

Abstract
Microbiome plays an important role during the tobacco aging process which was an indispensable link in the production and processing of cigarettes. However, the structure and functions of microbiome have not been clarified during the tobacco aging process. In this study, 16S rDNA and ITS amplicon sequencing techniques were used to analyze the core microbiome of 15 tobacco samples from five different aging stages. The whole bacterial microbiome was classified into 29 microbial phyla and 132 orders. Enterobacteriales (63%), Pseudomonadales (16%), Sphingomonadales (8%), Xanthomonadales (4%), Burkholderiales (4%), Rhizobiales (3%), and Bacillales (2%) comprised the core bacterial microbiome. The whole fungal microbiome was classified into five microbial phyla and 52 orders. Incertae_sedis_Eurotiomycetes (27%), Wallemiales (25%), Sporidiobolales (17%), Capnodiales (5%), Eurotiales (2%), an unclassified Ascomycota (12%), and an unidentified Eurotiomycetes (4%) comprised the core fungal microbiome. FAPROTAX function prediction suggested that the core microbiome has a substantial potential for the carbon cycle, nitrate metabolism, aromatic compound degradation, chitinolysis, cellulolysis, and xylanolysis, but simultaneously, the core microbiome is also a source of human pathogens. The dynamics of the bacterial community were primarily determined by the total nitrogen in tobacco leaves during the aging process, while those of the fungal microbiome were primarily determined by total organic carbon. This study indicated that the core microbiome activities may play an important role in regulating the loss of carbon organic compounds and enhancing the secondary metabolites during tobacco leaves aging process.
Keywords: community structure, core microbiome, function prediction, nutritional components, tobacco aging
This study indicated that the core microbiome was involved in the ecological activities of tobacco leaves during the aging process and may play an important role in regulating the loss of carbon and nitrogen organic compounds and enhancing the secondary metabolites of tobacco leaves.
![]()
1. INTRODUCTION
Flue‐cured tobacco is one of the important cash crops in the world, which requires high quality and security. However, unaged tobacco is unsuitable for cigarette products because it contains strong green miscellaneous gas and produces irritating smoke with an unacceptably harsh flavor (Zhao et al., 2007). In the cigarette industry, a process called natural aging is often used to improve the quality of flue‐cured tobacco. Tobacco aging is a process that changes the physical and chemical characteristics and significantly improves the aroma and flavor of tobacco leaves under certain temperature and humidity conditions. This process typically requires 24 to 30 months (Dixon, Darkis, Wolf, & Hall, 1936), following microbial (or/and enzymatic) actions and other chemical interactions in the leaves (Huang et al., 2010). Microbial activities throughout the aging process are likely to play important roles in improving the tobacco quality during the process.
The bacteria and fungi that inhabit tobacco and tobacco products are varied. The familiar bacterial populations in tobacco include Bacillus, Pseudomonas, Sphingomonas, Stenotrophomonas, Erwinia, Pantoea, Lactococcus, Clostridium, Staphylococcus, Enterobacter, and Paenibacillus (Chopyk et al., 2017; English, Bell, & Berger, 1967; Huang et al., 2010; Su et al., 2011; Ye et al., 2017; Zhao et al., 2007; Zhu et al., 2001 ). The fungal groups include Aspergillus, Penicillium, Phoma, Alternaria, Chaetomium, Cladosporium, Rhizopus, Fusarium, Trichoderma, Monographella, Rhodotorula, and Sporidiobolales (Chen, Wu, et al., 2018; Chen, Li, et al., 2018; Villemur, Lacasse, & Morin, 2009; Welty, 1972; Yang et al., 2008; Zhang et al., 2018). Microorganisms can not only act on the tobacco aging process by their own existence but also through metabolites and enzymes that promote the transformation of biomacromolecules in tobacco leaves, thereby reducing the harmful components, such as nicotine and nitrosamines, increasing the fragrance and improving the quality characteristics (Golias, Dumsday, Stanley, & Pamment, 2000; Liu et al., 2015; Maldonado‐Robledo, Rodriguez‐Bustamante, Sanchez‐Contreras, Rodriguez‐Sanoja, & Sanchez, 2003; Wei et al., 2014).
Tobacco aging is a complex ecological process, and the physical and chemical properties change significantly in tobacco leaves during the process. For example, the total organic acid and volatiles gradually increase, while the nicotine, volatile alkali, total sugar, reducing sugar, and pH values decrease (Sun et al., 2011). After farm curing, tobacco is usually shipped to different countries or regions where it is aged for several months or years, while the storage conditions may vary substantially depending on the location. The water content of tobacco is generally required to be in a controlled range from 10% to 13% during the aging process, but this is usually affected by the humidity of the storage location (Villemur et al., 2009). Moreover, microbial communities in nature are usually complex, and this complexity is exacerbated by interactions among the ecosystem members (Butler & O'Dwyer, 2018). These facts render the analysis of the microbial community structure and function challenging. In addition, the challenge will be further amplified when one considers transient or allochthonous community members, and even though they contribute little to the functionality of the system, they are still detected by the current sensitive molecular approaches (Astudillo‐García et al., 2017). An approach that only considers persistent (and sometimes abundant) members of a microbial community (Shade & Handelsman, 2012) provides a possible way to address these issues.
The core microbiome is considered to be a key component of the basic function of the holobionts, which is enriched, selected, and inherited through evolutionary processes (Lemanceau, Blouin, Muller, & Moënne‐Loccoz, 2017). The core microbiomes are commonly used to reveal microbial species that are closely related to the health, growth, and physiology of the host plant and are currently widely used to describe the key species in human, soil, plant, lake, and wastewater treatment system (Chen, Wu, et al., 2018; Chen, Li, et al., 2018; Ji, Parks, Edwards, & Pruden, 2015; Turnbaugh et al., 2007; Zarraonaindia et al., 2015). However, the core microbiomes and their function in the tobacco aging process still remain to be elucidated. In this study, we used an Illumina Hi‐Seq platform to sequence the 16S rRNA genes and ITS1 amplicons from DNA prepared from 15 tobacco samples that had been stored in three tobacco warehouses of China Tobacco Guizhou Industrial Co., Ltd. We analyzed the core fungal and bacterial microbiomes in tobacco leaves during the aging process. In addition, we also predicted the bacterial functional profiles of the core microbiome in the tobacco leaves using Functional Annotation of Prokaryotic Taxa (FAPROTAX) software.
2. MATERIALS AND METHODS
2.1. Tobacco storing environment and sampling
The tobacco leaves samples were collected from Baoshan, Yunnan Province, in China. The grade of all the samples was C3F, and the variety of Nicotiana tabacum was Yun 87. After harvesting and re‐drying, the tobacco leaves were immediately packed into a carton of 1 m3 (density: 200 kg/carton) and transported to Guiyang (GY), Tanchang (TC), and Ziyun (ZY) in Guizhou Province for their respective storage and aging. Each location contained at least three cartons of tobacco, which were stored in a warehouse at ambient conditions where the temperature was between 6°C and 32°C, and the humidity was between 49% and 94%. Sampling began in the first month after all the samples were in storage and continued every six months. A total of 15 samples were obtained with sampling and grouping numbers described respectively as follows: month_0 (GY‐0, TC‐0, ZY‐0), month_6 (GY‐6, TC‐6, ZY‐6), month_12 (GY‐12, TC‐12, ZY‐12), month_18 (GY‐18, TC‐18, ZY‐18), and month_24 (GY‐24, TC‐24, ZY‐24). A five‐point sampling method was used, and five hundred grams of tobacco was sampled at a minimum of 15 cm below the top surface of each tobacco carton (Sun et al., 2011; Villemur et al., 2009). Samples taken from three cartons in the same storage were mixed into one portion with each weighing 1.5 kg. To avoid contamination, sterile gloves and sterile bags were used to sample and seal the tobacco. The tobacco leaves from three locations were sampled within two days and then transported to the laboratory of Guizhou University for storage at −20ºC. In the following experiments, 500 g of each sample was used to determine the chemical components, 500 g was used for microbial analysis, and the remaining samples were used for unavoidable emergencies.
2.2. Determination of the nutritional components in tobacco leaves
The nutritional components of the tobacco leaves were determined using the analytical methods of the quality of tobacco chemistry described by Li and Mao (2007). The total organic carbon (TOC) was determined by potassium dichromate–sulfuric acid oxidation at high temperature. The total nitrogen (TN) was determined using concentrated sulfuric acid and the hydrogen peroxide digestion–semi‐microdistillation method. The water‐soluble sugar (WSS) was determined using the arsenomolybdate method. Nicotine was measured using alkaline distillation–ultraviolet spectrophotometry. The starch was determined using the dilute acid–molybdate colorimetric method. The protein was determined using the copper hydroxide precipitation method. The cellulose was treated with acid‐base alcohol ether.
