

Abstract
The gut microbiota plays an important role in animal health and is strongly affected by the environment. Captivity and human source food have been shown to influence drastically the gut microbiota composition and function of wild animals. Therefore, in the present study, the gut microbiota of provisioned and wild populations of limestone‐living rhesus macaques (Macaca mulatta) were compared using high‐throughput 16S rRNA sequencing and bioinformatic analyses. The results indicated that provisioned macaques had a higher microbial richness than wild macaques, but there was no significant difference in the evenness of the gut microbiota between the two populations. Provisioned macaques also showed a higher abundance of Firmicutes and a lower abundance of Bacteroidetes than wild macaques. Functional analysis revealed that wild macaques had enriched microbial pathways involved in glycan biosynthesis and metabolism, transport and catabolism, and the digestive and endocrine systems, while provisioned macaques were richer in pathways associated with signaling molecules and interaction, neurodegenerative diseases. These differences were likely due to modification of the gut microbiota of the provisioned macaques to enable the digestion of new foods.
Keywords: food provisioning, gut microbiota, limestone forest, rhesus macaque
Our results indicated that provisioned macaques had a higher microbial richness than wild macaques, but there was no significant difference in the evenness of the gut microbiota between the two populations. Provisioned macaques also showed a higher abundance of Firmicutes and a lower abundance of Bacteroidetes than wild macaques. These differences were likely due to modification of the gut microbiota of the provisioned macaques to enable the digestion of new foods.

1. INTRODUCTION
Animal intestines feature complex microbial ecosystems that play important roles in host digestive function, metabolism, immune regulation, and disease resistance (Cao et al., 2008; Marino, 2016; Murphy et al., 2010; Saxena et al., 2012; Wallace et al., 2011)—indeed, the gut microbiome is even considered the second genome in animals (Zhu, Wang, & Li, 2010). The gut microbiota is affected by multiple intrinsic and extrinsic factors. Previous studies have identified host genetics as a crucial determinant of the gut microbiota (Bonder et al., 2016; Kurilshikov, Wijmenga, Fu, & Zhernakova, 2017), which varies not only between species due to differences in the digestive tract characteristics and functions (Ley et al., 2008), but also within species, with genetically similar individuals having greater gut microbial similarities than genetically different individuals (Goodrich et al., 2016, 2014). However, recent studies have indicated that environmental factors play more crucial roles in shaping the gut microbiota than host genetics (Barelli et al., 2015; David et al., 2014; Nelson, Rogers, Carlini, & Brown, 2013; Rothschild et al., 2018; Vangay et al., 2018), with genetically unrelated individuals who live together in the long term having similar gut microbiota and relatives who live apart exhibiting significant differences in their gut microbiota (Rothschild et al., 2018).
The effects of the environment on the gut microbiota are strongly associated with diet (Amato et al., 2016; Barelli et al., 2015; Gomez et al., 2015; Scott, Gratz, Sheridan, Flint, & Duncan, 2013), with the gut microbiome of mammals exhibiting varied responses to altered dietary patterns (Angelakis et al., 2016; David et al., 2014; Muegge et al., 2011). For example, the gut microbiome of US immigrants from non‐Western countries is characterized by reduced diversity and functional losses compared with those of preimmigration and newly arrived individuals (Vangay et al., 2018). Furthermore, a study on the effects of the 1975 Japanese diet (a more diverse and healthy dietary pattern than the modern Japanese diet) on the gut microbiota revealed that after 28 days, the proportions of unclassified Lachnospiraceae, Parabacteroides spp., and unclassified Rikenellaceae had significantly decreased and the proportion of Sutterella spp. had markedly increased in the gastrointestinal tracts of 10 young adults compared with people consuming the modern Japanese diet (Kushida et al., 2018). In addition, the gut microbiota of black howler monkeys (Alouatta pigra) has been shown to vary within habitats in terms of microbial richness, diversity, and composition, most likely due to seasonal variations in diet (Amato et al., 2015).
Captivity has an important effect on the gut microbiota of mammals, with captive animals having a lower gut microbial diversity than wild animals. Captive environments differ from wild environments in terms of diet, lifestyle, and contact with other individuals, all of which could alter the structure of the gut microbiota for most mammals (McKenzie et al., 2017). In general, wild animals have a more varied diet than captive animals, which explains the difference in gut microbial diversity (McKenzie et al., 2017; Nelson et al., 2013; Uenishi et al., 2007). In addition, wild animals must adapt to seasonal variations in ecological factors, such as the availability of food resources and climate, which typically involves a change in feeding strategies (Hansen et al., 2010; Huang, Wu, Zhou, Li, & Cai, 2008; Zhou, Huang, Wei, & Huang, 2018). Therefore, diet is likely to be the main factor that causes changes in the gut microbiota in captive animals. Consequently, investigation of the gut microbiota may provide an insight into the effects of food provisioning on captive populations to help improve their management and conservation.
To explore the effect of food provisioning on the gut microbiota of rhesus macaques (Macaca mulatta), we compared the gut microbiota of food‐provisioned rhesus macaques from Guangxi Longhu Mountain Natural Reserve (hereafter “Longhu Mountain”) with that of completely wild rhesus macaques from Guangxi Chongzuo White‐headed Langur National Natural Reserve (hereafter “Chongzuo”). Limestone‐living rhesus macaques preferably feed on young leaves, which are supplemented with mature leaves when supplies of their preferred foods are sparse (Tang et al., 2016), whereas the food‐provisioned macaques in Longhu Mountain live in a natural environment but also receive a portion of their diet from reserve staff and tourists, such as corn and peanut. This food provisioning is likely leads to a high‐fat diet, which probably reduce the gut microbial diversity (Jami, White, & Mizrahi, 2014; Ley, Turnbaugh, Klein, & Gordon, 2006; Murphy et al., 2010). To date, there has been much research on the gut microbiota of rhesus macaques, including as laboratory animals for human gut microbiota research (Ardeshir et al., 2014; Martin et al., 2013; O'Sullivan et al., 2013), and the relationship between the gut microbiota and ecology of these monkeys (Cui, Wang, Yu, Ye, & Yang, 2019; Yasuda et al., 2015; Zhao et al., 2018), but the effect of partial food provisioning on the gut microbiota of rhesus macaques has not been investigated. Therefore, this research may provide advice for the management and protection of provisioned macaques.
We compared the gut microbial composition and diversity in rhesus macaques inhabiting Longhu Mountain and Chongzuo through the collection of fecal samples, as it has been shown that the composition of the gut microbiota in the large intestine is highly correlated with the composition in the feces (Yasuda et al., 2015). The gut microbiota in the fecal samples were then assessed using high‐throughput 16S rRNA sequencing. We predicted that (a) the gut microbial diversity would be lower in provisioned rhesus macaques than in wild rhesus macaques, reflecting the findings for captive versus wild mammals (McKenzie et al., 2017; Nelson et al., 2013; Uenishi et al., 2007); (b) food provisioning would cause the gut microbiota of the provisioned macaques to be richer in high‐fat diet bacteria; and (c) the gut microbiota of wild rhesus macaques would be richer in bacteria that contribute to cellulose degradation due to leaves being taken as a staple food (Tang et al., 2016).
2. MATERIALS AND METHODS
2.1. Study sites and fecal sample collection
Longhu Mountain is located in Long'an County in Guangxi Province (22°56′–23°00′N, 107°27′–107°41′E), and Chongzuo is located approximately 140 km away in Jiangzhou District and Fusui County in Guangxi Province (22°15′–22°17′N, 107°29′–107°32′E). Both reserves have limestone landscapes and vegetation that mainly comprises tropical and subtropical evergreen and deciduous forests (Yao et al., 2012; Zhang, Huang, & Huang, 2007). The habitat in Chongzuo has been fragmented by human activities, but the rhesus macaques that inhabit the reserve do not range close to or interact directly with humans. By contrast, animals in Longhu Mountain commonly range close to humans, and the reserve staff regularly feed them corn to attract tourists and also sell peanuts to tourists to feed these monkeys. Furthermore, the monkeys in Longhu Mountain also consume other foods provided by the tourists, such as bread, fruits, and drinks.
At Chongzuo, fecal samples were collected from a group of rhesus macaques comprising approximately 20 individuals at a stationary point to ensure that the fecal samples were collected from this group. At Longhu Mountain, fecal samples were collected at a stationary provisioned point at which a group of approximately 400 rhesus macaques resided. Within 20 min of defecation, samples of the fecal interiors, which do not contact the air or soil, were collected into sterile collection tubes using bamboo sticks while wearing polyethylene gloves. The samples were frozen in dry ice immediately after collection, transported to an ultralow‐temperature refrigerator in the laboratory, and stored at −80°C until DNA extraction.
In total, 35 fecal samples were collected from provisioned rhesus macaques from Longhu Mountain in October and November 2018, and 23 fecal samples were collected from wild rhesus macaques from Chongzuo in September, November, and December 2018. The age–sex classes of the sampled individuals were not known due to limitations of the current condition.
2.2. DNA extraction, 16S rRNA amplification, and sequencing
Total bacterial genomic DNA was extracted from all fecal samples using an E.Z.N.A.® soil DNA Kit (Omega Bio‐tek) according to the manufacturer's instructions. The DNA concentration and purity were determined using a NanoDrop 2000 UV–Vis spectrophotometer (Thermo Scientific), and DNA quality was assessed by 1% agarose gel electrophoresis. The V3–V4 hypervariable region of the 16S rRNA gene was amplified by polymerase chain reaction (PCR) (GeneAmp 9700; ABI) using the universal bacterial primers (338F, 5′‐ACTCCTACGGGAGGCAGCAG‐3′; 806R, 5′‐GGACTACHVGGGTWTCTAAT‐3′) (Mori et al., 2014). The initial PCR was conducted using TransGen AP221‐02: TransStart® Fastpfu DNA Polymerase with 20 μl of reaction mixture containing 10 ng of template DNA, 4 μl of 5 × FastPfu Buffer, 2 μl of 2.5 mM deoxyribonucleotide triphosphates (dNTPs), 0.8 μl of each primer (5 μM), and 0.2 μl of bovine serum albumin. The PCR conditions included initial denaturation at 95°C for 3 min, followed by 28 cycles of denaturation at 95°C for 30 s, annealing at 53°C for 30 s, and extension at 72°C for 45 s, and final extension at 72°C for 10 min, following which the samples were incubated at 10°C until the reaction stopped. The PCR products were eluted from a 2% agarose gel, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences), and quantified using QuantiFluor™‐ST (Promega) according to the manufacturer's protocol. The purified PCR fragments were pooled at equimolar concentrations, and paired‐end sequencing (2 × 300) was undertaken on an Illumina MiSeq platform (Illumina) by Majorbio Bio‐Pharm Technology Co. Ltd. (Shanghai).
