absorbance wavelength of 415 nm. Data are shown as mean with standard deviation from technical triplicate. All differences in complement deposition between non‐modified antibodies and their carbamylated counterparts were significant, as analyzed with t‐test (all P < 0·05). Shown is a representative experiment performed at least twice.
Figure 3.
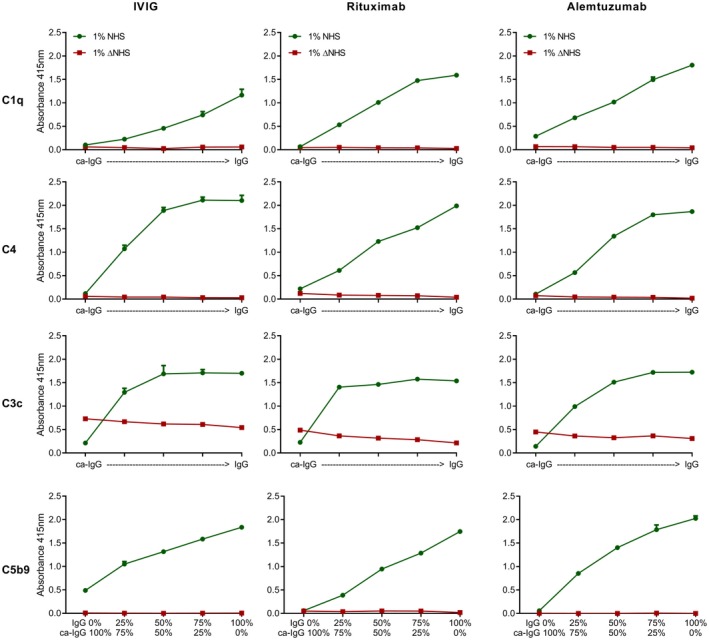
Deposition of different complement components is not affected by the presence of carbamylated (ca)‐immunoglobulin (Ig)G. Complement deposition enzyme‐linked immunosorbent assays (ELISAs) were performed using plate‐bound non‐modified or ca‐IVIg, rituximab and alemtuzumab, with a sum of 10 μg/ml coating mixed in different ratios, and for rituximab a sum of 2·5 μg/ml (C4) or 5 μg/ml (C1q, C3c, C5b9). From left to right, ca‐IgG versus non‐modified IgG: 100%: 0%, 75%: 25%, 50%: 50%, 25%: 75%, 0%: 100%. Deposition of complement components C1q, C4, C3c and C5b9 were measured at an absorbance wavelength of 415 nm. Green circles depict the complement deposition after incubation with 1% normal human serum (NHS), red squares depict complement deposition after incubation with 1% heat‐inactivated normal human serum (ΔNHS) for 1 h. Shown is a representative experiment which has been performed independently at least twice.
CDC is not affected by the presence of ca‐IgG
Because of the observed differences in complement binding and activation by plate‐immobilized IgGs, we next explored the effect of carbamylation on antibody binding and CDC using tumour cell lines. IgG hexamerization is required for optimal C1q binding and complement activation in this setting. Binding of non‐modified or carbamylated rituximab and alemtuzumab was analyzed using Daudi and Wien‐133 cell lines, respectively. The carbamylated variants of rituximab and alemtuzumab displayed a slightly lower binding compared to their non‐modified counterparts, which actually might reflect the lower binding efficiency of the anti‐IgG detection conjugate antibody to the ca‐IgG compared to the non‐modified IgG (Supporting information, Fig. S2). Next, we investigated whether carbamylated IgG affects the ability of non‐modified IgG to induce CDC. CDC assays were performed with different ratios of non‐modified or carbamylated rituximab and alemtuzumab (Fig. 4). No CDC was observed in the presence of carbamylated rituximab or alemtuzumab only, which cannot be solely attributed to the reduced binding of ca‐IgG to the surface. Non‐modified rituximab and alemtuzumab induced substantial CDC, while titrating in their carbamylated counterparts resulted in a decrease in killing capacity. For rituximab, the decrease in killing capacity was gradual with the titrating of the carbamylated variant, whereas for alemtuzumab there is a sharp decrease in CDC after the carbamylated variant exceeded the 50% ratio. These results indicate that at the chosen antibody concentrations and ratios there is no dominant negative effect of ca‐IgG presence on the capacity of non‐modified IgG to induce CDC.
Figure 4.
