

Abstract
Tissue fibrosis is characterized by uncontrolled deposition and diminished clearance of fibrous connective tissue proteins, ultimately leading to organ scarring. Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) have recently emerged as pivotal drivers of mesenchymal cell activation in human fibrosis. Therapeutic strategies inhibiting YAP and TAZ have been hindered by the critical role that these proteins play in regeneration and homeostasis in different cell types. Here we find that the Gαs-coupled dopamine receptor D1 (DRD1) is preferentially expressed in lung and liver mesenchymal cells relative to other resident cells of these organs. Agonism of DRD1 selectively inhibits YAP/TAZ function in mesenchymal cells and shifts their phenotype from pro-fibrotic to fibrosis-resolving, reversing in vitro extracellular matrix stiffening and in vivo tissue fibrosis in mouse models. Aromatic L-amino acid decarboxylase (DOPA decarboxylase, or DDC), the enzyme responsible for the final step in biosynthesis of dopamine, is decreased in the lungs of subjects with idiopathic pulmonary fibrosis, and its expression inversely correlates with disease severity, consistent with an endogenous protective role for dopamine signaling that is lost in pulmonary fibrosis. Together, these findings establish a pharmacologically-tractable and cell-selective approach to targeting YAP/TAZ via DRD1 that reverses fibrosis in mice.
One Sentence Summary: Pharmacologic stimulation of a G protein coupled receptor preferentially expressed on fibroblasts reverses experimental lung and liver fibrosis.
INTRODUCTION
Fibrosis contributes to 45% of all deaths in the developed world (1), with a central role in end stage organ failure in diseases such as idiopathic pulmonary fibrosis (IPF) and liver cirrhosis (2, 3). The activation of fibroblasts to a proliferative, matrix depositing, apoptosis resistant phenotype represents a final common pathway driving organ fibrosis (4), highlighting the need to identify effective targets for inhibition or reversal of this fibrogenic fibroblast state. YAP and TAZ are transcriptional co-activators and central effectors of the Hippo pathway (5) recently described to play pathological roles in mesenchymal cell activation and fibrosis in multiple organs, including in human lung and liver disease (6–11). YAP and TAZ respond to an array of mechanical and biochemical signals implicated in fibrosis, including matrix stiffness, metabolic reprogramming, transforming growth factor-beta (TGFβ), myocardin-related transcription factor (MRTF), and WNT signaling (12–14), challenging efforts to inhibit all potential upstream drivers of their activation. More broadly, YAP and TAZ are widely expressed across multiple cell types and tissue compartments, playing important roles in numerous processes including organ growth during tissue morphogenesis, regulation of epithelial and endothelial homeostasis, and physiological tissue regeneration (15–22). The widespread expression and pleiotropic roles of YAP and TAZ highlight the need to identify cell specific strategies for inhibiting their pathological functions in disease contexts. Supporting such a strategy, recent work has demonstrated that fibroblast-selective genetic deletion of YAP/TAZ is sufficient to attenuate kidney fibrosis (8). However, pharmacologically tractable approaches for cell specific YAP and TAZ inhibition are needed to generate more clinically-relevant approaches to treat fibrotic pathologies.
G protein-coupled receptors (GPCRs) make up the largest family of membrane receptors in the human genome, and have been prolific therapeutic targets, with their ligands accounting for >30% of all clinically approved drugs (23). In relation to fibrotic conditions, both inhibitors (24–27) and activators (28–30) of GPCRs have been explored for therapeutic benefit. GPCRs are linked to effector proteins from four main classes of G-proteins. Recent work has highlighted that activation of receptors that couple to Gα12/13, Gαq/11 and Gαi/o stimulate YAP/TAZ nuclear translocation and transcriptional activity; in contrast, receptors that couple to Gαs inhibit YAP/TAZ nuclear localization and activity via elevation of cyclic adenosine monophosphate (cAMP) (31). Importantly, GPCR expression varies across organs and even within adjacent cell types in the same tissue (32), raising the possibility that GPCR directed therapeutics could offer a strategy for cell-selective YAP/TAZ inhibition. In particular, discovery and agonism of a GPCR selectively expressed on fibroblasts could offer a targeted approach capable of overriding multiple pro-fibrotic stimuli through YAP/TAZ inhibition within the activated fibroblasts that drive disease progression.
Here we identified the dopamine receptor DRD1 as a fibroblast-selective target whose agonism inhibits YAP/TAZ-mediated fibroblast activation as well as extracellular matrix (ECM) deposition and stiffening, resulting in reversal of experimental lung and liver fibrosis. DRD1 selectively couples to Gαs to promote cAMP elevation (33), and overrides both mechanical and biochemical pro-fibrotic stimuli that promote fibroblast activation, effectively switching fibroblasts from a state that supports matrix deposition and stiffening to one that favors matrix degradation and softening. Although most commonly studied in the central nervous system, dopamine receptors are broadly expressed and play crucial roles in peripheral physiology (34, 35), though relatively little is known about the role of endogenous dopamine signaling in organs such as the lung (36). Building on our observation that DRD1 agonism promotes fibrosis resolution, we focused on DDC, the gene encoding the final step in the dopamine biosynthetic pathway, and demonstrate that its expression is decreased in the lungs of patients with IPF, and inversely correlates with physiologic measures of disease severity, consistent with a protective role for endogenous dopaminergic signaling. Together these findings identify fibroblast-selective DRD1 agonism, acting through YAP/TAZ inhibition, as an effective strategy to reverse fibroblast activation and experimental lung and liver fibrosis.
RESULTS
Non-cell-specific YAP/TAZ RNAi worsens lung injury and fibrosis in mice
Based on the identification of YAP and TAZ as pivotal regulators of fibroblast pro-fibrotic activation and tissue fibrosis (6–11), we first tested whether nonselective YAP and TAZ targeting is effective in ameliorating experimental pulmonary fibrosis. We administered YAP and TAZ small interfering RNA (siRNA) intratracheally to mice following bleomycin injury. Non-cell-specific targeting of YAP/TAZ in this context amplified fibrosis (measured by hydroxyproline assay) (Fig. 1A) and increased lung injury and vascular leakage assessed visually (Fig. 1B) and by measuring lung weight (Fig. 1C) and total protein content in bronchoalveolar lavage, illustrating the limits of such a nonselective approach. Recent work has demonstrated that YAP and TAZ regulate epithelial regeneration (17, 22) as well as endothelial homeostasis and barrier function (37, 38), raising the likelihood that YAP/TAZ function in these cellular compartments is essential to lung repair after injury. Motivated by these observations and the recent proof of concept that fibroblast-specific YAP/TAZ genetic targeting is beneficial in fibrosis (8), we sought to identify a pharmacological approach by which to target YAP and TAZ in a fibroblast-selective fashion and evaluate such an approach in preclinical models of fibrosis.
Fig.1. Intratracheal YAP/TAZ siRNA worsens bleomycin induced lung injury and fibrosis.
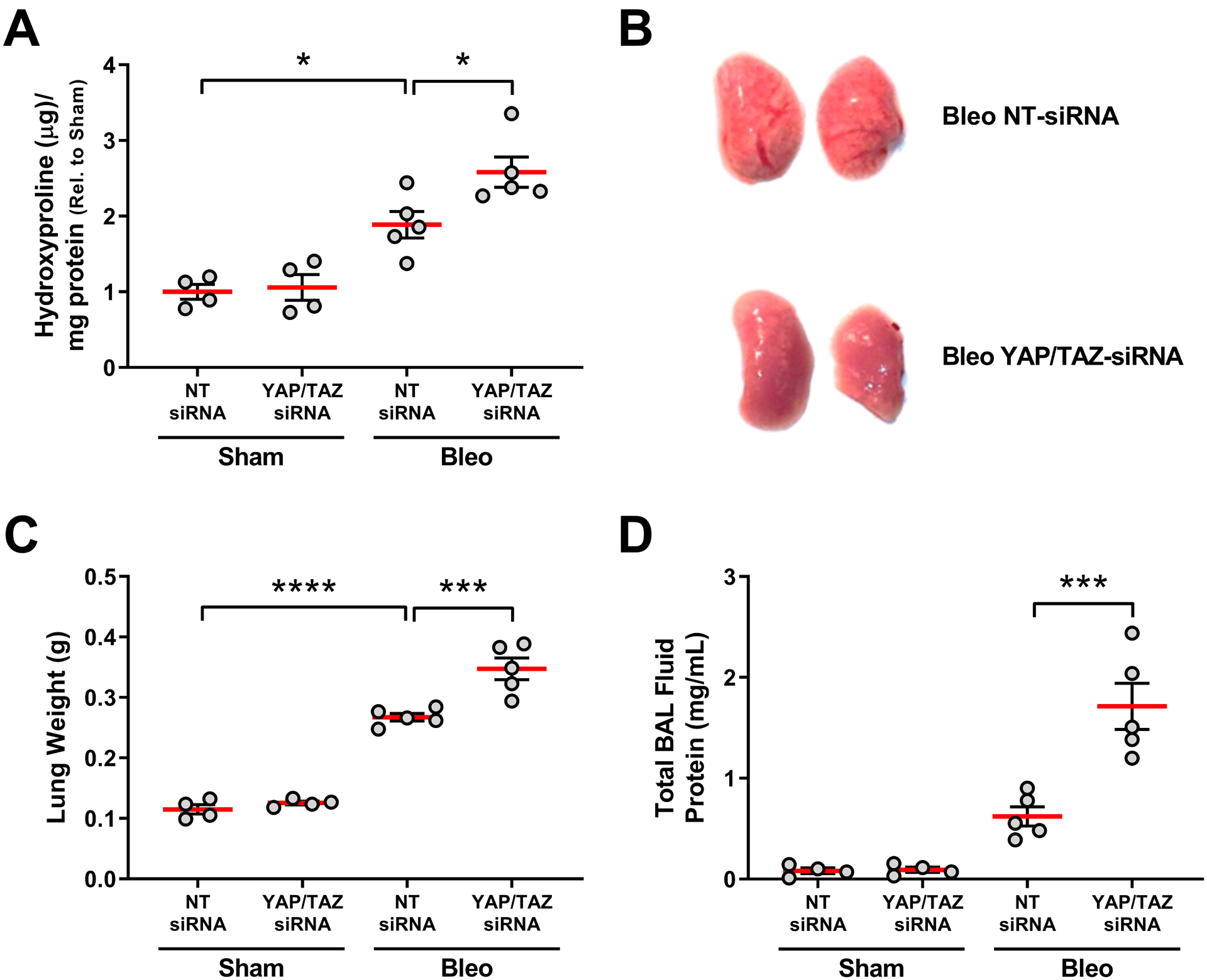
Mice were injured by intratracheal bleomycin exposure on day 0 and on day 14 treated with siRNAs targeting both YAP and TAZ. On day 21 lungs were assessed for fibrosis by measuring hydroxyproline (a surrogate for collagen deposition) (A), and injury assessed visually (B) or by measuring lung weight (C) and total protein in bronchoalveolar lavage (BAL) fluid as markers of vascular leak (D). N=4 mice/group for sham, n=5 mice/group for bleomycin. NT = non-targeting control (comparisons made using ANOVA, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs. indicated group).