2.3. DNA extraction, PCR amplification, and sequencing
The total microorganisms were collected as previously described by Zhao et al. (2007) and Su et al. (2011). The DNA was extracted using the E.Z.N.A.® Soil Kit (Omega Bio‐Tek), and the concentration of DNA and purity were monitored by electrophoresis on 1.0% (w/v) agarose gels. The primers 515F (5′‐GTGCCAGCMGCCGCGGTAA‐3′) and 806R(5′‐GGACTACHVGGGTWTCTAAT‐3′)were used to amplify the V4 region of the bacterial 16S rRNA gene. The primers ITS5‐1737F (5′‐GGAAGTAAAAGTCGTAACAAGG‐3′) and ITS2‐2043R(5′‐GCTGCGTTCTTCATCGATGC‐3′)were used to amplify the ITS1 fragment of the fungal ITS gene. All the PCR reactions were performed using Phusion® High‐Fidelity PCR Master Mix (New England Biolabs). After quantification, qualification, pooling, and purification, the PCR amplification products were sequenced on an Illumina Hi‐Seq 2,500 platform at Novogene.
2.4. Bioinformatics analysis
Raw reads from the original DNA fragments were merged and quality‐filtered using FLASH (Magoč & Salzberg, 2011) and QIIME (Caporaso et al., 2010). The chimeric sequences were subsequently identified and removed using a UCHIME algorithm (Edgar, Haas, Clemente, Quince, & Knight, 2011), and the effective reads were finally obtained. Operational taxonomic units (OTUs) were clustered with a ≥97% similarity cutoff using UParse (Edgar, 2013). The bacterial gene sequences were annotated with taxonomic information using the RDP classifier (Wang, Garrity, Tiedje, & Cole, 2007) against the Greengene database (DeSantis et al., 2006). The OTU taxonomic information of the fungi was obtained by aligning each representative sequence against the Unite ITS database (Kõljalg et al., 2013). OTUs abundance information was normalized using a standard of sequence number corresponding to the sample with the least sequences.
We submitted the OTU biome file to the Metagenomics Core Microbiome Exploration Tool (MetaCoMET), which is a web platform for the discovery and visualization of the core microbiome, and then, we selected the parameters and persistence methods to obtain the core microbiome in tobacco leaves during the aging process (Wang, Xu, Gu, & Coleman‐Derr, 2016; Wang, Lu, Shi, & Xu, 2016). The FAPROTAX was applied to predict the function of the bacterial community in the tobacco leaves. FAPROTAX had been constructed by integrating multiple culturable prokaryotic bacteria whose pronuclear functions had been reported and contained more than 7,600 functional annotations for more than 4,600 species (Louca, Parfrey, & Doebeli, 2016). Furthermore, the relationships between the core microbiome and chemical components were evaluated with CANOCO 5 during the tobacco aging according to the redundancy analysis (RDA).
3. RESULTS
3.1. Nutritional characterization of the tobacco leaves
Carbohydrates and nitrogenous compounds are important components of tobacco leaves and vital sources of nutrition for the tobacco microorganisms. Therefore, we determined the content of the TOC, TN, WSS, nicotine, protein, starch, and cellulose in the tobacco leaves. As shown in Figure 1, the content of the TOC changed significantly during the aging process (p = .002) with a decrease from 43.43 ± 0.13% at month_6 to 42.16 ± 0.14% at month_18. The WSS appeared to decrease by 2.63% during the whole process. The contents of the TN and nicotine were relatively low in the tobacco leaves (1.90%–2.33% and 1.69%–2.18%, respectively) and decreased noticeably after six months of aging. There were slight changes in protein, starch, and cellulose, but none were statistically significant.
Figure 1.
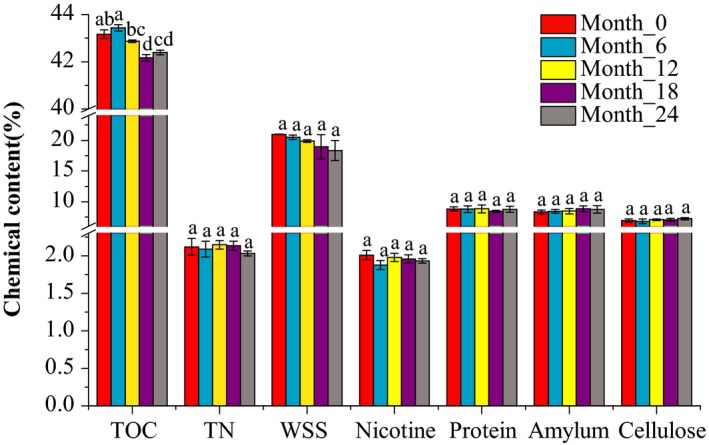
The changes in the chemical components in tobacco leaves during aging. The different small letters show the changes at the .05 level to be significant (p < .05)
3.2. Bacterial community composition
The bacterial 16S rRNA sequencing resulted in 1,242,236 raw reads with 1,191,887 of them passing the quality and length filtering parameters. A total of 2,652 OTUs were obtained by clustering with 97% similarity, including 29 phyla, 66 classes, 134 orders, 264 families, and 490 genera. At the phylum level, Proteobacteria had an absolute advantage, accounting for 84.1%–95.2% of the total bacterial communities. In addition, Firmicutes and Actinobacteria also accounted for 1.5%–10.9% and 1.1%–2.5%, respectively. The relative abundance of Proteobacteria decreased from 95.2% to 84.1% after 12 months, while Firmicutes increased from 1.5% to 10.9% (Figure A1a in Appendix 1). At the class level, Gammaproteobacteria (52.9%–84.8%), Alphaproteobacteria (8.3%–27.4%), Bacilli (1.2%–10.2%), and Betaproteobacteria (1.5%–5.9%) were the primary bacterial groups. The relative abundance of Gammaproteobacteria, Alphaproteobacteria, and Clostridia changed significantly during the aging process (Figure A1b in Appendix 1). At the order level, the bacterial microbiome was dominated by Enterobacteriales (19.9%–69.8%), Pseudomonadales (8.8%–21.5%), Sphingomonadales (3.9%–12.0%), Xanthomonadales (2.2%–10.2%), Rhizobiales (1.9%–7.2%), Rickettsiales (1.6%–6.8%), Bacillales (1.1%–9.8%), and Burkholderiales (1.4%–5.6%) (Figure 2a). The relative abundance of Enterobacteriales, Pseudomonadales, Sphingomonadales, and Rhizobiales changed significantly during the aging process. Enterobacteriales increased from 34.2% at the beginning to 69.8% at month_12 and then decreased to 19.9% at month_24. In contrast, the relative abundance of Pseudomonadales decreased from 18.4% to 8.8% during the first 18 months and then increased to 21.8% at month_24. During the first 12 months, Sphingomonadales and Rhizobiales decreased from 11.5% and 7.2% to 4.0% and 1.9%, respectively, but recovered to 12.0% and 5.1% by 24 months. In addition, the relative abundance of Xanthomonadales increased from 2.2% at month_6 to 10.2% at month_24. Bacillales increased from 1.1% at month_12 to 9.8% at month_24.
Figure 2.
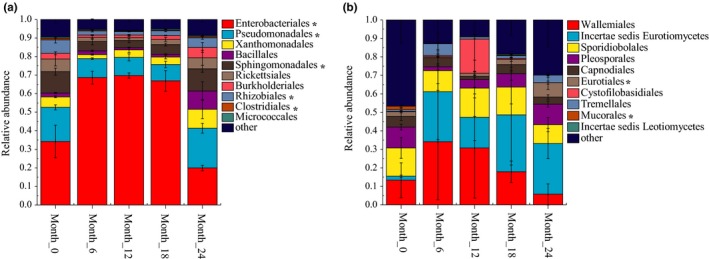
The composition and dynamics of the dominant bacterial (a) and fungal (b) community at the order level. Groups with a relative abundance of <.01 were assigned as “others.” * indicates a significant change (p value < .05) in relative abundance of the OTU of tobacco during aging process
In addition, Enterobacteriaceae (19.9%–69.8%), Pseudomonadaceae (7.0%–17.0%), Sphingomonadaceae (3.7%–11.4%), Xanthomonadaceae (1.9%–9.7%), Moraxellaceae (1.0%–4.5%), Bacillaceae (0.6%–6.9%), Comamonadaceae (0.4%–3.7%), and Methylobacteriaceae (0.6%–3.6%) were the predominant families (Figure A1c in Appendix 1). Pseudomonas (6.9%–15.0%), Sphingomonas (3.1%–10.1%), Stenotrophomonas (0.9%–8.2%), Acinetobacter (1.0%–4.4%), Bacillus (0.6%–6.7%), and Methylobacterium (0.5%–3.4%) were the dominant genera (Figure A1d in Appendix 1). Enterobacteriaceae (Gammaproteobacteria and Enterobacteriales) were the primary members of the Proteobacteria, and their relative abundance was significantly reduced after 18 months, while the Firmicutes represented by Bacillaceae (Bacilli, Bacillales, Bacillus) were significantly increased.
