

Abstract
CTNNB1, encoding β-catenin, is frequently mutated in hepatocellular carcinoma, the most rapidly growing solid cancer in the U.S., and activating mutations in this gene are associated with increased expression of glutamine synthetase. A new report by Adebayo Michael et al. identifies mTOR as a direct target of WNT/β-catenin signaling through increased production of glutamine, which is required for the carcinogenic effects of WNT/ β-catenin activity in the liver.
Hepatocellular carcinoma (HCC) incidence and mortality are rapidly rising in the U.S., and treatment options for advanced stage disease remain limited (Villanueva, 2019). Tightly associated with chronic liver disease and cirrhosis, HCC is characterized by a wide variety of genetic mutations, including those in CTNNB1, encoding β-catenin, as well as in AXIN2 and APC, whose gene products also affect the WNT/β-catenin signaling pathway. Prior work has demonstrated the important role of WNT/β-catenin signaling for liver development, zonation, regeneration and cancer formation (Russell and Monga, 2018), but the factors by which WNT/β-catenin directly contributes to carcinogenesis have been largely elusive.
In the mammalian liver, hepatocytes are arranged across the hepatic lobule from the portal vein to the central vein, with different metabolic and synthetic functions of these cells depending on their location. This heterogeneity is established and maintained by a gradient of WNT/β-catenin signaling activity that is highest around the pericentral region (Benhamouche et al., 2006). An established target of β-catenin and its nuclear effector T cell factor (TCF) in the liver is GLUL, which encodes glutamine synthetase (GS) (Sekine et al., 2006). This enzyme assimilates ammonia into glutamate to form glutamine, and GS overexpression directly contributes to nucleotide biosynthesis as a pre-requisite for cell proliferation (Cox et al., 2016). In this issue, Adebayo Michael and colleagues identify mTOR as a novel downstream target of WNT/β-catenin activity in the liver (Adebayo Michael et al., 2019). Utilizing elegant studies based on a rich set of diverse genetic mouse strains and a large collection of human liver tumors of different histology, they first show a correlation between CTNNB1 mutations, GS expression and phosphorylation of mTOR at Ser2448, indicative of activated, in both human and murine tissue, and then a requirement for GS and mTOR activity for carcinogenesis in Cttnb1-mutated murine models of HCC. Inhibition of GS or mTOR markedly reduces HCC formation in vivo.
While mTOR activation has been previously reported as a frequent event in HCC (Villanueva et al., 2008) and has been linked to pre-malignant changes in the cirrhotic liver (Chaturantabut et al., 2019), a clinical trial using the mTOR inhibitor everolimus as a 2nd line treatment for advanced HCC has been disappointing (Zhu et al., 2014). Treatment with an immunosuppressive regimen containing an mTOR inhibitor after liver transplant for HCC, however, did result in improved overall survival initially, indicating the possible relevance of mTOR inhibition clinically (Geissler et al., 2016). The current manuscript not only provides mechanistic insight how WNT/β-catenin activity directly contributes to hepatocarcinogenesis by connecting its known impact on GS expression with the previously revealed role of glutamine in activating mTOR, but this insight may also enable better patient stratification in future clinical trials, identifying patients with CTNNB1 mutations or other activators of GS, such as Yap (Cox et al., 2016) for appropriate selection.
The authors observe a thin rim of GS expression and associated mTOR activation in the pericentral hepatocytes in the normal liver. A recent study reveals the importance of mTOR for liver regeneration (Chaturantabut et al., 2019). So beyond cancer, these findings have potentially important implications for our understanding of the liver’s regenerative capacity and its metabolic role in homeostasis as currently the role of stem or progenitor cells, characterized by individual cell markers including Lgr5, Axin2, Sox9, and Tert, for liver regeneration is debated. What is undisputed, however, is the eminent role of WNT/β-catenin activity in the proliferative phase of liver repair. What if regenerative capacity is not an expression of (stem) cell identity, but a result of (pericentral) position and (metabolic) state? If peak WNT/β-catenin activity and GS expression co-localized with WNT targets Lgr5 and Axin2, and the now-identified mTOR in pericentral hepatocytes, glutamine may be the critical metabolite that has an important role in enabling cell proliferation and growth after non-zonal liver injury, such as partial hepatectomy. After an injury disrupting liver zonation, such as acetaminophen toxicity, which ablates the pericentral zone, other mechanisms could either re-establish a WNT/metabolic gradient or enable other hepatocytes or cholangiocytes to contribute to organ repair. It is unknown whether WNT signaling plays a role in maintaining liver metabolic function after injury in addition to regulating proliferation. Further, could pericentral hepatocytes, with the signaling and metabolic machinery poised towards proliferation, be particularly vulnerable to initiate tumor formation in cirrhotic patients? Careful fate-mapping studies, co-localizing all members of this signaling cascade, are needed to further investigate these questions.