Dopamine Receptor D1 is selectively expressed on fibroblasts
Drug discovery efforts to identify direct inhibitors of the YAP/TAZ transcriptional activation complex have had limited success (39, 40). However, a variety of upstream signaling pathways have been identified that regulate YAP/TAZ cellular localization, protein stability, and their activation/inactivation (21, 41–43). Among these pathways, GPCR signaling regulates YAP/TAZ inhibition or activation in a receptor class-specific fashion (31, 44) (Fig. 2A), which led us to hypothesize that GPCRs could be attractive therapeutic targets for anti-fibrotic therapy based on their potential cell-specific expression, and the availability of abundant pharmacologic tools to explore their function. Therefore, we profiled RNA expression of the GPCRome in multiple cell types involved in tissue fibrogenesis and repair, including: primary adult human lung fibroblasts, alveolar epithelial cells, and pulmonary microvascular endothelial cells (Fig. 2B, Fig. S1 and Supplementary Table S1). We focused on receptors that exclusively couple to Gαs (33) (highlighted in red, Fig. 2B and Fig. S1, and yellow in Supplementary Table S1) and are expressed preferentially in fibroblasts. Of the 28 Gαs coupled receptors, expression of the D1 dopamine receptor (DRD1) exhibited pronounced enrichment in lung fibroblasts compared to alveolar epithelial cells (Fig. 2B) and endothelial cells (Fig. S1). We confirmed abundant transcripts for DRD1 in cultured normal human lung fibroblasts and fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF), and undetectable expression of DRD1 in both primary human alveolar epithelial and microvascular endothelial cells (Fig. 2C). Preferential protein expression of DRD1was confirmed in IPF patient-derived lung fibroblasts compared to alveolar epithelial and endothelial cells by western blotting (Fig. 2D).To extend our findings to freshly isolated lung cell populations, we sorted mouse lung tissue into epithelial, endothelial, and mesenchymal enriched fractions (Fig. S2) at Day 10 post bleomycin or sham treatment and assessed dopamine receptor expression across cells types (Fig. 2E, and Fig. S3). As in cultured human cells we observed robust and stable expression of Drd1 (the murine homolog of DRD1) in freshly isolated mesenchymal cells, but undetectable expression in other lung cell populations.
Fig. 2. Gαs-coupled DRD1 is selectively expressed in pulmonary fibroblasts.
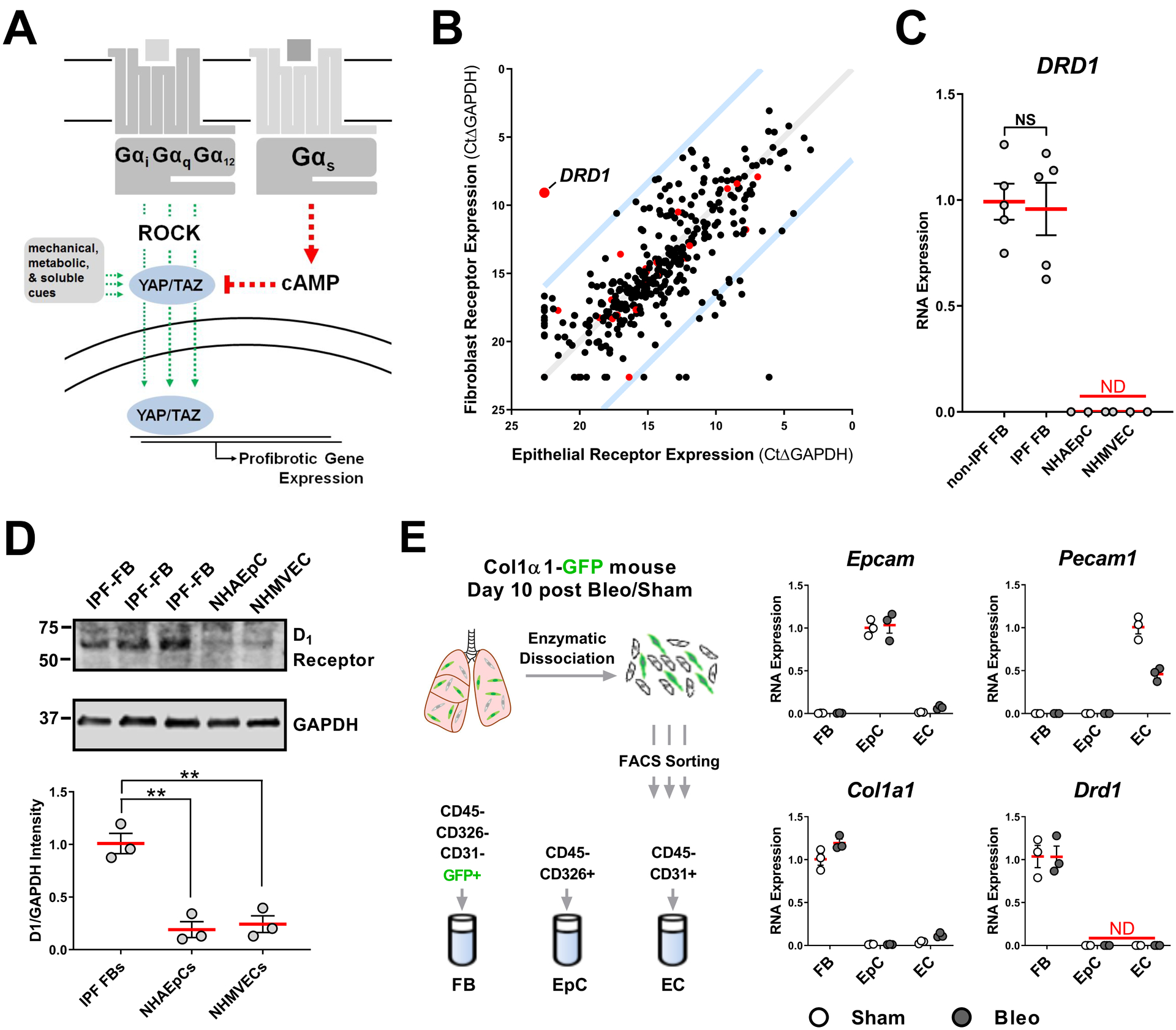
A. Receptors that couple to Gαs elevate cAMP and induce phosphorylation of YAP/TAZ, blocking nuclear localization. Receptors that couple to Galphαi/q/12 promote nuclear localization and activity of YAP/TAZ, as do other mechanical, metabolic and soluble cues, through Rho-kinase (ROCK) and other pathways. B. GPCR expression profile of primary cultured human alveolar epithelial cells and normal human pulmonary fibroblasts. Red points indicate GPCRs that selectively couple to Gαs. Blue lines indicate 100-fold preferential expression. C. DRD1 expression in cultured non-IPF associated fibroblasts (N=5 biologically independent samples), IPF patient-derived fibroblasts (N=5 biologically independent samples), normal human alveolar epithelial cells (NHAEpC)(N=3 biologically independent samples), and normal human microvascular endothelial cells (NHMVEC) (N= 3 biologically independent samples), all passage 6 or less. D. Western blot protein expression of the D1 dopamine receptor from IPF patient derived fibroblasts, normal human alveolar epithelial cells (NHAEpC), and normal human microvascular endothelial cells (N=3 biologically independent samples). E. Expression of Drd1 in freshly isolated mouse lung fibroblasts (FB), epithelial (EpC), and endothelial cells (EC). Lung from sham or Day 10 post bleomycin treated Col1a1-GFP expressing mouse was enzymatically digested then sorted for markers of epithelial cells (validated by Epcam expression), endothelial cells (validated by Pecam1 expression), and fibroblasts (validated by Col1a1 expression) followed by RNA isolation and qPCR for Drd1. N=3 mice/group (comparisons made using ANOVA, ** p < 0.01 vs. indicated group).
DRD1 agonism selectively inhibits YAP/TAZ nuclear localization in fibroblasts
Dopamine receptors have been studied for decades, providing a rich library of receptor ligands. Dopamine receptors are divided into two families: Gαs coupled D1-like (DR1 and DR5) and Gαi coupled D2-like (DR2–4). To identify an appropriate pharmacological lead, we tested thirty-three previously defined dopaminergic agonists with varying D1/D2 selectivity for their ability to inhibit YAP/TAZ nuclear localization in IPF fibroblasts (Fig. 3A). Cells plated on stiff matrix (plastic) and lacking cell contact inhibition exhibit abundant nuclear localization of YAP/TAZ due to mechanotransduction (6, 45). Of the agonists tested, all D1-selective, full agonists reduced YAP/TAZ nuclear localization, with dihydrexidine (DHX) the most effective, as did the non-selective biological ligand dopamine (Fig. 3A, Supplementary Table S2, Fig S4). DHX elevated cAMP (Fig. 3B) and promoted YAP serine 127 phosphorylation (Fig. 3C), consistent with the previously defined mechanism whereby YAP/TAZ nuclear localization is attenuated by cAMP-dependent phosphorylation of serine residues, promoting YAP/TAZ cytoplasmic retention or degradation (31, 44). DHX was effective at inhibiting YAP/TAZ nuclear localization across a panel of mesenchymal cell types, including cardiac and dermal fibroblasts and hepatic stellate cells (Fig. 3D), suggesting potentially broad relevance of this ligand for inhibiting mesenchymal cell YAP and TAZ activation. In contrast to these observations, DHX had no effect on YAP/TAZ localization in subconfluent pulmonary epithelial and endothelial cells (Fig. 3D,E and Fig. S5), consistent with the absence of detectable transcripts for DRD1 in these lung cell types. Additionally, DHX treatment selectively repressed expression of established YAP/TAZ target genes (46–49) in fibroblasts but not lung epithelial or endothelial cells (Fig. 3E and Fig. S6A). Conversely, agonism of the prostaglandin EP2 receptor, a Gαs activating receptor expressed in all three cell types (Fig. S7A), reduced localization of YAP/TAZ and repressed expression of YAP/TAZ target genes across all cell types (Fig. 3F, and Fig. S6B), highlighting the cell-selective nature of the DHX effects on fibroblasts. Although EP2 receptor stimulation is known to exert anti-fibrotic effects on lung fibroblasts, the potency of such effects is diminished in IPF(50). Importantly, DHX-mediated inhibition of YAP/TAZ nuclear localization was equally potent in normal lung fibroblasts and those derived from patients with IPF, unlike EP2 receptor activation (50) (Fig. S7B). Thus, DHX exhibited both cell selectivity and preserved potency in targeting YAP/TAZ in IPF fibroblasts.
Fig. 3. DRD1 agonism selectively blocks YAP/TAZ nuclear localization in fibroblasts.
