

Abstract
Objective
To determine the molecular characterization and disease-associated complications of beta-thalassemia intermedia (β-TI) patients in Sulaymaniyah province, northeastern Iraq.
Methods
A total of 159 β-TI) patients in Sulaymaniyah province, northeastern Iraq. β-TI) patients in Sulaymaniyah province, northeastern Iraq.
Results
Nineteen different β-globin gene mutations arranged in 37 various genotypes were determined. The most frequent were IVS-II-I (G>A) (47.2%), followed by IVS-I-6 (T>C) (23.3%) and IVS-I-110 (G>A) (5%). Among disease-related morbidities documented, bone disease amounted to 53% (facial deformity and osteoporosis), followed by endocrinopathies 17.6% (growth retardation and subclinical hypothyroidism), cholelithiasis 13.8%, pulmonary hypertension 11.3%, and abnormal liver function test 7.5%, whereas venous thrombosis, extramedullary hemopoiesis, and leg ulcer were less frequently observed. Age ≥ 35 and female sex were risk factors for cholelithiasis, while age was an independent risk for hypothyroidism and female sex was associated with increased risk for osteoporosis. Mean serum ferritin of ≥1000 μg/L was associated with an increased risk of osteoporosis, whereas chelation therapy was protective for a multitude of other complications. Transfusion, on the other hand, increased the risk of osteoporosis, yet it was protective for cholelithiasis and hypothyroidism. Moreover, splenectomy was protective for cholelithiasis, although it was an independent risk for hypothyroidism. Finally, hydroxyurea was associated with an increased risk of osteoporosis, while it was protective for cholelithiasis. Discussion and Conclusion. β+-thalassemia mutation had contributed to 41.25 of families with a less severe β-thalassemia phenotype in the northeastern part of Iraq, justifying the need to investigate the contribution of genetic modifiers in ameliorating disease severity. In addition, the substantial number of β-TI patients developed disease-related morbidities, which necessitates the need for more appropriate clinical management with earlier intervention.β-TI) patients in Sulaymaniyah province, northeastern Iraq. μg/L was associated with an increased risk of osteoporosis, whereas chelation therapy was protective for a multitude of other complications. Transfusion, on the other hand, increased the risk of osteoporosis, yet it was protective for cholelithiasis and hypothyroidism. Moreover, splenectomy was protective for cholelithiasis, although it was an independent risk for hypothyroidism. Finally, hydroxyurea was associated with an increased risk of osteoporosis, while it was protective for cholelithiasis. Discussion and Conclusion. β+-thalassemia mutation had contributed to 41.25 of families with a less severe β-thalassemia phenotype in the northeastern part of Iraq, justifying the need to investigate the contribution of genetic modifiers in ameliorating disease severity. In addition, the substantial number of β-TI patients developed disease-related morbidities, which necessitates the need for more appropriate clinical management with earlier intervention.Discussion and Conclusion. β+-thalassemia mutation had contributed to 41.25 of families with a less severe β-thalassemia phenotype in the northeastern part of Iraq, justifying the need to investigate the contribution of genetic modifiers in ameliorating disease severity. In addition, the substantial number of β-TI patients developed disease-related morbidities, which necessitates the need for more appropriate clinical management with earlier intervention.β-TI) patients in Sulaymaniyah province, northeastern Iraq. β-TI) patients in Sulaymaniyah province, northeastern Iraq.
1. Introduction
Beta-thalassemia (β-thal) is one of the most widely distributed autosomal recessive disorders which affects the β-globin gene of the hemoglobin with considerable predominance in the thalassemia belt area, including, Iraq [1]. The disease is characterized by diminished (β+, β++) or lack (β0) of generation of the β-globin chain [2].
All over the world, over 350 various mutations of the β-globin gene were recorded [3, 4]. The genetic pathophysiology of TI results from marked molecular heterogeneity of the β-globin gene with the inheritance of one or two mutations interacting with many genetic variables among which is concomitant α-thalassemia and polymorphism at the three major quantitate loci (QTL) to increase Hb F production [5].
Numerous serious long-term sequelae have been determined in β-TI related to the degree of anemia and iron deposition [6]. The severity of anemia stems from ineffective erythropoiesis and peripheral hemolysis of the mature red blood cells. The morbidities include facial deformity, pulmonary hypertension (PHT), thromboembolic complication, extramedullary hemopoiesis (EMH), and endocrine complications (hypothyroidism, diabetes mellitus, hypogonadism, and osteoporosis) [7, 8].
β-Thal is a major public health burden in the Kurdistan Region of Iraq with an estimated carrier frequency in Sulaymaniyah of 4.1% [9]. The current study had investigated the molecular characterization of β-TI in the northeastern part of Iraq and attempted to evaluate the disease-associated complications in this subgroup of thalassemia.
2. Patients and Methods
2.1. Patients
This study was conducted between July 2018 and August 2019 enrolling a total of 159 β-thalassemia intermedia patients from 114 registered families at the Sulaymaniyah Thalassemia Care Center through random selection from the targeted population to avoid bias. All medical files of these patients were reevaluated. The diagnostic criteria were based on (i) age at diagnosis or initiation of transfusion ≥ 2 years, (ii) maintain hemoglobin ≥ 7 − 7.5 g/dL, and (iii) intermittent transfusion requirements [10, 11].
The study was approved by the ethical committee at the College of Medicine, University of Sulaymaniyah, Iraq, and informed consent was obtained from all enrollees.
2.2. Clinical Assessment
A standardized comprehensive questionnaire was used to obtain detailed clinical information regarding demographic data, age at the initiation of transfusion and diagnosis, time interval of blood transfusion in the past year, family history of thalassemia, rate of consanguinity, and parenthood. All patients were physically examined regarding skeletal facial deformity, height (to detect growth retardation when height > 2SD below 3rd percentile for the mean age and gender), and recurrent leg ulceration. The sizes of the spleen (unless splenectomized) and liver were also determined, and gall bladder stones were screened by ultrasound. Details regarding pathological fractures, diabetes, documented venous thrombosis, and extramedullary hemopoiesis, with medications used for iron chelation and hydroxyurea were obtained from patients' files.
2.3. Laboratory Investigations
2.3.1. Hematological Tests
Five milliliters of venous blood sample was aspirated and transferred equally into an EDTA anticoagulated tube and a plain tube. The anticoagulated blood was used to perform red cell parameters using a fully automated hematology analyzer (Swelab, Spånga, Sweden), and Hb A2 and Hb F quantification was performed using high-performance liquid chromatography (HPLC) (D-10, Bio-Rad Laboratories, Hercules, CA, USA). These investigations were performed just before receiving the next blood transfusion, while for patients who were on regular transfusion sessions (every 3-4 months), the HPLC results were recorded at the time of first presentation or when diagnosis was documented. The remaining EDTA blood sample was centrifuged, and a buffy coat was separated and frozen at −20°C for later DNA extraction.