3.3. Fungal community composition
A total of 1,209,369 paired‐end reads were obtained from the 15 tobacco samples using Illumina sequence analysis. After filtering, optimizing, and removing the low‐quality sequences, the remaining 1,173,560 effective sequences were used for the subsequent analyses. A total of 656 OTUs were clustered at 97% similarity and classified into five phyla, 22 classes, 52 orders, 103 families, and 168 genera. Ascomycota (33.3%–79.4%) and Basidiomycota (20.4%–66.5%) were the dominant fungal phyla at the phylum level. The relative abundance of Basidiomycota gradually increased during the first 12 months but decreased gradually later in the process. The Ascomycota fungi reacted in an opposite manner. However, the proportion of other fungal phyla, including Zygomycota, Chytridiomycota, and Glomeromycota, was less than 1% (Figure A2a in Appendix 1). Eurotiomycetes (13.0%–43.9%), Wallemiomycetes (5.8%–34.2%), Microbotryomycetes (10.3%–15.7%), Dothideomycetes (6.9%–18.8%), and Tremellomycetes (1.2%–19.8%) were the dominant fungal classes (Figure 2b). At the order level, the fungal community was dominated by Incertae_sedis_Eurotiomycetes (2.2%–30.8%), Wallemiales (5.8%–34.2%), Sporidiobolales (10.3%–15.7%), and an unclassified Ascomycota (Figure 2b). In addition, Pleosporales, Cystofilobasidiales, Tremellales, Capnodiales, Eurotiales, and two unclassified Ascomycota also exceeded 1%. The relative abundance of Incertae_sedis_Eurotiomycetes rose rapidly from 2.2% to 27.0% in the first six months and declined slightly during the subsequent processing period but exceeded 16%. The relative abundance of Wallemiales decreased gradually after 6 months (34.2% to 5.8%). Sporidiobolales also showed a gradual decline after 12 months (15.7% to 5.8%).
In addition, Monascaceae (2.2%–30.8%), Wallemiaceae (13.3%–34.2%), Incertae sedis Sporidiobolales (10.3%–15.7%), Mycosphaerellaceae (1.7%–5.7%), Incertae sedis Pleosporales (0.8%–5.6%), Pleosporaceae (1.1%–5.9%), and Incertae sedis Tremellales (1.0%–6.4%) were the dominant fungal families (Figure A2c in Appendix 1). Additionally, Xeromyces (1.9%–30.7%), Wallemia (5.8%–34.1%), Rhodotorula (10.1%–15.5%), Cercospora (1.1%–5.2%), Alternaria (1.1%–5.6%), Cryptococcus (1.0%–6.4%), and Aspergillus (0.4%–7.4%) were the predominant genera at the genus level (Figure A2d in Appendix 1). Xeromyces increased from 1.9% to 27.0% during the first 6 months and was the primary fungal population (abundance over 27%) after 12 months. The relative abundance of Wallemiaceae and Wallemia decreased from 34.2% and 34.1% to 5.8% and 5.8% after 6 months, respectively, while that of Trichocomaceae and Aspergillus increased significantly from 0.5% and 0.4% to 7.8% and 7.4%, respectively. Wallemia (Wallemiomycetes, Wallemiales, Wallemiaceae), Rhodotorula (Microbotryomycetes, Sporidiobolales, Incertae sedis Sporidiobolales), and Cryptococcus (Tremellomycetes, Tremellales, Incertae sedis Tremellales) are members of the Basidiomycota. However, their relative abundance gradually decreased during late aging. Xeromyces (Eurotiomycetes, Incertae sedis Eurotiomycetes, Monascaceae), Aspergillus (Eurotiomycetes, Eurotiales, Trichocomaceae), and Alternaria (Dothideomycetes, Pleosporales, Pleosporaceae) are members of the Ascomycota, and their relative abundance gradually decreased during late aging.
3.4. Determination of the core bacterial microbiome in tobacco leaves during aging
We used the persistence method to identify the OTUs present across 15 tobacco samples and determine the core bacterial microbiome in tobacco leaves during the aging process. This core bacterial microbiome contained 38 OTUs and corresponded to 83.7% of the whole bacterial microbiome. Enterobacteriales was by far the most common and comprised up to 63% of the core bacterial microbiome in tobacco leaves at the order level (Figure 3). However, Pseudomonadales, Sphingomonadales, Xanthomonadales, Burkholderiales, Rhizobiales, and Bacillales also contributed to 16%, 8%, 4%, 4%, 3%, and 2%, respectively. At the genus level (Table 1), an unclassified Enterobacteriales, Pseudomonas, Sphingomonas, Stenotrophomonas, Acinetobacter, Methylobacterium, and an unclassified Burkholderiales occupied 63%, 14%, 7%, 4%, 3%, 2%, and 2% of the core bacteria microbiome, respectively. In addition, Comamonas, Bacillus, and Staphylococcus exceeded 1%.
Figure 3.
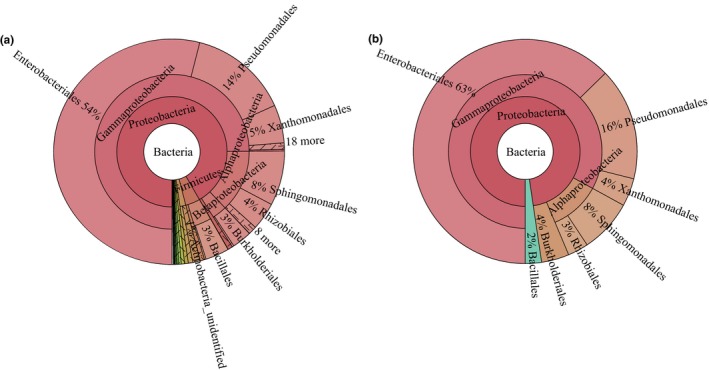
Taxonomic composition of the whole (a) and core (b) bacterial microbiome from tobacco leaves using MetaCoMET. Circles from inside to outside represent the community composition of the bacteria at different classification levels of kingdom, phylum, class, and order, respectively. The size of the fan represents the relative proportion of the annotation results of different bacterial OTU
Table 1.
The dominant bacterial genera (≥1%) within the core bacterial microbiome of tobacco during aging
| Number | Taxonomy | Proportion (%) |
|---|---|---|
| 1 | k_Bacteria; p_Proteobacteria; c_Gammaproteobacteria; o_Enterobacteriales; f_Enterobacteriaceae; g_unclassified | 63 |
| 2 | k_Bacteria; p_Proteobacteria; c_Gammaproteobacteria; o_Pseudomonadales; f_Pseudomonadaceae; g_Pseudomonas | 14 |
| 3 | k_Bacteria; p_Proteobacteria; c_Alphaproteobacteria; o_Sphingomonadales; f_Sphingomonadaceae; g_Sphingomonas | 7 |
| 4 | k_Bacteria; p_Proteobacteria; c_Gammaproteobacteria; o_Xanthomonadales; f_Xanthomonadaceae; g_Stenotrophomonas | 4 |
| 5 | k_Bacteria; p_Proteobacteria; c_Gammaproteobacteria; o_Pseudomonadales; f_Moraxellaceae; g_Acinetobacter | 3 |
| 6 | k_Bacteria; p_Proteobacteria; c_Alphaproteobacteria; o_Rhizobiales; f_Methylobacteriaceae; g_Methylobacterium | 2 |
| 7 | k_Bacteria; p_Proteobacteria; c_Betaproteobacteria; o_Burkholderiales; f_Alcaligenaceae; g_unclassified | 2 |
| 8 | k_Bacteria; p_Proteobacteria; c_Betaproteobacteria; o_Burkholderiales; f_Comamonadaceae; g_Comamonas | 1 |
| 9 | k_Bacteria; p_Firmicutes; c_Bacilli; o_Bacillales; f_Bacillaceae; g_Bacillus | 1 |
| 10 | k_Bacteria; p_Firmicutes; c_Bacilli; o_Bacillales; f_Staphylococcaceae; g_Staphylococcus | 1 |
3.5. Determination of the core fungal microbiome in tobacco leaves during aging
We interpreted the core fungal microbiome for tobacco leaves using the same persistence approach. The core fungal microbiome of the tobacco leaves contained seven OTUs and contributed to 78% of the total fungal microbiome abundance in the tobacco leaves during the aging process. At the order level (Figure 4), Incertae_sedis_Eurotiomycetes (27%), Wallemiales (25%), Sporidiobolales (17%), and an unclassified Ascomycota (12%) were the primary dominant fungal orders. Capnodiales, Eurotiales, and an unidentified Eurotiomycetes comprised 5%, 2%, and 4%, respectively. Xeromyces (27%), Wallemia (25%), a genus of Ascomycota (22%), and Rhodotorula (17%) were the dominant genera in the core fungal microbiome in the tobacco leaves during the aging process, whereas Cercospora, Aspergillus, and a genus of Eurotiomycetes also comprised 5%, 4%, and 2%, respectively (Table 2).