2.3. Data analysis
Raw FASTQ files were demultiplexed and filtered using Trimmomatic and merged using FLASH. Reads at any site that had an average quality score of < 20 over a 50‐bp sliding window were truncated. The primers’ barcodes were matched, allowing exactly two nucleotide mismatches, and any reads that contained ambiguous bases were removed. Sequences with overlaps of > 10 bp were merged according to their overlap sequence. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE (USEARCH version 7.1, http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. Taxonomic classification of each 16S rRNA gene sequence was performed through comparison against the 16S rRNA Greengenes 135 bacteria database (Release 13.5 http://greengenes.secondgenome.com/) using the RDP Classifier algorithm (http://rdp.cme.msu.edu/), with a confidence threshold of 70%.
The evenness and richness of the gut microbiota were assessed by calculating alpha diversity indices (Shannon index, Simpson index, abundance‐based coverage estimator [ACE], and Chao estimator) using the Mothur program (version v.1.30.1; http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity). Rank abundance, rarefaction, and diversity curves were also plotted to reflect the sequencing depth. For beta diversity analysis, weighted and unweighted UniFrac distance matrices were calculated and visualized using principal coordinate analysis (PCoA). Permutational multivariate analysis of variance (PERMANOVA) was used to further identify the differences in gut microbiota between the two populations, and a histogram of bacterial composition was plotted according to the results of the taxonomic analysis. The Wilcoxon rank‐sum test was used to identify differences in alpha diversity indices and community structure of the gut microbiota between the two populations, using false discovery rate (FDR)‐adjusted p‐values. These analyses were run with the R statistical software (version 3.2.2), using the plot function to plot the curves and histogram and the vegan package to undertake the PERMANOVA analysis. Differences in the structure of the gut microbial communities were further analyzed using the linear discriminant analysis effect size (LEfSe) (http://huttenhower.sph.harvard.edu/galaxy/root?tool_xml:id=lefse_upload). Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) (Langille et al., 2013) was then applied to predict the functional profiles of the gut microbial communities, and the Wilcoxon rank‐sum test was used to test for differences in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the gut microbiota of the two populations, using FDR‐adjusted p‐values. Differences in the KEGG pathways were analyzed using the IBM SPSS statistical software (version 23.0). All data were analyzed using the Majorbio I‐Sanger Cloud Platform (http://www.i-sanger.com).
3. RESULTS
3.1. Sequencing quality evaluation
In total, 3,108,909 sequences of the hypervariable V3–V4 region of the 16S rRNA gene were obtained from the 58 fecal samples, 2,291,877 of which were valid. These corresponded to an average of 53,601.9 ± 6,038.6 reads per sample, which were then subsampled to an equal sequencing depth (31,200 reads per sample). A total of 1,183 OTUs were clustered using a sequence similarity of 97%. The rank abundance, rarefaction, and alpha diversity curves that were constructed based on these OTUs revealed that the sequencing depth was sufficient (Figures A1, A2), while Good's coverage estimations revealed that approximately 99.5%–99.8% of the species were obtained for all of the samples (Table A1).
3.2. Alpha and beta diversity analyses
Alpha diversity analyses based on the 1,183 OTUs (Table A1) revealed that there were no significant differences in the Shannon or Simpson indices between the wild and provisioned populations (Figure 1a,b), indicating a similar evenness. However, the provisioned population had significantly higher ACE and Chao1 estimator values than the wild population (Figure 1c,d), indicating that there were differences in bacterial richness. PCoA based on unweighted and weighted UniFrac distances demonstrated that the gut microbes were strongly clustered by population, as indicated by the beta diversity (Figure 2, Figure A3), and the PERMANOVA based on unweighted and weighted UniFrac distances revealed significant differences between the two populations (R2 = 0.153, adjusted p < .001 for both analyses).
Figure 1.
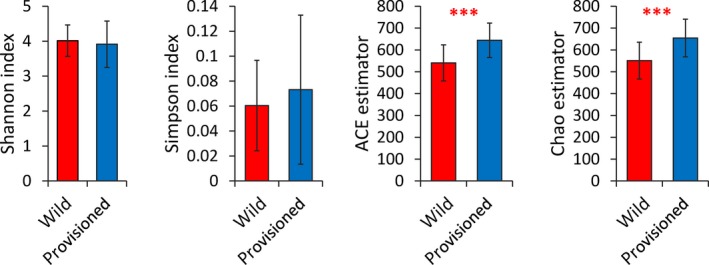
Alpha diversity of the gut microbiota of rhesus macaques from Chongzuo (wild) and Longhu Mountain (provisioned). The p‐value is represented by “*”; significant difference p < .001 is marked as “***”
Figure 2.
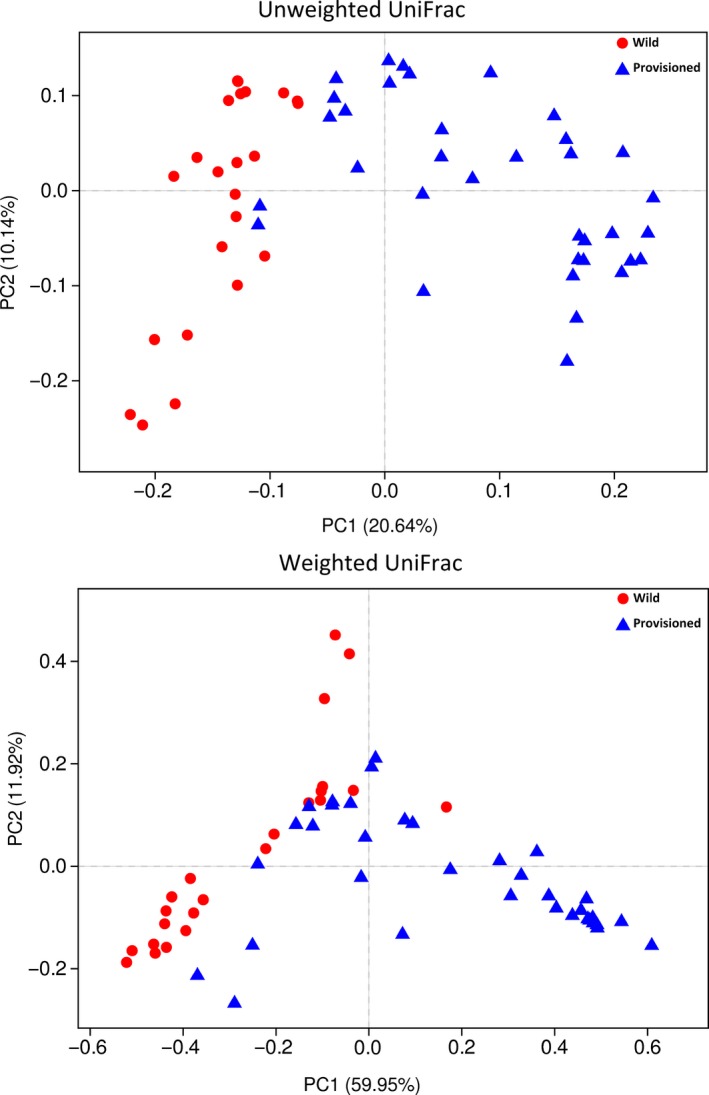
PCoA of structure differentiation and interindividual similarity on gut microbiota of rhesus macaques from Chongzuo (wild) and Longhu Mountain (provisioned)
3.3. Gut microbial community structure
Taxonomic analysis revealed that the 1,183 OTUs obtained from the fecal samples consisted of 14 classified bacterial phyla, 1 unclassified phylum, and 153 microbial genera. The most dominant phyla were Firmicutes (65.02% ± 20.63%) and Bacteroidetes (23.98% ± 17.49%), followed by Actinobacteria (3.46% ± 4.37%), Spirochetes (3.39% ± 8.32%), Proteobacteria (1.29% ± 1.97%), and Tenericutes (1.20% ± 0.96%) (Figure 3a). The dominant genus was Prevotella (18.28% ± 15.68%), followed by no‐rank Ruminococcaceae (9.99% ± 6.85%), and no‐rank Clostridiaceae (7.82% ± 11.41%) (Figure 3b). The proportions of other bacterial phyla and genera are shown in Tables A2 and A3.
Figure 3.
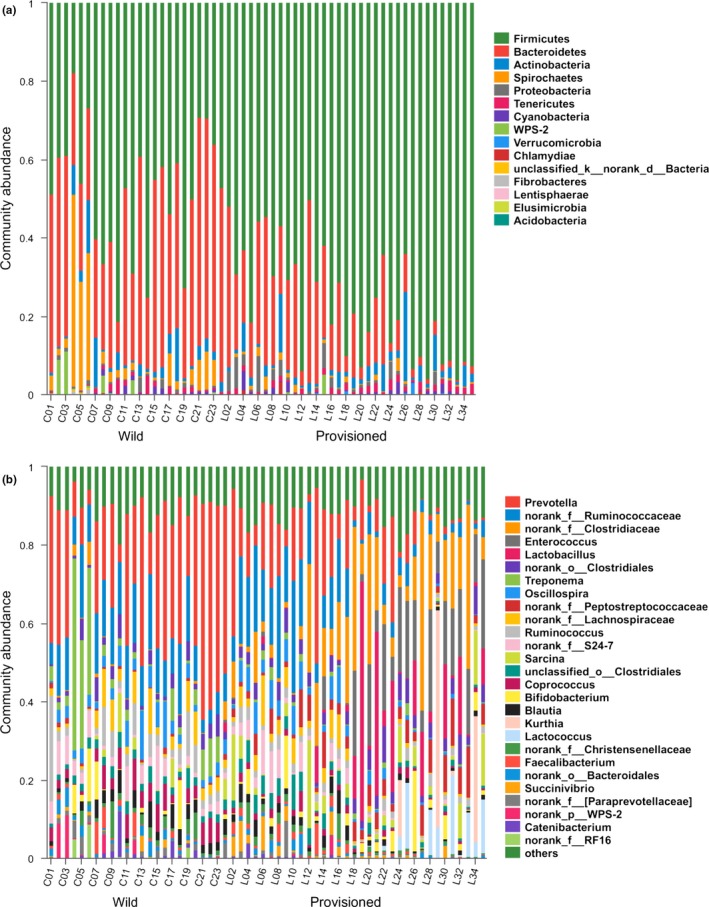
Stacked bar graphs illustrate the abundances of phyla and genus on gut microbiota from Chongzuo (wild) and Longhu Mountain (provisioned). (a) At phylum level; (b) At genus level
3.4. Differences in gut microbial composition
The proportions of gut bacteria significantly differed between the two populations according to Wilcoxon rank‐sum tests. At the phylum level, Bacteroidetes, Spirochetes, WPS‐2, and Fibrobacteres were more enriched in the wild population than in the provisioned population, whereas Firmicutes and Verrucomicrobia were more enriched in the provisioned population (Figure 4a; Table A2). At the genus level, Prevotella, Treponema, no‐rank S24–7, and Coprococcus were more enriched in the wild population, whereas no‐rank Clostridiaceae, Enterococcus, Lactobacillus, no‐rank Peptostreptococcaceae, Sarcina, Kurthia, and Lactococcus were more enriched in the provisioned population (Figure 4b). Other significant differences between the two populations at the genus level are presented in Table A3.