2.3.2. Biochemical and Other Tests
The serum was utilized for the estimation of ferritin by an ELISA method (Biokit, USA), as well as serum glucose (mg/dL) and serum alanine transaminase (ALT) (U/L). Virological screening included Hepatitis B surface antigen (HBsAg) (Plasmatec Laboratory Products, UK), antihepatitis C virus (HCV) antibodies (Plasmatec Laboratory Products, UK), and Human Immune Deficiency Virus (HIV) antibodies 1 and 2 (Plasmatec Laboratory Products, UK). Thyroid function tests including thyroid-stimulating hormone (TSH) and free T4 were estimated using enzyme immunoassay (TOSOH, Japan). Thyroid-stimulating hormone and free T4 were measured in all TI patients ≥10 years annually [12]. Patients with elevated TSH > 4.7 μIU/mL and low free T4 < 0.8 ng/dL were labeled as overt hypothyroidism, while subclinical hypothyroidism was diagnosed if patients had elevated high TSH > 4.7 μIU/mL in conjunction with normal free T4 > 0.8 ng/dL [13].
2.3.3. Molecular Tests
The genomic DNA was isolated from peripheral blood leukocytes which were collected in a 0.5 M K2EDTA anticoagulated tube based on the phenol-chloroform procedure [14]. The DNA samples were then amplified in a multiplex polymerase chain reaction (PCR), followed by reverse hybridization to specific oligonucleotide arrays fixed on test strips which are constructed to determine a panel of 22 relatively common Mediterranean β-thal mutations using the β-globin StripAssay MED® Kit (ViennaLab Diagnostics GmbH, Vienna, Austria). All steps were performed as recommended by the manufacturer's instructions. The mutations include -101 (C>T); -87 (C>G); -30 (T>A); codon 5 (-CT); codon 6 (G>A); codon 6 (A>T); codon 6 (-A); codon 8 (-AA); codon 8/9 (+G); codon 15 (G>A); codon 27 (G>T); IVS I-1 (G>A); IVS I-5 (G>C); IVS I-6 (T>C); IVS I-110 (G>A); IVS I-116 (T>G), IVS I-130 (G>C); codon 39 (C>T); codon 44 (-C); IVS II-1 (G>A); IVS II-745 (C>G); and IVS II-848 (C>A). In 17 cases, when no or one mutation was revealed by reverse hybridization, both EDTA blood and DNA samples were sent to Kariminejad-Najmabadi Pathology and Genetic Center in Tehran, Iran, for direct sequencing of the whole β-globin gene (HBB) using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction kit and the ABI Prism 377 DNA Automatic Sequencer (Perkin Elmer, Foster City, CA). Forward primer A (CTTAGGCTGCTGGTGGTCTACC) and reverse primers B (AGCACTTTCTTGCCATGAGCC) and C (ATGCACTGACCTCCCACATTCC) corresponded to positions 391, 469, and 1740, respectively. We screened for -α3.7, -α4.2, and the -Med-1α-thal deletions as well as α-gene triplication by gap PCR [15].
2.4. Echocardiography
All patients at their regular visiting dates were subjected to periodic echocardiographic evaluation. Those who had a pulmonary artery systolic pressure (PASP) ≥ 25 mmHg combined with exertional dyspnea at rest without evidence of left-side heart failure were regarded as pulmonary hypertension [16].
2.5. Bone Mineral Density Evaluation
Dual Energy X-ray Absorptiometry (DEXA) scan was performed annually for patients with ages of ≥10 years [12]. The diagnosis of osteoporosis was based on finding a decreased bone mass density with a T − score≤−2.5SD based on BMD measurement at the lumbar spine and hip [17, 18].
2.6. Statistical Methods
SPSS version 25.0 (Armonk, NY; IBM Corp., USA) for Windows was used for data analysis. Means and SD were calculated in continuous data and the frequency proportion for categorical data. ANOVA test for the statistical differences in means and chi-square for the relation of different genotype groups, complications, and other categories to different parameters was calculated. Multivariate logistic regression analysis was done for each complication as a dependent variable to determine the independent effect of study parameters. The P value was considered statistically significant at a level of <0.05.
3. Results
3.1. Patient Characteristics
One hundred fifty-nine β-thalassemia intermedia (β-TI) patients were enrolled, including 90 (56.6%) males and 69 (43.4%) females, with a male : female ratio of 1.3 : 1. The patients' ages ranged between 1.4 and 54 years with a median of 15 yrs. Age at diagnosis varied between 1 and 50 yrs with a median of 5 yrs.
3.2. Disease Characteristics
Ninety-three patients (58.5%) had splenomegaly, of which 32.7% had a splenectomy, while hepatomegaly and HCV were reported in 37.7% and 11.3%, respectively. The most common disease-related complications were bone disease, facial deformity (62.3%), and osteoporosis (28.3%), and 3 of those with osteoporosis had bone fractures. Endocrinopathies were second, including growth retardation (27.8%), subclinical hypothyroidism (16.8%), cholelithiasis (13.8%), PHT (11.3%), and abnormal liver function (7.5%) (Table 1). Thrombosis, EMH, and leg ulcers were less frequent, while diabetes mellitus and heart failure were not identified. Furthermore, the probability of developing the above morbidities increased significantly with age (Figure 1).
Table 1.
Patient and disease characteristics of the current study.
| Parameter | Frequency | Number of evaluated patients | Percent |
|---|---|---|---|
| Demographic data | |||
| Age (years) | |||
| <18 | 90 | 159 | 56.6 |
| 18-35 | 55 | 159 | 34.6 |
| >35 | 14 | 159 | 8.8 |
| Gender | |||
| Male | 90 | 159 | 56.6 |
| Female | 69 | 159 | 43.4 |
| Splenectomized | 52 | 159 | 32.7 |
| Serum ferritin (μg/L) | |||
| <1000 | 122 | 159 | 76.7 |
| >1000 | 37 | 159 | 23.3 |
| Treatment | |||
| None transfused | 33 | 159 | 20.7 |
| Occasional transfusion | 75 | 159 | 47.2 |
| Regular transfusion | 51 | 159 | 32.1 |
| Iron chelation | 63 | 159 | 39.6 |
| Hydroxyurea | 75 | 159 | 47.2 |
| Complications | |||
| Facial deformity | 99 | 159 | 62.3 |
| ∗Osteoporosis | 17 | 60 | 28.3 |
| ∗∗Growth retardation | 17 | 90 | 18.9 |
| ∗∗∗Subclinical hypothyroidism | 22 | 131 | 16.8 |
| ∗∗∗∗Cholelithiasis | 19 | 137 | 13.8 |
| Pulmonary hypertension | 18 | 159 | 11.3 |
| Abnormal liver function | 12 | 159 | 7.5 |
| Thrombosis | 2 | 159 | 1.3 |
| EMH | 1 | 159 | 0.6 |
| Leg ulcer | 1 | 159 | 0.6 |
∗Osteoporosis and ∗∗∗subclinical hypothyroidism were evaluated in patients ≥ 10 years old and/or symptomatic, ∗∗growth retardation (height > 2SD below 3rd percentile for the mean age and gender) in patients ≤ 18 years, and ∗∗∗∗cholelithiasis estimated in 137 patients excluding the 22 patients that underwent cholecystectomy.