Finally, neither WNT/β-catenin nor mTOR evolved to produce cancer in the liver. Why are GS expression and mTOR activity so exquisitely tightly regulated and spatially restricted in the normal liver? Why is carbamoyl phosphate synthase 1 (CPS1), which opposes the action of GS and deaminates glutamine, located in the periportal region of the liver lobule? This may simply be explained by metabolic necessity, with the need to remove ammonia from the nutrient-rich blood in the portal circulation, while preserving proliferative potential in pericentral hepatocytes. Cps1 and Glul reporter mouse strains should further elucidate function and fate of these diverse hepatocyte populations.
In summary, the current work provides a tight link between WNT/β-catenin activity, metabolic regulation and mTOR activity for cancer formation. But it also throws light on the possibility that the position of a hepatocyte within the liver lobule and its metabolic state matter more for destiny than predetermined identity.
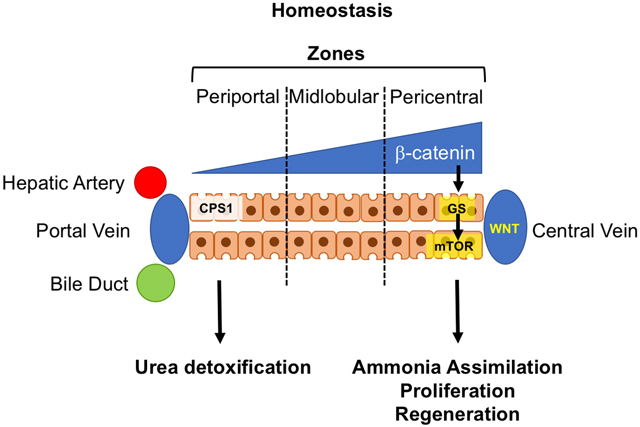
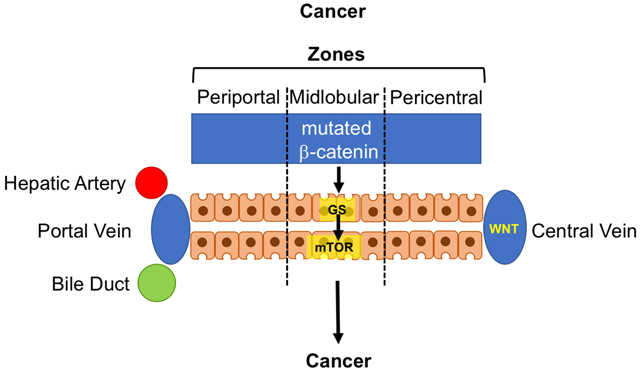
Schematic illustration of the impact of β-catenin gradient in homeostatic conditions on hepatic zonation in the hepatic lobule and associated CPS11 and GS expression and mTOR activity (top). In cancer (bottom) associated with mutated CTNNB1, the β-catenin gradient is replaced with overall elevated WNT/β-catenin activity leading to predominant mTOR signal outside the pericentral zone.
Acknowledgements:
W.G. is supported by NIH R24OD017870, R01DK090311 and R01DK105198, the Claudia Adams Barr Program in Cancer Research and the Pew Charitable Trusts Biomedical Sciences Scholars Program.
Footnotes
Declaration of Interests:
W.G. is a consultant and scientific advisory board member of Camp4 Therapeutics.
References:
- Adebayo Michael AO, Ko S, Tao J, Moghe A, Yang H, Xu M, Russell JO, Pradhan-Sundd T, Liu S, Singh S , et al. (2019). Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C , et al. (2006). Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell 10, 759–770. [DOI] [PubMed] [Google Scholar]
- Chaturantabut S, Shwartz A, Evason KJ, Cox AG, Labella K, Schepers AG, Yang S, Aravena M, Houvras Y, Mancio-Silva L , et al. (2019). Estrogen Activation of G-Protein-Coupled Estrogen Receptor 1 Regulates Phosphoinositide 3-Kinase and mTOR Signaling to Promote Liver Growth in Zebrafish and Proliferation of Human Hepatocytes. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Hwang KL, Brown KK, Evason K, Beltz S, Tsomides A, O’Connor K, Galli GG, Yimlamai D, Chhangawala S , et al. (2016). Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol 18, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler EK, Schnitzbauer AA, Zulke C, Lamby PE, Proneth A, Duvoux C, Burra P, Jauch KW, Rentsch M, Ganten TM , et al. (2016). Sirolimus Use in Liver Transplant Recipients With Hepatocellular Carcinoma: A Randomized, Multicenter, Open-Label Phase 3 Trial. Transplantation 100, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JO, and Monga SP (2018). Wnt/beta-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu Rev Pathol 13, 351–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Lan BY, Bedolli M, Feng S, and Hebrok M (2006). Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43, 817–825. [DOI] [PubMed] [Google Scholar]
- Villanueva A (2019). Hepatocellular Carcinoma. N Engl J Med 380, 1450–1462. [DOI] [PubMed] [Google Scholar]
- Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M , et al. (2008). Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 135, 1972–1983, 1983 e1971–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL , et al. (2014). Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 312, 57–67. [DOI] [PubMed] [Google Scholar]