Figure 4.
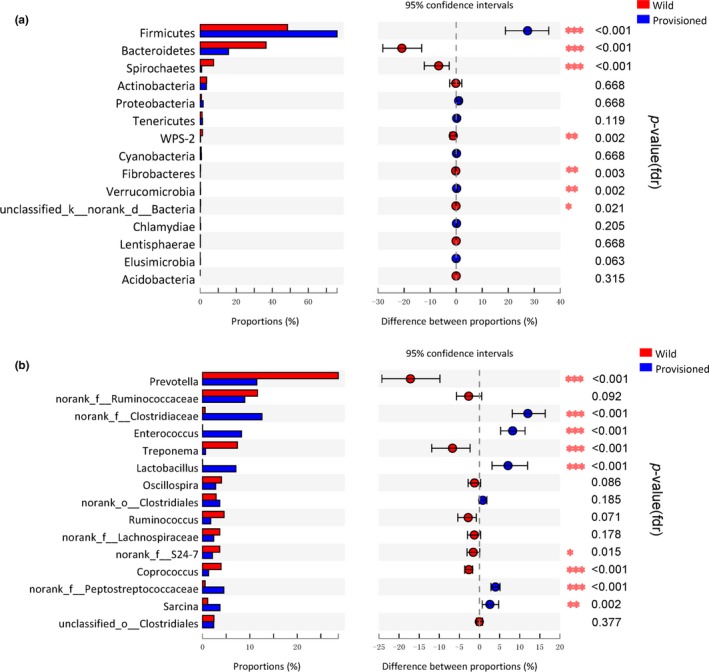
Abundance differences analysis (Wilcoxon rank‐sum test) of gut microbiota community on rhesus macaques from Chongzuo (wild) and Longhu Mountain (provisioned). (a) At phylum level. (b) At genus level, only the first 15 bacterial species with significant differences were showed
To further identify shifts in the gut microbial composition between the two populations, we used LEfSe to detect differences in the relative abundances of the bacterial taxa at the phylum, class, order, family, and genus levels. Differences in the abundances of bacterial phyla were caused by differences in the bacterial genera (Figure A3a), with similar results being observed to those described above for the Wilcoxon rank‐sum test only with more different genera being found. Thus, the wild population showed higher abundances of Prevotella, Treponema, Ruminococcus, Coprococcus, no‐rank S24–7, no‐rank Ruminococcaceae, and Oscillospira, whereas the provisioned population showed higher abundances of no‐rank Clostridiacese, Enterococcus, Lactobacillus, Lactococcus, Sarcina, and Kurthia (Figure A3b).
3.5. Differences in the functional profiles of the gut microbiota
To further explore the functions of the gut microbiota, we predicted the functional profiles of the gut microbial communities from the two populations of rhesus macaques using PICRUSt (mean nearest sequenced taxon index [NSTI]: overall = 0.13 ± 0.03; wild = 0.15 ± 0.03; and provisioned = 0.12 ± 0.03). We also investigated the effects of food provisioning on the functional profiles of the gut microbiota by examining differences in the KEGG pathways using Wilcoxon rank‐sum tests. There was no significant difference between the populations at KEGG pathway level 1 (Table A4). However, at KEGG pathway level 2, pathways associated with glycan biosynthesis and metabolism, transport and catabolism, and digestive and endocrine systems were significantly richer in the wild population, whereas pathways related to signaling molecules and their interaction and neurodegenerative diseases were richer in the provisioned population (Figure 5; Table A5).
Figure 5.
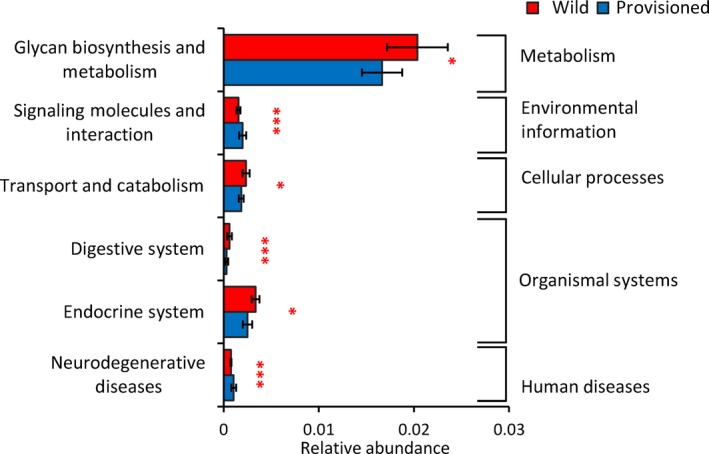
Predicted functional metagenomic on gut microbiota of rhesus macaques from Chongzuo (wild) and Longhu Mountain (provisioned). The p‐value is represented by “*”. Significant difference 0.01 < p< 0.05 is marked as “*” and p < .001 is marked as “***
4. DISCUSSION
Changes in the gut microbiota as a result of environmental variations are often strongly associated with diet (Amato et al., 2016; Barelli et al., 2015). In the present study, the gut microbiota of rhesus macaques inhabiting Longhu Mountain and Chongzuo formed two distinct clusters by population, which matches previous findings for rhesus macaques, humans, and other primates species inhabiting different environments (Amato et al., 2013; Gomez et al., 2015; Kohl, Varner, Wilkening, & Dearing, 2018; Rothschild et al., 2018; Vangay et al., 2018; Zhao et al., 2018). These differences may be explained by differences in dietary composition, as macaques in Chongzuo exclusively depend on natural foods, whereas those in Longhu Mountain are provided with additional foods. In general, wild animals have a higher gut microbial diversity than captive animals due to their more complex dietary composition (Amato et al., 2013; McKenzie et al., 2017; Nelson et al., 2013; Uenishi et al., 2007). However, our results did not support these findings or our first prediction that provisioned macaques would have a significantly lower gut microbial diversity. This may be because although rhesus macaques inhabiting Longhu Mountain are regularly provided with food, they also heavily depend on natural foods, such as leaves, flowers, and fruits (Wang, Jiang, Liu, & Feng, 1994), which may cause them to have similar digestion requirements as their wild counterparts. These results imply that the provisioned macaques in Longhu Mountain have maintained their ability to digest natural foods, which has undoubtedly improved their survival in this provisioned environment. However, more detailed comparisons are required in the future.
The gut microbiota of the rhesus macaques sampled in the present study was dominated by Firmicutes and Bacteroidetes, which is similar to the findings of previous studies on rhesus macaques and other primates species (Gomez et al., 2015; Su et al., 2016; Szekely et al., 2010; Trosvik, Rueness, Muinck, Moges, & Mekonnen, 2018; Zhao et al., 2018). In general, Firmicutes species can decompose various substances to help their host digest and absorb nutrients via digestive enzymes (Kaakoush, 2015), whereas Bacteroidetes species assist the host in degrading carbohydrates and proteins in foods (Fernando et al., 2010; Jami et al., 2014). However, the abundances of these bacteria varied greatly between the two populations, with rhesus macaques in Longhu Mountain having a higher ratio of Firmicutes/Bacteroidetes. Since an increased prevalence of Firmicutes and decreased prevalence of Bacteroidetes can improve the digestion and absorption of food energy (Bird & Conlon, 2015; Jami et al., 2014; Ley et al., 2006; Murphy et al., 2010), the ratio of Firmicutes/Bacteroidetes increases in response to the consumption of a high‐fat diet (Jami et al., 2014; Ley et al., 2006; Murphy et al., 2010) and has been linked to obesity (Ley et al., 2005; Turnbaugh et al., 2006; Vebo, Karlsson, Avershina, Finnby, & Rudi, 2016). Therefore, this result supports our second prediction that the gut microbial community structure of food‐provisioned rhesus macaques would be richer in high‐fat‐diet bacteria. This is likely due to rhesus macaques in Longhu Mountain being provided with corn and peanuts, both of which are rich in starch and fats, as well as rhesus macaques from Chongzuo spending more time foraging for food than those from Longhu Mountain, which may decrease the ratio of Firmicutes/Bacteroidetes in the gut (Denou, Marcinko, Surette, Steinberg, & Schertzer, 2016).
Rhesus macaques in the Chongzuo population were also found to have a higher proportion of cellulose‐degrading Fibrobacteres species in their gut microbiota, which may be explained by their greater consumption of cellulose‐rich food (Ransom‐Jones, Jones, McCarthy, & McDonald, 2012), supporting our third prediction. A previous study indicated that rhesus macaques inhabiting limestone forests tend to be folivorous and prefer young leaves as staple foods, supplementing their diet with mature leaves when required (Tang et al., 2016), which are rich in cellulose (Richard, 1985). Therefore, since the current study was conducted during the dry season when young leaves and fruits are scarce, macaques in Chongzuo may have had a more fiber‐rich diet than the provisioned macaques in Longhu Mountain, explaining why they hosted greater proportions of Fibrobacteres in their guts. Some bacterial genera were also present at significantly higher proportions in the Chongzuo population than in the Longhu Mountain population, likely due to differences in the dietary compositions of the two populations. These included the genera Prevotella and Treponema, which enable calories to be extracted from indigestible polysaccharides such as xylan and cellulose (De Filippo et al., 2010); Ruminococcus, which is important for cellulose and hemicellulose fermentation in ruminants (Ntaikou, Gavala, Kornaros, & Lyberatos, 2008; Pettipher & Latham, 1979); and Coprococcus and genera in the family Ruminococcaceae, which are commonly found in the guts of ruminants and other mammals and occur as fibrolytic communities that are likely associated with cellulose degradation (Biddle, Stewart, Blanchard, & Leschine, 2013; Henderson et al., 2015).