Figure 1.
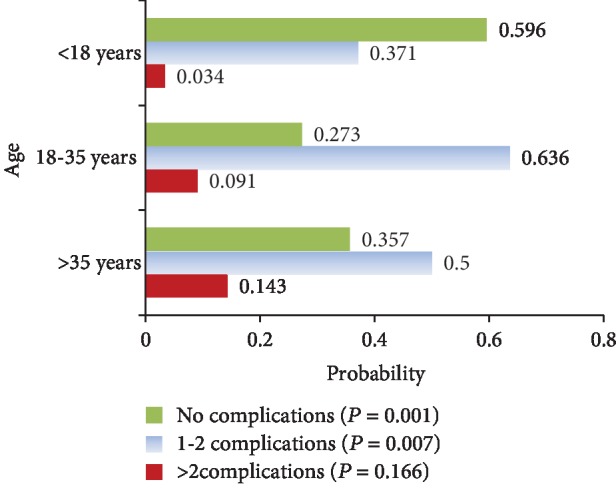
Probability of developing disease-related morbidities at different age intervals.
3.3. Transfusion History and Chelation Therapy
The median age at onset of transfusion was 4.7 years with a range between 1 and 50 yrs. Almost half of the patients (47.2%) received occasional transfusion sessions ranging from 0 to 3 per year (during severe infection, operation, or pregnancy), while just 32.1% of the patients received regular transfusion > 3/year, whereas 20.7% were never been transfused. Sixty-three patients (39.6%) received iron-chelating drugs for at least a one-year duration. Deferasirox was used in 85.7%, and deferoxamine was used in 14.3%, while 60.3% of the β-TI patients did not receive any type of chelating drugs. Hydroxyurea, on the other hand, was given to 47.2% of the patients.
3.4. Laboratory Investigations
The mean Hb at the time of enrollment was 8.9 ± 1.4 g/dL, with a range of 4.1-13.8 g/dL. The mean MCV and MCH were 72 ± 10.6 and 24.4 ± 4, respectively. The Hb F level at the time of the first presentation ranged between 4.6 and 99.5% with a mean of 65.7 ± 34.8%, while Hb A2 ranged from 0.4 to 8.4%, with a mean of 3.2 ± 2.2. Furthermore, the serum ferritin level ranged from 27 to 9882 μg/L with a mean of 853.3 ± 1192.7 (Table 2). One hundred twenty-two patients (76.7%) had a serum ferritin level < 1000 μg/L, while just 23.3% had a ferritin level ≥ 1000 μg/L. Elevated ALT ≥ 50 IU/L, on the other hand, was reported in 12 patients (7.5%), and 9 of them (75%) had a ferritin level ≥ 1000 μg/L) with a P value < 0.001, while none was HCV positive. HBV infection was not reported.
Table 2.
Laboratory investigations of 159 β-thalassemia intermedia patients in the current study.
| Laboratory tests | Mean ± SD | Range |
|---|---|---|
| Hb (g/dL) | 8.9 ± 1.4 | 4.1–13.8 |
| PCV (L/L) | 26.6 ± 4.4 | 10.5–42.7 |
| MCV (fL) | 72 ± 10.6 | 48.7–116.2 |
| MCH (pg) | 24.4 ± 4 | 15.8–38.2 |
| Hb A2 (%) | 3.2 ± 2.2 | 0.4–8.4 |
| Hb F (%) | 65.7 ± 34.8 | 4.6–99.5 |
| S. ferritin (μg/L) | 853.3 ± 1192.7 | 27–9882 |
Selected variables were studied in a logistic regression analysis to identify their role in the pathophysiology of disease-related complications, PHT, cholelithiasis, hypothyroidism, and osteoporosis (Table 3). Age ≥ 35 was an independent risk factor for cholelithiasis and hypothyroidism. Likewise, female sex was associated with an increased risk of cholelithiasis and osteoporosis. Whereas mean serum ferritin of ≥1000 μg/L was independently associated with an increased risk of osteoporosis, iron chelation therapy was protective for a multitude of other complications (PHT, cholelithiasis, hypothyroidism, and osteoporosis). Although transfusion was associated with an increased risk of osteoporosis, it was protective for cholelithiasis and hypothyroidism. Moreover, splenectomy was protective for cholelithiasis, while it was an independent risk for hypothyroidism. Finally, hydroxyurea was associated independently with increased risk of osteoporosis, though it was protective for cholelithiasis.
Table 3.
Multivariate analysis for determinants of complication rate.
| Complication/parameters | RR | 95% CI | P |
|---|---|---|---|
| PHT | |||
| Age ≥ 35 y | 0.798 | 0.13-4.64 | 0.801 |
| Splenectomy | 0.839 | 0.27-2.6 | 0.762 |
| Transfusion | 0.670 | 0.06-6.81 | 0.735 |
| Hydroxyurea | 0.600 | 0.19-1.82 | 0.369 |
| Iron chelation | 0.160 | 0.03-0.65 | 0.011 |
| Cholelithiasis | |||
| Age ≥ 35 y | 1.153 | 0.19-6.73 | 0.874 |
| Female | 1.693 | 0.53-5.34 | 0.369 |
| Splenectomy | 0.140 | 0.03-0.50 | 0.003 |
| Transfusion | 0.537 | 0.05-5.27 | 0.594 |
| Hydroxyurea | 0.179 | 0.05-0.60 | 0.006 |
| Iron chelation | 0.325 | 0.08-1.19 | 0.09 |
| Hypothyroidism | |||
| Age ≥ 35 y | 1.817 | 0.20-16.14 | 0.592 |
| Splenectomy | 2.178 | 0.70-6.75 | 0.178 |
| Transfusion | 0.251 | 0.05-1.24 | 0.091 |
| Hydroxyurea | 0.645 | 0.23-1.79 | 0.401 |
| Iron chelation | 0.552 | 0.241-1.265 | 0.149 |
| Osteoporosis | |||
| Age ≥ 35 y | 0.616 | 0.235-1.62 | 0.364 |
| Female | 4.139 | 0.87-19.58 | 0.073 |
| Ferritin ≥ 1000 μg/L | 6.86 | 1.09-42.97 | 0.040 |
| Splenectomy | 0.994 | 0.24-4.11 | 0.994 |
| Transfusion | 14.352 | 0.86-221.12 | 0.063 |
| Hydroxyurea | 9.004 | 1.67-48.41 | 0.010 |
| Iron chelation | 0.443 | 0.09-2.08 | 0.302 |
3.5. Molecular Investigations
A total of 19 different β-thalassemia mutations were determined among 159 TI patients; 12 (63.2%) were identified by reverse hybridization with IVS-II-I (G>A) as the most prevalent mutation, followed by IVS-I-6 (T>C) and IVS-I-110 (G>A). Other mutations were less prevalent or sporadic as shown in Table 4. On the other hand, 7 mutations were revealed by direct sequencing: CAP +1 (A>C), IVS-I-128, +20/IVS-II.745, codon 36-37, codon 82-83, IVS-II.850, and codon 127 (Table 4).