Figure 4.
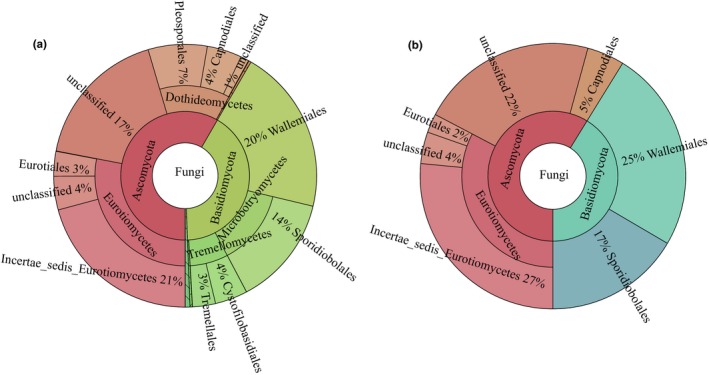
Taxonomic composition of the whole (a) and core (b) fungal microbiome from tobacco leaves using MetaCoMET. Circles from inside to outside represent the community composition of the fungi at different classification levels of kingdom, phylum, class, and order, respectively. The size of the fan represents the relative proportion of annotation results of different fungal OTU
Table 2.
The dominant fungal genera (≥1%) within the core fungal microbiome of tobacco during aging
| Number | Taxonomy | Proportion (%) |
|---|---|---|
| 1 | k_Fungi; p_Ascomycota; c_Eurotiomycetes; o_Incertae_sedis_Eurotiomycetes; f_Monascaceae; g_Xeromyces | 27 |
| 2 | k_Fungi; p_Basidiomycota; c_Wallemiomycetes; o_Wallemiales; f_Wallemiaceae; g_Wallemia | 25 |
| 3 | k_Fungi; p_Ascomycota; c_unclassified; o_unclassified; f_unclassified; g_unclassified | 22 |
| 4 | k_Fungi; p_Basidiomycota; c_Microbotryomycetes; o_Sporidiobolales; f_Incertae_sedis_Sporidiobolales; g_Rhodotorula | 17 |
| 5 | k_Fungi; p_Ascomycota; c_Dothideomycetes; o_Capnodiales; f_Mycosphaerellaceae; g_Cercospora | 5 |
| 6 | k_Fungi; p_Ascomycota; c_Eurotiomycetes; o_unclassified; f_unclassified; g_unclassified | 4 |
| 7 | k_Fungi; p_Ascomycota; c_Eurotiomycetes; o_Eurotiales; f_Trichocomaceae; g_Aspergillus | 2 |
3.6. Functional prediction of the 16S genes using FAPROTAX
The 16S sequencing results were selected for additional analysis of their functional prediction because the abundance of bacterial communities obtained from tobacco leaves was far greater than that of the fungal communities. Functional annotation of the OTUs was conducted using FAPROTAX. A total of 68 functional assignments for 703 OTUs (26.5% of the total OTUs) were obtained. Of these, 71.7% and 54.8% OTUs were associated with chemoheterotrophy and aerobic chemoheterotrophy, respectively, indicating that the microorganisms primarily obtained their nutrition by decomposing organic matter in the tobacco leaves. Figure 5a shows that the functional groups, such as denitrification, nitrate denitrification, nitrite denitrification, photoheterotrophy, phototrophy, methanol oxidation, methylotrophy, and methanol oxidation, were the most abundant at the initiation of aging but gradually decreased with the increase in the aging time. Functions, such as hydrocarbon degradation, fermentation, and predation or exoparasitism, were the most abundant at six months, and their proportion was gradually reduced during the subsequent processing. Sulfate respiration and sulfite respiration and aromatic compound degradation accounted for the highest proportion at 12 months. At 24 months, chitinolysis, invertebrate parasites, and animal parasites or symbionts were significant. In addition, the nitrate respiration, nitrate reduction, and nitrogen respiration associated with the nitrogen cycle gradually increased after 12 months, and this change could be related to the accumulation of tobacco‐specific nitrosamines during tobacco aging. In short, with the increase in the aging time, the functional abundance related to carbon metabolism gradually decreased, and nitrate action and parasitism or symbiosis gradually increased after 12 months.
Figure 5.
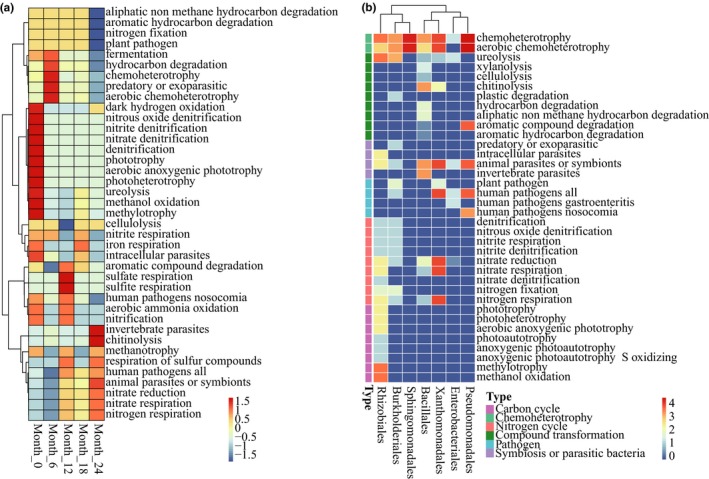
Functional composition of the bacterial community in tobacco leaves using FAPROTAX. (a) The changes of the top predictive gene families. (b) Association of the functional groups with members of the core bacterial microbiome
Figure 5b shows the relationship between the core microbiome and the functional group. Rhizobiales played an important role in the carbon and nitrogen cycle. Burkholderiales promoted nitrogen metabolism and also promoted urea decomposition. Bacillales could effectively degrade xylitol, urea, cellulose, chitosan, aliphatic nonmethane hydrocarbons, and aromatic compounds and also participated in nitrate respiration, nitrate reduction, and nitrogen respiration. Xanthomonadales had a substantial potential for functions, including nitrate respiration, nitrate reduction, nitrogen respiration, human pathogenicity, and animal parasitism or serving as symbionts. Most Pseudomonadales were potential animal parasites or symbionts and human pathogens, but they also played an important role in the degradation of aromatic compounds.
3.7. Effects of nutrient substances on the core bacterial and fungal microbiome in tobacco leaves
To further explore the relationship between the nutritional components and microbiome in the tobacco leaves, seven factors, including the TOC, TN, WSS, nicotine, protein, starch, and cellulose, were used in the RDA analysis. As shown in Figure 6, all the chemicals explained 47.3% of the core bacterial microbiome variance and 35.5% of the core fungal microbiome variance, respectively. Among them, the TN influenced most of the core bacterial microbiome and explained 20.5% of the variation in the bacterial composition. The TOC influenced most of the core fungal microbiome and explained 11.4% of the variation of the fungal component. Therefore, we believed that the nutritional component had an important effect on the changes of microbiome in tobacco leaves during the aging process, in which TN and TOC played an important role in the change in the microbiome.