Another phylum that was detected in the gut of the rhesus macaques was WPS‐2, which has rarely been detected in the primate gut microbiota previously. This lesser‐known bacterial phylum was first detected in polychlorinated biphenyl‐polluted soil from Wittenberg, Germany (Nogales et al., 2001), and was single cloned from the canine oral microbiome (Dewhirst et al., 2012). However, the functional roles of members of this phylum in the host remain unclear. The occurrence of WPS‐2 in the gut microbiota of rhesus macaques in the present study may be attributed to geophagy, which is a common behavior in most primates. Primates obtain minerals by licking rocks or eating soil (Hsu, Agoramoorthy, & Lin, 2001; Li et al., 2014; Pebsworth, Bardi, & Huffman, 2012), which may result in bacteria from the soil colonizing the gut. However, the specific reasons for the presence of WPS‐2 in the guts of these rhesus macaques warrants further research.
The gut microbiota is closely associated with host health, and certain metabolites of the gut microbiota play important roles in host metabolism, digestion, and immunity (Castellazzi et al., 2017; Miani et al., 2018; Million et al., 2018; Rooks & Garrett, 2016; Tamburini & Clemente, 2017). In the present study, we used PICRUSt to predict the functional profiles of the gut microbiota of the two populations of rhesus macaques. However, it should be noted that this tool has some limitations in its capacity to predict functions, as only 16S marker gene sequences corresponding to bacterial and archaeal genomes are currently included and the accuracy is not high in the case of the reference genome pool (Langille et al., 2013). NSTI was used to quantify the availability of nearby genome representatives for each microbiome sample, and the accuracy of PICRUSt decreases as NSTI increases (Langille et al., 2013). However, the mean NSTI value was 0.13 ± 0.03, which is within the range of that previously reported for mammals (NSTI = 0.14 ± 0.06; (Langille et al., 2013)), indicating that our results are interpretable. The enriched functional profiles of the digestive and endocrine systems in rhesus macaques from Chongzuo may be attributable to their completely natural diet. Individuals in the wild population spend more time and energy on foraging than those in the provisioned population, and natural food items, such as leaves, contain greater levels of relatively indigestible fibers and toxic compounds (Richard, 1985). Thus, the enrichment of functional flora for digestion may facilitate the adaptation of animals to environmental variations. Furthermore, the gut microbiota of rhesus macaques from Chongzuo was enriched with Fibrobacteres, Prevotella, Treponema, Coprococcus, and Ruminococcus, which facilitate cellulose digestion (De Filippo et al., 2010; Ntaikou et al., 2008; Pettipher & Latham, 1979; Ransom‐Jones et al., 2012) and so may explain the enrichment of the digestive system.
In conclusion, although there was no significant difference in the diversity of gut microbes between provisioned and wild rhesus macaques, there was great variation in the richness and bacterial community structure of the two populations, indicating that food provisioning alters the gut microbiota of this species. In particular, food provisioning increased the ratio of Firmicutes/Bacteroidetes, which helped the rhesus macaques from Longhu Mountain to digest high‐fat foods more easily while maintaining a similar gut microbiota diversity. This suggests that an excessive reliance on provisioned feeding may increase the risk of obesity and gradually reduce the ability of these monkeys to survive in the wild. Therefore, a balance of provisioned and wild feeding is crucial to sustain the ability of rhesus macaques to digest a range of foods, which will allow them to effectively adapt to environmental variations.
5. ETHICS STATEMENT
We were permitted to enter the study site and collect samples under the Guangxi Chongzuo White‐Headed Langur National Nature Reserve ticket code 145141911048–00879801 and Guangxi Longhu Mountain Nature Reserve ticket code 1450119M0030‐0000986. This study did not involve any animal tissues. All fecal samples were collected after the animals left to avoid a stress reaction.
CONFLICT OF INTERESTS
None declared.
AUTHORS’ CONTRIBUTIONS
Ting Chen: Formal analysis‐Equal, Writing‐original draft‐Equal; Yuhui Li: Investigation‐Equal; Jipeng Liang:Investigation‐Equal; Youbang Li:Writing‐review & editing‐Equal; Zhonghao Huang: Conceptualization‐Equal, Writing‐review & editing‐Equal.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No 31660616, 31960106, 31960104), the Guangxi Natural Science Foundation, China (2017GXNSFAA198046; 2018GXNSFAA281029), the Doctoral Scientific Research Foundation of Guangxi Normal University, China (2017BQ016), and the Innovation Project of Guangxi Graduate Education, China (XYCSZ2019078). We are very grateful to the Guangxi Chongzuo White‐headed Langur National Nature Reserve and the Guangxi Longhu Mountain Nature Reserve for permitting us to conduct research in the study site. We thank Dr. Gang Hu from Nanning Normal University, Dr. Chuangbin Tang, and Mr. Shoufu Feng from Guangxi Normal University for Nationalities for their helps in fecal samples collecting and conserving. We also thank Mr. Feiyun He, Mr. Xianfeng Huang, and Mr. Da Li for their assists in the field.
APPENDIX 1.
Table A1.
Alpha diversity index of rhesus macaques gut microbiota
| Wild | Provisioned | p‐value (fdr) | |
|---|---|---|---|
| Shannon | 4.02 ± 0.45 | 3.91 ± 0.66 | .763 |
| Simpson | 0.06 ± 0.04 | 0.07 ± 0.06 | .763 |
| Ace | 540.64 ± 82.81 | 644.14 ± 78.65 | <.001 |
| Chao | 551.29 ± 84.30 | 654.73 ± 86.12 | <.001 |
| Coverage | 0.997 ± 0.000 | 0.996 ± 0.001 | <.001 |
Table A2.
The proportion differences of gut microbiota community of rhesus macaques from two different environments (at phylum level)
| Species name | Wild (%) | Provisioned (%) | p‐value (fdr) |
|---|---|---|---|
| Firmicutes | 48.49 ± 16.71 | 75.93 ± 15 | <.001 |
| Bacteroidetes | 36.58 ± 14.67 | 15.73 ± 14.05 | <.001 |
| Spirochaetes | 7.45 ± 12.25 | 0.73 ± 0.89 | <.001 |
| Actinobacteria | 3.54 ± 4.21 | 3.4 ± 4.46 | .668 |
| Proteobacteria | 0.7 ± 0.86 | 1.68 ± 2.37 | .668 |
| Tenericutes | 1.07 ± 1.21 | 1.28 ± 0.75 | .119 |
| WPS‐2 | 1.3 ± 3 | 0.17 ± 0.84 | .002 |
| Cyanobacteria | 0.55 ± 0.65 | 0.69 ± 1.08 | .668 |
| Fibrobacteres | 0.18 ± 0.33 | 0.01 ± 0.02 | .003 |
| Verrucomicrobia | 0.01 ± 0.01 | 0.18 ± 0.54 | .002 |
| Unclassified Bacteria | 0.13 ± 0.14 | 0.05 ± 0.04 | .021 |
| Chlamydiae | <0.01 | <0.01 | .205 |
| Lentisphaerae | <0.01 | <0.01 | .668 |
| Elusimicrobia | <0.01 | <0.01 | .063 |
| Acidobacteria | <0.01 | 0 | .315 |
Table A3.