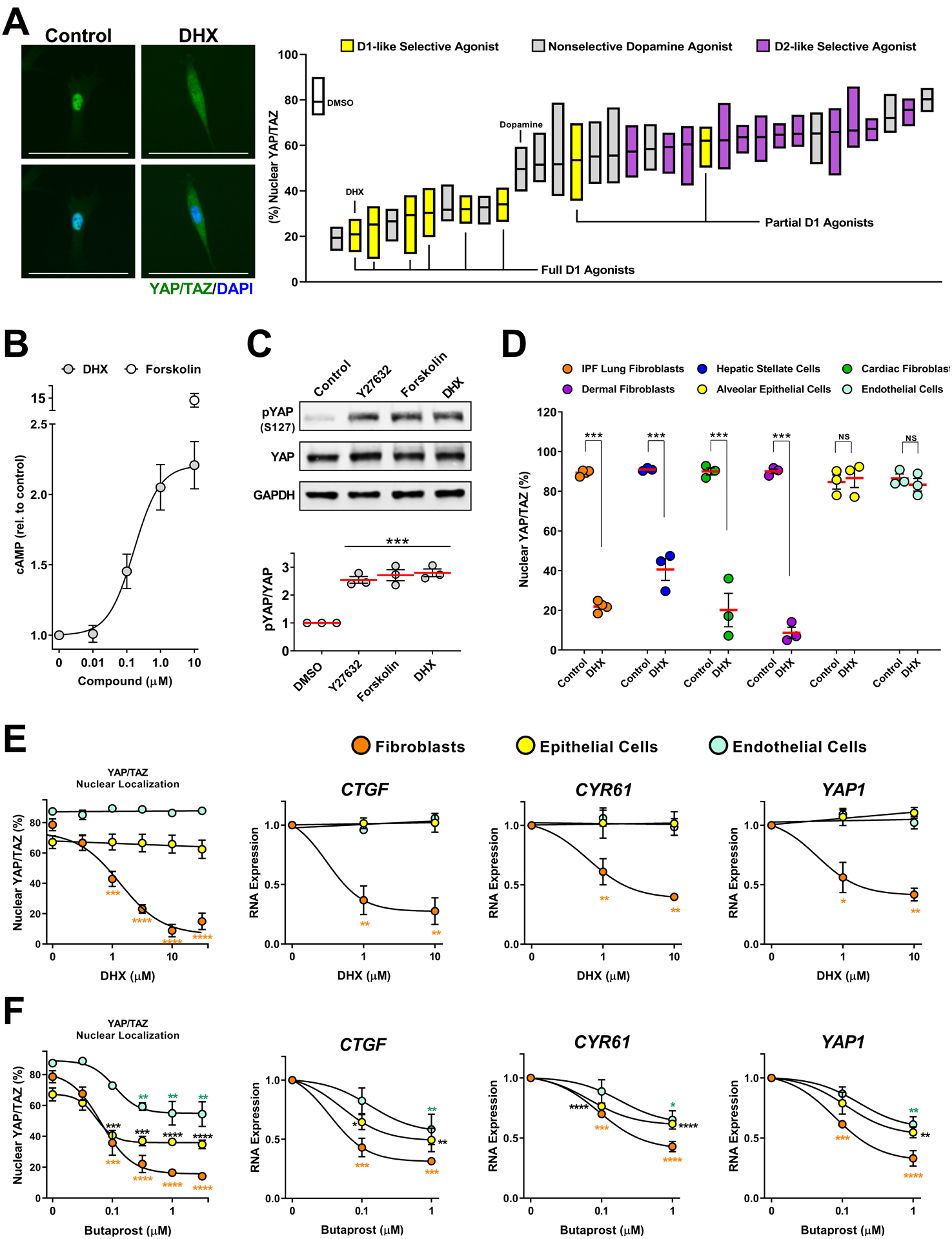
A. IPF patient-derived lung fibroblasts cells treated 2 hours prior to fixation with a library of diverse, mixed selectivity, dopaminergic agonists (10μM). N=4 biologically independent samples. Representative image: 10μM dihydrexidine (DHX). %nuclear localization of YAP/TAZ was determined using automated imaging software. Scale bar represents 100μm. Box plots represent range and mean. B. cAMP measured in IPF patient-derived fibroblasts treated for 20 minutes with the indicated concentration of DHX or forskolin (10μM). N=3 biologically independent samples. C. Total and phospho-ser127 YAP responses to the Rho-kinase inhibitor Y27632 (20μM), Forskolin (10μM), or DHX (10μM). IMR-90 lung fibroblasts N=3 independent experiments. (comparisons made using ANOVA, *** p < 0.001 vs. 0.1% DMSO vehicle control) D. YAP/TAZ nuclear localization response to DHX in fibroblasts from multiple organs: IPF patient-derived lung fibroblasts, hepatic stellate cells, human adult cardiac fibroblasts, and human dermal fibroblasts, and human lung alveolar epithelial or human pulmonary microvascular endothelial cells. N=3 biologically independent samples (comparisons made using t-test, *** p < 0.001 vs. 0.1% DMSO vehicle control). E and F. IPF derived lung fibroblasts, lung alveolar epithelial (NHAEp) and endothelial (NHMVE) cells treated with DHX (E) or with butaprost (EP2 receptor agonist, F). YAP/TAZ localization was determined after 2 hours (left), and expression of YAP/TAZ target genes at 24 hours (right). N=3 biologically independent samples (comparisons made using ANOVA, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs. the corresponding control treated cells. Orange * relate to fibroblast data, black * relate to epithelial cell data, and blue * relate to endothelial cell data).
To confirm the receptor specificity of DHX effects, we employed both RNAi and receptor antagonist approaches. We found that the inhibition of YAP/TAZ nuclear localization, elevation of cAMP, and repression of YAP/TAZ target genes by DHX could all be attenuated using two structurally independent D1 receptor selective antagonists, as well as by treating cells with DRD1-siRNA (Fig. S8).
DRD1 agonism reverses profibrotic phenotypes and promotes matrix degradation
Fibrosis is thought to occur through a combination of fibroblast activities, including activation to a contractile myofibroblast state, as well persistent imbalances in both proliferation/apoptosis and matrix deposition/degradation. To test whether dopamine receptor agonism alters contractile activation, we treated IPF-patient derived lung fibroblasts with the same library of thirty-three dopaminergic agonists in the presence of TGFβ for 72 hours (Fig. 4A, Supplementary Table S3) and quantified expression of the contractile myofibroblast marker, α-smooth muscle actin (αSMA). In agreement with the results observed for inhibition of YAP/TAZ nuclear localization (Fig. 3A), D1 receptor full agonists, including DHX, were highly effective in blocking αSMA expression, as was the non-selective biological ligand dopamine. Functionally, traction force microscopy confirmed that DHX dose-dependently reduced the contractile forces generated by fibroblasts (Fig. 4B).
Fig. 4. DHX reverses fibroblast matrix deposition, contraction and stiffening.
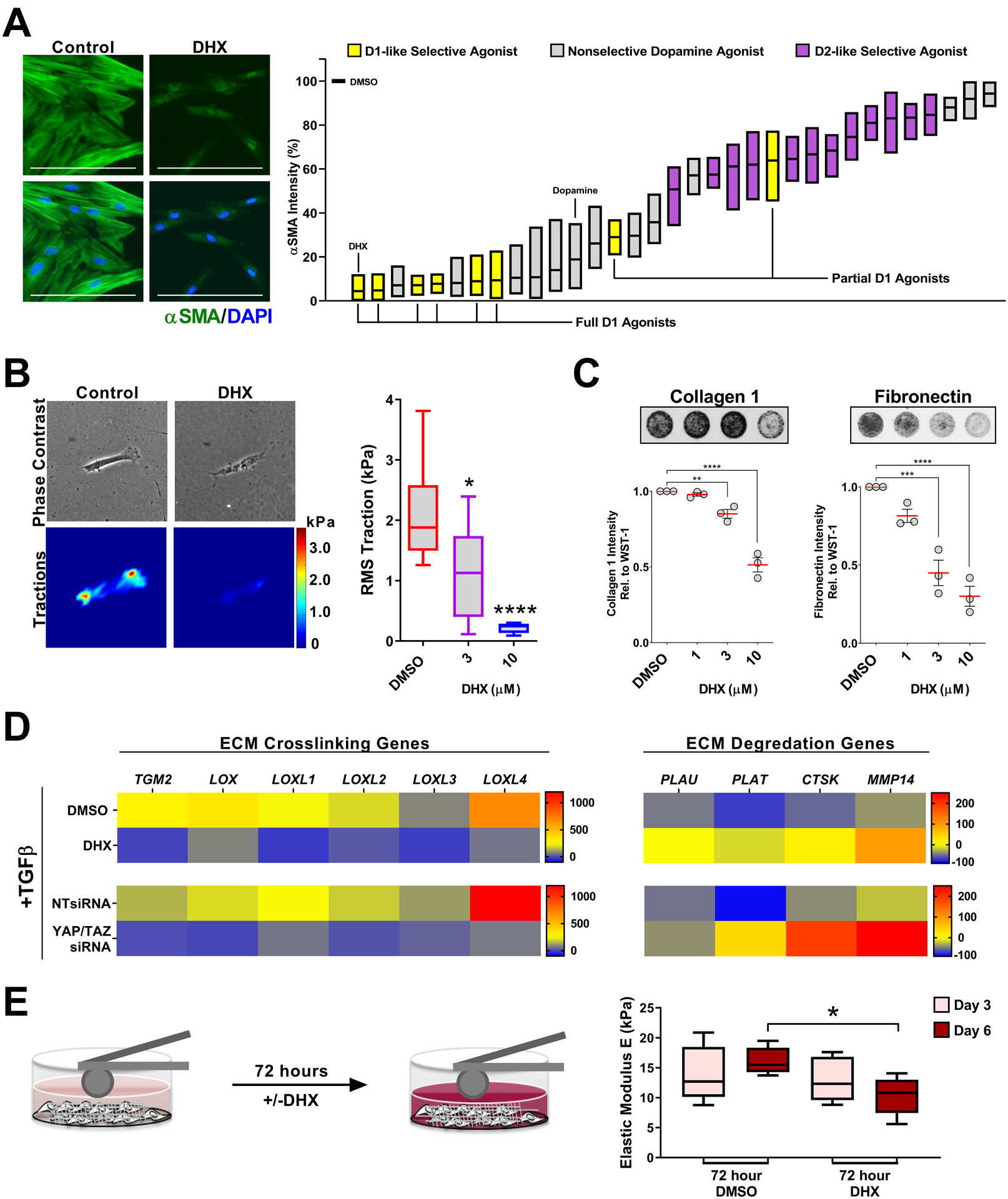
A. IPF patient-derived lung fibroblasts cells treated for 72 hours prior to fixation with a library of diverse, mixed selectivity dopaminergic agonists (1μM) + TGFβ. N=4 biologically independent samples different patient samples. Representative image: 1μM dihydrexidine (DHX). αSMA intensity was determined using automated imaging software. Box plots represent range and mean. Scale bar represents 100μm. B. IPF fibroblast traction responses to DHX. N=10 cells per condition. Shown are the min to max, quartile range, and median of cell tractions (comparisons made using ANOVA, **** p < 0.0001, * p < 0.05 vs. 0.1% DMSO vehicle control). C. ECM deposited by IPF patient-derived fibroblasts pre-stimulated with 2ng/mL TGFβ for 48 hours, then treated with DHX +2ng/mL TGFβ for additional 24 hours. N=3 biologically independent samples (comparisons made using ANOVA, **** p < 0.0001, *** p < 0.001, ** p < 0.01 vs. 0.1% DMSO vehicle control). D. ECM crosslinking and degradation associated gene programs measured in IPF fibroblasts treated 24 hours with 2ng/mL TGFβ +/− 10μM DHX or YAP and TAZ siRNA (>90% knockdown). N=3 biologically independent samples. Heat map indicates % change relative to unstimulated controls. E. Stiffness of IPF patient derived fibroblasts and their cell-derived matrices measured by AFM microindentation after 72 hours, then again after treatment +/− 10μM DHX for additional 72 hours. N=5 biologically independent samples (comparisons made using t-test, * p < 0.05 vs. 0.1% DMSO vehicle control).
Focusing next on proliferation/apoptosis, we sought to assess the potential power of D1 receptor activation as a means to broadly counteract proliferative signals. We tested DHX in parallel with multiple stimuli, including GPCR ligands and growth factors known to promote fibrosis (51, 52). Under confluent culture conditions we observed low YAP/TAZ nuclear localization that was elevated in response to endothelin-1 (ET-1), lysophosphatidic acid (LPA) and serotonin (5-HT), all GPCR ligands implicated in promotion of fibrosis (53–56), as well as pro-fibrotic growth factors TGFβ and Connective Tissue Growth Factor (CTGF).DHX blocked nuclear localization of YAP/TAZ in response to all of these ligands (Fig. S9A). Functionally, we observed that DHX broadly inhibited the proliferative effects of LPA, Et-1, TGFβ and CTGF in IPF fibroblasts (Fig. S9B–C), but was not toxic at any concentration tested (Fig. S9D), as measured by two different cell viability assays. These results confirm that a variety of pro-fibrotic GPCR ligands and growth factors positively influence both YAP/TAZ nuclear localization and fibroblast proliferation, and identify DHX as an effective strategy for broadly inhibiting proliferative effects downstream of these diverse stimuli.