Figure 6.
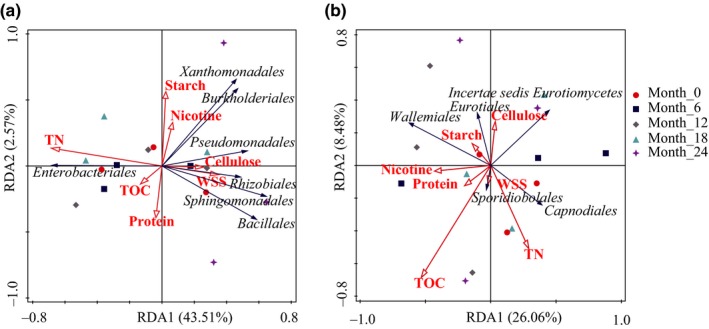
Redundancy analysis (RDA) plot showing the relationship of chemical substances to the core bacterial (a) and fungal (b) microbiomes in the tobacco leaves. TN, total nitrogen; TOC, total organic carbon; WSS, water‐soluble sugars
4. DISCUSSION
With the rapid development of molecular biology and bioinformatics, the core microbiome has attracted a substantial amount of attention in the fields of microbiology and ecology. Currently, research on the core microbiome has been successively reported. It was found that different ecosystems existed within different core microbiomes, and they played an important role in maintaining the sustainability and stability of the ecosystem. Rui et al. (2015) reported that the core microbiome consisting of Clostridium, Clostridium XI, Syntrophomonas, Cloacibacillus, Sedimentibacter, and Turicibacter could enhance the resistance against environmental stress and maintain digestion efficiency in the household biogas digesters. Acetobacter, Lactobacillus, Enhydrobacter, Lactococcus, Gluconacetobacer, Bacillus, and Staphylococcus were functional core microbiota for the production of flavors in Zhenjiang aromatic vinegar (Wang, Xu, et al., 2016; Wang, Lu, et al., 2016). The core bacterial microbiomes were composed of Gammaproteobacteria, Alphaproteobacteria, Betaproteobacteria, Sphingobacteria, Bacilli, and Actinobacteria, and the core fungal microbiome included Dothideomycetes, Leotiomycetes, and Tremellomycetes. They were important components of the plant microbiome and also a gene reservoir of secondary metabolism in Salvia miltiorrhiza seeds (Chen, Wu, et al., 2018; Chen, Li, et al., 2018). This study suggested that the core bacterial microbiome in tobacco leaves during the aging process included Enterobacteriales, Pseudomonadales, Sphingomonadales, Xanthomonadales, Burkholderiales, Rhizobiales, and Bacillales and included Incertae_sedis_Eurotiomycetes, Wallemiales, Sporidiobolales, Capnodiales, Eurotiales, an unclassified Ascomycota, and an unidentified Eurotiomycetes form the core fungal microbiome. These core microbiomes play important roles in improving the quality of leaves during tobacco aging.
A large amount of literature showed that microorganisms can reduce the harmful components, regulate the content of chemical components, and increase the aroma substances in tobacco leaf aging. For example, Pseudomonas and Aspergillus can effectively degrade nicotine. Examples include P. putida (Civilini, Domenis, Sebastianutto, & Bertoldi, 1997), Pseudomonas sp. Nic22 (Chen et al., 2008), P. stutzeri ZCJ (Zhao et al., 2012), Pseudomonas sp. HF‐1 (Wang et al., 2013), and A. oryzae 11282 (Meng, Lu, Gu, & Xiao, 2010). Wei et al. (2014) found that Bacillus amyloliquefaciens DA9 could reduce 32% of the nitrite content and 47% of the tobacco‐specific nitrosamine (TSNA) content in the tobacco leaves during the air‐curing process of burley drying. Maldonado‐Robledo et al. (2003) reported that Bacillus sp. could be responsible for the reduction of norisoprenoid to produce 7,8‐dihydro‐ß‐ionone and 7,8‐dihydro‐ß‐ionol. Burkholderiales, Rhizobiales, Xanthomonadales, Enterobacteriales, Pseudomonadales, and Sphingomonadales are important microorganisms in the degradation of furan and/or aromatic compounds in nature (Baraniecki, Aislabie, & Foght, 2002; Wierckx, Koopman, Ruijssenaars, & Winde, 2011). In addition, the Sporidiobolales could provide an important pathway for carotenoid synthesis (Frengova & Beshkova, 2009).
Microorganisms could play important roles in the transformation of compounds in tobacco leaves during the aging process. A study by Li (2015) showed that the Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Basidiomycota in tobacco leaves before and after storage at different sites were significantly correlated with chemical indicators, including total nitrogen, alkaloids, water‐soluble sugar, and total potassium and chlorine. In this study, the RDA analysis further showed that the changes in the microbial community were closely related to the changes of nutritional components in tobacco leaves (Figure 6). In addition, the FAPROTAX analysis revealed that the microorganisms had the extensive potential to decompose or degrade carbon and nitrogen compounds in tobacco leaves (Figure 5). For example, Bacillales, an identified core microbe of tobacco, was isolated the most frequently from tobacco leaves in the aging system, and it could promote the degradation of protein, starch, cellulose, and other compounds, increase the total and reducing sugars, while reducing the amounts of nicotine and total nitrogen (Chen et al., 2016; Li et al., 2015). In addition, three strains of ammonia‐degrading bacteria, including Stenotrophomonas maltophilia, Lysinibacillus fusiformis, and Brevibacillus parabrevis, were also isolated from tobacco leaves and demonstrated a strong ability to degrade organic nitrogen (Zhou et al., 2016). We hypothesized that this process is similar to the degradation of litter in forest ecosystems and that tobacco would be completely decomposed by the microorganisms if given enough time. Therefore, these results indicate that the dominant microbiomes in tobacco aging play key roles in the formation of the quality of tobacco leaves.
However, most fungi were observed in the form of molds and included Aspergillus, Penicillium, Alternaria, Cladosporium, and Chaetomium. They could be primarily responsible for the postharvest rots and deterioration of tobacco, and they could significantly reduce the quality and use value of tobacco (Welty & Vickroy, 1975). More worryingly, previous studies had shown that some microorganisms (bacteria and fungi) and/or microbial toxins (endotoxins and mycotoxins) in tobacco might lead to the development of diseases, such as chronic inflammation and cancer (Huuskonen et al., 1984; Lander, Jepsen, & Gravesen, 1988; Pauly & Paszkiewicz, 2011; Rooney, Swezey, Wicklow, & McAtee, 2005). Recently, a study of the bacterial metagenomics on four brands of cigarettes showed that there was a broad range of potentially pathogenic organisms in cigarettes, and Acinetobacter, Bacillus, Burkholderia, Clostridium, Klebsiella, Pseudomonas, and Serratia were the most notable and accounted for ≥90% of all the cigarette samples. Other pathogenic bacteria detected included Campylobacter, Enterococcus, Proteus, and Staphylococcus (Sapkota, Berger, & Vogel, 2010). FAPROTAX analysis in this study found that approximately 7.3% of the microorganisms comprised human pathogenic bacteria in tobacco during the aging process, including Rhodospirillales, Legionellales, Pseudomonadales, Xanthomonadales and Enterobacteriales. These findings strongly suggest that tobacco or cigarettes themselves could be the direct source of exposure to a substantial amount of potentially pathogenic microbes for smokers and other people exposed to secondhand smoke.
Therefore, the ability of the core microbiome to change has important value for the quality of tobacco aging and the safety of tobacco products. Coordinating the relationship between the beneficial microorganisms and harmful microorganisms will have a significant impact on tobacco producers and smokers in the production and processing of tobacco.
In addition, Bacillales (39%), Lactobacillales (21%), Pseudomonadales (17%), Enterobacteriales (6%), Sphingomonadales (4%), Xanthomonadales (3), Rhizobiales (3%), Betaproteobacteriales (2%), Flavobacteriales (2%), and Sphingobacteriales (1%) are important components of the core bacterial microbiome in tobacco leaves of the variety Zhongyan 100 (Figure A3 in Appendix 1). Additionally, Pseudomonadales (42%), Enterobacteriales (37%), Bacillales (8%), Rhizobiales (3%), Xanthomonadales (2%), Micrococcales (2%), Betaproteobacteriales (1%), Sphingomonadales (1%), and Sphingobacteriales (1%) were the dominant orders of the core bacterial microbiome in the leaves from burley tobacco(Figure A4 in Appendix 1), a type of tobacco, which was air‐cured under various temperature and relative humidity levels. The result of our study was significantly different from those of the study referenced, suggesting that there are differences in the core microbiome of tobacco leaves between different varieties. However, it was found that some microflora, such as Bacillales, Pseudomonadales, Enterobacteriales, Xanthomonadales, Sphingomonadalesetc, and Rhizobiales, seemed to be an important component of the core microbiome of each variety of tobacco, but more evidence is needed to confirm this hypothesis.