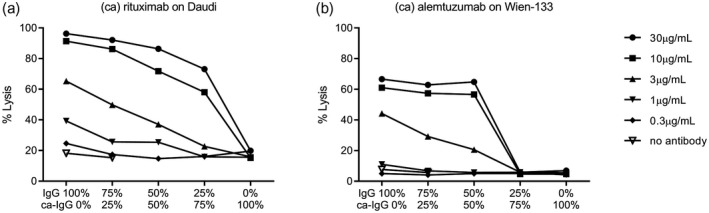
Complement‐dependent cytotoxicity is not affected by the presence of carbamylated (ca)‐immunoglobulin (Ig)G. Complement‐dependent cytotoxicity (CDC) assays were performed with Daudi (a) and Wien‐133 (b) cells opsonized with various concentrations of non‐modified and/or ca‐rituximab or alemtuzumab, respectively, in the presence of 20% pooled normal human serum (NHS). CDC assays were performed using different ratios of mixed non‐modified (IgG) and ca‐IgG antibody variants. Ratios of ca‐IgG versus non‐modified IgG from left to right are: 100%: 0%, 75%: 25%, 50%: 50%, 25%: 75%, 0%: 100%, each depicted for a total of five different final antibody concentrations. Shown is a representative experiment which has been performed independently twice.
Monomeric and hexameric IgG
CDC activity is highly dependent upon IgG1 hexamer formation, and in the CDC assay it was unclear whether ca‐IgGs form hexamers in the presence of non‐modified IgGs. Therefore, we explored whether the effect of reduced complement activation by ca‐IgG is also present when using pre‐formed ca‐IgG‐hexamers. DNP antibodies were produced containing three point mutations in the Fc domain (DNP‐RGY), which causes the IgGs to form stable hexamers in solution 14 (Fig. 5a). Next, these preparations were also carbamylated and the ability of IgG‐RGY mutants to hexamerize in solution was analyzed using size exclusion chromatography (HP‐SEC). Control IgG1‐DNP‐RGY primarily existed as a hexamer: an early eluting peak was observed for hexameric IgG (58·6%) and a second peak for monomeric IgG (39·4%). In solution, both species are in equilibrium, with a relatively high fraction forming hexameric complexes. Carbamylated IgG clearly only showed a monomeric IgG peak (98%) (Fig. 5b), indicating that hexamerization was abrogated. First, we tested the effect of carbamylation on monomeric IgG or on hexameric IgG regarding complement activation in a liposome lysis assay, a setting in which the bound antibodies can move freely and form hexamers on the surface. In a liposome lysis assay, complement activation is measured by sulforhodamine B release from liposomes as a consequence of MAC formation 22. In non‐modified conditions, both anti‐DNP antibody preparations are able to lyse liposomes (the lysis caused by DNP‐RGY is higher, as pre‐formed hexamers enhance complement activation) 14. Carbamylation of both these antibodies leads to a complete loss of liposome lysis (Fig. 5c,d).
Figure 5.
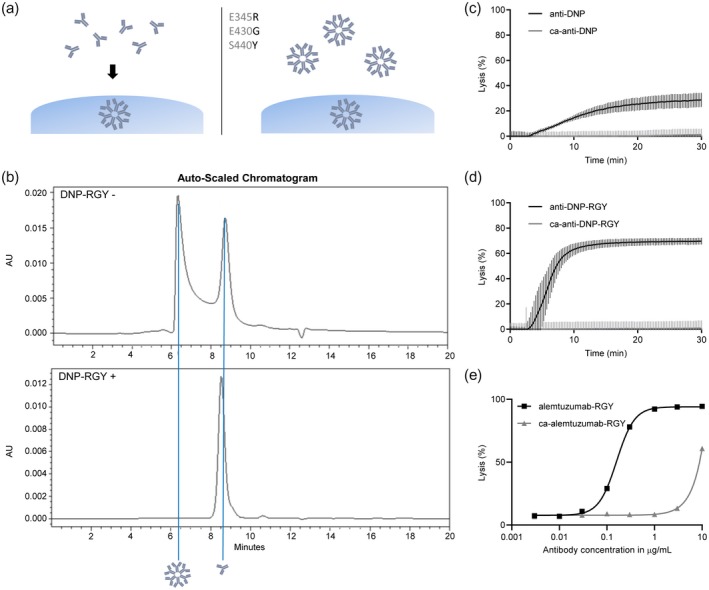
Monomeric and hexameric immunoglobulin (Ig)G both lose liposomal lysis capacity upon carbamylation (ca). (a) Schematic representation of monomeric IgG1‐dinitrophenyl (DNP), which forms hexamers on the cell surface (left) and IgG1‐DNP‐E345R, E430G and S440Y (RGY), which forms hexamers in both solution and on the cell surface (right). (b) The effect of ca of IgG1‐DNP‐E345R, E430G and S440Y (RGY) on hexamerization in solution was analyzed with high‐pressure size exclusion chromatography (HP‐SEC). (c,d) Liposomes expressing DNP were lysed in the presence of E345R, E430G and S440Y normal human serum (NHS) and 4·35 µg/ml (ca) anti‐DNP(RGY) antibodies. Fluorescence of sulforhodamine B is normalized to total‐lysis by addition of ethanol, which is set at 100%. Data shown are the mean with standard deviation from technical triplicate and results are representative of three independent experiments. (e) Alemtuzumab‐RGY was carbamylated and complement‐dependent cytotoxicity (CDC) assays on Wien‐133 were performed. Cells were opsonized with various concentrations of non‐modified and/or (ca)‐alemtuzumab‐RGY in the presence of 20% pooled NHS.