The proportion differences of gut microbiota community of rhesus macaques from two different environments (at genus level)
| Species name | Wild (%) | Provisioned (%) | p‐value (fdr) |
|---|---|---|---|
| Prevotella | 28.66 ± 15.12 | 11.47 ± 12.01 | <.001 |
| No‐rank Ruminococcaceae | 11.64 ± 4.68 | 8.94 ± 7.87 | .092 |
| No‐rank Clostridiaceae | 0.56 ± 1.29 | 12.58 ± 12.58 | <.001 |
| Enterococcus | <0.01 | 8.24 ± 9.14 | <.001 |
| Treponema | 7.4 ± 12.25 | 0.68 ± 0.86 | <.001 |
| Lactobacillus | <0.01 | 7.09 ± 13.37 | <.001 |
| Oscillospira | 4.03 ± 2.99 | 2.79 ± 2.78 | .086 |
| No‐rank Clostridiales | 2.87 ± 1.88 | 3.68 ± 1.95 | .185 |
| Ruminococcus | 4.58 ± 5.79 | 1.77 ± 1.01 | .071 |
| No‐rank Lachnospiraceae | 3.68 ± 3.47 | 2.38 ± 2.88 | .178 |
| No‐rank S24‐7 | 3.66 ± 3.07 | 2.11 ± 2.99 | .015 |
| Coprococcus | 3.95 ± 2.35 | 1.29 ± 0.99 | <.001 |
| No‐rank Peptostreptococcaceae | 0.56 ± 0.47 | 4.53 ± 3.34 | <.001 |
| Sarcina | 1.12 ± 2.52 | 3.7 ± 5.42 | .002 |
| Unclassified Clostridiales | 2.42 ± 1.03 | 2.37 ± 1.99 | .377 |
| Blautia | 2.74 ± 1.99 | 1.78 ± 1.49 | .14 |
| Bifidobacterium | 2.08 ± 4.27 | 2.26 ± 4.18 | .113 |
| No‐rank Christensenellaceae | 1.96 ± 2.23 | 1.13 ± 1.4 | .126 |
| Kurthia | 0 | 2.95 ± 10.87 | .029 |
| Lactococcus | <0.01 | 2.59 ± 5.41 | .016 |
| Unclassified Ruminococcaceae | 1.4 ± 0.81 | 1.11 ± 0.99 | .178 |
| Faecalibacterium | 1.33 ± 2.19 | 1.04 ± 1.63 | .739 |
| No‐rank Bacteroidales | 1.52 ± 1.37 | 0.83 ± 1.05 | .042 |
| No‐rank RF39 | 1.02 ± 1.17 | 1.26 ± 0.75 | .116 |
| No‐rank Coriobacteriaceae | 1.25 ± 1.29 | 0.89 ± 0.58 | .859 |
| Succinivibrio | 0.49 ± 0.9 | 1.4 ± 2.29 | .212 |
| No‐rank [Paraprevotellaceae] | 1.23 ± 1.3 | 0.57 ± 1.63 | <.001 |
| No‐rank WPS‐2 | 1.3 ± 3 | 0.17 ± 0.84 | .003 |
| Unclassified Lachnospiraceae | 0.78 ± 0.71 | 0.64 ± 0.55 | .8 |
| Catenibacterium | 1.1 ± 2.53 | 0.28 ± 0.64 | .167 |
| Butyrivibrio | 0.73 ± 1.04 | 0.35 ± 0.33 | 1 |
| Bulleidia | 0.26 ± 0.24 | 0.66 ± 0.53 | <.001 |
| No‐rank RF16 | 0.85 ± 2.1 | 0.06 ± 0.07 | <.001 |
| No‐rank [Mogibacteriaceae] | 0.33 ± 0.25 | 0.44 ± 0.35 | .422 |
| Unclassified Lactobacillales | 0 | 0.73 ± 0.97 | <.001 |
| No‐rank Streptophyta | 0.14 ± 0.21 | 0.5 ± 0.98 | .091 |
| Dorea | 0.46 ± 0.5 | 0.15 ± 0.14 | .006 |
| No‐rank YS2 | 0.41 ± 0.58 | 0.18 ± 0.24 | .6 |
| Dialister | 0.47 ± 1.18 | 0.09 ± 0.17 | .06 |
| [Ruminococcus] | 0.33 ± 0.39 | 0.21 ± 0.19 | .398 |
| Clostridium | 0.26 ± 0.26 | 0.25 ± 0.37 | .179 |
| Unclassified Bacteroidales Bacteroidia | 0.15 ± 0.17 | 0.31 ± 0.66 | .859 |
| No‐rank Rikenellaceae | 0.15 ± 0.41 | 0.16 ± 0.41 | .364 |
| [Prevotella] | 0.19 ± 0.32 | 0.06 ± 0.14 | .023 |
| Unclassified Firmicutes | <0.01 | 0.25 ± 0.38 | <.001 |
| Unclassified Planococcaceae | 0 | 0.24 ± 0.76 | .0633 |
| No‐rank Streptococcaceae | 0 | 0.23 ± 0.42 | <.001 |
| Roseburia | 0.15 ± 0.22 | 0.08 ± 0.1 | .645 |
| Rummeliibacillus | 0 | 0.22 ± 0.71 | .063 |
| Collinsella | 0.08 ± 0.1 | 0.11 ± 0.11 | .314 |
| Fibrobacter | 0.18 ± 0.33 | 0.01 ± 0.02 | .005 |
| No‐rank Erysipelotrichaceae | 0.12 ± 0.15 | 0.06 ± 0.07 | .232 |
| No‐rank RFP12 | <0.01 | 0.18 ± 0.54 | <.001 |
| Unclassified no‐rank Bacteria | 0.13 ± 0.14 | 0.05 ± 0.04 | .029 |
| Lachnospira | 0.03 ± 0.04 | 0.13 ± 0.11 | <.001 |
| Pediococcus | <0.01 | 0.16 ± 0.76 | .173 |
| Unclassified Bacilli | <0.01 | 0.16 ± 0.2 | <.001 |
| Unclassified Clostridia | 0.14 ± 0.3 | 0.01 ± 0.02 | .003 |
| Candidatus Rhabdochlamydia | <0.01 | 0.14 ± 0.54 | .232 |
| Slackia | 0.09 ± 0.07 | 0.06 ± 0.04 | .276 |
| No‐rank Enterobacteriaceae | 0 | 0.14 ± 0.67 | <.001 |
| Unclassified Clostridiaceae | 0 | 0.14 ± 0.2 | <.001 |
| Unclassified Prevotellaceae | 0.09 ± 0.07 | 0.04 ± 0.06 | .003 |
| RFN20 | 0.07 ± 0.1 | 0.03 ± 0.08 | .009 |
| Anaerostipes | 0.1 ± 0.22 | <0.01 | .006 |
| YRC22 | 0.05 ± 0.1 | 0.05 ± 0.08 | .536 |
| [Eubacterium] | 0.02 ± 0.04 | 0.08 ± 0.06 | <.001 |
| No‐rank Dehalobacteriaceae | 0.08 ± 0.24 | 0.01 ± 0.02 | .655 |
| Adlercreutzia | 0.03 ± 0.03 | 0.07 ± 0.05 | <.001 |
| Sphaerochaeta | 0.04 ± 0.04 | 0.05 ± 0.05 | .461 |
| No‐rank GMD14H09 | 0.05 ± 0.12 | 0.03 ± 0.09 | .614 |
| Flexispira | 0.05 ± 0.05 | 0.03 ± 0.06 | .221 |
| L7A E11 | 0.03 ± 0.07 | 0.04 ± 0.08 | .487 |
| No‐rank Rickettsiales | 0.06 ± 0.07 | 0.02 ± 0.02 | .002 |
| p‐75‐a5 | 0.03 ± 0.03 | 0.03 ± 0.03 | .607 |
| Mogibacterium | 0.02 ± 0.02 | 0.04 ± 0.04 | .009 |
| Lachnobacterium | 0.04 ± 0.06 | <0.01 | .44 |
| Phascolarctobacterium | 0.03 ± 0.06 | 0.01 ± 0.02 | .31 |
| Desulfovibrio | 0.04 ± 0.05 | <0.01 | .003 |
| Unclassified Enterococcaceae | 0 | 0.04 ± 0.05 | <.001 |
| Unclassified Bacillales | 0 | 0.04 ± 0.15 | .063 |
| CF231 | 0.01 ± 0.01 | 0.03 ± 0.05 | .652 |
| Streptococcus | 0.01 ± 0.02 | 0.02 ± 0.07 | .533 |
| Parabacteroides | 0.02 ± 0.06 | 0.01 ± 0.02 | .001 |
| Unclassified Tenericutes | 0.02 ± 0.05 | <0.01 | .022 |
| Unclassified Coriobacteriaceae | <0.01 | 0.02 ± 0.02 | .487 |
| No‐rank p‐2534‐18B5 | 0 | 0.02 ± 0.05 | <.001 |
| 02d06 | <0.01 | 0.02 ± 0.02 | <.001 |
| No‐rank Lactobacillaceae | <0.01 | 0.02 ± 0.1 | .967 |
| No‐rank ML615J‐28 | 0.02 ± 0.02 | <0.01 | .063 |
| No‐rank Leuconostocaceae | 0 | 0.02 ± 0.04 | <.001 |
| Anaerofustis | <0.01 | 0.01 ± 0.01 | <.001 |
| Unclassified Alphaproteobacteria | <0.01 | 0.01 ± 0.03 | .14 |
| Dehalobacterium | <0.01 | <0.01 | .801 |
| No‐rank mitochondria | <0.01 | <0.01 | .271 |
| Megasphaera | <0.01 | 0.01 ± 0.05 | .015 |
| Coprobacillus | <0.01 | <0.01 | .138 |
| Brachyspira | <0.01 | <0.01 | .377 |
| No‐rank Mycoplasmataceae | <0.01 | 0 | .001 |
| Anaeroplasma | <0.01 | <0.01 | .615 |
| No‐rank M2PT2‐76 | <0.01 | <0.01 | .001 |
| Anaerovibrio | <0.01 | <0.01 | .479 |
| No‐rank Veillonellaceae | <0.01 | <0.01 | .888 |
| No‐rank RF32 | <0.01 | <0.01 | .314 |
| Actinobacillus | <0.01 | <0.01 | .406 |
| Oxalobacter | <0.01 | <0.01 | .536 |
| Unclassified Burkholderiales | 0 | <0.01 | .091 |
| Solibacillus | 0 | <0.01 | .372 |
| Unclassified Spirochaetes | <0.01 | <0.01 | .698 |
| Bacillus | <0.01 | <0.01 | .232 |
| No‐rank Prevotellaceae | <0.01 | <0.01 | .254 |
| Akkermansia | <0.01 | <0.01 | .194 |
| No‐rank Alphaproteobacteria | <0.01 | <0.01 | 1 |
| Acinetobacter | <0.01 | <0.01 | .126 |
| No‐rank Victivallaceae | <0.01 | <0.01 | .652 |
| No‐rank Anaeroplasmataceae | <0.01 | <0.01 | .828 |
| Veillonella | <0.01 | <0.01 | .795 |
| Rickettsiella | 0 | <0.01 | .262 |
| No‐rank Xenococcaceae | 0 | <0.01 | .043 |
| Unclassified Erysipelotrichaceae | <0.01 | 0 | .003 |
| Unclassified Rickettsiales | <0.01 | 0 | .006 |
| No‐rank Elusimicrobiaceae | <0.01 | <0.01 | .081 |
| Butyricimonas | <0.01 | <0.01 | .022 |
| No‐rank Caulobacteraceae | <0.01 | <0.01 | .406 |
| Unclassified Bradyrhizobiaceae | <0.01 | <0.01 | .003 |
| Rubellimicrobium | 0 | <0.01 | .063 |
| Kocuria | <0.01 | <0.01 | .859 |
| Sphingomonas | <0.01 | <0.01 | .336 |
| Alloscardovia | <0.01 | <0.01 | .958 |
| Unclassified Micrococcaceae | 0 | <0.01 | .184 |
| Dysgonomonas | <0.01 | 0 | .077 |
| No‐rank Peptococcaceae | <0.01 | <0.01 | .917 |
| No‐rank Planococcaceae | 0 | <0.01 | .536 |
| Curtobacterium | <0.01 | <0.01 | .025 |
| No‐rank Bifidobacteriaceae | 0 | <0.01 | .536 |
| No‐rank Pseudonocardiaceae | <0.01 | <0.01 | .822 |
| Candidatus Phytoplasma | 0 | <0.01 | .536 |
| No‐rank Bacillaceae | <0.01 | 0 | .035 |
| Anaerococcus | <0.01 | <0.01 | .346 |
| rc4‐4 | <0.01 | <0.01 | .129 |
| Sutterella | <0.01 | <0.01 | .614 |
| Ruminobacter | 0 | <0.01 | .262 |
| Unclassified Erythrobacteraceae | 0 | <0.01 | .184 |
| No‐rank R4‐45B | <0.01 | <0.01 | 1 |
| Unclassified Proteobacteria | <0.01 | <0.01 | .629 |
| Brevundimonas | <0.01 | <0.01 | .751 |
| Epulopiscium | 0 | <0.01 | .372 |
| Actinomyces | 0 | <0.01 | .184 |
| No‐rank 32‐20 | <0.01 | 0 | .346 |
| Arcanobacterium | 0 | <0.01 | .372 |
| Bacteroides Bacteroidaceae | <0.01 | 0 | .346 |
| Unclassified Acholeplasmatales | 0 | <0.01 | .536 |
| Aerococcus | 0 | <0.01 | .536 |
Table A4.