5. CONCLUSIONS
In summary, microbial diversity is an important component of the tobacco aging ecosystem, and the core microbiome drives the composition and function of the community. In this study, 38 core bacterial OTUs and seven core fungal OTUs were obtained using MetaCoMET software, respectively. The core microbiome has a significant potential application in tobacco carbon metabolism, nitrate and nitrite action, aromatic compound degradation and the decomposition of xylitol, cellulose, and butanol. However, there is a wide range of potentially pathogenic bacteria in tobacco leaves. Therefore, the coordination mechanism between beneficial regulation and the pathogenicity of these microorganisms in tobacco leaves during the aging process merits further study.
CONFLICT OF INTERESTS
None declared.
AUTHOR CONTRIBUTION
Jiaxi Zhou: Conceptualization‐Equal, Formal analysis‐Equal, Investigation‐Equal, Writing‐original draft‐Equal, Writing‐review & editing‐Equal; Lifei Yu: Formal analysis‐Equal, Writing‐review & editing‐Equal; Jian Zhang: Investigation‐Equal, Writing‐review & editing‐Equal; Xiaomin Zhang: Investigation‐Equal; Yuan Xue: Resources‐Equal; Jing Liu: Resources‐Equal; Xiao Zou: Conceptualization‐Equal, Formal analysis‐Equal, Investigation‐Equal, Project administration‐Equal, Supervision‐Equal, Writing‐review & editing‐Equal.
ETHICS STATEMENT
None required.
ACKNOWLEDGMENTS
This work was supported by the Construction Program of Biology First‐class Discipline in Guizhou (GNYL [2017]009), Science and Technology Project of the China Tobacco Guizhou Industry Limited Liability Company (Qian Yan Gong Ji [2015]8), Science and Technology Project of the Anshun Tobacco Company (201712), and Science and Technology Project of the Zunyi Tobacco Company (2018‐02).
APPENDIX 1.
Figure A1.
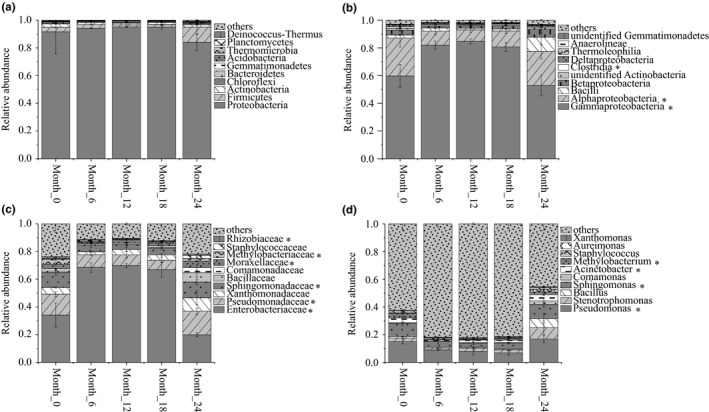
Composition and dynamics of the top 10 bacterial communities at different levels of phyla A, class B, family C and genus D. The relative abundance of other groups with the exception of the top 10 was assigned as “others”. * indicates a significant change (p value < .05) in relative abundance of the OTU of tobacco during aging process.
Figure A2.
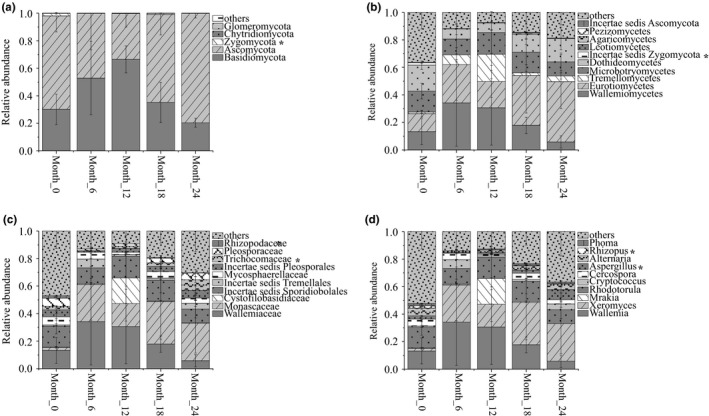
Composition and dynamics of the top 10 fungal communities at different levels of phylum A, class B, family C and genus D. The relative abundance of the other groups with the exception of the top 10 was assigned as “others”. * indicates a significant change (p value < .05) in relative abundance of the OTU of tobacco during aging process.
Figure A3.
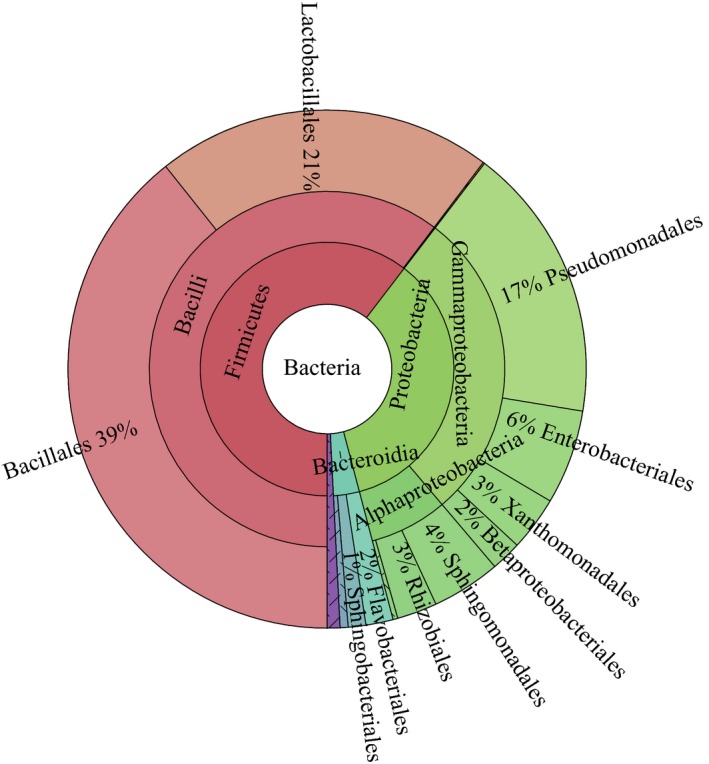
Core bacterial microbiome of tobacco leaves of the variety Zhongyan 100 and grade C3F obtained from Henan Province, China. Data were obtained from the NCBI Sequence Read Archive database (accession number: SRP082278) and had been published by Ye et al. (2017). In our study, we used a total of two tobacco samples for unaged re‐dried (after threshing and redrying, SRR4042109) and aged re‐dried (after 1 year of fermentation, SRR4041956).
Figure A4.
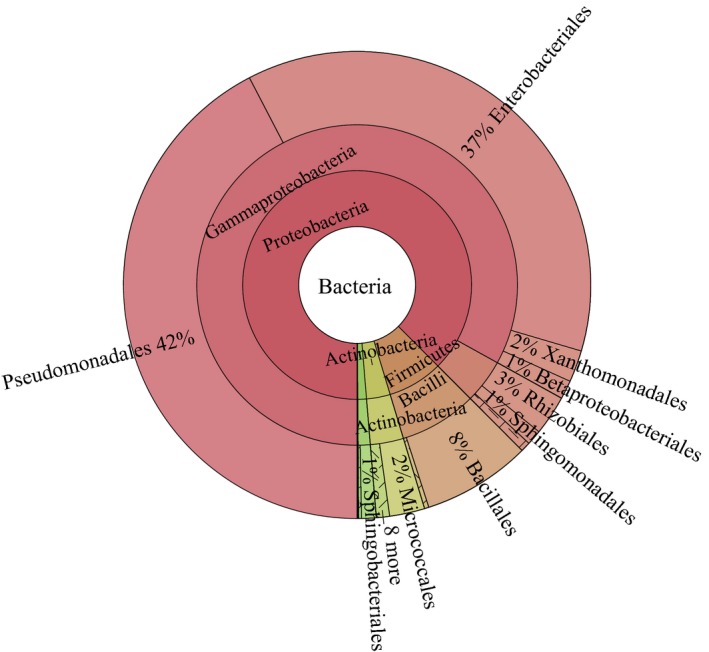
Core bacterial microbiome of the leaves from burley tobacco line TN90H collected from the University of Kentucky's Spindletop Farm. Data were obtained from the NCBI Sequence Read Archive database (accession number: SRP055959) and had been published by Law et al. (2016). In our study, we used a total of 21 tobacco samples, which were air‐cured under conditions of varying heat (15.6–30°C) and relative humidity (60%–90%) in constructed curing chambers.