Next, the RGY variant of alemtuzumab was generated and carbamylated (or treated with PBS as control) and CDC assays were performed. Again, carbamylation of RGY resulted in loss of CDC capacity (Fig. 5e). Collectively, the data on the size exclusion chromatography, the liposome lysis assay and the CDC assays indicate that carbamylation removes the capacity of IgG‐RGY mutants to spontaneously from hexamers in solution.
Discussion
Several carbamylated proteins are known to be present in vivo; carbamylated albumin was identified in patients suffering from renal failure 23, 24, and carbamylated fibrinogen and alpha‐1 antitrypsin (A1AT) were discovered in samples from RA patients. In addition, ca‐IgG was present in synovial tissue of RA patients 10. It has also been shown that carbamylation of IgG1 occurs in vivo, resulting in the loss of C1q binding 10, 11 and a subsequently reduced complement activation. These observations may have interesting implications. In RA, anti‐carbamylated protein (CarP) antibodies that target carbamylated proteins are present 3, 6, possibly including ca‐IgG, which in such a scenario would have rheumatoid factor‐like properties. Furthermore, ca‐IgG will behave differently in the inflamed joint of the patient. For several therapeutic antibodies, such as the CD20‐specific antibody rituximab, the mechanism of action involves CDC. When therapeutic antibodies become compromised by carbamylation in vivo their functionality may be affected, as well as their half‐life. Therefore, we were interested to reproduce the previously known inhibitory effects of ca‐IgG on the complement system and CDC, and specifically analyze ca‐IgG in the presence of non‐modified IgG and variants of IgG that form hexamers in solution.
We confirmed that carbamylation of IgG results in a decrease in complement activation, both in ELISA and in CDC assays. For plate‐bound assays, we observed that the effect on the level of C3c deposition of ca‐IgG was less pronounced, which could be related to some alternative pathway activation of the plastic wells at high serum concentrations. The binding of rituximab and alemtuzumab to cell surfaces appeared reduced upon carbamylation; however, this could also be the result of reduced detection efficiency by the anti‐IgG detection antibodies used, as the detection of plate‐bound IgG, a condition not depending on the antigen binding by the IgG, was similarly impaired. Nevertheless, when the carbamylated variants were mixed with the non‐modified IgG there was no dominant inhibitory effect on complement activation. However, for alemtuzumab there is a sharp decrease in CDC once the ca‐IgG exceeds the 50% ratio. This is in agreement with other published data, where a less potent CDC antibody had negative effect when exceeding the 1 : 1 ratio 25. These data on alemtuzumab suggest that the ability of non‐modified IgG to form hexamers and consequently bind C1q was not affected by the presence of ca‐IgG at ratios below 50%. To investigate whether carbamylation interferes with antibody hexamer formation (Fc–Fc interactions) we used DNP antibodies and the DNP‐RGY mutant, the latter of which forms spontaneous hexamers in solution, and analyzed the ability of these antibodies to lyse DNP‐coated liposomes. Upon carbamylation, the IgG1‐DNP‐RGY was no longer able to hexamerize in solution, indicating that carbamylation negatively affects Fc–Fc interaction resulting in the loss of C1q binding by IgG.