The differences of KEGG Pathways Level 1
| Pathway in level 1 | Relative abundance (%) | p‐value (fdr) | |
|---|---|---|---|
| Wild | Provisioned | ||
| Cellular processes | 3.55 ± 0.52 | 3.02 ± 0.55 | .6 |
| Environmental information processing | 13.82 ± 1 | 15.09 ± 0.82 | .578 |
| Genetic information processing | 20.96 ± 0.46 | 20.59 ± 0.8 | .945 |
| Human diseases | 0.7 ± 0.03 | 0.74 ± 0.05 | .656 |
| Metabolism | 46.22 ± 0.92 | 45.64 ± 0.6 | .81 |
| None | 0.21 ± 0.02 | 0.19 ± 0.03 | .824 |
| Organismal systems | 0.78 ± 0.07 | 0.63 ± 0.08 | .072 |
| Unclassified | 13.77 ± 0.13 | 14.1 ± 0.29 | .781 |
Table A5.
The differences of KEGG Pathways Level 2
| Pathway in level 2 | Relative abundance (%) | p‐value (fdr) | |
|---|---|---|---|
| Wild | Provisioned | ||
| Amino acid metabolism | 9.4 ± 0.19 | 9.12 ± 0.38 | .938 |
| Biosynthesis of other secondary metabolites | 0.86 ± 0.04 | 0.79 ± 0.06 | .543 |
| Carbohydrate metabolism | 10.2 ± 0.37 | 10.3 ± 0.35 | 1 |
| Energy metabolism | 5.82 ± 0.22 | 5.47 ± 0.29 | .658 |
| Enzyme families | 2.23 ± 0.06 | 2.22 ± 0.09 | 1 |
| Glycan biosynthesis and metabolism | 2.04 ± 0.32 | 1.67 ± 0.21 | .031 |
| Lipid metabolism | 2.73 ± 0.17 | 2.82 ± 0.17 | 1 |
| Metabolism | 2.37 ± 0.07 | 2.6 ± 0.18 | .543 |
| Metabolism of cofactors and vitamins | 4.27 ± 0.21 | 4.21 ± 0.2 | 1 |
| Metabolism of other amino acids | 1.34 ± 0.07 | 1.45 ± 0.08 | .567 |
| Metabolism of terpenoids and polyketides | 1.64 ± 0.1 | 1.61 ± 0.08 | .89 |
| Nucleotide metabolism | 4.18 ± 0.19 | 4.26 ± 0.27 | 1 |
| Xenobiotics biodegradation and metabolism | 1.6 ± 0.1 | 1.8 ± 0.21 | .456 |
| Folding, sorting and degradation | 2.46 ± 0.1 | 2.33 ± 0.1 | .656 |
| Genetic information processing | 2.77 ± 0.06 | 2.79 ± 0.11 | 1 |
| Replication and repair | 9.39 ± 0.29 | 9.17 ± 0.46 | .824 |
| Transcription | 2.95 ± 0.13 | 3.15 ± 0.12 | .683 |
| Translation | 6.2 ± 0.19 | 5.98 ± 0.32 | .677 |
| Membrane transport | 12.24 ± 0.94 | 13.39 ± 0.75 | .656 |
| Signal transduction | 1.45 ± 0.1 | 1.53 ± 0.14 | .839 |
| Signaling molecules and interaction | 0.16 ± 0.02 | 0.2 ± 0.04 | 0 |
| Cell communication | 0 | 0 | 1 |
| Cell growth and death | 0.56 ± 0.03 | 0.51 ± 0.03 | .533 |
| Cell motility | 2.76 ± 0.51 | 2.33 ± 0.55 | .539 |
| Cellular processes and signaling | 3.81 ± 0.15 | 3.85 ± 0.19 | 1 |
| Transport and catabolism | 0.23 ± 0.04 | 0.18 ± 0.03 | .031 |
| Circulatory system | <0.01 | <0.01 | .444 |
| Digestive system | 0.06 ± 0.02 | 0.03 ± 0.02 | 0 |
| Endocrine system | 0.34 ± 0.04 | 0.25 ± 0.05 | .031 |
| Environmental adaptation | 0.17 ± 0.01 | 0.15 ± 0.01 | .617 |
| Excretory system | 0.02 ± 0.01 | 0.02 ± 0.01 | .399 |
| Immune system | 0.1 ± 0.01 | 0.08 ± 0.02 | .1 |
| Nervous system | 0.11 ± 0 | 0.1 ± 0.01 | .658 |
| Sensory system | 0 | 0 | 1 |
| Cancers | 0.1 ± 0.01 | 0.08 ± 0.01 | .092 |
| Cardiovascular diseases | <0.01 | <0.01 | .631 |
| Immune system diseases | 0.04 ± 0.01 | 0.05 ± 0.01 | .549 |
| Infectious diseases | 0.37 ± 0.02 | 0.4 ± 0.03 | .456 |
| Metabolic diseases | 0.11 ± 0.01 | 0.1 ± 0.01 | .695 |
| Neurodegenerative diseases | 0.08 ± 0.01 | 0.1 ± 0.03 | 0 |
| Poorly characterized | 4.84 ± 0.08 | 4.88 ± 0.11 | 1 |
Figure A1.
(a) Rank abundance distribution curves and rarefaction curves; (b) Alpha diversity curves.
Figure A2.
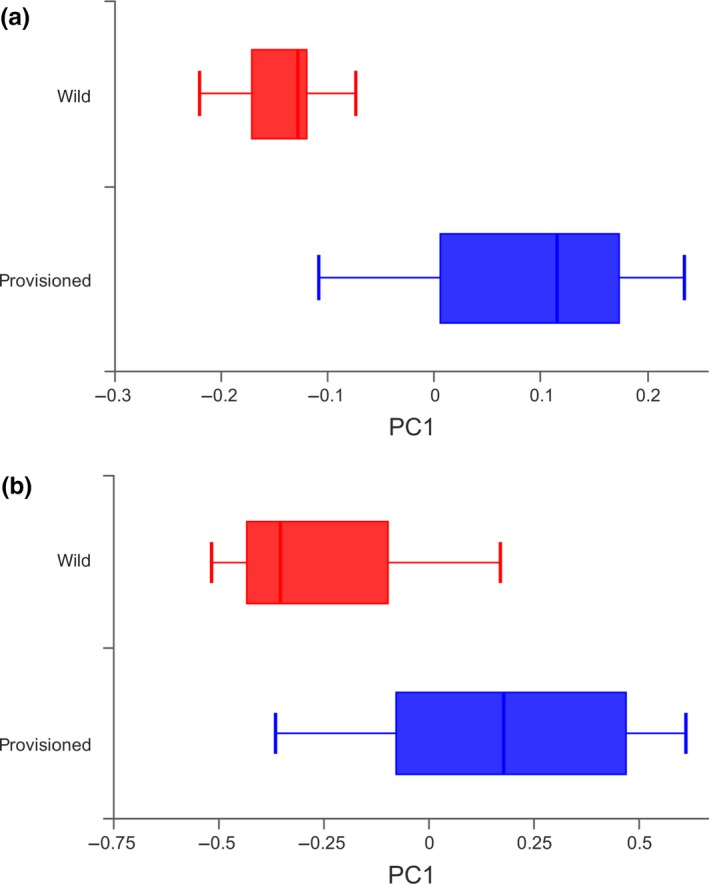
The dispersion degree box plot of PCoA of structure differentiation and inter‐individual similarity on gut microbiota of wild and provisioned rhesus macaques; (a) based on unweighted UniFrac distance; (b) based on weighted UniFrac distance.
Figure A3.
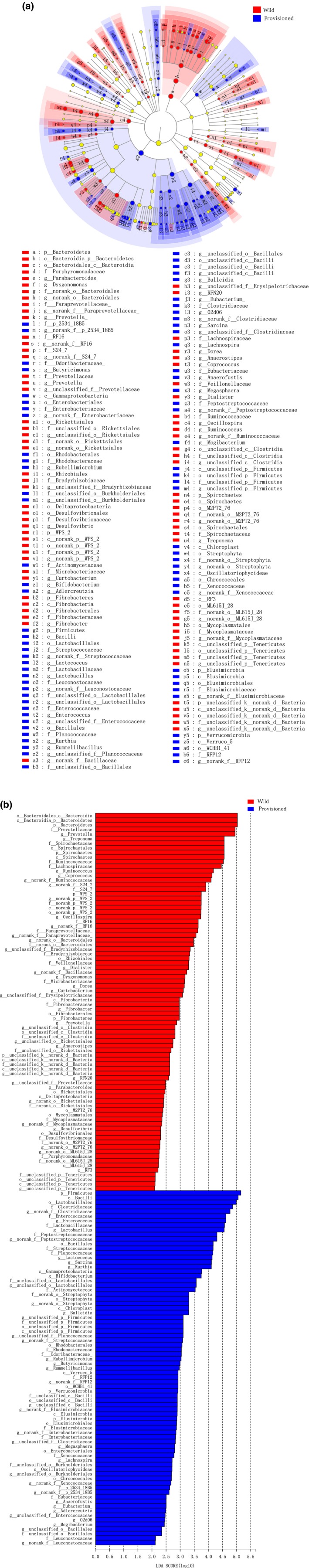
Linear discriminant analysis effect size (LEfSe) analysis on gut microbiota composition of wild and provisioned rhesus macaques (LDA > 2, p < .05).
Chen T, Li Y, Liang J, Li Y, Huang Z. Gut microbiota of provisioned and wild rhesus macaques (Macaca mulatta) living in a limestone forest in southwest Guangxi, China. MicrobiologyOpen. 2020;9:e981 10.1002/mbo3.981
DATA AVAILABILITY STATEMENT
The raw sequencing data from the current study are available in the NCBI repository at https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA590350, and the SRA accession number is PRJNA590350.