Turning to matrix deposition, we found that a 24 hour treatment with DHX dose-dependently reduced TGFβ-stimulated accumulation of collagen I and fibronectin (Fig. 4C, and Fig. S10). To assess whether this effect depended on inhibition of YAP/TAZ, we employed NIH-3T3 cells that stably express a doxycycline-inducible, constitutively active, mutant TAZ (TAZ4SA)(6). In these cells, DHX failed to affect pro-fibrotic gene expression or extracellular matrix accumulation (Fig. S11), demonstrating that YAP/TAZ serine phosphorylation and inhibition is essential to the anti-fibrotic effects of DHX.
We and others have recently shown that activated fibroblasts self-amplify the fibrotic response by increasing matrix stiffness, which promotes mechano-activation of quiescent fibroblasts in a positive-feedback mechanism (57–59). We thus asked whether DHX or YAP/TAZ knockdown influenced expression of key matrix crosslinking and degradation genes (Fig. 4D, Fig. S12). Both DHX and YAP/TAZ siRNAs broadly reversed TGFβ-mediated expression of matrix crosslinking genes and increased expression of key genes associated with matrix degradation and clearance in IPF fibroblasts. To directly test the effects of DHX on matrix deposition and stiffening, we developed an in vitro cell-derived matrix remodeling assay. We first plated IPF patient derived fibroblasts at confluence (Fig.4E) and stimulated with TGFβ and ascorbic acid to promote matrix synthesis and deposition. After 72 hours we measured the stiffness of the cells and their cell-derived matrix using atomic force microscopy (AFM). To test the ability of DHX to induce cell-mediated matrix remodeling, we maintained the TGFβ and ascorbic acid +/− DHX for an additional 72 hours before probing the matrix again with AFM. In the absence of DHX treatment the cells and matrix continued to stiffen over time. In contrast, DHX treatment reversed this trend and reduced the observed stiffness (Fig. 4E). Together these results demonstrate that DRD1 agonism results in a shift in fibroblast program away from a contractile, proliferative and matrix depositing state toward a matrix degrading and softening state potentially linked to fibrosis resolution.
DRD1 agonism reverses experimental lung and liver fibrosis
To test the therapeutic efficacy of DHX treatment in models of experimental tissue fibrosis we again employed the bleomycin model of pulmonary fibrosis. Mice were administered bleomycin intratracheally at Day 0. We then waited until Day 10 for injury and inflammation to subside and fibrosis to be ongoing before randomizing into two groups, one receiving DHX for 14 days (5 mg/kg once daily i.n.) and the other, vehicle control. The DHX-treated group lost less weight than the vehicle control group (Fig. 5A). Histologically, the DHX-treated group exhibited nearly complete reversal of established lung fibrosis compared to the vehicle control group, as assessed by a pathologist blinded to the treatments (Fig. 5B,C). Total collagen in the lungs of DHX-treated mice was not different from sham treated mice, and reduced compared to vehicle control mice (Fig. 5D). Bleomycin exposure enhanced transcripts for pro-fibrotic genes, all of which were attenuated by DHX treatment (Fig. 5E). Bleomycin also increased staining for αSMA in the lungs and this was reversed by DHX treatment (Fig. 5F). To assess whether DHX adversely affected lung remodeling in the absence of fibrosis, we exposed control mice to DHX following an identical time course and route of exposure. The lungs of Sham DHX mice did not differ from those of control mice using any of these measurements (Fig. S13).
Fig. 5. DHX therapeutically reverses bleomycin-induced pulmonary fibrosis.
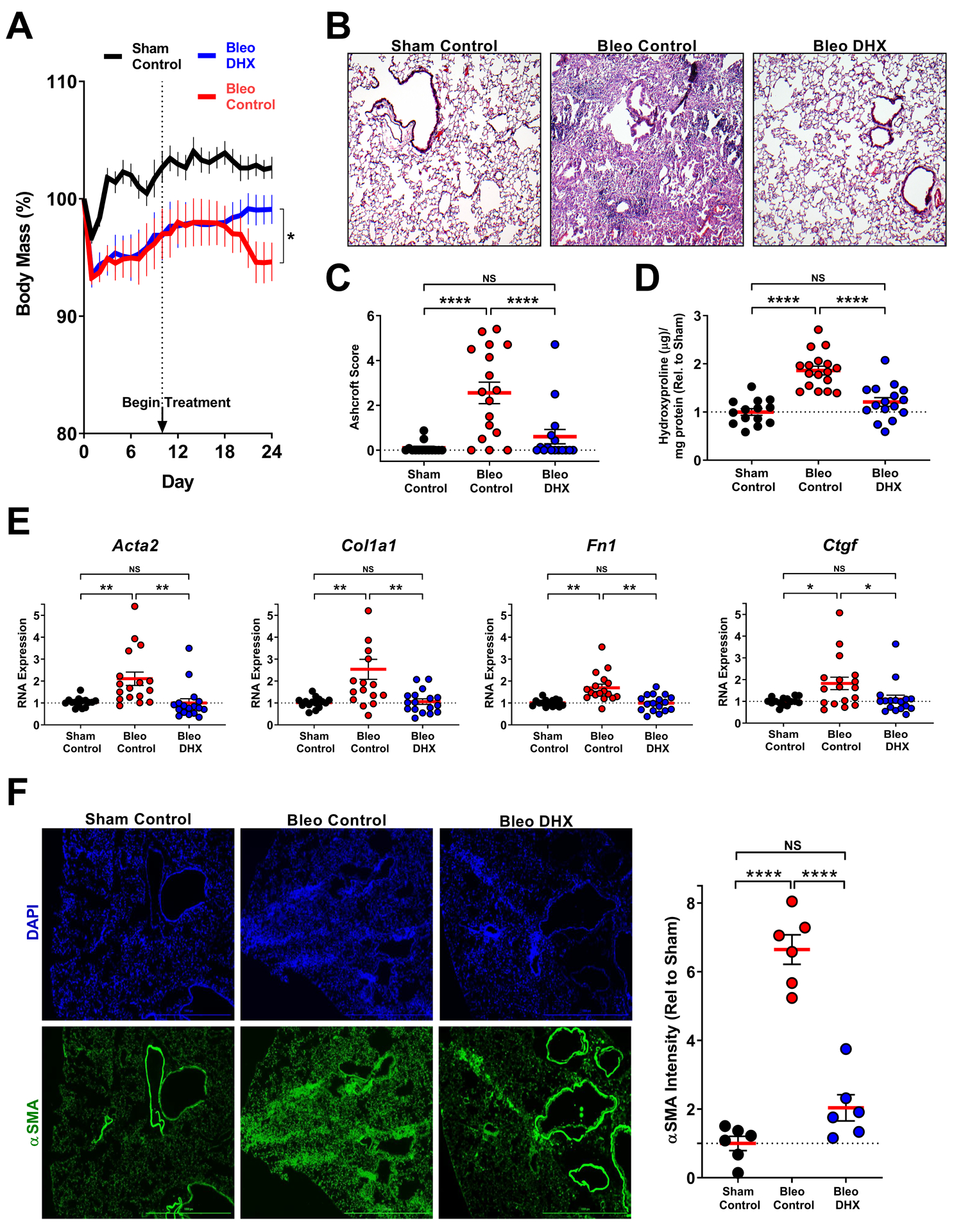
A. Weight change as a result of bleomycin-induced lung injury +/− DHX. Female mice were intratracheally administered Bleomycin on Day 0; DHX treatment was initiated on Day 10 (5mg/kg DHX i.n., daily) and continued until day 24 (comparisons made using t-test, * p < 0.05 vs. bracket indicated group). B,C. H&E staining to visualize architectural changes (40X objective). Paraffin embedded lung sections were stained and scored using the Ashcroft method. D. Lung tissue was biochemically analyzed for collagen abundance using the hydroxyproline assay. E. Changes in pro-fibrotic gene expression in whole lung homogenates. F. Immunofluorescence imaging of lung sections for αSMA. Representative section from each group (4X objective) shown. αSMA intensity was quantified by automated image analysis. Scale bar represents 1000μM. Sham Control N=15, Bleo Control N=17, and Bleo DHX N=17, from two independent experiments (C-F comparisons made using ANOVA, **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05 vs. bracket indicated group).
Our original selection of DRD1 as a target was based on the goal of selectively targeting YAP/TAZ in lung fibroblasts. To assess the cell specificity of in vivo DHX treatment, we administered bleomycin intratracheally to mice at Day 0, then waited until Day 10 for injury and inflammation to subside and fibrosis to be ongoing before randomizing into two groups, one receiving DHX for short treatment (5 mg/kg i.n.) and the other, vehicle control. Mice were treated 24 and 2 hours prior to either cell isolation from the lungs by FACS or immunofluorescence imaging (Fig 6A). We implemented the same FACS strategy as in Fig. 2, and measured changes in well-established YAP/TAZ target genes as in our in vitro studies (Fig. 3E, F and Fig. S6). Freshly sorted lung fibroblasts, but not lung epithelial or endothelial cells exhibited DHX-dependent decreases in Yap1, Ccne1, Axl and Cyr61 (Fig. 6A,B). To further confirm DHX-mediated YAP/TAZ inhibition in lung fibroblasts in situ, we assessed YAP/TAZ nuclear localization in Col1aGFP-positive cells by confocal microscopy. Fibroblasts in DHX-treated lungs exhibited reduced YAP/TAZ nuclear localization relative to vehicle treated controls, further supporting the D1 receptor as a mediator of YAP/TAZ activation specifically targetable on lung-resident fibroblasts (Fig. 6C).
Fig. 6. DHX selectively blocks expression of YAP/TAZ target genes in lung fibroblasts in vivo.
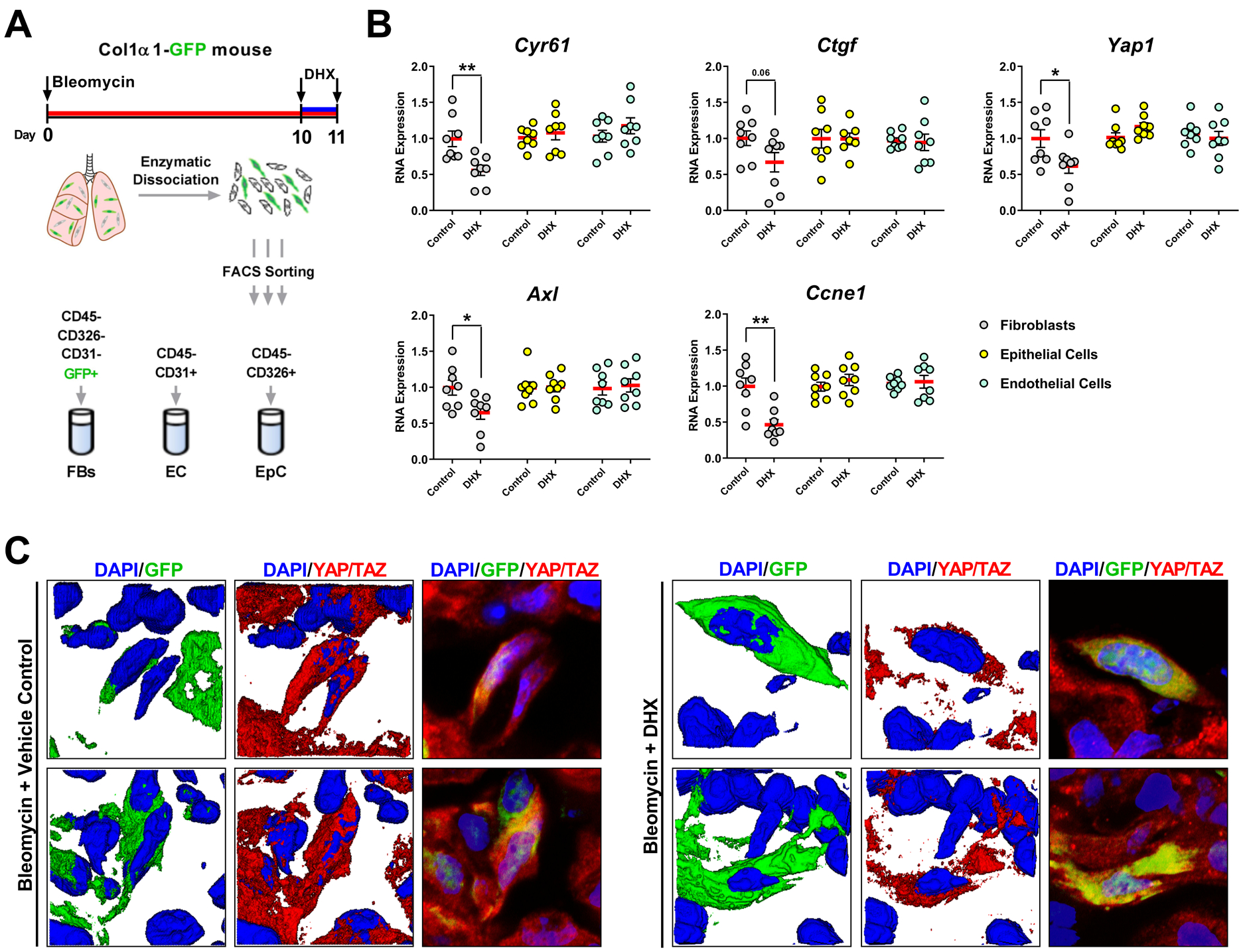
A. Mice received bleomycin intratracheally at day 0 and two doses of DHX or vehicle control 2 and 24 hours prior to collecting lungs at day 11 for flow sorting of fibroblasts, epithelial, and endothelial cells. B. Expression of YAP/TAZ target genes from freshly isolated cells. N=8 mice/group (comparisons made using t-test, ** p < 0.01, * p < 0.05, vs. indicated group). C. Representative confocal microscopy images of YAP/TAZ nuclear localization in GFP-positive lung fibroblasts shown in both 2D and 3D projections.