Zhou J, Yu L, Zhang J, et al. Characterization of the core microbiome in tobacco leaves during aging. MicrobiologyOpen. 2020;9:e984 10.1002/mbo3.984
Funding information
Construction Program of Biology First‐class Discipline in Guizhou (GNYL [2017]009), Science and Technology Project of the China Tobacco Guizhou Industry Limited Liability Company (Qian Yan Gong Ji [2015]8), Science and Technology Project of the Anshun Tobacco Company (201712), and Science and Technology Project of the Zunyi Tobacco Company (2018‐02).
DATA AVAILABILITY STATEMENT
All data are provided in full in the results section of this paper apart from the Illumina 16S rRNA, and ITS gene sequences data, which were deposited in the NCBI Sequence Read Archive (SRA) database under the BioProject accession number PRJNA560468, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA560468.
REFERENCES
- Astudillo‐García, C. , Bell, J. J. , Webster, N. S. , Glasl, B. , Jompa, J. , Montoya, J. M. , & Taylor, M. W. (2017). Evaluating the core microbiota in complex communities: A systematic investigation. Environmental Microbiology, 19(4), 1450–1462. 10.1111/1462-2920.13647 [DOI] [PubMed] [Google Scholar]
- Baraniecki, C. A. , Aislabie, J. , & Foght, J. M. (2002). Characterization of Sphingomonas sp. Ant 17, an aromatic hydrocarbon‐degrading bacterium isolated from antarctic soil. Microbial Ecology, 43(1), 44–54. [DOI] [PubMed] [Google Scholar]
- Butler, S. , & O'Dwyer, J. P. (2018). Stability criteria for complex microbial communities. Nature Communications, 9, 2970 10.1038/s41467-018-05308-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7(5), 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Li, X. , Yang, J. , Gong, X. , Li, B. , & Zhang, K. Q. (2008). Isolation of nicotine‐degrading bacterium Pseudomonas sp. Nic22, and its potential application in tobacco processing. International Biodeterioration & Biodegradation, 62(3), 226–231. 10.1016/j.ibiod.2008.01.012 [DOI] [Google Scholar]
- Chen, H. , Wu, H. , Yan, B. , Zhao, H. , Liu, F. , Zhang, H. , … Liang, Z. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: A rich reservoir of beneficial microbes for secondary metabolism? International Journal of Molecular Sciences, 19(3), 672 10.3390/ijms19030672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Li, J. , Lin, J. , Bao, K. , Fan, J. , Zhang, R. , … He, W. (2018). High‐throughput sequencing fungal community structures in aging tobacco strips from different growing areas and stalk positions. Tobacco Science & Technology, 51(4), 12–19. [Google Scholar]
- Chen, X. , Zhang, T. , Dang, L. , Wang, X. , Chen, W. , Mo, M. , & Duan, Y. (2016). Study on improving tobacco quality by using Bacillus siamensis . Journal of Yunnan Agricultural University (Natural Science), 31(2), 322–327. [Google Scholar]
- Chopyk, J. , Chattopadhyay, S. , Kulkarni, P. , Smyth, E. M. , Hittle, L. E. , Paulson, J. N. , … Sapkota, A. R. (2017). Temporal variations in cigarette tobacco bacterial community composition and tobacco‐specific nitrosamine content are influenced by brand and storage conditions. Frontiers in Microbiology, 8, 358 10.3389/fmicb.2017.00358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civilini, M. , Domenis, C. , Sebastianutto, N. , & de Bertoldi, M. (1997). Nicotine decontamination of tobacco agro‐industrial waste and its degradation by micro‐organisms. Waste Management & Research, 15(4), 349–358. 10.1177/0734242X9701500403 [DOI] [Google Scholar]
- Desantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes: Chimera‐checked 16S rRNA gene database and workbench compatible in ARB. Applied & Environmental Microbiology, 72(7), 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, L. F. , Darkis, F. R. , Wolf, F. A. E. , & Hall, J. A. (1936). Flue‐cured tobacco, natural ageing of flue‐cured cigarette tobaccos. Industrial & Engineering Chemistry, 28(2), 180–189. [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. , Haas, B. J. , Clemente, J. C. , Quince, C. , & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16), 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, C. F. , Bell, E. J. , & Berger, A. J. (1967). Isolation of thermophiles from broadleaf tobacco and effect of pure culture inoculation on cigar aroma and mildness. Applied Microbiology, 15(1), 117–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frengova, G. I. , & Beshkova, D. M. (2009). Carotenoids from Rhodotorula and Phaffia: Yeasts of biotechnological importance. Journal of Industrial Microbiology & Biotechnology, 36(2), 163–180. 10.1007/s10295-008-0492-9 [DOI] [PubMed] [Google Scholar]
- Golias, H. , Dumsday, G. J. , Stanley, G. A. , & Pamment, N. B. (2000). Characteristics of cellulase preparations affecting the simultaneous saccharification and fermentation of cellulose to ethanol. Biotechnology Letters, 22(7), 617–621. [Google Scholar]
- Huang, J. , Yang, J. , Duan, Y. , Gu, W. , Gong, X. , Zhe, W. , … Zhang, K.‐Q. (2010). Bacterial diversities on unaged and aging flue‐cured tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 88(2), 553–562. 10.1007/s00253-010-2763-4 [DOI] [PubMed] [Google Scholar]
- Huuskonen, M. S. , Husman, K. , Järvisalo, J. , Korhonen, O. , Kotimaa, M. , Kuusela, T. , … Mäntyjärvi, R. (1984). Extrinsic allergic alveolitis in tobacco industry. British Journal of Industrial Medicine, 41(1), 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, P. , Parks, J. , Edwards, M. A. , & Pruden, A. (2015). Impact of water chemistry, pipe material and stagnation on the building plumbing microbiome. PLoS ONE, 10(10), e0141087 10.1371/journal.pone.0141087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kõljalg, U. , Nilsson, R. H. , Abarenkov, K. , Tedersoo, L. , Taylor, A. F. S. , Bahram, M. , … Larsson, K.‐H. (2013). Towards a unified paradigm for sequence‐based identification of fungi. Molecular Ecology, 22(21), 5271–5277. 10.1111/mec.12481 [DOI] [PubMed] [Google Scholar]
- Lander, F. , Jepsen, J. R. , & Gravesen, S. (1988). Allergic alveolitis and late asthmatic reaction due to molds in the tobacco industry. Allergy, 43(1), 74–76. 10.1111/j.1398-9995.1988.tb02047.x [DOI] [PubMed] [Google Scholar]
- Law, A. D. , Fisher, C. , Jack, A. , & Moe, L. A. (2016). Tobacco, microbes, and carcinogens: Correlation between tobacco cure conditions, tobacco-specific nitrosamine content, and cured leaf microbial community. Microbial Ecology, 72(1), 120–129. [DOI] [PubMed] [Google Scholar]
- Lemanceau, P. , Blouin, M. , Muller, D. , & Moënne‐Loccoz, Y. (2017). Let the core microbiota be functional. Trends in Plant Science, 22(7), 583–595. 10.1016/j.tplants.2017.04.008 [DOI] [PubMed] [Google Scholar]
- Li, C. , & Mao, S. (2007). Chemical Constituents and analysis of tobacco leaves (pp. 158–195). Kunming: Yunnan University Press. [Google Scholar]
- Li, N. , Cui, Y. , Ji, X. , Wei, Y. , Yu, H. , Ma, Y. , … Tang, X. (2015). Isolation and identification of protease‐producing bacterial strains and their effect on quality of tobacco. Chinese Journal of Microecology, 27(12), 1376–1380. [Google Scholar]
- Li, S. H. (2015). Study on the relation between microbial community structure and the quality in cigarette processing and storage process (pp. 49–72). Kunming: Yunnan University. [Google Scholar]
- Liu, J. , Ma, G. , Chen, T. , Hou, Y. , Yang, S. , Zhang, K. , & Yang, J. (2015). Nicotine‐degrading microorganisms and their potential applications. Applied Microbiology and Biotechnology, 99(9), 3775–3785. 10.1007/s00253-015-6525-1 [DOI] [PubMed] [Google Scholar]