Table 4.
β-Globin gene mutations in 159 thalassemia intermedia patients in the current study.
| β-Thalassemia mutations | Frequency of allele | Percent |
|---|---|---|
| Very mild β++ | ||
| (1) CAP +1 | 2 | (0.6) |
| (2) -101 | 1 | (0.3) |
| Mild β+ | ||
| (1) IVS I.6 | 74 | (23.3) |
| (2) IVS I.128 | 8 | (2.5) |
| (3) IVS II.745 | 3 | (0.9) |
| Severe β+ | ||
| (1) IVS I.110 | 16 | (5) |
| (2) IVS I.5 | 1 | (0.3) |
| β 0 | ||
| (1) IVS II.1 | 150 | (47.2) |
| (2) IVS I.1 | 15 | (4.7) |
| (3) Cod 8 | 15 | (4.7) |
| (4) Cod 8/9 | 11 | (3.5) |
| (5) Cod 5 | 6 | (1.9) |
| (6) Cod 39 | 3 | (0.9) |
| (7) Cod 36/37 | 2 | (0.6) |
| (8) Cod 44 | 2 | (0.6) |
| (9) Cod 82-83 | 1 | (0.3) |
| (10) IVS II.850 | 1 | (0.3) |
| (11) Cod 15 | 1 | (0.3) |
| Wild | 5 | (1.6) |
| Dominant like β-thalassemia Cod 127 | 1 | (0.3) |
The current work had determined 37 genotypes; the most frequent was a homozygous IVS-II-1, followed by a homozygous IVS-I-6 and IVS-II-1/IVS-I-6 (Table 5). Twenty-four families (21.1%) had inherited a homozygous or compound heterozygous β+/β+ mutation, while 20.2% had the β0/β+ genotype and 56.1% had the β0/β0 genotype. Among our patients, 52.8% were the results of a consanguineous marriage. Moreover, those who inherited homozygous mutations were significantly associated with consanguinity (45.3% of patients) with a P value < 0.001.
Table 5.
Genotypes of 159 thalassemia intermedia patients in the current study.
| Genotypes | Frequency | Percent |
|---|---|---|
| β 0/β0 | ||
| (1) IVS II.1/IVS II.1 | 57 | 35.9 |
| (2) IVS II.1/Cod 8/9 | 5 | 3.2 |
| (3) IVS II.1/IVS I.1 | 4 | 2.5 |
| (4) IVS II.1/Cod 8 | 4 | 2.5 |
| (5) Cod 8/9/Cod 8 | 3 | 1.9 |
| (6) Cod 8/Cod 8 | 2 | 1.3 |
| (7) IVS I.1/Cod 8 | 2 | 1.3 |
| (8) IVS II.1/Cod 5 | 2 | 1.3 |
| (9) IVS II.1/Cod 36/37 | 2 | 1.3 |
| (10) IVS II.1/Cod 39 | 2 | 1.3 |
| (11) IVS I.1/IVS I.1 | 2 | 1.3 |
| (12) Cod 5/Cod 5 | 1 | 0.6 |
| (13) IVS II.1/Cod 15 | 1 | 0.6 |
| (14) IVS II.1/Cod 82-83 | 1 | 0.6 |
| (15) Cod 44/Cod 44 | 1 | 0.6 |
| β +/β+ | ||
| (1) IVS I.6/IVS I.6 | 30 | 18.9 |
| (2) IVS I.110/IVS I.110 | 4 | 2.5 |
| (3) IVS I.128/IVS I.128 | 2 | 1.3 |
| (4) IVS I.110/IVS I.128 | 1 | 0.6 |
| (5) IVS I.6/IVS II.745 | 1 | 0.6 |
| β 0/β+ | ||
| (1) IVS II.1/IVS I.6 | 7 | 4.4 |
| (2) IVS II.1/IVS I.110 | 4 | 2.5 |
| (3) Cod 5/IVS I.128 | 2 | 1.3 |
| (4) IVS I.1/IVS I.6 | 2 | 1.3 |
| (5) Cod 8/9/IVS I.110 | 2 | 1.3 |
| (6) IVS I.1/CAP +1 | 2 | 1.3 |
| (7) Cod 8/IVS I.6 | 1 | 0.6 |
| (8) IVS II.1/IVS I.5 | 1 | 0.6 |
| (9) Cod 8/9/IVS I.6 | 1 | 0.6 |
| (10) Cod 39/IVS I.6 | 1 | 0.6 |
| (11) IVS II.850/IVS I.6 | 1 | 0.6 |
| (12) Cod 8/IVS I.110 | 1 | 0.6 |
| (13) IVS II.1/IVS I.128 | 1 | 0.6 |
| (14) IVS I.1/-101 | 1 | 0.6 |
| β 0/wt IVS II.1/wt (αααanti3.7) | 2 | 1.25 |
| β +/wt +20, IVS II.745/wt | 2 | 1.25 |
| Dominant like β-thalassemia Cod 127/wt |
1 | 0.6 |
4. Discussion
The current study had evaluated the largest cohort of β-TI patients in Iraq, including 159 patients from 114 families to investigate the molecular defect of β-TI in an attempt to improve the available diagnostic tests and current management protocols.
The inheritance of β+ mutation (β+/β+ or β0/β+) had contributed to 40.9% of β-TI genotypes (41.2% of the families), a result that is lower than figures from other parts of Kurdistan, Iraq (Dohuk, 54.9%; Erbil, 60.2%) [19, 20], while it is approaching figures reported from those of Baghdad (49%) [21]. Our results are also comparable to some extent to studies from India and Iran, where the inheritance of β+ alleles was not responsible for the majority of the milder β-thal phenotypes [22]. β0-Thal mutation, on the other hand, amounted to 56.1% of the β-TI family genotype, which highly suggests the coinheritance of ameliorating factors including α-thalassemia and/or coinheritance of a single nucleotide polymorphism (SNP) in the three major quantitative trait loci (QTLs) for the continuous synthesis of Hb F to modify the α : β chain imbalance and to reduce ineffective hemopoiesis [23]. This has been particularly emphasized in studies from Iran, where β0 mutations are more frequent in β-TI patients and XmnI polymorphism was found to be a considerable ameliorating factor [24, 25].
The relative frequency and distribution of different mutations vary in different geographical locations, and in the current study, the three most common mutations identified were IVS II-1 (47.2%), IVS I-6 (23.3%), and IVS I-110 (5%). This high prevalence of IVS II-1, a Mediterranean β0-thal mutation, is in accordance with an earlier study performed on couples attending a Sulaymaniyah premarital screening clinic, where the IVS-II-1 mutation was the most common (25.2%), although IVS I-6 amounted to 4.1% of the β-thal-detected mutation [26]. The later high frequency is rather expected as this study enrolled β-TI patients. Furthermore, the sequence of the three common mutations in this study agreed with that of Baghdad, Central Iraq [21], whereas previous studies from other parts of Kurdistan, Iraq revealed that IVS I-6 was the most frequent β-thal mutation, detected at around 33% [19, 20], which probably explains their higher reported frequency of β+β+ or β0β+ genotypes. Moreover, our results are consistent with that seen in different studies from Iran [24, 25, 27], including Iranian Kurds [28]. In contrast, IVS-I-6 was the most frequent β-thal mutation in Turkey [29], Lebanon [11], Egypt [30], Cyprus [22], and Italy [31].