Based on the efficacy of DHX in experimental lung fibrosis, and in attenuating YAP/TAZ nuclear localization across an array of cultured mesenchymal cell types including hepatic stellate cells (Fig. 3D), we sought to extend our findings to liver fibrosis. Previous studies have demonstrated a pro-fibrotic role for hepatocyte TAZ activation (60), and limited beneficial effects of global YAP/TAZ inhibition in experimental liver fibrosis (7, 9), However, YAP/TAZ are essential to liver regeneration (20), and nonspecific YAP knockdown in the liver promotes hepatocyte necrosis (61), highlighting again the need to interrogate complementary targets upstream from YAP/TAZ such as the D1 dopamine receptor. We profiled the GPCRome of cultured hepatic stellate cells (HSCs) and hepatocytes (Heps) and confirmed preferential expression of DRD1 in HSCs (Fig. 7A, B and Supplementary Table S1), echoing our findings in the lung. We then confirmed the ability of DHX to reduce baseline and TGFβ-mediated fibrogenic activation in HSCs (Fig. 7C). Finally, we tested the efficacy of DHX in the bile duct ligation (BDL) model of cholestatic liver fibrosis. BDL was performed at Day 0 and treatment with DHX or vehicle begun at Day 7 and continued until day 21. DHX improved histological fibrosis caused by BDL, reduced αSMA and exhibited a trend toward reduced hydroxyproline (Fig. 7D, E), extending the efficacy of our GPCR-based approach to experimental liver fibrosis. As anticipated, DHX had no effect on indices of liver injury highlighting its HSC targeted mode of action (Fig S14).
Fig. 7. DHX reverses hepatic stellate cell activation and hepatic fibrosis.
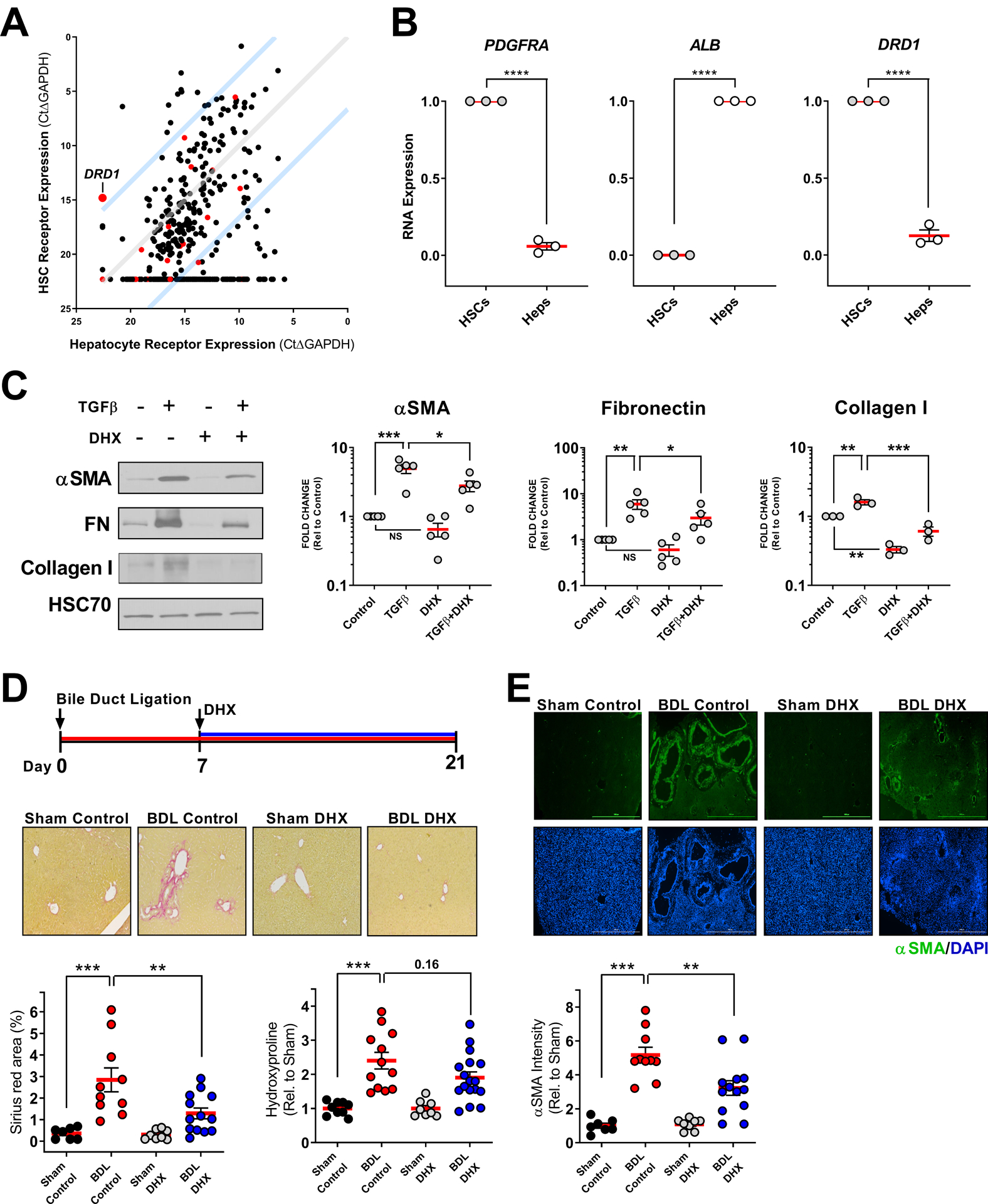
A. GPCR expression profile of primary cultured human hepatic stellate cells (HSCs) and hepatocytes. Red points indicate GPCRs that selectively couple to Gαs. Blue lines indicate 100-fold preferential expression. B. DRD1 expression in hepatic stellate cells (validated by PDGFRA expression) relative to hepatocytes (Heps, validated by ALB expression). N=3 biologically independent samples (comparisons made using t-test, **** p < 0.0001 vs. indicated group). C. DHX effects on TGFβ-mediated hepatic stellate cell activation in vitro measured by αSMA, fibronectin, and collagen I western blots. 2ng/mL TGFβ 48 hours +/− 10μM DHX final 24 hours, N=3–5 independent experiments (comparisons made using ANOVA, *** p < 0.001, ** p < 0.01, * p < 0.05, vs. indicated group). D and E. On day 7 following bile duct ligation surgery, male and female mice began receiving DHX (5mg/kg, i.p. once daily) until day 21 when livers were assessed for fibrosis. Shown arerepresentative Sirius red (D) and αSMA (E) stained sections (4X objective) and quantification of collagen by Sirius red area and hydroxyproline (D) and αSMA by immunofluorescence (E). αSMA intensity was quantified by automated image analysis. Scale bar represents 1000μM. For αSMA and Sirius red, Sham Control N=7, BDL Control N=10, Sham DHX=8, and BDL DHX N=13. For hydroxyproline, Sham Control N=9, BDL Control N=12, Sham DHX=8, and BDL DHX N=17, both collected from two independent replicate experiments (comparisons made using ANOVA, *** p < 0.001, ** p < 0.01 vs. indicated group).
DOPA decarboxylase expression is diminished in IPF
As introduced above, the dopamine receptor family has 5 members from two classes: D1-like (DRD1 and DRD5) coupling to Gαs, and D2-like (DRD2-DRD4) coupling to Gαi. DRD1 is the dominant receptor in lung fibroblasts (Fig. S3 and Supplementary Table S1), hence the fibroblast response to the nonselective biological ligand dopamine is similar to that with DHX (Fig. 3A, 4A, S15), suggesting local dopamine production in the lung could provide an endogenous anti-fibrotic signal. DOPA decarboxylase is the enzyme responsible for the final biosynthesis of dopamine from L-3,4-dihydroxyphenylalanine (L-DOPA) precursor and is classically known to be diminished in brain regions of patients with Parkinson’s disease (62). To determine if genes associated with dopaminergic signaling are differentially expressed in the lungs of IPF patients compared to controls, we analyzed expression of DDC (encoding human DOPA decarboxylase) and DRD1 (encoding human dopamine receptor D1). Using the Lung Genomics Research Consortium (http://www.lung-genomics.org/) tissue microarray dataset, we compared expression from 134 IPF and 108 controls. The demographic and clinical characteristics of these subjects have been previously described in detail (63) and are summarized in Supplementary Table S4. The relative expression of DDC transcripts in IPF lungs were decreased compared to the control lungs (Fig. 8A), whereas DRD1 transcripts were not different between IPF and control lungs (Fig. 8B). To confirm altered expression of DOPA decarboxylase in IPF lung tissue at protein level, we compared DDC protein expression using explanted lung tissues from IPF patients and donor lungs that were not suitable for transplantation. As shown in Figure 8C, D the expression of DDC protein was reduced in IPF lungs compared to donor lungs. In addition to the 52 KD predicted full length protein, the predominant form of DDC protein expressed in the lung tissues was ~47 KD, potentially reflecting alternative splicing (64). The expression of both the 52 KD and the lower molecular weight variant in IPF were reduced (53% and 60% compared to donor lungs) (Fig. 8D). Therefore, we conclude that both DDC transcripts and DDC protein are reduced in IPF lungs. We next tested whether the changes in DDC gene expression in IPF correlate with pulmonary function. Decreased DDC transcripts were correlated with worsening of both forced vital capacity (FVC, a measure of lung compliance) and the lung’s gas diffusing capacity (as measured by the diffusing capacity of the lungs for carbon monoxide, DLCO, Fig. 8E and 8F). Finally, we confirmed that DDC transcripts also inversely correlated with the GAP score (65), a validated mortality prediction tool in IPF (Fig. S16). To extend these observations to liver fibrosis, we analyzed a smaller published dataset of transcriptional profiles from liver samples of 7 controls and 15 individuals with alcoholic hepatitis (66). However, in this cohort, DDC expression was not statistically different between controls and individuals with alcoholic hepatitis (Fig. S17). Together these results demonstrate that expression of DDC, encoding the final enzyme responsible for dopamine synthesis, is diminished in the lungs of patients with IPF, and decreased DDC expression correlates with worsened lung function and higher predicted mortality. Connecting these results to the beneficial effects of DRD1 agonism seen in our experimental fibrosis models supports the potential physiological relevance of the dopaminergic system as a therapeutic target in human fibrotic pathologies.