- Louca, S. , Parfrey, L. W. , & Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science, 353(6305), 1272–1277. [DOI] [PubMed] [Google Scholar]
- Magoč, T. , & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado‐Robledo, G. , Rodriguez‐Bustamante, E. , Sanchez‐Contreras, A. , Rodriguez‐Sanoja, R. , & Sanchez, S. (2003). Production of tobacco aroma from lutein. Specific role of the microorganisms involved in the process. Applied Microbiology and Biotechnology, 62(5–6), 484–488. [DOI] [PubMed] [Google Scholar]
- Meng, X. , Lu, L. , Gu, G. , & Xiao, M. (2010). A novel pathway for nicotine degradation by Aspergillus oryzae 112822 isolated from tobacco leaves. Research in Microbiology, 161(7), 626–633. 10.1016/j.resmic.2010.05.017 [DOI] [PubMed] [Google Scholar]
- Pauly, J. L. , & Paszkiewicz, G. (2011). Cigarette smoke, bacteria, mold, microbial toxins, and chronic lung inflammation. Journal of Oncology, 2011, 819129 10.1155/2011/819129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney, A. P. , Swezey, J. L. , Wicklow, D. T. , & McAtee, M. J. (2005). Bacterial species diversity in cigarettes linked to an investigation of severe pneumonitis in US military personnel deployed in Operation Iraqi Freedom. Current Microbiology, 51(1), 46–52. 10.1007/s00284-005-4491-z [DOI] [PubMed] [Google Scholar]
- Rui, J. , Li, J. , Zhang, S. , Yan, X. , Wang, Y. , & Li, X. (2015). The core populations and co‐occurrence patterns of prokaryotic communities in household biogas digesters. Biotechnology for Biofuels, 8(1), 158 10.1186/s13068-015-0339-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota, A. R. , Berger, S. , & Vogel, T. M. (2010). Human pathogens abundant in the bacterial metagenome of cigarettes. Environmental Health Perspectives, 118(3), 351–356. 10.1289/ehp.0901201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade, A. , & Handelsman, J. (2012). Beyond the Venn diagram: The hunt for a core microbiome. Environmental Microbiology, 14(1), 4–12. 10.1111/j.1462-2920.2011.02585.x [DOI] [PubMed] [Google Scholar]
- Su, C. , Gu, W. , Zhe, W. , Zhang, K. , Duan, Y. , & Yang, J. (2011). Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Applied Microbiology and Biotechnology, 92(5), 1033–1044. 10.1007/s00253-011-3367-3 [DOI] [PubMed] [Google Scholar]
- Sun, J. , He, J. , Wu, F. , Tu, S. , Yan, T. , Si, H. , & Xia, H. (2011). Comparative analysis on chemical components and sensory quality of aging flue‐cured tobacco from four main tobacco areas of China. Agricultural Sciences in China, 10(8), 1222–1231. 10.1016/S1671-2927(11)60113-2 [DOI] [Google Scholar]
- Turnbaugh, P. J. , Ley, R. E. , Hamady, M. , Fraser‐Liggett, C. M. , Knight, R. , & Gordon, J. I. (2007). The human microbiome project. Nature, 449(7164), 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemur, R. , Lacasse, M. , & Morin, A. (2009). Monitoring the bacterial and fungal biota of eleven Tobacco grades stored at three different locations. Beiträge zur Tabakforschung International/Contributions to . Tobacco Research, 23(6), 368–376. 10.2478/cttr-2013-0871 [DOI] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16), 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Tang, L. , Yao, Y. , Wang, H. , Min, H. , & Lu, Z. (2013). Bioremediation of the tobacco waste‐contaminated soil by Pseudomonas sp. HF‐ 1: Nicotine degradation and microbial community analysis. Applied Microbiology and Biotechnology, 97(13), 6077–6088. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Xu, L. , Gu, Y. Q. , & Coleman‐Derr, D. (2016). MetaCoMET: A web platform for discovery and visualization of the core microbiome. Bioinformatics, 32(22), 3469–3470. 10.1093/bioinformatics/btw507 [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Lu, Z. , Shi, J. , & Xu, Z. (2016). Exploring flavour‐producing core microbiota in multispecies solid‐state fermentation of traditional Chinese vinegar. Scientific Reports, 6, 26818 10.1038/srep26818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X. , Deng, X. , Cai, D. , Ji, Z. , Wang, C. , Yu, J. , … Chen, S. (2014). Decreased tobacco‐specific nitrosamines by microbial treatment with Bacillus amyloliquefaciens DA9 during the air‐curing process of burley tobacco. Journal of Agricultural and Food Chemistry, 62(52), 12701–12706. [DOI] [PubMed] [Google Scholar]
- Welty, R. E. (1972). Fungi isolated from flue‐cured tobacco sold in Southeast United States, 1968–1970. Applied and Environmental Microbiology, 24(3), 518–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welty, R. E. , & Vickroy, D. G. (1975). Evaluations of cigarettes made with mold‐damaged and nondamaged flue‐cured tobacco. Beiträge zur Tabakforschung/Contributions to . Tobacco Research, 8(2), 102–106. 10.2478/cttr-2013-0363 [DOI] [Google Scholar]
- Wierckx, N. , Koopman, F. , Ruijssenaars, H. J. , & de Winde, J. H. (2011). Microbial degradation of furanic compounds: Biochemistry, genetics, and impact. Applied Microbiology and Biotechnology, 92(6), 1095–1105. 10.1007/s00253-011-3632-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Duan, Y. , Chen, C. , Li, Q. , Huang, J. , & Zhang, K. (2008). Identification and phylogenesis analysis of cultivable microorganisms on tobacco leaf surface during aging. Tobacco Science & Technology, 11, 51–55. [Google Scholar]
- Ye, J. , Yan, J. I. , Zhang, Z. , Yang, Z. , Liu, X. , Zhou, H. , … Yang, X. (2017). The effects of threshing and redrying on bacterial communities that inhabit the surface of tobacco leaves. Applied Microbiology and Biotechnology, 101(10), 4279–4287. 10.1007/s00253-017-8143-6 [DOI] [PubMed] [Google Scholar]
- Zarraonaindia, I. , Owens, S. M. , Weisenhorn, P. , West, K. , Hampton‐Marcell, J. , Lax, S. , … Gilbert, J. A. (2015). The soil microbiome influences grapevine‐associated microbiota. MBio, 6(2), e02527–e2614. 10.1128/mBio.02527-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Zhang, G. , Chen, J. , Wei, B. , Lan, B. , & Du, J. (2018). Diversity of culturable fungi on surface of tobacco leaves during aging. Tobacco Science & Technology, 51(12), 23–30. [Google Scholar]
- Zhao, L. , Zhu, C. , Gao, Y. , Wang, C. , Li, X. , Shu, M. , … Zhong, W. (2012). Nicotine degradation enhancement by Pseudomonas stutzeri ZCJ during aging process of tobacco leaves. World Journal of Microbiology and Biotechnology, 28(5), 2077–2086. 10.1007/s11274-012-1010-9 [DOI] [PubMed] [Google Scholar]
- Zhao, M. , Wang, B. , Li, F. , Qiu, L. , Li, F. , Wang, S. , & Cui, J. (2007). Analysis of bacterial communities on aging flue‐cured tobacco leaves by 16S rDNA PCR–DGGE technology. Applied Microbiology and Biotechnology, 73(6), 1435–1440. 10.1007/s00253-006-0625-x [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Zhang, X. , Hu, D. , Hui, J. , Yu, T. , & Zou, X. (2016). Isolation and identification of ammonifying bacteria in the flue‐cured tobacco and characteristics of degrading organic nitrogen. Chinese Journal of Ecology, 35(11), 3005–3011. [Google Scholar]
- Zhu, D. , Chen, R. , Chen, Z. , Zhou, Y. , Han, J. , Jin, B. , … Yu, J. (2001). The relationship between microorganisms and enzyme activities in flue‐cured tobacco during aging and fermentation. Acta Tabacaria Sinica, 7(2), 26–30. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are provided in full in the results section of this paper apart from the Illumina 16S rRNA, and ITS gene sequences data, which were deposited in the NCBI Sequence Read Archive (SRA) database under the BioProject accession number PRJNA560468, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA560468.