Three new β-thal mutations were identified for the first time in Iraq, although they have been reported by earlier studies from other parts of the world. The first was the +20 (C>T) β+ mutation in the 5′ untranslated region (5′ UTR) coinherited with another β+ mutation IVS II-745 (C>T), most probably in transposition leading to the β-TI phenotype; otherwise, coinheritance of +20 in the cis position would have resulted in a β-thal minor phenotype [32]. The second was CAP +1 (A>C), a silent β++ Asian-Indian mutation that interacts with IVS I-1 (β0-thal mutation) resulting in a mild clinical phenotype. Finally, a dominant-like β-thalassemia, Hb Houston (codon 127 A>G), was also reported for the first time in Iraq (a missense mutation at codon 127 in exon III that produces an unstable hemoglobin and thalassemia intermedia phenotype in the heterozygous state) [33]. Only 5 patients (3.1%) had inherited a single β-thal mutation, namely, +20, IVS II-745/wt (2 patients), codon 127/wt (one patient), and IVS II-1/wt αααanti3.7 in another 2 patients. The later genotype had resulted in increased α : β imbalance, hemolysis, and ineffective erythropoiesis [34]. We have noticed that patients with β0/β+ and β0/β0 genotypes were diagnosed at an earlier age and transfused earlier in comparison to other reported genotypes. Also, Hb F levels were the highest while Hb A2 levels were the lowest in β0/β0 patients. Likewise, β0/β+ and β0/β0 patients showed a higher frequency of PHT in comparison to other genotypes (Table 6).
Table 6.
Relations of different parameters in all genotype groups.
| Parameters |
β
0/β0 N (%) |
β
0/β+ N (%) |
β
+/β+ N (%) |
Dominant Hb Houston/wt |
β
+
/wt N (%) |
β
0
/wt N (%) |
P value |
|---|---|---|---|---|---|---|---|
| Age at diagnosis | |||||||
| Mean ± SD | 6.7 ± 5.6 | 5.2 ± 2.6 | 8.6 ± 9.7 | 10 | 20.0 ± 2.8 | 12.5 ± 3.5 | 0.019 |
| Age at first transfusion | |||||||
| Mean ± SD | 5.3 ± 4.4 | 4.8 ± 2.9 | 7.6 ± 10.8 | 8 | 19.0 ± 2.8 | 12.5 ± 3.5 | 0.020 |
| Transfusion | |||||||
| No | 19 (21.3) | 5 (18.5) | 9 (23.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0.900 |
| Yes | 70 (78.7) | 22 (81.5) | 29 (76.3) | 1 (100.0) | 2 (100.0) | 2 (100.0) | |
| Iron chelation | |||||||
| No | 48 (53.9) | 19 (70.4) | 28 (73.7) | 0 (0.0) | 0 (0.0) | 1 (50.0) | 0.071 |
| Yes | 41 (46.1) | 8 (29.6) | 10 (26.3) | 1 (100.0) | 2 (100.0) | 1 (50.0) | |
| Hb A2 % | |||||||
| <3.5 | 52 (92.9) | 14 (73.7) | 3 (10.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | <0.0001 |
| ≥3.5 | 4 (7.1) | 5 (26.3) | 26 (89.7) | 1 (100.0) | 1 (100.0) | 2 (100.0) | |
| Hb F % | |||||||
| <50 | 4 (6.9) | 3 (15.8) | 26 (89.7) | 1 (100.0) | 1 (100.0) | 2 (100.0) | <0.0001 |
| ≥50 | 54 (93.1) | 16 (84.2) | 3 (10.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Splenectomy | |||||||
| No | 55 (62.5) | 21 (77.8) | 27 (73.0) | 0 (0.0) | 0 (0.0) | 2 (100.0) | 0.079 |
| Yes | 33 (37.5) | 6 (22.2) | 10 (27.0) | 1 (100.0) | 2 (100.0) | 0 (0.0) | |
| PHT | |||||||
| No | 77 (86.5) | 23 (85.2) | 37 (97.4) | 0 (0.00 | 2 (100.0) | 2 (100.0) | 0.036 |
| Yes | 12 (13.5) | 4 (14.8) | 1 (2.6) | 1 (100.0) | 0 (0.0) | 0 (0.0) | |
| Hepatomegaly | |||||||
| No | 51 (57.3) | 18 (66.7) | 27 (71.1) | 1 (100.0) | 1 (50.0) | 1 (50.0) | 0.659 |
| Yes | 38 (42.7) | 9 (33.3) | 11 (28.9) | 0 (0.0) | 1 (50.0) | 1 (50.0) | |
| Hepatitis C | |||||||
| No | 77 (86.5) | 25 (92.1) | 35 (92.1) | 0 (0.0) | 2 (100.0) | 2 (100.0) | 0.087 |
| Yes | 12 (13.5) | 2 (7.4) | 3 (7.9) | 1 (100.0) | 0 (0.0) | 0 (0.0) | |
| Osteoporosis | |||||||
| No | 21 (61.8) | 10 (90.9) | 10(83.3) | 1 (100.0) | 1 (50.0) | — | 0.257 |
| Yes | 13 (38.2) | 1 (9.1) | 2 (16.7) | 0 (0.0) | 1 (50.0) | — | |
| Cholelithiasis | |||||||
| No | 78 (87.6) | 25 (92.6) | 32 (84.2) | 1 (100.0) | 2 (100.0) | 2 (100.0) | 0.882 |
| Yes | 11 (12.4) | 2 (7.4) | 6 (15.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Hypothyroidism | |||||||
| No | 67 (87.0) | 18 (72.0) | 31 (86.1) | 1 (100.0) | 2 (100.0) | 2 (100.0) | 0.349 |
| Yes | 10 (13.0) | 7 (28.0) | 5 (13.9) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Growth retardation | |||||||
| No | 31 (60.8) | 17 (89.5) | 16 (84.2) | — | — | 1 (100.0) | 0.323 |
| Yes | 20 (39.2) | 2 (10.5) | 3 (15.8) | — | — | 0 (0.0) | |
| ALT | |||||||
| ≤50 | 81 (91.0) | 24 (88.9) | 37 (97.4) | 1 (100.0) | 2 (100.0) | 2 (100.0) | 0.779 |
| >50 | 8 (9.0) | 3 (11.1) | 1 (2.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| S. ferritin | |||||||
| <1000 | 65 (74.7) | 22 (81.5) | 31 (83.) | 1 (100.0) | 1 (50.0) | 2 (100.0) | 0.663 |
| ≥1000 | 22 (25.3) | 5 (18.5) | 6 (16.2) | 0 (0.0) | 1 (50.0) | 0 (0.0) | |
| Facial deformity | |||||||
| No | 22 (24.7) | 13 (48.1) | 22 (57.9) | 1 (100.0) | 0 (0.0) | 2 (100.0) | 0.001 |
| Yes | 67 (75.3) | 14 (51.9) | 16 (42.1) | 0 (0.0) | 2 (100.0) | 0 (0.0) |
When we compared our results with practices in TI management outlined by one of the first landmark studies “OPTIMAL CARE study” [35], which highlighted the management approaches in several Mediterranean and Middle Eastern countries (Table 7), we found that our patients were less regularly transfused with lesser numbers of splenectomized and chelated patients, whereas hydroxyurea therapy was prominently implemented at our center. Furthermore, our patients were more regularly transfused than other thalassemia centers in Iraq (Dohuk and Basra) [19, 36], Iran [37], and Italy [38] with a much higher rate of splenectomy than recent reports from Sri Lanka (12%) [39] and Qatar (7%) [40]. Despite the lower frequency of chelation therapy used in this study, 76.6% of our patients had their ferritin value < 1000 μg/L, a figure that is higher than the reported value of the OPTIMAL CARE study [35] and those from previous figures from Iraq [19, 36] (Table 7). Such a finding could be attributed to the use of hydroxyurea among a higher proportion of our patients, which had improved the α : β chain imbalance and subsequently improved the ineffective hemopoiesis [12].