Fig. 8. DOPA decarboxylase is decreased in IPF, and correlates with worsening disease severity.
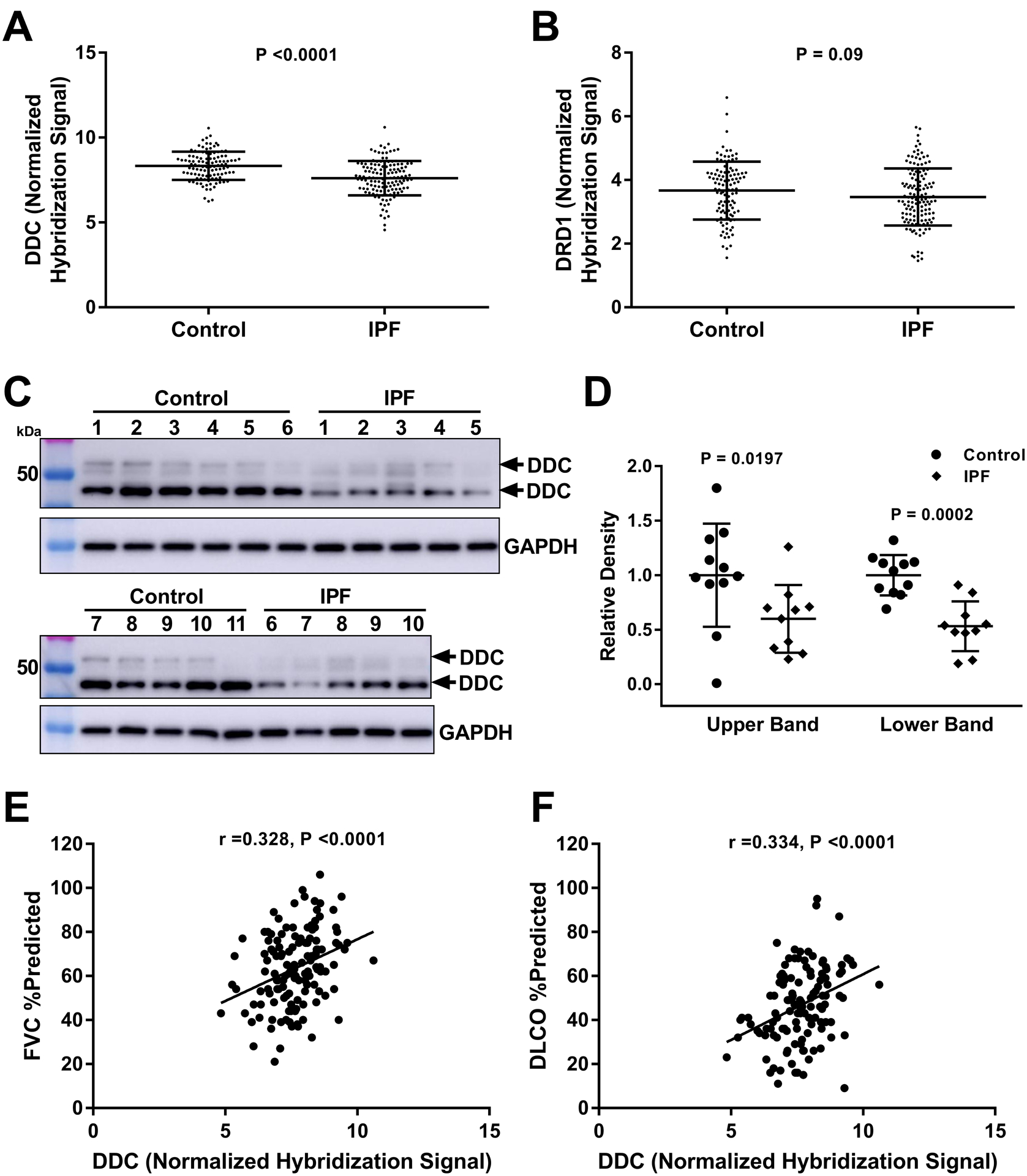
A and B. Expression of DDC and DRD1 were queried from microarray analyses of IPF (n=134) and control (n=108) lungs. Each data point represents expression from an individual. Bars indicate mean and standard deviation (comparisons made using t-test, p value indicated in the figure panel). C. Western blotting to detect DDC protein expression in whole lung homogenates from IPF (n=10) and control donor (n=11) lungs. D. Quantification of upper and lower bands from western blots, normalized to GAPDH. Bars indicate mean and standard deviation (comparisons made using t-test, p value indicated in the figure panel).. E and F. Univariate analysis of the correlation of DDC expression with forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DLCO) assessed using the Pearson’s correlation coefficient (r). Each data point represents expression and lung function (expressed a percent predicted based on age, sex and ideal body weight) from an individual (p values indicated in the figure panels).
DISCUSSION
Fibrotic diseases remain a major medical burden with limited treatment options (2). Our work highlights DRD1 as a potential therapeutic target for lung and liver fibrosis. DRD1 agonism inactivates YAP/TAZ and switches activated fibroblasts from a contractile, proliferative and matrix depositing fibrogenic state to a pro-resolving matrix degrading state. Importantly, DRD1 is preferentially expressed on lung and liver mesenchyme relative to other structural cells of these organs, providing a cell-selective approach that preserves YAP/TAZ function in epithelial and endothelial compartments important for injury resolution and tissue repair (15–21). Whereas our focus on DRD1 emerged from primary studies in lung fibroblasts, comparison of the GPCRome in hepatocytes and hepatic stellate cells identified additional attractive targets, including the serotonin receptor 5-HT7, and limited published evidence suggests that 5-HT7 agonism is also beneficial in liver fibrosis (67). Thus our approach may be expandable to include additional GPCR candidates and consideration of a similar approach in other organs, allowing identification of therapeutic targets specific to fibroblasts in other tissues affected by fibrotic pathologies, such as kidney, heart, pancreas and skin.
Activated GPCRs signal through a multitude of downstream pathways including altered YAP/TAZ activation (68). In our studies overexpression of mutant TAZ4SA was sufficient to override the anti-fibrotic effects of DRD1 agonism, indicating that YAP/TAZ inactivation is an essential downstream component of fibroblast signaling via this receptor, an observation echoed in recent observations of an essential role for YAP/TAZ inactivation downstream of the prostacyclin receptor (69). Abundant published evidence also supports the concept that GPCR responses, both those that stimulate and those that inhibit pro-fibrotic fibroblast activation, mirror effects on YAP/TAZ activation or inhibition (27, 70, 71). For example, LPA, ET-1 and serotonin have all been implicated in fibroblast activation and fibrosis, and all are competent to enhance fibroblast YAP/TAZ nuclear localization. In contrast, stimulation of numerous Gαs coupled GPCRs ablate YAP/TAZ activation (31), and several have emerged as potential anti-fibrotic targets, including relaxin, prostaglandin and adenosine receptors (72–74). Accumulating evidence suggests that Gαs coupled receptor loss may be a common occurrence in pathological fibrosis, as both prostaglandin EP2 (75, 76) and relaxin RXFP1 (77) receptors have been reported to decline in expression in IPF patients. In the case of DRD1, our data demonstrate that the receptor remains expressed in the lung and on fibroblasts, but we see decreased expression of DOPA decarboxylase, the enzyme responsible for dopamine production, at both the transcript and protein level in human IPF, suggesting a loss of local endogenous dopaminergic Gαs signaling. Our findings specifically highlight DRD1 agonism as a highly attractive therapeutic modality in IPF, as the receptor remains expressed in diseased lung fibroblasts and available for agonist targeted therapy.
Endogenous dopaminergic signaling in the lung and liver have received relatively little attention, though local dopamine production has been implicated in fluid clearance during lung injury (78, 79). Efforts to measure local dopamine in the lung and liver may be helpful as biomarkers to understand fibrosis resolution and patients who might benefit from DRD1 targeted therapeutics, as exemplified by the variability in DDC expression amongst individuals with IPF and alcoholic hepatitis. As a circulating biomarker, dopamine may provide less useful information, as dopamine is rapidly sulfonated by SULT1A3 in multiple tissues and circulates in a predominantly inactive form (80). SULT1A3 expression is dramatically reduced during development toward adulthood in liver and lung, supporting the plausibility of dopamine acting as a local paracrine mediator of fibroblast biology in these tissues (81). Our work highlights the need to further verify the local action and cellular source(s) of endogenous dopamine production during tissue repair and fibrosis, while clearly establishing the therapeutic potential of exogenously targeting this pathway.
Finally, our results raise the intriguing possibility that reprogramming of activated fibrotic fibroblasts into a matrix-degrading phenotype may represent a major cellular mechanism during normal resolution of fibrosis, particularly in response to signaling through Gαs coupled receptors and YAP/TAZ inactivation. Although the focus of fibrosis resolution research has largely centered on macrophages (82, 83), fibroblasts also express key genes implicated in matrix degradation, and are competent to degrade fibrillar collagens (82, 84). Treatment with either DHX or YAP/TAZ siRNA reduced expression of matrix cross-linking genes and enhanced expression of genes which code for collagen degrading enzymes cathepsin K and MMP14, both of which have been shown to reduce collagen accumulation in experimental fibrosis models (85–88). These findings, and the accompanying change in cell/ECM stiffness we observed after DRD1 agonist treatment, raise the possibility that fibroblasts may be active participants in normal matrix resorption after tissue injury. Whereas recent efforts to target fibroblasts have focused on removing senescent or activated cells through targeted clearance (89, 90), our findings add another attractive possibility: eliciting fibroblast’s native fibrosis-resolving capacities through receptor-mediated stimulation.
An important limitation and question raised by our study is whether such matrix degrading capacities of fibroblasts can be effective in the setting of established human fibrotic pathologies, particularly as the extent of collagen crosslinking found in human disease is not likely captured in the relatively short term models tested here. An additional limitation to our study is that we have not directly measured endogenous dopamine production or signaling in the lung or liver. Hence, further effort will be needed to clarify the functional roles of dopamine within the context of tissue injury, repair and fibrosis. Lastly, our analysis of dopamine receptor expression was limited to cells sorted by classical, but relatively broad, lineage markers. Additional consideration of the cellular subpopulations within repairing and fibrotic tissues will be important to further define the cellular targets of D1 agonism in the lung and liver.
Taken together our findings demonstrate that GPCR agonism can be used to pharmacologically target YAP and TAZ in select cell populations to exert beneficial effects on tissue fibrosis. The safety and therapeutic window for DHX and other dopaminergic therapies in human CNS indications are well known (91, 92), with the most prevalent side effect being hypotension, an effect largely ameliorated in later studies (93). Development of peripherally restricted and organ targeted approaches should be prioritized so that D1 receptor agonism could be safely evaluated for the treatment of tissue fibrosis in the lung, liver and other organs. This approach might represent a significant step forward in developing effective therapies for patients with fibrotic diseases.