REFERENCES
- Amato, K. R. , Leigh, S. R. , Kent, A. , Mackie, R. I. , Yeoman, C. J. , Stumpf, R. M. , … Garber, P. A. (2015). The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microbial Ecology, 69, 434–443. 10.1007/s00248-014-0554-7 [DOI] [PubMed] [Google Scholar]
- Amato, K. R. , Martinez‐Mota, R. , Righini, N. , Raguet‐Schofield, M. , Corcione, F. P. , Marini, E. , … Leigh, S. R. (2016). Phylogenetic and ecological factors impact the gut microbiota of two Neotropical primate species. Oecologia, 180(3), 717–733. 10.1007/s00442-015-3507-z [DOI] [PubMed] [Google Scholar]
- Amato, K. R. , Yeoman, C. J. , Kent, A. , Righini, N. , Carbonero, F. , Estrada, A. , … Leigh, S. R. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. International Society for Microbial Ecology, 7(7), 1344–1353. 10.1038/ismej.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelakis, E. , Yasir, M. , Bachar, D. , Azhar, E. I. , Lagier, J. C. , Bibi, F. , … Raoult, D. (2016). Gut microbiome and dietary patterns in different Saudi populations and monkeys. Scientific Reports, 6, 32191 10.1038/srep32191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardeshir, A. , Narayan, N. R. , Mendez‐Lagares, G. , Lu, D. , Rauch, M. , Huang, Y. , … Hartigan‐O'Connor, D. J. (2014). Breast‐fed and bottle‐fed infant rhesus macaques develop distinct gut microbiotas and immune systems. Science Translational Medicine, 6(252), 252ra120 10.1126/scitranslmed.3008791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelli, C. , Albanese, D. , Donati, C. , Pindo, M. , Dallago, C. , Rovero, F. , … De Filippo, C. (2015). Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Scientific Reports, 5, 14862 10.1038/srep14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddle, A. , Stewart, L. , Blanchard, J. , & Leschine, S. (2013). Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity, 5(3), 627–640. 10.3390/d5030627 [DOI] [Google Scholar]
- Bird, A. R. , & Conlon, M. A. (2015). Obesity, diet and the gut microbiota. Current Nutrition Reports, 4(4), 340–347. 10.1007/s13668-015-0146-2 [DOI] [Google Scholar]
- Bonder, M. J. , Kurilshikov, A. , Tigchelaar, E. F. , Mujagic, Z. , Imhann, F. , Vila, A. V. , … Zhernakova, A. (2016). The effect of host genetics on the gut microbiome. Nature Genetics, 48(11), 1407–1412. 10.1038/ng.3663 [DOI] [PubMed] [Google Scholar]
- Cao, S. , Wang, M. , Cheng, A. , Qi, X. , Yang, X. , Deng, S. , … Chen, X. (2008). Comparative analysis of intestinal microbial community diversity between healthy and orally infected ducklings with Salmonella enteritidis by ERIC‐PCR. World Journal of Gastroenterology, 14(7), 1120–1125. 10.3748/wjg.14.1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellazzi, A. , Tagliacarne, S. C. , Soldi, S. , Perna, S. , Ziviani, L. , Milleri, S. , … Valsecchi, C. (2017). Stress and immune function: There is a role for the gut microbiota? Paper presented at the The 9th Probiotics, Prebiotics and New Foods, Nutraceuticals and Botanicals for Nutrition & Human and Microbiota Health Meeting, Rome, Italy. [Google Scholar]
- Cui, Y. , Wang, F. , Yu, L. , Ye, H. , & Yang, G. (2019). Metagenomic comparison of the rectal microbiota between rhesus macaques (Macaca mulatta) and cynomolgus macaques (Macaca fascicularis). Zoological Research, 40(2), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, L. A. , Maurice, C. F. , Carmody, R. N. , Gootenberg, D. B. , Button, J. E. , Wolfe, B. E. , … Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505(7484), 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo, C. , Cavalieri, D. , Di Paola, M. , Ramazzotti, M. , Poullet, J. B. , Massart, S. , … Lionetti, P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America, 107(33), 14691–14696. 10.1073/pnas.1005963107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denou, E. , Marcinko, K. , Surette, M. G. , Steinberg, G. R. , & Schertzer, J. D. (2016). High‐intensity exercise training increases the diversity and metabolic capacity of the mouse distal gut microbiota during diet‐induced obesity. American Journal of Physiology‐endocrinology and Metabolism, 310(11), E982–E993. 10.1152/ajpendo.00537.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst, F. E. , Klein, E. A. , Thompson, E. C. , Blanton, J. M. , Chen, T. , Milella, L. , … Marshall‐Jones, Z. V. (2012). The canine oral microbiome. PLoS ONE, 7(4), e36067 10.1371/journal.pone.0036067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando, S. C. , Purvis, H. T. II , Najar, F. Z. , Sukharnikov, L. O. , Krehbiel, C. R. , Nagaraja, T. G. , … Desilva, U. (2010). Rumen microbial population dynamics during adaptation to a high‐grain diet. Applied and Environmental Microbiology, 76(22), 7482–7490. 10.1128/AEM.00388-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, A. , Petrzelkova, K. , Yeoman, C. J. , Vlckova, K. , Mrazek, J. , Koppova, I. , … Leigh, S. R. (2015). Gut microbiome composition and metabolomic profiles of wild western lowland gorillas (Gorilla gorilla gorilla) reflect host ecology. Molecular Ecology, 24(10), 2551–2565. [DOI] [PubMed] [Google Scholar]
- Goodrich, J. K. , Davenport, E. R. , Beaumont, M. , Jackson, M. A. , Knight, R. , Ober, C. , … Ley, R. E. (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host and Microbe, 19(5), 731–743. 10.1016/j.chom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich, J. K. , Waters, J. L. , Poole, A. C. , Sutter, J. L. , Koren, O. , Blekhman, R. , … Ley, R. E. (2014). Human genetics shape the gut microbiome. Cell, 159(4), 789–799. 10.1016/j.cell.2014.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, R. L. , Carr, M. M. , Apanavicius, C. J. , Jiang, P. , Bissell, H. A. , Gocinski, B. L. , … Kouba, A. J. (2010). Seasonal shifts in giant panda feeding behavior: Relationships to bamboo plant part consumption. Zoo Biology, 29(4), 470–483. [DOI] [PubMed] [Google Scholar]
- Henderson, G. , Cox, F. , Ganesh, S. , Jonker, A. , Young, W. , & Global_Rumen_Census_Collaborators & Janssen PH,, (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Scientific Reports, 5, 14567 10.1038/srep14567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, M. J. , Agoramoorthy, G. , & Lin, J. (2001). Geophagy amongst formosan macaques at Mount Longevity. Taiwan. Folia Primatologica, 72, 339–341. 10.1159/000052751 [DOI] [PubMed] [Google Scholar]
- Huang, C. , Wu, H. , Zhou, Q. , Li, Y. , & Cai, X. (2008). Feeding strategy of François' langur and white‐headed langur at Fusui. China. American Journal of Primatology, 70(4), 320–326. 10.1002/ajp.20490 [DOI] [PubMed] [Google Scholar]
- Jami, E. , White, B. A. , & Mizrahi, I. (2014). Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE, 9(1), e85423 10.1371/journal.pone.0085423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakoush, N. O. (2015). Insights into the role of Erysipelotrichaceae in the human host. Frontiers in Cellular and Infection Microbiology, 5, 84 10.3389/fcimb.2015.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, K. D. , Varner, J. , Wilkening, J. L. , & Dearing, M. D. (2018). Gut microbial communities of American pikas (Ochotona princeps): Evidence for phylosymbiosis and adaptations to novel diets. Journal of Animal Ecology, 87(2), 323–330. [DOI] [PubMed] [Google Scholar]
- Kurilshikov, A. , Wijmenga, C. , Fu, J. , & Zhernakova, A. (2017). Host genetics and gut microbiome: Challenges and perspectives. Trends in Immunology, 38(9), 633–647. 10.1016/j.it.2017.06.003 [DOI] [PubMed] [Google Scholar]
- Kushida, M. , Sugawara, S. , Asano, M. , Yamamoto, K. , Fukuda, S. , & Tsuduki, T. (2018). Effects of the 1975 Japanese diet on the gut microbiota in younger adults. Journal of Nutritional Biochemistry, 64, 121–127. 10.1016/j.jnutbio.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Langille, M. G. , Zaneveld, J. , Caporaso, J. G. , McDonald, D. , Knights, D. , Reyes, J. A. , … Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31(9), 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Bӓckhed, F. , Turnbaugh, P. , Lozupone, C. A. , Knight, R. D. , & Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America, 102(31), 11070–11075. 10.1073/pnas.0504978102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Hamady, M. , Lozupone, C. , Turnbaugh, P. J. , Ramey, R. R. , Bircher, J. S. , … Gordon, J. I. (2008). Evolution of mammals and their gut microbes. Science, 320, 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Turnbaugh, P. J. , Klein, S. , & Gordon, J. I. (2006). Microbial ecology: Human gut microbes associated with obesity. Nature, 444(7122), 1022–1023. 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- Li, D. , Ren, B. , Hu, J. , Zhang, Q. , Yang, Y. , Grueter, C. C. , … Li, M. (2014). Geophagy of Yunnan snub‐nosed monkeys (Rhinopithecus bieti) at Xiangguqing in the Baimaxueshan Nature Reserve. China. North‐western Journal of Zoology, 10(2), 293–299. [Google Scholar]