Table 7.
Comparison of some parameters, treatment options, and disease-related complications between the current study and some other related studies.
| Parameter | Current study (n = 159) % | Lebanon (n = 73) 2000 [28] | Optimal care (n = 584) 2010 [34] | Iran (n = 153) 2011 [36] | Basra (n = 80) 2013 [35] | Dohuk (n = 74) 2014 [17] | Italy (n = 70) 2014 [37] | Sri Lanka (n = 50) 2019 [38] |
|---|---|---|---|---|---|---|---|---|
| Splenectomized | 32.7 | 59 | 55.7 | 46.9 | ∗ | 23.0 | 49 | 12.0 |
| Serum ferritin (μg/L) | ||||||||
| <1000 | 76.7 | ∗ | 64.4 | ∗ | 55 | 67.6 | ∗ | ∗ |
| >1000 | 23.3 | 35.6 | 45 | 32.4 | ||||
| Treatment | ||||||||
| Transfusion | ||||||||
| Never transfused | 20.8 | 28.8 | 23.8 | 27.5 | 21.2 | 32.4 | 53 | 4.0 |
| Occasionally | 47.2 | 58.9 | 24.5 | 45.5 | 78.8 | 51.4 | 34 | 42 |
| Regularly | 32.1 | 12.3 | 51.7 | 27 | 16.2 | 13 | 44 | |
| Iron chelation | 39.6 | ∗ | 47.5 | ∗ | ∗ | 14.9 | 56 | 46 |
| Hydroxyurea | 47.2 | ∗ | 34.6 | ∗ | ∗ | 2.7 | 16 | ∗ |
| Complications | ||||||||
| Facial deformity | 62.3 | 44 | ∗ | ∗ | ∗ | 73 | ∗ | ∗ |
| Osteoporosis | 28.3 | ∗ | 22.9 | 53 | 30.0 | ∗ | 49 | ∗ |
| Growth retardation (height < 3rd percentile) | 27.8 | ∗ | ∗ | ∗ | 42.5 | 31.3 | ∗ | 26.7 |
| Subclinical hypothyroidism | 16.8 | ∗ | ∗ | ∗ | ∗ | ∗ | 33.3 | ∗ |
| Cholelithiasis | 13.8 | ∗ | 17.1 | 9.8 | 2.5 | ∗ | ∗ | 10.0 |
| PHT | 11.3 | ∗ | 11 | 23.5 | 5.0 | 20.4 | ∗ | 33.3 |
| Abnormal liver function | 7.5 | ∗ | 9.8 | 29.3 | 13.5 | ∗ | ∗ | |
∗Not mentioned in the study.
β-TI patients has had many clinical complications reported in this study despite their independence from frequent transfusion. The pathophysiology is multifactorial due to the interaction of ineffective erythropoiesis, iron overload, and chronic tissue hypoxia (chronic hemolytic anemia) [7]. The discrepancy in the rates of disease morbidities reported in various studies (Table 7) had resulted from the difference in management approaches (limited or regular transfusion, the extent of chelation, more frequent splenectomy, or use of fetal hemoglobin modulating therapy) in thalassemia centers in Iraq and worldwide [12].
This study had reported bone abnormalities as the most prevalent morbidity. Less transfusion, ineffective erythropoiesis, and eventually bone marrow expansion were directly implicated in addition to splenectomy, as well as low fetal hemoglobin [7, 38]. Osteoporosis, a well-recognized complication in β-TI, was correlated in this study with iron overload, female gender, transfusion, and hydroxyurea as risk factors, while lower rates were observed in patients on iron chelation therapy, in accordance with previous studies [7, 35]. Hydroxyurea was shown to be a risk factor rather than a protective factor in our study, though the clinical benefits of hydroxyurea therapy are still to be systematically established [12].
Limited data are available on endocrinopathies in β-TI which are reported to be less commonly seen in TI in comparison to β-thalassemia major [38]. The incidence of subclinical hypothyroidism, an iron-overload-related morbidity, was in accordance with previous studies from Egypt (16.7%) [41] and from Iran (19%) [42], while it was much lower than an Italian figure of 33.3% [38]. Regarding growth retardation, 27.8% had a height > 2SDbelow the 3rd percentile for the mean age and gender, a figure that is lower than previous reports from Iraq [19, 36] and Sri Lanka [39] (Table 7). This might be attributed to younger age patients and a lower proportion of splenectomized TI patients in this study as intact spleen might be a reservoir of excess body iron in addition to its scavenging effect on iron-free fraction, including nontransferrin bound iron [43].
Splenectomy in TI had been considered a significant risk factor for many disease-related complications, in particular, thrombosis and PHT [44]. In our group, two patients (1.9%) had thrombosis, and both were splenectomized. The later low incidence may be explained by younger age and the possibility of nondocumented asymptomatic cases of thrombosis. Furthermore, chronic thromboembolism in splenectomized TI patients was linked with a high frequency of PHT [45]. This study did not reveal an increased risk of PHT in splenectomized patients. Our 18 patients were diagnosed with PHT; half were splenectomized, 8/9 patients were ≤35 years, 4 used either HU therapy or chelation therapy, while another 4 used both. All of the above parameters are proposed protective factors for the development of PHT in splenectomized patients [7]. In light of the above morbidities associated with splenectomy and despite the advantage of splenectomy in maintaining higher Hb levels, clinical practice is gradually shifting to restrict indications, growth retardation, hypersplenism with symptomatic leukopenia, and/or thrombocytopenia or symptomatic hypersplenism [7, 46].
Disease-related morbidities among our β-TI patients potentially increased after adulthood, a result which had been suggested by a few previous studies [7, 35]. Such finding justifies an earlier intervention to curtail substantial long-term sequelae.