MATERIALS AND METHODS
Study design
The goal of this study was to identify a strategy to selectively inhibit YAP and TAZ activity in resident mesenchymal cells associated with lung and liver fibrosis. We identified the Dopamine Receptor D1 as a putative target to treat these diseases and tested the D1 receptor agonist dihydrexidine in vitro and in vivo. In cell based assays, we treated lung fibroblasts and hepatic stellate cells with dihydrexidine and observed a pronounced shift in their phenotype from pro-fibrotic to fibrosis-resolving. For in vivo mouse studies, intratracheal bleomycin and bile duct ligation were chosen as well established and relevant models of experimental lung and liver fibrosis, respectively. Sample sizes were calculated by power analysis based on previous experience and feasibility. For lung fibrosis experiments testing dihydrexidine, n ≥ 15–17 mice per group were used to achieve statistical significance and for the liver fibrosis experiments, n ≥ 7–13 mice per group were used. Mice were randomly assigned to treatment groups. Biochemical and histological outcomes were analyzed with the investigator blinded to the treatment groups and no animals were excluded as outliers from the reported dataset. All in vitro and in vivo experiments were performed in 2–4 technical replicates. The number of biologically independent samples and/or biologically independent experiments is identified in each figure legend. Detailed methods for in vitro and tissue analyses are provided in the Supplemental Materials.
Mice
Eight weeks old female and male C57/BL6 mice were purchased from Jackson Laboratories. Col1a1-GFP transgenic mice were generated as previously described (94). All animal experiments testing DHX were carried out under protocols approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). In the initial in vivo siRNA study (Fig. 1), adult male age-matched C57BL/6N mice at 6–8 weeks of age were purchased from the National Cancer Institute (NCI)-Frederick Mouse Repository. These experiments were performed in accordance with National Institute of Health guidelines and protocols approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Bleomycin Mouse Studies
6–8 weeks old mice were anesthetized with ketamine and xylazine before exposure of the trachea. Lung fibrosis was induced by intratracheal injection of bleomycin (50 μL at 1.2 U/kg) or phosphate buffered saline (PBS; as control) on day 0. After 14 days Small interfering RNA (siRNA) duplexes targeting mouse Yap (L-046247–01–0005) or Taz (L-058248–01-–) mRNA (Dharmacon) or nontargeting control siRNA were administered in vivo by intratracheal instillation at a single dose of 25 μg (each siRNA) per mouse, 50μg of nontargeting. On day 21 mice were sacrificed and lungs harvested for collagen determination and biochemical analyses. To obtain BAL samples for total protein concentration determination, lungs were lavaged with six 0.5-mL aliquots of PBS. BAL samples were centrifuged at 3,000g for 20 min at 4°C and transferred the supernatants to siliconized low-binding Eppendorf tubes (PGC Scientifics) for subsequent analysis. Total protein concentration of the BAL fluid was determined by BCA Protein Assay Kit (Pierce). In the dihydrexidine treatment studies, eight week female C57/BL6 mice (Fig. 5) or eight week old female Col1a1-GFP transgenic mice (Fig. 6), lung fibrosis was induced with bleomycin (BLEO; Fresenius Kabi) delivered intratracheally with 1.2 U/kg to the lungs using MicroSprayer® Aerosolizer (Penn-Century). The Sham mice received sterile 0.9% saline instead using identical methods. Mice were weighed every 24 hours, and both groups were then randomized at day 10 into DHX and Control treatment groups, matching for the degree of weight change. DHX (5mg/kg) was administered everyday intranasally (i.n.) dissolved in surfactant (infasurf) which has previously shown to aid in spreading to pulmonary aveoli (104) for 14 days (Fig. 5) or 1 day, dosed 24 and 2 hours prior to lung collection (Fig. 6). The control groups of mice received the equivalent vehicle dose of surfactant. Following the final DHX treatment, mice were sacrificed and the right lungs were inflated with 4% paraformaldehyde (PFA) and further incubated in 4% PFA for 24 hours prior processing for paraffin embedding. The left lobe of the lung was snap frozen in liquid nitrogen for RNA isolation and hydroxyproline assay (Fig. 5) or used for FACS (Fig. 6).
Bile Duct Ligation
BDL was performed as previously described(105). Briefly, 8–10 weak old male and female C57BL/6N underwent either BDL or sham surgery. Mice were anesthetized on Day 0 following IACUC protocol, and the bile duct was ligated using sterile 3/0 silk ligatures. Sham surgery was performed by passing a silk ligature under the bile duct. Starting on Day 7, DHX (5mg/kg) or vehicle control was administered everyday intraperitoneally (i.p.) for 14 days. Following the final DHX treatment, mice were sacrificed and the livers harvested for analysis of fibrosis. Liver enzymes were analyzed by loading 100 uL of serum in a Mammalian Liver Enzymes Rotor (Abaxis) and read by Vetscan 2.0.
Lung Tissue Gene Expression Analysis
Publicly available data from the Lung Genomics Research Consortium (http://www.lung-genomics.org/) were queried for expression of dopamine signaling-related genes from the lungs of 134 patients with IPF and from 108 donor controls. RNA isolation and microarray procedures have been described previously (107, 108). These data and methods are available in the Gene Expression Omnibus database (accession number GSE47460). The clinical characteristics of the patients have been published previously (63).
Statistics
In experiments comparing groups of three or more, groups were compared by one-way analysis of variance with Tukey’s post-hoc comparison after confirming that data displayed a normal distribution. In experiments comparing two groups, groups were compared using unpaired t-test with Welch’s correction. Results are expressed throughout as the mean ± standard error of the mean (SEM) or with box and whisker plots showing min to max, quartile, and median. Fig. 1–7 statistical tests were carried out using GraphPad Prism 7 with statistical significance defined as P<0.05. For human lung tissue gene expression analyses (Fig. 8), we used the Mann-Whitney test to determine the difference in expression of DDC and DRD1 between IPF and controls and DDC protein between explanted IPF and donor lungs. Univariate analysis was performed by using the Pearson’s correlation coefficient of gene expression data with FVC and diffusing capacity of the lung for carbon monoxide (DLCO). These analyses were performed by using STATA 14.0 (StataCorp). Gender-Age-Pulmonary function (GAP) scoring was performed based on previously established criteria (65).
Supplementary Material
Fig S1. GPCR expression profile of primary cultured human pulmonary microvascular endothelial cells and normal human pulmonary fibroblasts.
Fig. S2. FACs gating strategy to isolate mouse pulmonary fibroblasts, epithelial and endothelial cells from Col1a1-GFP transgenic mice.
Fig. S3. Expression of Drd1-4 in freshly isolated mouse lung fibroblasts (FB), epithelial (EpC), and endothelial cells (EC).
Fig. S4 DHX inhibits nuclear localization of YAP and TAZ.
Fig. S5 Representative YAP/TAZ localization in IPF derived fibroblasts, alveolar epithelial cells, and pulmonary microvascular endothelial cells treated with DHX.
Fig. S6 Expression of additional YAP/TAZ target genes in fibroblasts, epithelial, and endothelial cells treated with D1 and EP2 receptor agonists.
Fig. S7 PGE2 receptor expression in human lung fibroblasts, epithelial cells, endothelial cells and loss of potency in IPF patient-derived lung fibroblasts.
Fig. S8. DHX activity is dependent on DRD1 receptor.
Fig. S9 DHX represses lung fibroblast proliferation without acute cytotoxicity.
Fig.S10 DHX reduces collagen I and fibronectin protein expression.
Fig. S11 DHX inhibits pro-fibrotic gene expression and matrix deposition through inhibition of YAP/TAZ.
Fig. S12. DHX and YAP/TAZ siRNA modulate matrix crosslinking and degradation gene programs.
Fig. S13 DHX alone does not affect lung matrix content or profibrotic gene expression.
Fig. S14 Liver function tests in BDL experiments.
Fig. S15 Dopamine promotes anti-fibrotic effects.
Fig. S16. DDC transcripts inversely correlate with the Stage of Gender-Age-Pulmonary function (GAP) score, a clinically validated tool to predict one-year mortality.
Fig. S17. DDC expression in subjects with alcoholic hepatitis.
Supplementary Table S1 GPCR expression profiles from primary human lung fibroblasts, epithelial cells, endothelial cells, hepatic stellate cells and hepatocytes.
Supplementary Table S2 Dopaminergic agonist library effects on YAP/TAZ localization
Supplementary Table S3 Dopaminergic agonist library effects on αSMA expression.
Supplementary Table S4 Clinical and Demographic Characteristics of the Lung Genomics Research Consortium (LGRC) Cohort
Supplementary Table S5 PCR Primers used in this study
Datafile S1: Raw data (provided as supplementary excel file)
Acknowledgments:
The authors acknowledge the guidance and assistance provided by the Mayo Clinic Office of Translation to Practice, housed within the Center for Clinical and Translational Science, supported by CTSA Grant Number UL1 TR002377 from the National Center for Advancing Translational Sciences.
Funding: Support provided by an ALA Senior Research Training Fellowship and Catalyst Award (A.J.H), ALF and the AASLD Pinnacle Award (E.K.), NIH HL092961 (D.J.T.), HL133320 (D.J.T. and V.H.S.), HL126990 (D.J.K.), a Scleroderma Foundation New Investigator Grant and an American Thoracic Society Foundation/Pulmonary Fibrosis Foundation Research Grant (D.L.), and a Scleroderma Research Foundation Investigator-Initiated Research Grant and NIH HL108975 (A.M.T).
Footnotes
Competing interests: A.J.H. and D.J.T. are co-inventors of a patent application (“Methods of Treating Fibrotic Pathologies” PCT/US2019/016178) based on some of the findings described in this manuscript.
Data Availability: All the data used for the preparation of the manuscript are present in the main text or in the supplementary material.