- Marino, E. (2016). The gut microbiota and immune‐regulation: The fate of health and disease. Clinical and Translational Immunology, 5(11), e107 10.1038/cti.2016.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. E. , Bhatnagar, S. , George, M. D. , Paster, B. J. , Canfield, D. R. , Eisen, J. A. , & Solnick, J. V. (2013). The impact of Helicobacter pylori infection on the gastric microbiota of the rhesus macaque. PLoS ONE, 8(10), e76375 10.1371/journal.pone.0076375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie, V. J. , Song, S. J. , Delsuc, F. , Prest, T. L. , Oliverio, A. M. , Korpita, T. M. , … Knight, R. (2017). The effects of captivity on the mammalian gut microbiome. Integrative and Comparative Biology, 57(4), 690–704. 10.1093/icb/icx090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miani, M. , Le Naour, J. , Waeckel‐Enee, E. , Verma, S. C. , Straube, M. , Emond, P. , … Diana, J. (2018). Gut microbiota‐stimulated innate lymphoid cells support beta‐defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metabolism, 28(4), 557–572 e556. [DOI] [PubMed] [Google Scholar]
- Million, M. , Tomas, J. , Wagner, C. , Lelouard, H. , Raoult, D. , & Gorvel, J.‐P. (2018). New insights in gut microbiota and mucosal immunity of the small intestine. Human Microbiome Journal, 7–8, 23–32. 10.1016/j.humic.2018.01.004 [DOI] [Google Scholar]
- Mori H., Maruyama F., Kato H., Toyoda A., Dozono A., Ohtsubo Y., … Kurokawa K. (2014). Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Research, 21(2), 217-227. 10.1093/dnares/dst052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge, B. D. , Kuczynski, J. , Knights, D. , Clemente, J. C. , González, A. , Fontana, L. , … Gordon, J. I. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science, 332, 970–974. 10.1126/science.1198719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, E. F. , Cotter, P. D. , Healy, S. , Marques, T. M. , O'Sullivan, O. , Fouhy, F. , … Shanahan, F. (2010). Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut, 59(12), 1635–1642. 10.1136/gut.2010.215665 [DOI] [PubMed] [Google Scholar]
- Nelson, T. M. , Rogers, T. L. , Carlini, A. R. , & Brown, M. V. (2013). Diet and phylogeny shape the gut microbiota of Antarctic seals: A comparison of wild and captive animals. Environmental Microbiology, 15(4), 1132–1145. 10.1111/1462-2920.12022 [DOI] [PubMed] [Google Scholar]
- Nogales, B. , Moore, E. R. , Llobet‐Brossa, E. , Rossello‐Mora, R. , Amann, R. , & Timmis, K. N. (2001). Combined use of 16S ribosomal DNA and 16S rRNA to study the bacterial community of polychlorinated biphenyl‐polluted soil. Applied and Environmental Microbiology, 67(4), 1874–1884. 10.1128/AEM.67.4.1874-1884.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntaikou, I. , Gavala, H. N. , Kornaros, M. , & Lyberatos, G. (2008). Hydrogen production from sugars and sweet sorghum biomass using Ruminococcus albus . International Journal of Hydrogen Energy, 33(4), 1153–1163. 10.1016/j.ijhydene.2007.10.053 [DOI] [Google Scholar]
- O'Sullivan, A. , He, X. , McNiven, E. M. , Hinde, K. , Haggarty, N. W. , Lonnerdal, B. , & Slupsky, C. M. (2013). Metabolomic phenotyping validates the infant rhesus monkey as a model of human infant metabolism. Journal of Pediatric Gastroenterology and Nutrition, 56(4), 355–363. 10.1097/MPG.0b013e31827e1f07 [DOI] [PubMed] [Google Scholar]
- Pebsworth, P. A. , Bardi, M. , & Huffman, M. A. (2012). Geophagy in chacma baboons: Patterns of soil consumption by age class, sex, and reproductive state. American Journal of Primatology, 74(1), 48–57. [DOI] [PubMed] [Google Scholar]
- Pettipher, G. L. , & Latham, M. J. (1979). Production of enzymes degrading plant cell walls and fermentation of cellobiose by Ruminococcus flavefaciens in batch and continuous culture. Journal of General Microbiology, 110, 29–38. 10.1099/00221287-110-1-29 [DOI] [Google Scholar]
- Ransom‐Jones, E. , Jones, D. L. , McCarthy, A. J. , & McDonald, J. E. (2012). The Fibrobacteres: An important phylum of cellulose‐degrading bacteria. Microbial Ecology, 63(2), 267–281. 10.1007/s00248-011-9998-1 [DOI] [PubMed] [Google Scholar]
- Richard, A. F. (1985). Primates in nature. New York, NY: W. H. Freeman. [Google Scholar]
- Rooks, M. G. , & Garrett, W. S. (2016). Gut microbiota, metabolites and host immunity. Nature Reviews Immunology, 16(6), 341–352. 10.1038/nri.2016.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild, D. , Weissbrod, O. , Barkan, E. , Kurilshikov, A. , Korem, T. , Zeevi, D. , … Segal, E. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature, 555(7695), 210–215. 10.1038/nature25973 [DOI] [PubMed] [Google Scholar]
- Saxena, D. , Li, Y. , Yang, L. , Pei, Z. , Poles, M. , Abrams, W. R. , & Malamud, D. (2012). Human microbiome and HIV/AIDS. Current HIV/AIDS Reports, 9(1), 44–51. 10.1007/s11904-011-0103-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, K. P. , Gratz, S. W. , Sheridan, P. O. , Flint, H. J. , & Duncan, S. H. (2013). The influence of diet on the gut microbiota. Pharmacological Research, 69(1), 52–60. 10.1016/j.phrs.2012.10.020 [DOI] [PubMed] [Google Scholar]
- Su, C. , Zuo, R. , Liu, W. , Sun, Y. , Li, Z. , Jin, X. , … Zhang, H. (2016). Fecal bacterial composition of Sichuan snub‐nosed monkeys (Rhinopithecus roxellana). International Journal of Primatology, 37(4–5), 518–533. 10.1007/s10764-016-9918-9 [DOI] [Google Scholar]
- Szekely, B. A. , Singh, J. , Marsh, T. L. , Hagedorn, C. , Werre, S. R. , & Kaur, T. (2010). Fecal bacterial diversity of human‐habituated wild chimpanzees (Pan troglodytes schweinfurthii) at Mahale Mountains National Park, Western Tanzania. American Journal of Primatology, 72(7), 566–574. [DOI] [PubMed] [Google Scholar]
- Tamburini, S. , & Clemente, J. C. (2017). Neonatal gut microbiota induces lung immunity against pneumonia. Nature Reviews Gastroenterology and Hepatology, 14(5), 263–264. 10.1038/nrgastro.2017.34 [DOI] [PubMed] [Google Scholar]
- Tang, C. , Huang, L. , Huang, Z. , Krzton, A. , Lu, C. , & Zhou, Q. (2016). Forest seasonality shapes diet of limestone‐living rhesus macaques at Nonggang. China. Primates, 57(1), 83–92. 10.1007/s10329-015-0498-7 [DOI] [PubMed] [Google Scholar]
- Trosvik, P. , Rueness, E. K. , de Muinck, E. J. , Moges, A. , & Mekonnen, A. (2018). Ecological plasticity in the gastrointestinal microbiomes of Ethiopian Chlorocebus monkeys. Scientific Reports, 8(1), 20 10.1038/s41598-017-18435-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P. J. , Ley, R. E. , Mahowald, M. A. , Magrini, V. , Mardis, E. R. , & Gordon, J. I. (2006). An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature, 444(7122), 1027–1131. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- Uenishi, G. , Fujita, S. , Ohashi, G. , Kato, A. , Yamauchi, S. , Matsuzawa, T. , & Ushida, K. (2007). Molecular analyses of the intestinal microbiota of chimpanzees in the wild and in captivity. American Journal of Primatology, 69(4), 367–376. 10.1002/ajp.20351 [DOI] [PubMed] [Google Scholar]
- Vangay, P. , Johnson, A. J. , Ward, T. L. , Al‐Ghalith, G. A. , Shields‐Cutler, R. R. , Hillmann, B. M. , … Knights, D. (2018). US immigration westernizes the human gut microbiome. Cell, 175(4), 962–972. 10.1016/j.cell.2018.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vebo, H. C. , Karlsson, M. K. , Avershina, E. , Finnby, L. , & Rudi, K. (2016). Bead‐beating artefacts in the Bacteroidetes to Firmicutes ratio of the human stool metagenome. Journal of Microbiological Methods, 129, 78–80. 10.1016/j.mimet.2016.08.005 [DOI] [PubMed] [Google Scholar]
- Wallace, T. C. , Guarner, F. , Madsen, K. , Cabana, M. D. , Gibson, G. , Hentges, E. , & Sanders, M. E. (2011). Human gut microbiota and its relationship to health and disease. Nutrition Reviews, 69(7), 392–403. 10.1111/j.1753-4887.2011.00402.x [DOI] [PubMed] [Google Scholar]
- Wang, J. , Jiang, H. , Liu, Z. , & Feng, M. (1994). Feeding habits of Macaca mulatta in tropical and subtropical forests. Chinese Journal of Applied Ecology, 5(2), 167–171. (in Chinese). [Google Scholar]
- Yao, M. , Yin, L. , Zhang, L. , Liu, L. , Qin, D. , & Pan, W. (2012). Parturitions in wild white‐headed langurs (Trachypithecus leucocephalus) in the Nongguan Hills, China. International Journal of Primatology, 33(4), 888–904. 10.1007/s10764-012-9625-0 [DOI] [Google Scholar]
- Yasuda, K. , Oh, K. , Ren, B. , Tickle, T. L. , Franzosa, E. A. , Wachtman, L. M. , … Morgan, X. C. (2015). Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host and Microbe, 17(3), 385–391. 10.1016/j.chom.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Huang, J. , & Huang, Q. (2007). Biodiversity status quo and protection countermeasures for Guangxi Longhushan Nature Reserve. Central South Forest Inventory and Planning, 26(4), 65–67. (in Chinese). [Google Scholar]
- Zhao, J. , Yao, Y. , Li, D. , Xu, H. , Wu, J. , Wen, A. , … Xu, H. (2018). Characterization of the gut Microbiota in six geographical populations of Chinese rhesus macaques (Macaca mulatta), implying an adaptation to high‐altitude environment. Microbial Ecology, 76(2), 565–577. 10.1007/s00248-018-1146-8 [DOI] [PubMed] [Google Scholar]
- Zhou, Q. , Huang, Z. , Wei, H. , & Huang, C. (2018). Variations in diet composition of sympatric Trachypithecus francoisi and Macaca assamensis in the limestone habitats of Nonggang, China. Zoological Research, 39(4), 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, B. , Wang, X. , & Li, L. (2010). Human gut microbiome: The second genome of human body. Protein and Cell, 1(8), 718–725. 10.1007/s13238-010-0093-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw sequencing data from the current study are available in the NCBI repository at https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA590350, and the SRA accession number is PRJNA590350.