Using MS, we identified several peptides of human immunoglobulins to be carbamylated in vivo in the synovial fluid of RA patients. These peptides were not found in all samples analyzed, which may suggest an accumulation in a specific disease condition, but may also be the result of limited sensitivity to observed these modified peptides. Although the number of peptides containing homocitrulline was limited, we observed similar peptides in several patients. Whether or not therapeutic antibodies may also become carbamylated in vivo in, e.g. the inflamed joint or the tumour micro‐environment is currently unknown. We still expect the majority of IgG to be unmodified even in an inflammatory environment. Based on our titration experiments, used to mimic the biological scenario, it is unlikely that the in vivo carbamylation of IgG would have a major impact on the complement activating potential of these therapeutic antibodies. It is interesting to note that by carbamylating a therapeutic IgG preparation it is possible to completely avoid any risk of complement activation by these antibodies. This is especially relevant for antibodies that should bind to cell surfaces, where they should modify receptor ligand interactions without killing or activating the cell such as, for example, with checkpoint blockade in anti‐tumour therapy.
In conclusion, the inability of ca‐IgG to activate the complement system is the combined effect of both decreased binding of C1q to the modified Fc of IgG and the reduced capacity of ca‐IgG to form hexamers.
Disclosures
P. A. v. V and L. A. T. are listed as inventors on a patent application on the detection of anti‐CarP antibodies in RA.
Author contributions
R. L., S. O., F. B. and L. T, designed the study and interpreted the data. R. L., S. O., D. D., M. V., L. A., A. R., G. J., E. B., B. B. and B. K. collected and assembled the data. R. L., S. O., F. B., D. D., M. V., L. A., A. R., G. J., E. B., B. B. and B. K. analyzed the obtained data. R. L., P. P., T. S., P. V., F. B. and L. T. critically evaluated the data and P. V. and L. T. obtained the funding. All authors critically revised and approved the manuscript.
Supporting information
Fig. S1. Coating of (ca‐)IgG to the plate. Antibody binding to the plate of carbamylated Igs (ca‐IgG) and their non‐modified counterpart (IgG) was performed by directly detecting the coated antibodies with anti‐human IgG‐HRP labelled antibody.
Fig. S2. Cellular binding (ca‐)IgG. Antibody binding was measured using flow cytometry for non‐modified or carbamylated rituximab and alemtuzumab, on Daudi and Wien‐133 cells, respectively. Data shown are representative for two experiments.
Acknowledgements
We acknowledge the financial support from the Netherlands Proteomics Center and the Center for Medical Systems Biology as part of the Netherlands Genomics Initiative. This work was supported by a Zon‐Mw Vidi grant (91712334) awarded to L. A. T. and by a research grant from the Leiden University Medical Center, Leiden, the Netherlands. L. A. T. has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 724517). This work was supported by an Investment Grant NWO Medium (project number 91116004, partly financed by ZonMw) to P. A. v. V. We would like to thank Andreea Ioan‐Facsinay from the Rheumatology department at Leiden University Medical Center for providing the synovium and synovial fluid samples analyzed in this study.