References:
- 1.Wynn TA, Cellular and molecular mechanisms of fibrosis. J Pathol 214, 199–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL, Sheppard D, Duffield JS, Violette S, Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 5, 167sr161 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Iozzo RV, Gubbiotti MA, Extracellular matrix: The driving force of mammalian diseases. Matrix Biol, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klingberg F, Hinz B, White ES, The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229, 298–309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccolo S, Dupont S, Cordenonsi M, The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94, 1287–1312 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Liu F et al. , Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol-Lung C 308, L344–L357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin K et al. , PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang M et al. , Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J Am Soc Nephrol 28, 3277–3289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannaerts I et al. , The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 63, 679–688 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Noguchi S et al. , TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci Rep 7, 42595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertero T et al. , A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep 5, 18277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piersma B, Bank RA, Boersema M, Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med (Lausanne) 2, 59 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panciera T, Azzolin L, Cordenonsi M, Piccolo S, Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 18, 758–770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enzo E et al. , Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J 34, 1349–1370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J et al. , YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest 127, 3447–3467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lange AW et al. , Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J Mol Cell Biol 7, 35–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z et al. , MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep 16, 1810–1819 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Totaro A et al. , YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L et al. , Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579.-+ (2016). [DOI] [PubMed] [Google Scholar]
- 20.Yimlamai D et al. , Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu FX, Zhao B, Guan KL, Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu C et al. , The Hippo-YAP Pathway Regulates the Proliferation of Alveolar Epithelial Progenitors after Acute Lung Injury. Cell Biol Int, (2019). [DOI] [PubMed] [Google Scholar]
- 23.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swigris JJ, Brown KK, The Role of Endothelin-1 in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Biodrugs 24, 49–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tager A et al. , The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Inflamm Res 56, S347–S347 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Ikeda H, Yatomi Y, Autotaxin in liver fibrosis. Clin Chim Acta 413, 1817–1821 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Pascual F, Busnadiego O, Gonzalez-Santamaria J, The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci 118, 156–164 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Polat B et al. , Liver 5-HT7 receptors: A novel regulator target of fibrosis and inflammation-induced chronic liver injury in vivo and in vitro. Int Immunopharmacol 43, 227–235 (2017). [DOI] [PubMed] [Google Scholar]
- 29.McBride A et al. , In search of a small molecule agonist of the relaxin receptor RXFP1 for the treatment of liver fibrosis. Sci Rep-Uk 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M, Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. American journal of physiology. Lung cellular and molecular physiology 292, L405–413 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Yu FX et al. , Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Insel PA et al. , GPCR expression in tissues and cells: Are the optimal receptors being used as drug targets? Brit J Pharmacol 165, 1613–1616 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flock T et al. , Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tayebati SK, Lokhandwala MF, Amenta F, Dopamine and vascular dynamics control: present status and future perspectives. Curr Neurovasc Res 8, 246–257 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Arreola R et al. , Immunomodulatory Effects Mediated by Dopamine. J Immunol Res 2016, 3160486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciarka A, Vincent JL, van de Borne P, The effects of dopamine on the respiratory system: friend or foe? Pulm Pharmacol Ther 20, 607–615 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Neto F et al. , YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J et al. , YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest 127, 3441–3461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zanconato F, Battilana G, Cordenonsi M, Piccolo S, YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol 29, 26–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu-Chittenden Y et al. , Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Gene Dev 26, 1300–1305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen CG, Moroishi T, Guan KL, YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol 25, 499–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plouffe SW, Hong AW, Guan KL, Disease implications of the Hippo/YAP pathway. Trends Mol Med 21, 212–222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varelas X, The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141, 1614–1626 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Kim M et al. , cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J 32, 1543–1555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dupont S et al. , Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Xu MZ et al. , AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 30, 1229–1240 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao B et al. , TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LaQuaglia MJ et al. , YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma. Sci Rep 6, 30238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Smolen GA, Haber DA, Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 68, 2789–2794 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Bozyk PD, Moore BB, Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol 45, 445–452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahluwalia N, Shea BS, Tager AM, New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med 190, 867–878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nanthakumar CB et al. , Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov 14, 693–720 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Ebrahimkhani MR et al. , Stimulating healthy tissue regeneration by targeting the 5-HT(2)B receptor in chronic liver disease. Nat Med 17, 1668–1673 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fonseca C, Abraham D, Renzoni EA, Endothelin in pulmonary fibrosis. Am J Respir Cell Mol Biol 44, 1–10 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Tager AM et al. , The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med 14, 45–54 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Lagares D et al. , Endothelin 1 contributes to the effect of transforming growth factor beta1 on wound repair and skin fibrosis. Arthritis Rheum 62, 878–889 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Liu F et al. , Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190, 693–706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH, Mechanosensing and fibrosis. Journal of Clinical Investigation 128, 74–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santos A, Lagares D, Matrix Stiffness: the Conductor of Organ Fibrosis. Curr Rheumatol Rep 20, (2018). [DOI] [PubMed] [Google Scholar]
- 60.Wang XB et al. , Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai H et al. , Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 56, 1097–1107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lloyd K, Hornykiewicz O, Parkinson’s disease: activity of L-dopa decarboxylase in discrete brain regions. Science 170, 1212–1213 (1970). [DOI] [PubMed] [Google Scholar]
- 63.Tan J et al. , Expression of RXFP1 Is Decreased in Idiopathic Pulmonary Fibrosis. Implications for Relaxin-based Therapies. Am J Respir Crit Care Med 194, 1392–1402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kokkinou I, Nikolouzou E, Hatzimanolis A, Fragoulis EG, Vassilacopoulou D, Expression of enzymatically active L-DOPA decarboxylase in human peripheral leukocytes. Blood cells, molecules & diseases 42, 92–98 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Ley B et al. , A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 156, 684–691 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Affo S et al. , Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 62, 452–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Polat B et al. , Liver 5-HT7 receptors: A novel regulator target of fibrosis and inflammation-induced chronic liver injury in vivo and in vitro. Int Immunopharmacol 43, 227–235 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Park HW, Guan KL, Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci 34, 581–589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zmajkovicova K et al. , The Antifibrotic Activity of Prostacyclin Receptor Agonism is Mediated through Inhibition of YAP/TAZ. Am J Respir Cell Mol Biol, (2018). [DOI] [PubMed] [Google Scholar]
- 70.Pyne NJ, Dubois G, Pyne S, Role of sphingosine 1-phosphate and lysophosphatidic acid in fibrosis. Biochim Biophys Acta 1831, 228–238 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Mann DA, Oakley F, Serotonin paracrine signaling in tissue fibrosis. Biochim Biophys Acta 1832, 905–910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bennett RG, Relaxin and its role in the development and treatment of fibrosis. Transl Res 154, 1–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castelino FV, Lipids and eicosanoids in fibrosis: emerging targets for therapy. Curr Opin Rheumatol 24, 649–655 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Shaikh G, Cronstein B, Signaling pathways involving adenosine A2A and A2B receptors in wound healing and fibrosis. Purinergic Signal 12, 191–197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore BB et al. , Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J Immunol 174, 5644–5649 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Huang SK et al. , Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol 177, 2245–2255 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan JN et al. , Expression of RXFP1 Is Decreased in Idiopathic Pulmonary Fibrosis Implications for Relaxin-based Therapies. Am J Resp Crit Care 194, 1392–1402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saldias FJ, Comellas AP, Pesce L, Lecuona E, Sznajder JI, Dopamine increases lung liquid clearance during mechanical ventilation. Am J Physiol-Lung C 283, L136–L143 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Chua BA, Perks AM, The effect of dopamine on lung liquid production by in vitro lungs from fetal guinea-pigs. J Physiol-London 513, 283–294 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eisenhofer G, Coughtrie MWH, Goldstein DS, Dopamine sulphate: An enigma resolved. Clin Exp Pharmacol P 26, S41–S53 (1999). [PubMed] [Google Scholar]
- 81.Richard K et al. , Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocr Metab 86, 2734–2742 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Glasser SW et al. , Mechanisms of Lung Fibrosis Resolution. American Journal of Pathology 186, 1066–1077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wynn TA, Vannella KM, Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKleroy W, Lee TH, Atabai K, Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am J Physiol-Lung C 304, L709–L721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buhling F et al. , Pivotal role of cathepsin K in lung fibrosis. American Journal of Pathology 164, 2203–2216 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Srivastava M et al. , Overexpression of cathepsin K in mice decreases collagen deposition and lung resistance in response to bleomycin-induced pulmonary fibrosis. Resp Res 9, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zigrino P et al. , Fibroblast-Derived MMP-14 Regulates Collagen Homeostasis in Adult Skin. Journal of Investigative Dermatology 136, 1575–1583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duarte S, Saber J, Fujii T, Coito AJ, Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biology 44–46, 147–156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schafer MJ et al. , Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lagares D et al. , Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Science Translational Medicine 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.George MS et al. , A single 20 mg dose of dihydrexidine (DAR-0100), a full dopamine D1 agonist, is safe and tolerated in patients with schizophrenia. Schizophr Res 93, 42–50 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Stowe RL et al. , Dopamine agonist therapy in early Parkinson’s disease. Cochrane Database Syst Rev, CD006564 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Rosell DR et al. , Effects of the D1 dopamine receptor agonist dihydrexidine (DAR-0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology 40, 446–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yata Y et al. , DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology 37, 267–276 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Marinkovic A, Mih JD, Park JA, Liu F, Tschumperlin DJ, Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF-beta responsiveness. American journal of physiology. Lung cellular and molecular physiology 303, L169–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Devor DC, Bridges RJ, Pilewski JM, Pharmacological modulation of ion transport across wild-type and DeltaF508 CFTR-expressing human bronchial epithelia. American journal of physiology. Cell physiology 279, C461–479 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Richards TJ et al. , Allele-specific transactivation of matrix metalloproteinase 7 by FOXA2 and correlation with plasma levels in idiopathic pulmonary fibrosis. American journal of physiology. Lung cellular and molecular physiology 302, L746–754 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haak AJ et al. , Phenylpyrrolidine structural mimics of pirfenidone lacking antifibrotic activity: A new tool for mechanism of action studies. Eur J Pharmacol 811, 87–92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beacham DA, Amatangelo MD, Cukierman E, Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr Protoc Cell Biol Chapter 10, Unit 10 19 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Thundat T, Warmack RJ, Chen GY, Allison DP, Thermal and Ambient-Induced Deflections of Scanning Force Microscope Cantilevers. Appl Phys Lett 64, 2894–2896 (1994). [Google Scholar]
- 101.Sicard D et al. , Aging and anatomical variations in lung tissue stiffness. American journal of physiology. Lung cellular and molecular physiology 314, L946–L955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robinson BK, Cortes E, Rice AJ, Sarper M, Del Rio Hernandez A, Quantitative analysis of 3D extracellular matrix remodelling by pancreatic stellate cells. Biol Open 5, 875–882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dimitriadis EK, Horkay F, Maresca J, Kachar B, Chadwick RS, Determination of elastic moduli of thin layers of soft material using the atomic force microscope. Biophys J 82, 2798–2810 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Massaro D, Massaro GD, Clerch LB, Noninvasive delivery of small inhibitory RNA and other reagents to pulmonary alveoli in mice. Am J Physiol-Lung C 287, L1066–L1070 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Maiers JL et al. , The Unfolded Protein Response Mediates Fibrogenesis and Collagen I Secretion Through Regulating TANGO1 in Mice. Hepatology 65, 983–998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ashcroft T, Simpson JM, Timbrell V, Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 41, 467–470 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bauer Y et al. , A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 52, 217–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herazo-Maya JD et al. , Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med 5, 205ra136 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. GPCR expression profile of primary cultured human pulmonary microvascular endothelial cells and normal human pulmonary fibroblasts.
Fig. S2. FACs gating strategy to isolate mouse pulmonary fibroblasts, epithelial and endothelial cells from Col1a1-GFP transgenic mice.
Fig. S3. Expression of Drd1-4 in freshly isolated mouse lung fibroblasts (FB), epithelial (EpC), and endothelial cells (EC).
Fig. S4 DHX inhibits nuclear localization of YAP and TAZ.
Fig. S5 Representative YAP/TAZ localization in IPF derived fibroblasts, alveolar epithelial cells, and pulmonary microvascular endothelial cells treated with DHX.
Fig. S6 Expression of additional YAP/TAZ target genes in fibroblasts, epithelial, and endothelial cells treated with D1 and EP2 receptor agonists.
Fig. S7 PGE2 receptor expression in human lung fibroblasts, epithelial cells, endothelial cells and loss of potency in IPF patient-derived lung fibroblasts.
Fig. S8. DHX activity is dependent on DRD1 receptor.
Fig. S9 DHX represses lung fibroblast proliferation without acute cytotoxicity.
Fig.S10 DHX reduces collagen I and fibronectin protein expression.
Fig. S11 DHX inhibits pro-fibrotic gene expression and matrix deposition through inhibition of YAP/TAZ.
Fig. S12. DHX and YAP/TAZ siRNA modulate matrix crosslinking and degradation gene programs.
Fig. S13 DHX alone does not affect lung matrix content or profibrotic gene expression.
Fig. S14 Liver function tests in BDL experiments.
Fig. S15 Dopamine promotes anti-fibrotic effects.
Fig. S16. DDC transcripts inversely correlate with the Stage of Gender-Age-Pulmonary function (GAP) score, a clinically validated tool to predict one-year mortality.
Fig. S17. DDC expression in subjects with alcoholic hepatitis.
Supplementary Table S1 GPCR expression profiles from primary human lung fibroblasts, epithelial cells, endothelial cells, hepatic stellate cells and hepatocytes.
Supplementary Table S2 Dopaminergic agonist library effects on YAP/TAZ localization
Supplementary Table S3 Dopaminergic agonist library effects on αSMA expression.
Supplementary Table S4 Clinical and Demographic Characteristics of the Lung Genomics Research Consortium (LGRC) Cohort
Supplementary Table S5 PCR Primers used in this study
Datafile S1: Raw data (provided as supplementary excel file)