The current study lacked the estimation of different genetic modifiers including concomitant α-thalassemia and polymorphism at QTL due to lack of financial support. In addition, ferritin measurement was used instead of liver R2 magnetic imaging to determine the iron burden and PASP was estimated through Doppler echocardiography instead of right heart catheterization to diagnose PHT which may falsely raise the rate of positive results. Finally, hypogonadism was not evaluated at the time of enrollment.
5. Conclusions
The current study, the largest from Iraq and Kurdistan on β-TI, revealed that β0-thal mutation underlies 56.1% of families with milder thalassemia phenotypes in the northeastern part of Iraq, and accordingly, it is prudent to assess genetic modifiers of disease severity in β0 homozygous or compound heterozygous. Furthermore, just over half of our β-TI patients suffered bone diseases as a result of limited transfusion and chelation therapy. The shift towards earlier and more regular blood transfusion with iron chelation therapy to avoid serious long-term sequelae is prudent in this aspect. Finally, the impact of age on morbidities needs to be better addressed.
Acknowledgments
The authors would like to extend their deepest gratitude and appreciation to the staff of Thalassemia and Congenital Blood Disorders Center in the General Medical Hospital in Sulaymaniyah, for their contribution to this study. We are also grateful to our participants.
Data Availability
The data supporting this clinical study are from previously reported studies and datasets, which have been cited. The processed data are available as PDFs once required from the corresponding author.
Conflicts of Interest
The authors declare no competing interests, which might be perceived as posing a conflict or bias.
References
- 1.Hamamy H. A., Al-Allawi N. A. S. Epidemiological profile of common haemoglobinopathies in Arab countries. Journal of Community Genetics. 2013;4(2):147–167. doi: 10.1007/s12687-012-0127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karim M. F., Ismail M., Hasan A. M., Shekhar H. U. Hematological and biochemical status of beta-thalassemia major patients in Bangladesh: a comparative analysis. International Journal of Hematology-Oncology and Stem Cell Research. 2016;10(1):7–12. [PMC free article] [PubMed] [Google Scholar]
- 3.De Sanctis V. β-thalassemia distribution in the old world: an ancient disease seen from a historical standpoint. Mediterranean Journal of Hematology and Infectious Diseases. 2017;9(1, article e2017018) doi: 10.4084/mjhid.2017.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kountouris P., Kousiappa I., Papasavva T., et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: a 20-year retrospective study. Scientific Reports. 2016;6(1, article 26371) doi: 10.1038/srep26371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farashi S., Bayat N., Faramarzi Garous N., et al. Interaction of an α-globin gene triplication with β-globin gene mutations in Iranian patients with β-thalassemia intermedia. Hemoglobin. 2015;39(3):201–206. doi: 10.3109/03630269.2015.1027914. [DOI] [PubMed] [Google Scholar]
- 6.Taher A., Isma'eel H., Cappellini M. D. Thalassemia intermedia: revisited. Blood Cells, Molecules, and Diseases. 2006;37(1):12–20. doi: 10.1016/j.bcmd.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Sleiman J., Tarhini A., Bou-Fakhredin R., Saliba A. N., Cappellini M. D., Taher A. T. Non-transfusion-dependent thalassemia: an update on complications and management. International Journal of Molecular Sciences. 2018;19(1):p. 182. doi: 10.3390/ijms19010182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inati A., Noureldine M. H. A., Mansour A., Abbas H. A. Endocrine and bone complications in β-thalassemia intermedia: current understanding and treatment. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/813098.813098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jalal S. D., Al-Allawi N. A., AH F., NH A. Prevalence of haemoglobinopathies in Sulaimani–Iraq. Duhok Medical Journal. 2008;2(1):71–79. [Google Scholar]
- 10.Borgna-Pignatti C., Marsella M., Zanforlin N. The natural history of thalassemia intermedia. Annals of the New York Academy of Sciences. 2010;1202(1):214–220. doi: 10.1111/j.1749-6632.2010.05550.x. [DOI] [PubMed] [Google Scholar]
- 11.Qatanani M., Taher A., Koussa S., et al. β-thalassaemia intermedia in Lebanon. European Journal of Haematology. 2000;64(4):237–244. doi: 10.1034/j.1600-0609.2000.90087.x. [DOI] [PubMed] [Google Scholar]
- 12.Taher A., Vichinsky E., Musallam K., Cappellini M.-D., Viprakasit V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT) Nicosia, Cyprus: Thalassaemia International Federation; 2013. [PubMed] [Google Scholar]
- 13.Baskin H. J., Cobin R. H., Duick D. S., et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocrine Practice. 2002;8(6):457–469. [PubMed] [Google Scholar]
- 14.Sambrook J., Russell D. W. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 15.Oron-Karni V., Filon D., Oppenheim A., Rund D. Rapid detection of the common mediterranean α-globin deletions/rearrangements using PCR. American Journal of Hematology. 1998;58(4):306–310. doi: 10.1002/(sici)1096-8652(199808)58:4<306::aid-ajh10>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Greiner S., Jud A., Aurich M., et al. Reliability of noninvasive assessment of systolic pulmonary artery pressure by Doppler echocardiography compared to right heart catheterization: analysis in a large patient population. Journal of the American Heart Association. 2014;3(4, article e001103) doi: 10.1161/jaha.114.001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanis J. A. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group. Osteoporosis International. 1994;4(6):368–381. doi: 10.1007/BF01622200. [DOI] [PubMed] [Google Scholar]
- 18.World Health Organization. Assessment of Fracture Risk and Its Application to Screening for Postmenopausal Osteoporosis: Report of a WHO Study Group [Meeting Held in Rome from 22 to 25 June 1992] Geneva: World Health Organization; 1994. [PubMed] [Google Scholar]
- 19.Al-Allawi N. A. S., Jalal S. D., Mohammad A. M., Omer S. Q., Markous R. S. D. β-Thalassemia Intermedia in Northern Iraq: A Single Center Experience. BioMed Research International. 2014;2014:9. doi: 10.1155/2014/262853.262853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamoon R. P., Al-Allawi N. A. S., Cappellini M. D., Di Pierro E., Brancaleoni V., Granata F. Molecular basis of β-thalassemia intermedia in Erbil Province of Iraqi Kurdistan. Hemoglobin. 2015;39(3):178–183. doi: 10.3109/03630269.2015.1032415. [DOI] [PubMed] [Google Scholar]