References
- 1. Liu J, Qian C, Cao X. Post‐translational modification control of innate immunity. Immunity 2016; 45:15–30. [DOI] [PubMed] [Google Scholar]
- 2. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochem Biophys Acta 2004; 1695:55–72. [DOI] [PubMed] [Google Scholar]
- 3. Trouw LA, Rispens T, Toes REM. Beyond citrullination: other post‐translational protein modifications in rheumatoid arthritis. Nat Rev Rheumatol 2017; 13:331–9. [DOI] [PubMed] [Google Scholar]
- 4. Anderton SM. Post‐translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol 2004; 16:753–8. [DOI] [PubMed] [Google Scholar]
- 5. Gajjala PR, Fliser D, Speer T, Jankowski V, Jankowski J. Emerging role of post‐translational modifications in chronic kidney disease and cardiovascular disease. Nephrol Dial Transplant 2015; 30:1814–24. [DOI] [PubMed] [Google Scholar]
- 6. Shi J, Knevel R, Suwannalai P et al Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA 2011; 108:17372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi J, van Veelen PA, Mahler M et al Carbamylation and antibodies against carbamylated proteins in autoimmunity and other pathologies. Autoimmun Rev 2014; 13:225–30. [DOI] [PubMed] [Google Scholar]
- 8. Nakabo S, Hashimoto M, Ito S et al Carbamylated albumin is one of the target antigens of anti‐carbamylated protein antibodies. Rheumatology (Oxf) 2017; 56:1217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verheul MK, Yee A, Seaman A et al Identification of carbamylated alpha 1 anti‐trypsin (A1AT) as an antigenic target of anti‐CarP antibodies in patients with rheumatoid arthritis. J Autoimmun 2017; 80:77–84. [DOI] [PubMed] [Google Scholar]
- 10. Koro C, Bielecka E, Dahl‐Knudsen A et al Carbamylation of immunoglobulin abrogates activation of the classical complement pathway. Eur J Immunol 2014; 44:3403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hutchinson D, Clarke A, Heesom K, Murphy D, Eggleton P. Carbamylation/citrullination of IgG Fc in bronchiectasis, established RA with bronchiectasis and RA smokers: a potential risk factor for disease. ERJ Open Res 2017; 3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology 2000; 49:159–70. [DOI] [PubMed] [Google Scholar]
- 13. Wang G, de Jong RN, van den Bremer ET et al Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol Cell 2016; 63:135–45. [DOI] [PubMed] [Google Scholar]
- 14. Diebolder CA, Beurskens FJ, de Jong RN et al Complement is activated by IgG hexamers assembled at the cell surface. Science (NY) 2014; 343:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ugurlar D, Howes SC, de Kreuk BJ et al Structures of C1‐IgG1 provide insights into how danger pattern recognition activates complement. Science (NY) 2018; 359:794–7. [DOI] [PubMed] [Google Scholar]
- 16. Sharp TH, Boyle AL, Diebolder CA, Kros A, Koster AJ, Gros P. Insights into IgM‐mediated complement activation based on in situ structures of IgM‐C1‐C4b . Proc Natl Acad Sci USA 2019; 116:11900–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez ML, Frank MB, Ramsland PA, Hanas JS, Waxman FJ. Structural analysis of IgG2A monoclonal antibodies in relation to complement deposition and renal immune complex deposition. Mol Immunol 2003; 40:307–17. [DOI] [PubMed] [Google Scholar]
- 18. Dekkers JS, Verheul MK, Stoop JN et al Breach of autoreactive B cell tolerance by post‐translationally modified proteins. Ann Rheum Dis 2017; 76:1449–57. [DOI] [PubMed] [Google Scholar]
- 19. Verheul MK, van Veelen PA, van Delft MAM et al Pitfalls in the detection of citrullination and carbamylation. Autoimmun Rev 2018; 17:136–41. [DOI] [PubMed] [Google Scholar]
- 20. Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6:359–62. [DOI] [PubMed] [Google Scholar]
- 21. de Jong RN, Beurskens FJ, Verploegen S et al A novel platform for the potentiation of therapeutic antibodies based on antigen‐dependent formation of IgG hexamers at the cell surface. PLOS Biol 2016; 14:e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharp TH, Koster AJ, Gros P. Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase‐plate cryo‐electron tomography. Cell Rep 2016; 15:1–8. [DOI] [PubMed] [Google Scholar]
- 23. Berg AH, Drechsler C, Wenger J et al Carbamylation of serum albumin as a risk factor for mortality in patients with kidney failure. Sci Transl Med 2013; 5:175ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaisson S, Pietrement C, Gillery P. Carbamylation‐derived products: bioactive compounds and potential biomarkers in chronic renal failure and atherosclerosis. Clin Chem 2011; 57:1499–505. [DOI] [PubMed] [Google Scholar]
- 25. van den Bremer ET, Beurskens FJ, Voorhorst M et al Human IgG is produced in a pro‐form that requires clipping of C‐terminal lysines for maximal complement activation. mAbs 2015; 7:672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Coating of (ca‐)IgG to the plate. Antibody binding to the plate of carbamylated Igs (ca‐IgG) and their non‐modified counterpart (IgG) was performed by directly detecting the coated antibodies with anti‐human IgG‐HRP labelled antibody.
Fig. S2. Cellular binding (ca‐)IgG. Antibody binding was measured using flow cytometry for non‐modified or carbamylated rituximab and alemtuzumab, on Daudi and Wien‐133 cells, respectively. Data shown are representative for two experiments.