- 21.Al-Allawi N. A. S., Puehringer H., Raheem R. A., Oberkanins C. Genetic modifiers in β-thalassemia intermedia: a study on 102 Iraqi Arab patients. Genetic Testing and Molecular Biomarkers. 2015;19(5):242–247. doi: 10.1089/gtmb.2014.0310. [DOI] [PubMed] [Google Scholar]
- 22.Verma I. C., Kleanthous M., Saxena R., et al. Multicenter study of the molecular basis of thalassemia intermedia in different ethnic populations. Hemoglobin. 2007;31(4):439–452. doi: 10.1080/03630260701641245. [DOI] [PubMed] [Google Scholar]
- 23.Taher A. T., Musallam K. M., Cappellini M. D. Thalassaemia intermedia: an update. Mediterranean Journal of Hematology and Infectious Diseases. 2009;1(1, article e2009004) doi: 10.4084/mjhid.2009.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arab A., Karimipoor M., Rajabi A., Hamid M., Arjmandi S., Zeinali S. Molecular characterization of β-thalassemia intermedia: a report from Iran. Molecular Biology Reports. 2011;38(7):4321–4326. doi: 10.1007/s11033-010-0557-5. [DOI] [PubMed] [Google Scholar]
- 25.Maryami F., Azarkeivan A., Fallah M. S., Zeinali S. A large cohort study of genotype and phenotype correlations of beta-thalassemia in Iranian population. International Journal of Hematology-Oncology and Stem Cell Research. 2015;9(4):198–202. [PMC free article] [PubMed] [Google Scholar]
- 26.Jalal S. D., al-Allawi N. A. S., Bayat N., Imanian H., Najmabadi H., Faraj A. β-thalassemia mutations in the Kurdish population of northeastern Iraq. Hemoglobin. 2010;34(5):469–476. doi: 10.3109/01676830.2010.513591. [DOI] [PubMed] [Google Scholar]
- 27.Rahimi Z. Genetic epidemiology, Hematological and Clinical Features of Hemoglobinopathies in Iran. BioMed Research International. 2013;2013:10. doi: 10.1155/2013/803487.803487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haghi M., Khorshidi S., Hosseinpour Feizi M. A., Pouladi N., Hosseinpour Feizi A. A. β-Thalassemia mutations in the Iranian Kurdish population of Kurdistan and West Azerbaijan Provinces. Hemoglobin. 2009;33(2):109–114. doi: 10.1080/03630260902862020. [DOI] [PubMed] [Google Scholar]
- 29.Altay Ç., Gürgey A. β-Thalassemia intermedia in Turkey. Annals of the New York Academy of Sciences. 1990;612:81–89. doi: 10.1111/j.1749-6632.1990.tb24293.x. [DOI] [PubMed] [Google Scholar]
- 30.Elmezayen A. D., Kotb S. M., Sadek N. A., Abdalla E. M. β-Globin mutations in Egyptian patients with β-thalassemia. Laboratory Medicine. 2015;46(1):8–13. doi: 10.1309/LM1AYKG6VE8MLPHG. [DOI] [PubMed] [Google Scholar]
- 31.Camaschella C., Mazza U., Roetto A., et al. Genetic interactions in thalassemia intermedia: analysis of β-mutations, α-genotype, γ-promoters, and β-LCR hypersensitive sites 2 and 4 in Italian patients. American Journal of Hematology. 1995;48(2):82–87. doi: 10.1002/ajh.2830480203. [DOI] [PubMed] [Google Scholar]
- 32.Ropero P., González F. A., Cela E., et al. Association inCisof the mutations +20 (C>T) in the 5′ untranslated region and IVS-II-745 (C>G) on the β-globin gene. Hemoglobin. 2013;37(2):112–118. doi: 10.3109/03630269.2013.766620. [DOI] [PubMed] [Google Scholar]
- 33.Kazazian H. H., Jr., Dowling C. E., Hurwitz R. L., Coleman M., Stopeck A., Adams J. G., 3rd. Dominant thalassemia-like phenotypes associated with mutations in exon 3 of the beta-globin gene. Blood. 1992;79(11):3014–3018. [PubMed] [Google Scholar]
- 34.Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Reviews. 2012;26:S12–S15. doi: 10.1016/S0268-960X(12)70005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taher A. T., Musallam K. M., Karimi M., et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892. doi: 10.1182/blood-2009-09-243154. [DOI] [PubMed] [Google Scholar]
- 36.Abdulwahid D. A., Hassan M.’a. K. β- and α-thalassemia intermedia in Basra, Southern Iraq. Hemoglobin. 2013;37(6):553–563. doi: 10.3109/03630269.2013.825841. [DOI] [PubMed] [Google Scholar]
- 37.Rafsanjani K. A., Mafi N., Tafreshi R. I. Complications of β-thalassemia intermedia in Iran during 1996–2010 (single-center study) Pediatric Hematology and Oncology. 2011;28(6):497–508. doi: 10.3109/08880018.2011.572144. [DOI] [PubMed] [Google Scholar]
- 38.Baldini M., Marcon A., Cassin R., et al. Beta-thalassaemia intermedia: evaluation of endocrine and bone complications. BioMed Research International. 2014;2014:5. doi: 10.1155/2014/174581.174581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perera S., Allen A., Silva I., et al. Genotype-phenotype association analysis identifies the role of α globin genes in modulating disease severity of β thalassaemia intermedia in Sri Lanka. Scientific Reports. 2019;9(1, article 10116) doi: 10.1038/s41598-019-46674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yassin M. A., Soliman A. T., De Sanctis V., Yassin K. S., Abdulla M. A. Final height and endocrine complications in patients with β-thalassemia intermedia: our experience in non-transfused versus infrequently transfused patients and correlations with liver iron content. Mediterranean Journal of Hematology and Infectious Diseases. 2019;11(1, article e2019026) doi: 10.4084/MJHID.2019.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdel-Razek A.-R. A., Abdel-Salam A., El-Sonbaty M. M., Youness E. R. Study of thyroid function in Egyptian children with β-thalassemia major and β-thalassemia intermedia. Journal of the Egyptian Public Health Association. 2013;88(3):148–152. doi: 10.1097/01.EPX.0000436490.10201.28. [DOI] [PubMed] [Google Scholar]
- 42.Karamifar H., Karimi M., Amirhakimi G. H., Badiei M. Endocrine function in thalassemia intermedia. International Journal of Biomedical Sciences. 2006;2(3):236–240. [PMC free article] [PubMed] [Google Scholar]
- 43.Tavazzi D., Duca L., Graziadei G., Comino A., Fiorelli G., Cappellini M. D. Membrane-bound iron contributes to oxidative damage of β-thalassaemia intermedia erythrocytes. British Journal of Haematology. 2001;112(1):48–50. doi: 10.1046/j.1365-2141.2001.02482.x. [DOI] [PubMed] [Google Scholar]
- 44.Taher A., Isma’eel H., Mehio G., et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thrombosis and Haemostasis. 2006;96(4):488–491. [PubMed] [Google Scholar]
- 45.Atichartakarn V., Likittanasombat K., Chuncharunee S., et al. Pulmonary arterial hypertension in previously splenectomized patients with β-thalassemic disorders. International Journal of Hematology. 2003;78(2):139–145. doi: 10.1007/BF02983382. [DOI] [PubMed] [Google Scholar]
- 46.Taher A. T., Musallam K. M., Cappellini M. D., Weatherall D. J. Optimal management of β thalassaemia intermedia. British Journal of Haematology. 2011;152(5):512–523. doi: 10.1111/j.1365-2141.2010.08486.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting this clinical study are from previously reported studies and datasets, which have been cited. The processed data are available as PDFs once required from the corresponding author.